
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(I)-catalysed dehydrative formation of ethers from benzylic alcohols and phenols†
Richard M. P.
Veenboer
a and
Steven P.
Nolan
*ab
aEaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: snolan@st-andrews.ac.uk
bChemistry Department, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 8th May 2015
Abstract
We report the cross-dehydrative reaction of two alcohols to form unsymmetrical ethers using NHC–gold(I) complexes (NHC = N-heterocyclic carbene). Our progress in developing this reaction into a straightforward procedure is discussed in detail. The optimised methodology proceeds under mild reaction conditions and produces water as the sole by-product. The synthetic utility of this environmentally benign methodology is exemplified by the formation of a range of new ethers from readily available phenols bearing electron withdrawing substituents and secondary benzylic alcohols with various substituents. Finally, we present experimental results to account for the chemoselectivity obtained in these reactions.
Introduction
The formation of C–O bonds by means of alkylation and arylation reactions has emerged as a major objective in the construction of pharmaceutical compounds.1 While the traditional Williamson ether synthesis represents a widely used approach, this multi-step procedure usually generates a stoichiometric amount of waste (Scheme 1).2 When starting from two alcohol molecules, it requires the conversion of one alcohol into a halide or pseudo-halide leaving group. Once this group is eliminated in the substitution step, it needs to be separated from the product. These disadvantages have challenged chemists to develop novel greener procedures to effect the direct activation of alcohols for nucleophilic substitution.3 Such an approach would form water as the only by-product and would reduce the operational effort in accordance with Wender and Miller's guidelines for the “ideal synthesis” of new molecules.4 Consequently, hydroxide activation for nucleophilic substitution has been recognised as a key area for green chemistry research.5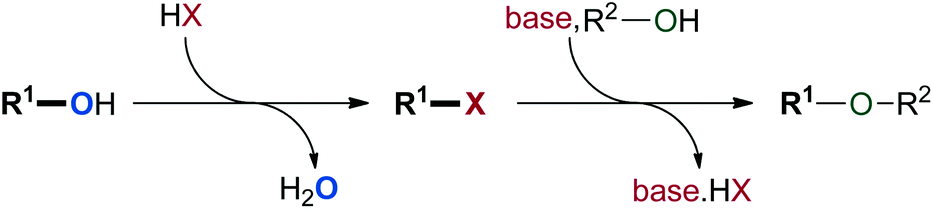 | ||
| Scheme 1 Conventional formation of ethers with poor atom-economy. | ||
Because of the inherent difficulty of activating alcohols for SN1 reactions, π-activated alcohols, such as propargylic and allylic alcohols were most commonly used in early reports.6–8 Benzylic alcohols were later recognised as suitable substrates for this type of reaction.9 The structural arrangement of these alcohols eases the activation of the C–O bond, as the positively charged intermediates are stabilised by the π-electron cloud through resonance.9 Those substrates are now commonly used as sources of “proto-electrophiles” for substitution reactions, and various catalytic methodologies have been developed for the formation of C–C,10,11 C–N,12–14 C–O,15–21 and C–S22 bonds (Scheme 2).
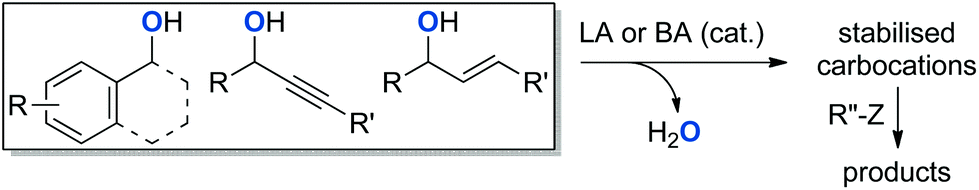 | ||
| Scheme 2 Dehydrative bond formation from π-activated alcohols. LA = Lewis acid. BA = Brønsted acid. Z = CH, N, NH2, OH, SH. | ||
Although Brønsted acids have been successfully used as catalysts in both homogeneous and heterogeneous procedures,23–26 the use of Lewis acids as catalysts constitutes the majority of the reports in literature.27,28 Cationic homogeneous gold complexes have been used as versatile catalysts for a plethora of organic transformations.29,30 More specifically, their Lewis acidic nature has permitted their utilisation in dehydrative reactions with alcohols.31,32 For example, simple chloride salts of gold(III)33,34 or phosphine–gold(I) complexes,35,36 have been used as catalysts for the formation of C–O and C–N bonds.
We previously reported that [Au(NHC)(CH3CN)][BF4] complexes (Fig. 1) catalyse the formation of symmetrical ethers from secondary benzylic alcohols, albeit as a side-reaction (Scheme 3).37 This observation prompted us to explore the capabilities of these well-defined complexes in catalysing the dehydrative formation of ethers.
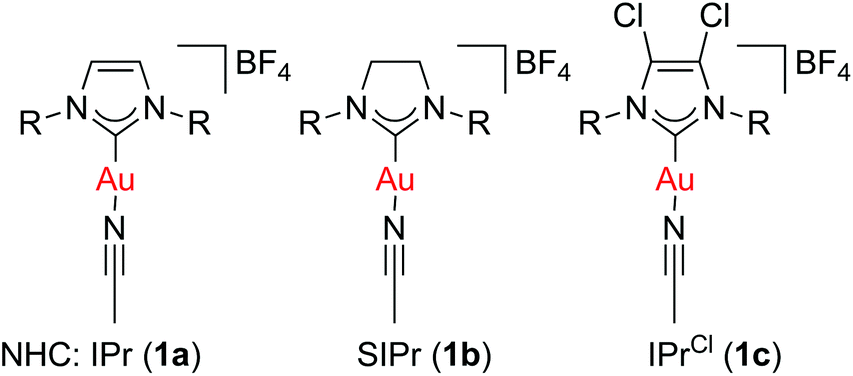 | ||
| Fig. 1 Complexes used as catalysts in this study. R = 2,6-diisopropylphenyl. IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. SIPr = 1,3-bis(2,6-diisopropyl-phenyl)imidazolin-2-ylidene. IPrCl = 4,5-dichloro-1,3-bis(2,6-diisopropyl-phenyl)-imidazol-2-ylidene. | ||
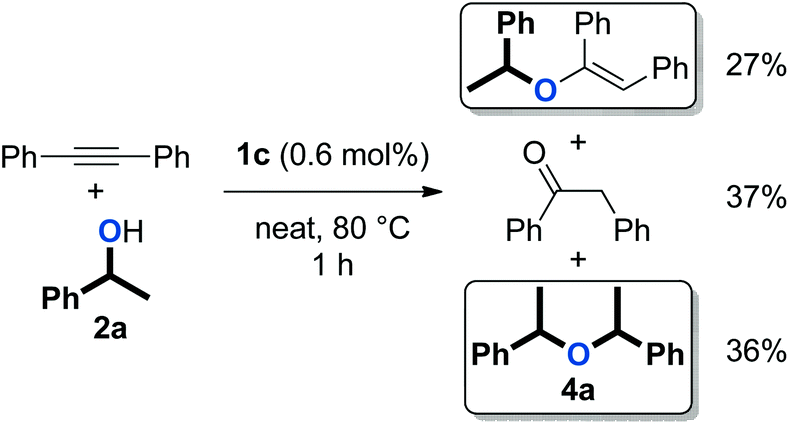 | ||
| Scheme 3 Formation of symmetrical ether 4a as a side-product in hydroalkoxylation (1.1 equiv. of 2a were used).37 | ||
We herein describe that complex 1c can be used to effectively form unsymmetrical ethers through a cross-dehydrative transformation of benzylic alcohols and phenols. These ethers are also accessible from procedures that use PhenoFluor/TMS-imidazole38 or catalytic amounts of Bi,39 or Ru40 complexes (Scheme 4). These reactions proceed via activation of the phenol (via formation of an imidazolium adduct, an oxocarbenium ion, and C–H activation respectively) instead of via elimination of water from the benzylic alcohol.
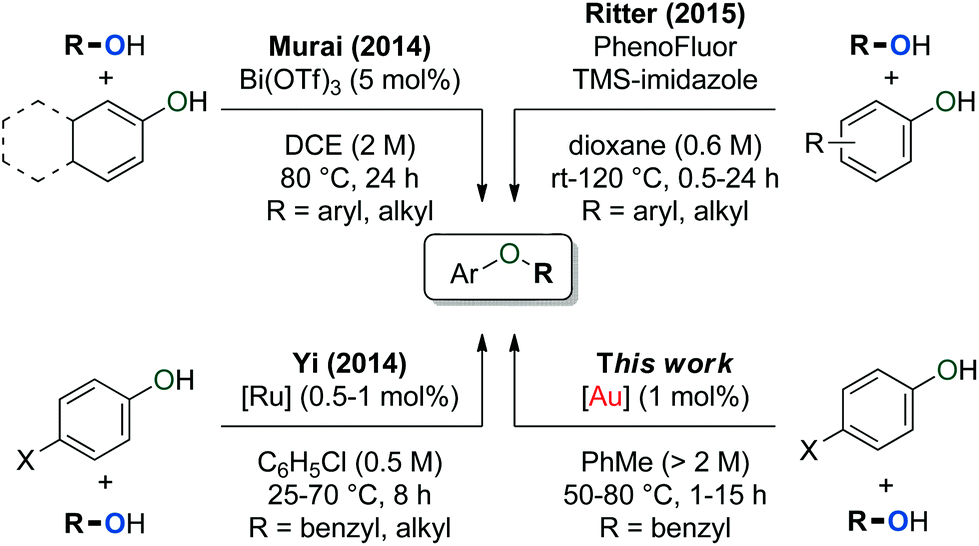 | ||
| Scheme 4 Catalytic procedures for the formation of aryl ethers. [Ru] = [Ru(C6H6)(PCy3)(CO)(H)][BF4]. [Au] = [Au(IPrCl)(CH3CN)][BF4]. DCE = CH2ClCH2Cl. | ||
Results and discussion
Optimisation studies
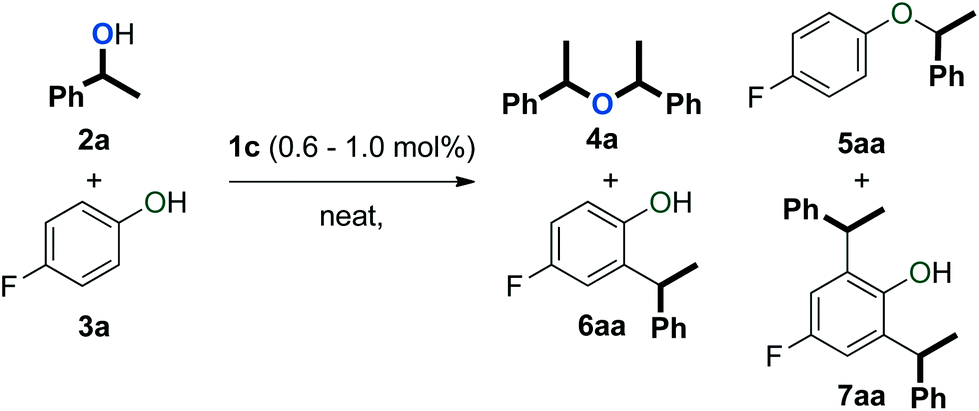
An initial evaluation of the reactivity of various alcohol combinations in the presence of 1c under neat conditions revealed that unsymmetrical ether 5aa could be formed from 1-phenylethanol (2a) and p-fluorophenol (3a) (see ESI†). This ether, however, was observed among a range of other products (Fig. 2) and optimisation was essential for the selective formation of ether 5aa.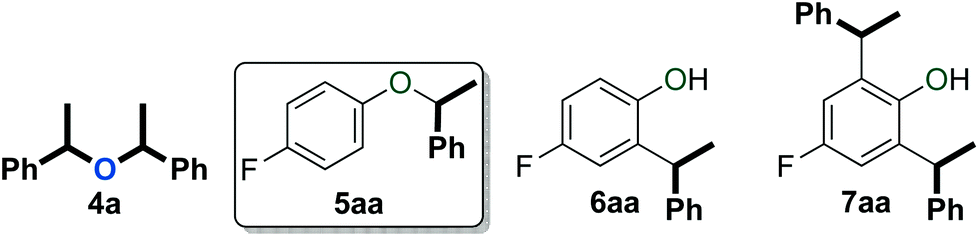 | ||
| Fig. 2 Products observed in the dehydrative reaction of 2a and 3a. | ||
When alcohols 2a and 3a were heated in the absence of a catalyst and solvent, no conversion was observed after 15 minutes (Table 1, entry 1). After 96 hours, 60% of 2a was converted into a 56/44 mixture of ethers 4a and 5aa. This outcome hints at an equilibrium process that is catalysed by weak Brønsted acids such as 3a. Consequently, we continued by testing both gold complexes and Brønsted acids for catalytic activity.
|
|
||
|---|---|---|
| Entry | Catalyst (mol%) | Conversionb (%) (4a/5aa/6aa) |
| a Reaction condition: 2a (0.25 mmol), 3a (0.25 mmol), neat, in air. Cat. = catalytic amount, approximately 1–10 mol%. b Determined by GC analysis, with respect to 2a. Product distribution is given in brackets. | ||
| 1 | None | 0 |
| 2 | [Au(IPr)(CH3CN)][BF4] 1a (0.6) | 0 |
| 3 | [Au(SIPr)(CH3CN)][BF4] 1b (0.6) | 19 (42/58/0) |
| 4 | [Au(IPrCl)(CH3CN)][BF4] 1c (0.6) | >95 (38/62/0) |
| 5 | [Au(IPr)(NTf2)] (1.0) | 0 |
| 6 | [Au(IPrCl)(NTf2)] (1.0) | 0 |
| 7 | [Au(PPh3)(NTf2)] (1.0) | 60 (57/43/0) |
| 8 | [{Au(IPr)}2(μ-OH)][BF4] (0.3) | 0 |
| 9 | [{Au(SIPr)}2(μ-OH)][BF4] (0.3) | 0 |
| 10 | [{Au(IPrCl)}2(μ-OH)][BF4] (0.3) | 0 |
| 11 | [Au(IPrCl)(OH)] (0.6) | 0 |
| 12 | HBF4 (cat.) | >95 (0/23/77) |
| 13 | H2SO4 (cat.) | >95 (0/0/100) |
| 14 | p-TsOH (cat.) | >95 (0/0/100) |
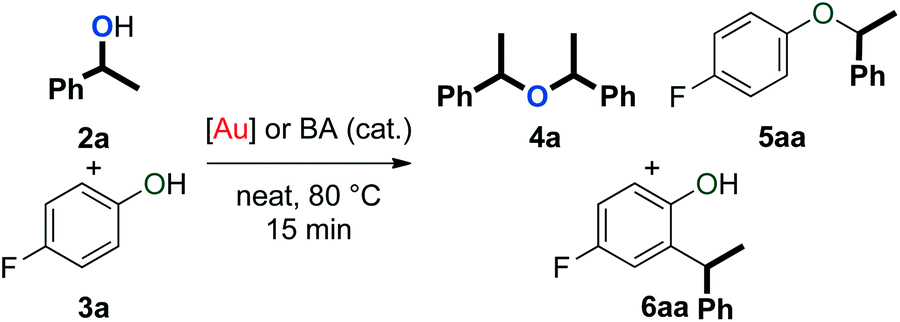
Among the series of gold complexes of the type [Au(NHC)(CH3CN)][BF4] (1a–c, Fig. 1), those bearing NHC ligands SIPr (1b) and IPrCl (1c) were particularly active and mixtures of ethers 4a and 5aa were produced (Table 1, entries 3–4). Gratifyingly, the formation of styrene from 2a was not observed despite its formation when using other catalyst systems.41–43 No reaction occurred with NHC-bearing Gagosz-type complexes, [Au(NHC)(NTf2)]44 (Table 1, entries 5 and 6). In contrast, the reaction using [Au(PPh3)(NTf2)]45 gave rapid conversion of the starting alcohols (Table 1, entry 7), but ethers 4a and 5aa were converted to a mixture of side-products 6aa and 7aa (Fig. 2) when the reaction was continued for another 45 minutes to reach complete conversion of 2a. No reaction occurred with complexes [{Au(NHC)}2(μ-OH)][BF4] (Table 1, entries 8–10) or [Au(IPrCl)(OH)] (Table 1, entry 11) as catalysts.46,47 This lack of reactivity can be attributed to the formation of gold phenoxide complexes that are inert under the reaction conditions.48 Reactions with catalytic amounts of different Brønsted acids led to rapid conversion of 2a, but arylakane 6aa was obtained instead of the targeted ether 5aa (Table 1, entries 12–14). Trace amounts of the corresponding di-alkylated product 7aa were observed as well.
These results demonstrate that secondary benzylic alcohols and phenols can be converted to various products (e.g. ethers 4 and 5 and arylalkanes 6 and 7) using either cationic gold complexes or Brønsted acids as catalysts. Control over reactivity appeared to be a challenge that could most elegantly be overcome by the use of gold complex 1c.
We continued our optimisation studies by evaluating product distributions from reactions in solvent (see ESI†), but no improvement was observed over the previous solvent-free conditions.
Next, we tested whether the use of an excess of phenol or a change in the reaction temperature could shift the product distribution towards ether 5aa (Table 2). The amount of phenol present was found to influence the rate of the reaction and we therefore evaluated the product distributions both after 15 minutes and after 1 hour. Gratifyingly, with 5 equivalents of 3a the product distribution shifted from symmetrical ether 4aa to the desired ether 5aa (Table 2, entries 7 and 8).49 Of note, the use of an excess of phenol should not be considered as a major disadvantage from an atom-economic point of view because it can be recycled.50 Of some concern was the observation of side-products 6aa and 7aa when reactions were performed using an excess of phenol (Table 2, entries 1–8). Additionally, extended reaction times led to the formation of these side-products under these reaction conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3a (equiv.) | 1c (mol%) | T (°C) | t (h) | Conversionb (%) (4a/5aa/6aa + 7aa)c |
| a Reaction conditions: 2a (0.25 mmol), 3a, neat, in air. b Determined by 1H NMR spectroscopy. Product distribution is given in brackets. c Ratio includes sum of amount of 6aa and 7aa.51 | |||||
| 1 | 2 | 0.6 | 80 | 0.25 | 61 (21/76/4) |
| 2 | 2 | 0.6 | 80 | 1 | >95 (19/74/7) |
| 3 | 3 | 0.6 | 80 | 0.25 | 90 (7/85/8) |
| 4 | 3 | 0.6 | 80 | 1 | >95 (9/80/11) |
| 5 | 4 | 0.6 | 80 | 0.25 | >95 (13/83/4) |
| 6 | 4 | 0.6 | 80 | 1 | >95 (7/87/6) |
| 7 | 5 | 0.6 | 80 | 0.25 | >95 (9/84/7) |
| 8 | 5 | 0.6 | 80 | 1 | >95 (2/87/11) |
| 9 | 5 | 1.0 | 80 | 15 | >95 (0/0/100) |
| 10 | 5 | 1.0 | 70 | 15 | >95 (0/0/100) |
| 11 | 5 | 1.0 | 60 | 15 | >95 (0/19/80) |
| 12 | 5 | 1.0 | 50 | 15 | >95 (7/86/7) |
In an attempt to suppress the detrimental formation of 6aa and 7aa, reactions were examined at lower temperatures (Table 2, entries 9–12). To compensate for potentially lower reaction rates, the catalyst loading was increased to 1 mol% and the reaction time was extended to 15 hours. A control reaction at 80 °C demonstrated clearly that ether 5aa transformed to arylalkanes 6aa and 7aa upon this extended reaction time (Table 2, entry 9). Reactions at 60 °C or 70 °C also led to the formation of mixtures of 6aa and 7aa (Table 2, entries 10 and 11). However, when the reaction was performed at 50 °C, the desired ether 5aa was the major product (Table 2, entry 12).
We had thus established that the use of excess phenol was required to favour the formation of unsymmetrical ether 5aa over symmetrical ether 4a, but that the reaction temperature had to be lowered to suppress the formation of arylalkanes 6aa and 7aa. In order to simplify the procedure, slower reactions that produce the desired product predominantly were chosen over lower catalyst loadings and higher temperatures that requires the optimisation of the reaction time to give the most favourable product distribution. We concluded that ether 5aa could thus be formed most effectively from 2a and 5 equivalents of 3a under solvent-free conditions at 50 °C using 1 mol% 1c as catalyst (Table 2, entry 12). Furthermore, we considered the addition of toluene as a means to lower the rate of the reaction.52
Determination of substrate scope and limitations
The scope of this procedure was evaluated by performing reactions with various secondary benzylic alcohols (2a–i) and phenols (3a–b) (Scheme 5). Reactions with p-fluorophenol (3a) and benzylic alcohols bearing alkyl or phenyl substituents in the α′-position (2a–d) produced the corresponding ether products 5aa–ad in good yields. The etherification reaction of model substrates 2a and 3a could also be performed on a 2.0 mmol scale to obtain ether 5aa again in 82% yield (355.9 mg).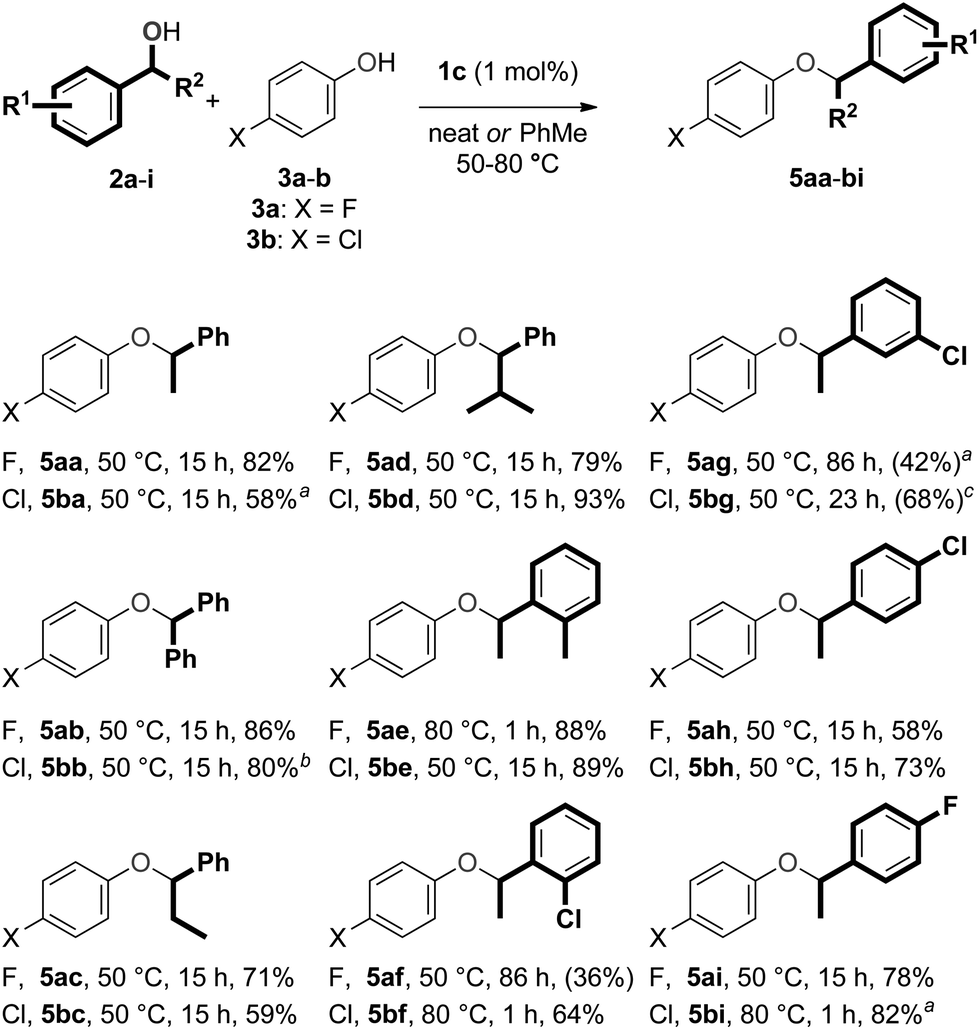 | ||
| Scheme 5 Scope for etherification with secondary benzylic alcohols and phenols. Reaction condition: 2a–i (0.25 mmol), 3a (1.25 mmol), in air. For each entry, the substituent on the phenol (X), the label of the final product, the reaction temperature, the reaction time and the yield are given. aIn PhCH3 (5 M). bIn PhCH3 (2.5 M). cPredominant formation of the corresponding diarylalkane was observed. | ||
Benzylic alcohols bearing CO2Me, CN or CF3 substituents in the α′-position, did not react (even at 80 °C). This lack of reactivity can be attributed to the deactivating electron-withdrawing nature of these functional groups. 2-Furylethanol was found to decompose under the reaction conditions.37 While secondary benzylic alcohols bearing substituents on the phenyl ring (2e–g) were reactive, we were only able to form the corresponding ethers (5ae–ag) selectively by modifying the reaction conditions slightly. Ether 5ae was obtained from 1-(o-tolyl)ethanol (2e) in very high yield by performing the reaction at 80 °C, but the reaction of 1-(o-chlorophenyl)ethanol (2f) produced a mixture of ether 5af and the corresponding aryl alkane 6af at this temperature. Ether 5af could be formed selectively at 50 °C, but complete conversion of alcohol 2f was not obtained, even after 86 hours. The reaction of 1-(m-chlorophenyl)ethanol (2g) proceeded in a similar fashion to that of alcohol 2f, but toluene had to be added to avoid the formation of the corresponding arylalkane product at 50 °C. Unfortunately, this reaction did not reach completion, even after 86 hours. The standard reaction conditions could be applied for secondary benzylic alcohols with chloro or fluoro substituents in the para position of the phenyl ring (2h–i), and the corresponding ethers (5ah–ai) were obtained in modest and good yields, respectively.
Reactions with p-chlorophenol (3b) were then examined. Once again, reactions with secondary benzylic alcohols bearing substituents in the α′-position (2a–d) proceeded smoothly and the corresponding ethers 5ba–bd were obtained in high yields. Interestingly, for the reaction of 1-phenylethanol (2a), toluene had to be added to the reaction mixture to avoid formation of the corresponding arylalkane. This trend in reactivity suggests that the process that converts ethers 5 to arylalkanes 6 and 7 is faster for ethers derived from p-chlorophenol (3b) compared to those derived from p-fluorophenol (3a), and that the size of the substituent in the α′-position of the benzylic alcohol has a significant influence on this process. Ether 5be could be obtained from 1-(o-tolyl)ethanol (2e) using the standard reaction conditions, but ether 5bf from 1-(o-chloro)ethanol was obtained most effectively at 80 °C. Unfortunately, reaction conditions could not be found which led to complete conversion of 1-(m-chlorophenyl)ethanol (2g) while avoiding the formation of the corresponding arylalkane. The desired ethers 5bh–bi could be formed from alcohols 2h–i and phenol 3b. Finally, the formation of unsymmetrical ethers from 1-phenylethanol (2a) and different phenols was tested as well. Both the use of phenol, and phenols bearing substituents in either the ortho (Cl), meta (F, Cl) or para-position (Br, Me, OMe, CF3) were evaluated. Unsatisfyingly, reactions with these substrates all gave mixtures of the desired unsymmetrical ethers and corresponding arylalkanes.53
Catalytic conversion of symmetrical ether
The observed catalytic of the formation of both symmetrical and unsymmetrical ethers (4 and 5) as well as arylalkanes (6 and 7) prompted us to investigate the origins of these compounds. Thus, we examined whether symmetrical ether 4a could be converted to ether 5aa and arylalkanes 6aa and 7aa (Table 3).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | 3a (equiv.) | T (°C) | t (h) | Conversionb (%) (5aa/6aa)c |
| a Reaction condition: 4a (0.10 mmol), neat, in air. b Determined by 1H-NMR or 19F{1H}-NMR spectroscopy with respect to 4a or 3a, respectively. Product distribution is given in brackets. c Reaction performed in absence of catalyst. | ||||
| 1 | 1 | 50 | 22 | 29 (91/9) |
| 2 | 2 | 50 | 22 | 68 (79/21) |
| 3 | 1 | 80 | 0.25 | 37 (91/9) |
| 4 | 2 | 80 | 0.25 | 81 (41/59) |
| 5 | 10 | 80 | 0.25 | >99 (16/84) |
| 6 | 10 | 80 | 1 | >99 (14/86) |
| 7 | 10 | 80 | 22 | >99 (0/100) |
| 8c | 10 | 80 | 1 | 71 (94/6) |
| 9c | 10 | 80 | 22 | >99 (0/100) |
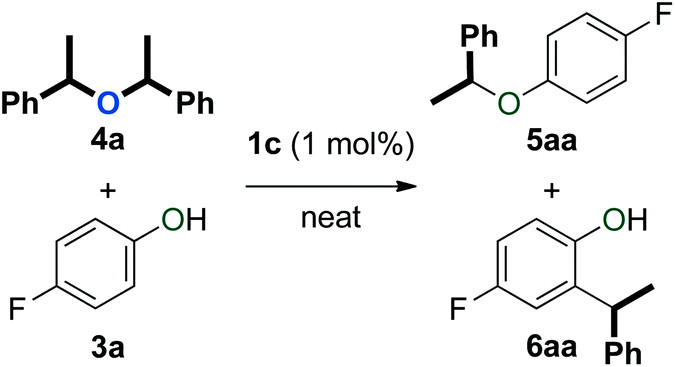
Symmetrical ether 4a was first subjected to different amounts of phenol 3a under the standard catalytic conditions (Table 3, entries 1 and 2). While the reaction using 1 equivalent of 3a was particularly slow, it produced unsymmetrical ether 5aa and a small amount of 6aa (Table 3, entry 1). When 2 equivalents of 3a were used, the reaction was significantly faster and the product distribution shifted towards arylalkane 6aa (Table 3, entry 2). These results are consistent with previous reports that describe acid-catalysed and thermal rearrangements of phenolic ethers to arylalkanes.54–56 As expected from our catalyst screening (Table 1), the transformation of 4a to mixtures of 5aa and 6aa was much more rapid at 80 °C (Table 3, entries 3 and 4). With 2 equivalents of 3a, the formation of arylalkane 7aa was also observed. The influence of phenol 3a in this transformation was further evaluated by using a ten-fold excess with respect to 4a (Table 3, entry 5). Ether 4a showed complete conversion to arylalkane 6aa upon extended reaction time (Table 3, entries 6 and 7). In this case, only trace amounts of arylalkane 7aa could be observed. When the experiment was repeated in the absence of 1c, we obtained similar results (Table 3, entries 8 and 9). This observation provides another hint that the Brønsted acidity of phenol 3a enables its role as a catalyst in this transformation.
Catalytic conversion of unsymmetrical ether
Finally, we evaluated the stability of unsymmetrical ether 5aa (Table 4). This product did not transform to arylalkanes 6aa and 7aa in the absence of phenol 3a (Table 4, entry 1). In the presence of 1 equivalent of phenol 3a, however, slow conversion to arylalkane 6aa was observed (Table 4, entry 2). This reaction reached completion when 5 equivalents of phenol 3a were used (Table 4, entry 3). Repeating the latter reaction in the absence of catalyst gave a much lower conversion (Table 4, entry 4), thereby demonstrating that gold complex 1c assists this transformation.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | 3a (equiv.) | t (h) | Conversionb (%) (6aa/7aa) |
| a Reaction condition: 5aa (0.05 mmol), neat, in air. b Determined by 1H-NMR or 19F{1H}-NMR spectroscopy with respect to 5aa. Product distribution is given in brackets. | ||||
| 1 | 1c (1) | — | 15 | — |
| 2 | 1c (1) | 1 | 15 | 33 (86/14) |
| 3 | 1c (1) | 5 | 15 | >99 (96/4) |
| 4 | — | 5 | 15 | 22 (96/4) |
| 5 | p-TsOH | — | 1 | 93 (32/68) |
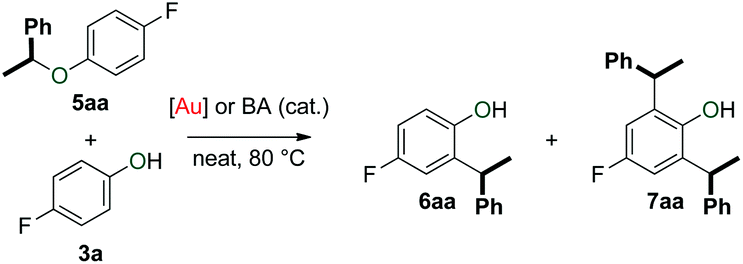
The formation of mixtures of 6aa and 7aa, especially in the reaction with 1 equivalent of phenol 3a (Table 4, entry 2) is intriguing. This result suggests the existence of a pathway that delivers a benzyl-fragment from 5aa that subsequently reacts with 6aa to form 7aa.
Reactions with enantiopure alcohol
Various mechanistic proposals have suggested the formation of a carbocation intermediate in dehydrative reactions with π-activated alcohols.33–35 This planar intermediate should give racemic products upon reaction with a nucleophile. Thus, the observation of racemic products from reactions with enantiopure alcohols would support such a mechanism. To test this, (S)-1-phenylethanol ((S)-2a) was subjected to the catalytic conditions with and without the addition of 5 equivalents of phenol 3a (eqn (1) and (2)). As expected, only racemic 4a and 5aa were observed by chiral HPLC analysis.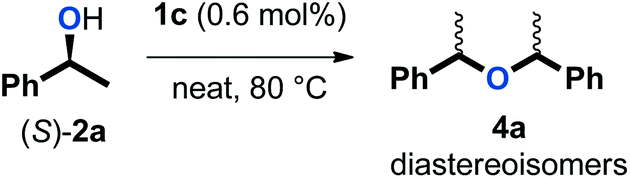 | (1) |
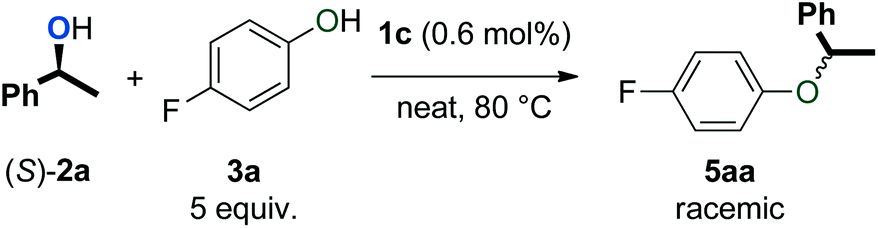 | (2) |
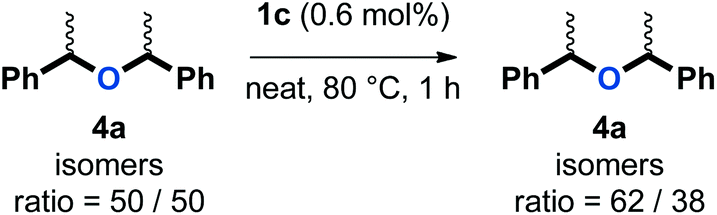 | (3) |
The ratio of isomers of 4a evolved upon extended reaction time in the reaction depicted in eqn (1). Therefore, a control experiment was conducted in which an equal mixture of racemic and meso isomers of 4a was subjected to the catalytic conditions (eqn (3)). The ratio of isomers evolved also in this reaction, indicating that an equilibrium process was operative. Of note, this phenomenon was not observed in the absence of a catalyst under otherwise identical conditions. This result is consistent with our observations that symmetrical ether 4 is not merely a side-product in our targeted etherification reaction, but rather a kinetic intermediate that can be converted to the more stable ether 5. The need for an excess of phenol (3) in our procedure can then be justified by the necessity to displace this equilibrium and to drive the reaction to the desired unsymmetrical ether (5).
Mechanistic proposal
Altogether, we propose a plausible mechanism to account for our observations (Scheme 6). It must be noted that while the gold catalyst is likely to serve as Lewis acid, we have not established its exact role in these transformations. The carbocation intermediate I that forms from formal gold-assisted elimination of hydroxide from 2 can be trapped by either the benzylic alcohol (2) or the phenol (3) to give symmetrical ether 4 or unsymmetrical ether 5. Since dehydration of 2via protonation would be more favourable than the direct release of hydroxide, phenol 3 is likely to be involved in this step.6 The formation of 4 is reversible under the reaction conditions employed, while 5 can be subsequently converted to thermodynamic products 6 and 7. Because of the low electron density on the arenes (3) used in this study, direct transformation from I or 4 to 6via aromatic substitution seems less favourable than a Fries-type rearrangement that transforms ether 5 to arylalkane 6 instead.56–58 As such, a pathway might be operative in which the ether is converted to the starting alcohols which then form the side-products via a Friedel–Crafts reaction.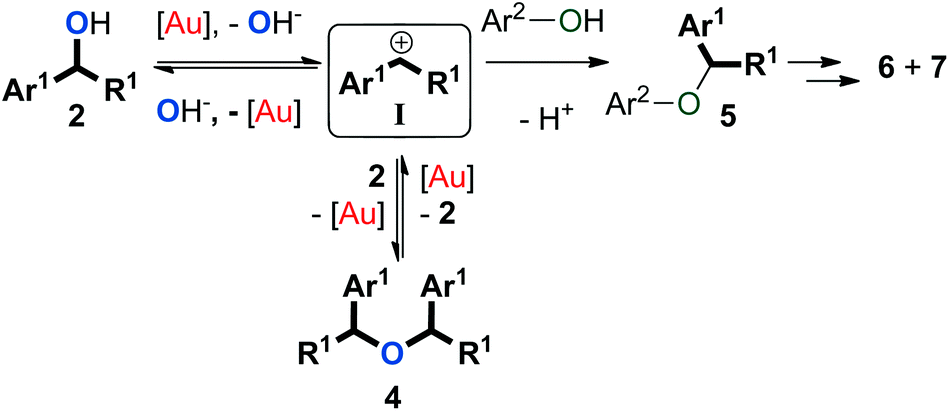 | ||
| Scheme 6 Plausible reaction mechanism. Ar2 = 4-X-phenyl. X = F, Cl. | ||
Conclusions
In summary, we have demonstrated that ethers can be prepared from readily available benzylic alcohols and phenols under mild and environmentally benign conditions. Besides giving access to new products, it provides another example of remarkable chemoselectivity that can be obtained by employing an appropriate NHC–gold(I) complex as catalyst. Investigations to use secondary benzylic alcohols as proto-electrophiles to react with nucleophiles other than phenols are currently ongoing in our laboratories.Experimental
General information
All reagents were obtained through commercial suppliers and were used as received. Unless otherwise stated, all alcohols were used as their racemate. [Au(NHC)(CH3CN)][BF4], [{Au(NHC)}2(μ-OH)][BF4] (NHC = IPr, SIPr and IPrCl) and [Au(L)(NTf2)] (L = IPr, IPrCl, PPh3) were synthesised according to previous reports.44,46,47,59,60 All reactions were set up on the benchtop in screw cap vials with Teflon seal inserts and carried out under an atmosphere of air. Flash column chromatography was performed using silica gel.General procedure for formation of ethers
To [Au(IPrCl)(MeCN)][BF4] (1c) (1.0 mol%) were added benzylic alcohol 2 (0.25 mmol), phenol 3 (1.25 mmol, 5 equiv.) and toluene (0–100 μL). The reaction mixture was stirred at 50 °C or 80 °C. After the reaction mixture was cooled down, the crude product was purified by flash column chromatography on silica gel (petroleum ether/diethyl ether = 9/1).61Acknowledgements
The EPSRC and ERC are gratefully acknowledged for support. KAUST is gratefully acknowledged for partial support of this work. Umicore AG is acknowledged for their generous gift of materials. The EPSRC National Mass Spectrometry Service Centre (NMSSC) is gratefully acknowledged for HRMS analyses. S.P.N. is a Royal Society Wolfson Research Merit Award holder.Notes and references
- J. Blacker and M. T. Williams, Pharmaceutical Process Development: Current Chemical and Engineering Challenges, Royal Society of Chemistry, 2011 Search PubMed.
- A. W. Q. Williamson, J. Chem. Soc., 1852, 4, 229–239 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 2000 Search PubMed.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. Leazer, L. Johnnie, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 647–666 CrossRef CAS PubMed.
- M. Bandini and M. Tragni, Org. Biomol. Chem., 2009, 7, 1501–1507 CAS.
- Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 CrossRef CAS PubMed.
- R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC.
- O. Debleds, E. Gayon, E. Vrancken and J.-M. Campagne, Beilstein J. Org. Chem., 2011, 7, 866–877 CrossRef CAS PubMed.
- X.-W. Yan, Q. Zhang, W. Wei and J.-X. Ji, Tetrahedron Lett., 2014, 55, 3750–3752 CrossRef CAS PubMed.
- V. Terrasson, S. Marque, M. Georgy, J.-M. Campagne and D. Prim, Adv. Synth. Catal., 2006, 348, 2063–2067 CrossRef CAS PubMed.
- X. Giner, P. Trillo and C. Nájera, J. Organomet. Chem., 2011, 696, 357–361 CrossRef CAS PubMed.
- L. Li, A. Zhu, Y. Zhang, X. Fan and G. Zhang, RSC Adv., 2013, 4, 4286–4291 RSC.
- Z. Zhu and J. H. Espenson, J. Org. Chem., 1996, 61, 324–328 CrossRef CAS.
- P. Salehi, N. Iranpoor and F. Kargar Behbahani, Tetrahedron, 1998, 54, 943–948 CrossRef CAS.
- Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11020 CrossRef CAS.
- G. V. M. Sharma, T. Rajendra Prasad and A. K. Mahalingam, Tetrahedron Lett., 2001, 42, 759–761 CrossRef CAS.
- K. J. Miller and M. M. Abu-Omar, Eur. J. Org. Chem., 2003, 1294–1299 CrossRef CAS PubMed.
- K. Iwanami, K. Yano and T. Oriyama, Synthesis, 2005, 2669–2672 CAS.
- Y. Liu, R. Hua, H.-B. Sun and X. Qiu, Organometallics, 2005, 24, 2819–2821 CrossRef CAS.
- L. Herkert, S. L. J. Green, G. Barker, D. G. Johnson, P. C. Young, S. A. Macgregor and A.-L. Lee, Chem. – Eur. J., 2014, 20, 11540–11548 CrossRef CAS PubMed.
- R. Sanz, A. Martínez, J. M. Álvarez-Gutiérrez and F. Rodríguez, Eur. J. Org. Chem., 2006, 1383–1386 CrossRef CAS PubMed.
- R. Sanz, A. Martínez, D. Miguel, J. M. Álvarez-Gutiérrez and F. Rodríguez, Adv. Synth. Catal., 2006, 348, 1841–1845 CrossRef CAS PubMed.
- K. Motokura, N. Nakagiri, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukawa and K. Kaneda, Org. Lett., 2006, 8, 4617–4620 CrossRef CAS PubMed.
- J.-L. Yu, H. Wang, K.-F. Zou, J.-R. Zhang, X. Gao, D.-W. Zhang and Z.-T. Li, Tetrahedron, 2013, 69, 310–315 CrossRef CAS PubMed.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 302–304 CrossRef PubMed.
- M. Hellal, F. C. Falk, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 CAS.
- H. Schmidbaur and A. Schier, Z. Naturforsch. B: Chem. Sci., 2011, 66, 329–350 CrossRef CAS PubMed.
- H. G. Raubenheimer and H. Schmidbaur, J. Chem. Educ., 2014, 91, 2024–2036 CrossRef CAS.
- J. Muzart, Tetrahedron, 2008, 64, 5815–5849 CrossRef CAS PubMed.
- B. Biannic and A. Aponick, Eur. J. Org. Chem., 2011, 6605–6617 CrossRef CAS PubMed.
- A. B. Cuenca, G. Mancha, G. Asensio and M. Medio-Simón, Chem. – Eur. J., 2008, 14, 1518–1523 CrossRef CAS PubMed.
- M. Georgy, V. Boucard and J.-M. Campagne, J. Am. Chem. Soc., 2005, 127, 14180–14181 CrossRef CAS PubMed.
- N. Ibrahim, A. S. K. Hashmi and F. Rominger, Adv. Synth. Catal., 2011, 353, 461–468 CrossRef CAS PubMed.
- Y. Liu, X. Wang, Y. Wang, C. Du, H. Shi, S. Jin, C. Jiang, J. Xiao and M. Cheng, Adv. Synth. Catal., 2015, 357, 1029–1036 CrossRef CAS PubMed.
- R. M. P. Veenboer, S. Dupuy and S. P. Nolan, ACS Catal., 2015, 5, 1330–1334 CrossRef CAS.
- X. Shen, C. N. Neumann, C. Kleinlein, N. W. Goldberg and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 5662–5665 CrossRef CAS PubMed.
- M. Murai, K. Origuchi and K. Takai, Org. Lett., 2014, 16, 3828–3831 CrossRef CAS PubMed.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS PubMed.
- S. Podder, J. Choudhury and S. Roy, J. Org. Chem., 2007, 72, 3129–3132 CrossRef CAS PubMed.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613–1616 CrossRef CAS PubMed.
- H. Wang, X. Zhu, Y. Lu, Y. Li and X. Gao, Chin. J. Chem., 2011, 29, 1180–1184 CrossRef CAS PubMed.
- L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS.
- J. Liu, E. Muth, U. Flörke, G. Henkel, K. Merz, J. Sauvageau, E. Schwake and G. Dyker, Adv. Synth. Catal., 2006, 348, 456–462 CrossRef CAS PubMed.
- R. S. Ramón, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2011, 17, 1238–1246 CrossRef PubMed.
- A. Gómez-Suárez, Y. Oonishi, S. Meiries and S. P. Nolan, Organometallics, 2013, 32, 1106–1111 CrossRef.
- Y. Oonishi, A. Gómez-Suárez, A. R. Martin and S. P. Nolan, Angew. Chem., Int. Ed., 2013, 52, 9767–9771 CrossRef CAS PubMed.
- When this reaction was repeated without catalyst, no conversion was observed after 1 hour. This result demonstrates that phenol 3a is no effective in catalyst for the targeted etherification reaction (cf.Table 1, entry 1).
- The phenol could be recycled after recovery from the products by flash column chromatography on silica gel.
- The ratio of 6aa and 7aa can be determined by 19F NMR spectroscopy or GC analysis. In these reaction, the amount of 7aa constitutes <10% of the total amount of product. As such, the exact amount can not be determined with high accuracy and the data is omitted.
- The addition of toluene could potentially enhance the homogeneity of reaction mixtures of substrates with high melting points or retard the formation of side-products from reactions with activated substrates.
- Attempts to alter the reaction conditions did not lead to clean conversions to the targeted ethers.
- F. A. Luzzio and J. Chen, J. Org. Chem., 2009, 74, 5629–5632 CrossRef CAS PubMed.
- G. A. Kraus and D. Chaudhary, Tetrahedron Lett., 2012, 53, 7072–7074 CrossRef CAS PubMed.
- S. R. Bandatmakuru, S. R. Amasa, V. R. Arava and M. C. Subha, Pharma Chem., 2014, 6, 299–311 Search PubMed.
- B. Sreedhar, V. Swapna and C. Sridhar, Synth. Commun., 2004, 34, 1433–1440 CrossRef CAS.
- We presume that di-alkylated product 7 forms via etherification of 6 with a subsequent rearrangement.
- S. R. Patrick, A. Gómez-Suárez, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 33, 421–424 CrossRef.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC.
- Alternative to flash column chromatography, excess phenol can be removed from the crude reaction mixture by filtration after crystallisation from pentane and cooling to −20 °C.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data and NMR spectra. See DOI: 10.1039/c5gc00684h |
| This journal is © The Royal Society of Chemistry 2015 |