
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral Rh phosphine–phosphite catalysts immobilized on ionic resins for the enantioselective hydrogenation of olefins in water†
P.
Kleman
a,
P.
Barbaro
*b and
A.
Pizzano
*a
aInstituto de Investigaciones Químicas (IIQ) and Centro de Innovación en Química Avanzada (ORFEO-CINQA), CSIC and Universidad de Sevilla, Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: pizzano@iiq.csic.es
bIstituto di Chimica dei Composti Organometallici (ICCOM-CNR), Area di Ricerca CNR di Firenze, via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy. E-mail: pierluigi.barbaro@iccom.cnr.it
First published on 13th May 2015
Abstract
The asymmetric hydrogenation of prochiral enamides with Rh complexes bearing chiral phosphine–phosphite ligands (P–OP) supported on sulphonated polystyrene resins has been studied. The complexes have been supported by simple treatment of preformed [Rh(diolefin)(P–OP)](BF4) with the Li salt of resins. In MeOH, high enantioselectivities were observed in the hydrogenation of methyl α-N-acetamido acrylate (4a), while they decreased for less reactive methyl α-N-acetamido cinnamate (4b). Moreover, a significant Rh leaching was observed in these reactions. In contrast, a low Rh leaching was observed in reactions performed in water. Catalyst optimization enabled by the highly modular structure of P–OP ligands led to efficient catalysts for the hydrogenation of 4a and 4b in water, with enantioselectivities over 95% ee and S/C values up to 2300 and 500, respectively. To investigate the synthetic potential of the present catalytic system, the hydrogenation of a set of β-aryl α-dehydroaminoacids (4c–4j) in water has also been studied. For these reactions good catalytic activity and high enantioselectivity (87–97% ee) were also observed. In the case of brominated substrates 4i and 4j an unexpected debromination reaction in water has been observed. This phenomenon seems associated with the use of the supported catalyst and water and could be minimized by the use of a catalyst formed from 3f. In addition, gel-phase 31P{1H} NMR studies performed with a representative supported complex show an analogous reactivity to that displayed by its soluble tetrafluoroborate counterpart. Finally, a selective deuterium labelling of the product has been observed in hydrogenations of 4a and 4b in D2O, similarly to that observed in the homogeneous hydrogenation of these enamides with water soluble catalysts.
Introduction
Rhodium catalyzed enantioselective hydrogenation of olefins is a highly practical and versatile tool for the synthesis of chiral compounds.1 Due to the broad scope and outstanding performance that the corresponding catalysts have reached, a plethora of optically active compounds have efficiently been prepared by this hydrogenation.2 This transformation has frequently been included in multi-step synthesis by the fine-chemical and pharmaceutical industries.3 This widespread use has therefore increased the interest in environmentally related and practical aspects of this transformation, often relegated by other fundamental aspects. In connection with this, the challenge that the mentioned industrial sectors face to decrease the environmental impact of their production should be recalled.4Hydrogenations of this kind are usually run with homogeneous catalysts in methylene chloride or methanol solutions, while the catalyst separation step is frequently done in a less polar organic solvent. Accordingly, the development of catalytic systems operating in water constitutes a highly interesting goal as water is a cheap, non-toxic and non-flammable solvent.5 Moreover, the generally low solubility of hydrogenated products in water may also enable a straightforward product separation.6
The attainment of efficient asymmetric olefin hydrogenations in water is, however, a rather challenging goal and only very rare cases of highly enantioselective reactions in neat water have been described in the literature.7 This fact is in clear contrast with the outstanding results that these hydrogenations have often shown in conventional organic solvents. However, the use of water as a reaction medium may introduce deep changes in the catalytic system. Hydrophobic effects and hydrogen bonding interaction with the substrate and catalytic intermediates caused by water,8,9 as well as the low solubility of hydrogen in this solvent, may have an important influence on the reaction. Moreover, reaction of water with catalytic intermediates (e.g. by coordination or by protonation/deprotonation steps)10 may provide alternative reaction pathways to the catalytic cycle operating in organic solvents,11 with a potential erosion of enantioselectivity.
Extensive research pursuing efficient hydrogenation reactions in water following different approaches has been described in the literature.12 Among them, those based on water soluble catalysts, either by ligand modification with suitable functional polar groups13 or by attaching them to a water soluble carrier,14 have been covered thoroughly. However, these catalysts generally display a worse performance than the parent non-water-soluble ones in organic solvents.15 Moreover, a perusal of the literature depicts a rather narrow scope of water-soluble catalysts. Thus, high enantioselectivities have been achieved in the reduction of water soluble substrates like methyl α-N-acetamido acrylate (A, 99% ee)7c or α-N-acetamido cinnamic acid (B, 94% ee),7a while lower values have been reported for less soluble substrates.15,16 For instance, in the hydrogenation of prototypical methyl α-N-acetamido cinnamate (C, Ar = Ph), the highest value obtained in neat water is 88% ee,16c while 89% ee has been reported for a CH2Cl2/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) system.16a In contrast, addition of surfactants allows the reaction to occur in micelles, which may produce a dramatic increase in catalyst activity and enantioselectivity even in the case of low soluble olefins.17 For instance, the group of Oehme has reported enantioselectivities up to 97% ee in the hydrogenation of methyl α-N-acetamido cinnamate derivatives C (Scheme 1).17c
:1) system.16a In contrast, addition of surfactants allows the reaction to occur in micelles, which may produce a dramatic increase in catalyst activity and enantioselectivity even in the case of low soluble olefins.17 For instance, the group of Oehme has reported enantioselectivities up to 97% ee in the hydrogenation of methyl α-N-acetamido cinnamate derivatives C (Scheme 1).17c
 | ||
| Scheme 1 Structures A–C. | ||
An appealing approach to perform these reactions in water is the immobilization of homogeneous catalysts on a hydrophilic support. Moreover, the heterogeneous nature of the system should enable a simple product isolation, with potential advantages over procedures based on biphasic systems or surfactants. However, immobilized catalysts have mostly been studied in conventional organic solvents,18 while the information about their performance in water is very limited.19 A remarkable study in this context, reported by Sheldon and coworkers, describes high activities and enantioselectivities up to 96% ee in the hydrogenation in water of A using a Rh phosphoramidite catalyst immobilized on different supports.19g Particularly good results were obtained with diverse inorganic supports (TOF up to 750 h−1 and 96% ee), while the performance decreased for catalysts supported on Na-Nafion (TOF up to 270 h−1 and 92% ee), which was attributed to a poor swelling of the resin in water. In this regard, immobilization on ion exchange resins is an approach of great interest due to the high swelling in water of these materials.20 Diverse precedents of Rh catalysts with chiral P-ligands immobilized onto exchange resins, as well as their successful application to the asymmetric hydrogenation of olefins in MeOH, can be found in the literature.21 However, the relevance of such supported catalysts for reactions performed in water has not been substantiated yet. Interestingly, a study about the application of water soluble Ir catalysts supported on exchange resins in the non-asymmetric hydrogenation of N-heterocyclic substrates in water has recently been reported.22
In this contribution we present a study on the immobilization of cationic rhodium complexes based on chiral phosphine–phosphite ligands23 on an ion exchange resin. Enabled by a high swelling of the resin in water, the corresponding catalysts show good catalyst activity in this medium, while a highly enantioselective catalyst, efficient for both solutions of A and slurries of C in water, was found by a modular approach. Finally, complementary information obtained by gel-phase 31P{1H} NMR about the reactivity of supported complexes under hydrogenation conditions is also presented.
Results and discussion
Preparation of supported catalysts
Rhodium catalysts based on phosphine–phosphite ligands have become an important class of catalysts for the asymmetric hydrogenation of olefins. Thus, these catalysts have provided excellent results in the reduction of diverse types of substrates like unsaturated phosphonates and enamides of enol-esters.24 Considering this synthetic utility, the development of supported versions of rhodium phosphine–phosphite catalysts appears to be a particularly interesting goal. Towards this, a simple approach that does not require a further ligand modification is highly desirable. In this respect, non-covalent immobilization on an ionic resin is ideal as it can directly be performed with the homogeneous catalyst precursor by a simple anion exchange.20 It should also be stressed that this approach is limited to solvents that produce a good swelling of the resin, typically alcohols and water. Thus, by stirring a solution of the complex [Rh(diolefin)(P–OP)](BF4) (diolefin = COD, NBD; P–OP = 1a–g)23c,24a,b over a sulfonated gel-type resin (Dowex 50WX2-100, 2% cross-linked, 4.8 mequiv. g−1 exchange capacity) as its lithium salt (herein abbreviated as Li-resin, Scheme 2), the corresponding supported catalyst precursor was prepared [diolefin = COD, P–OP = 1d (2d); NBD, P–OP = 1a (3a), 1b (3b), 1c (3c), 1e (3e), 1f (3f), 1g (3g), 1h (3h)]. It is worth noting that the incorporation of the Rh complex into the resin was moderate to high and ranged from 61 to 87% (See the ESI† for details). This accounts for values between 1.6 and 2.2% of the sulphonic groups of the resin bound to [Rh(diolefin)(P–OP)]+ cations.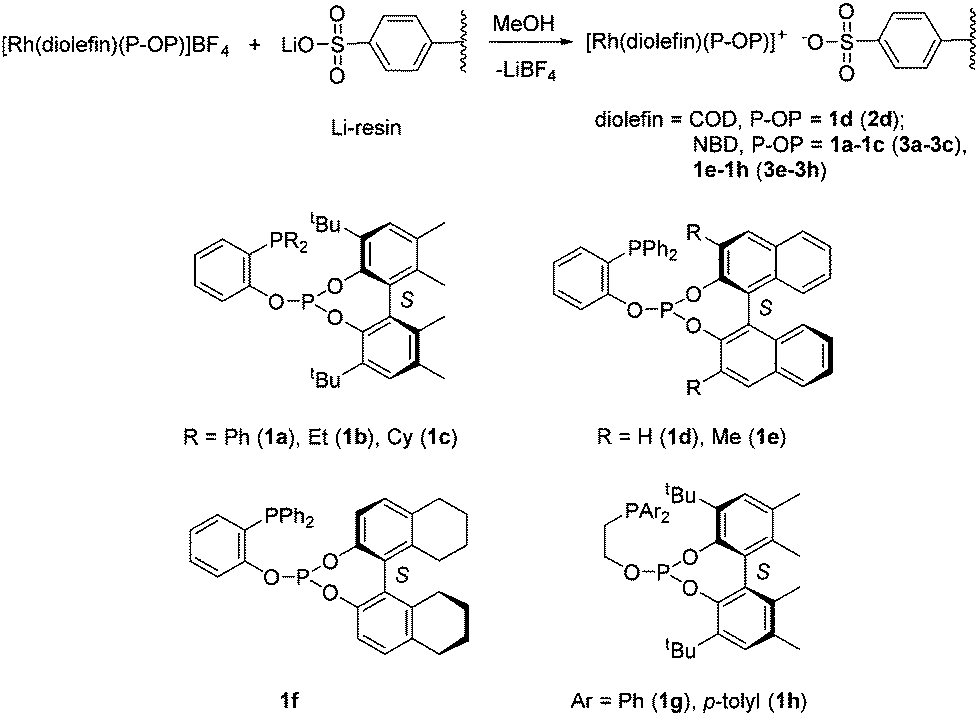 | ||
| Scheme 2 Preparation of immobilized complexes. | ||
Hydrogenations in methanol
In the initial stage of the study, we examined the performance of representative catalyst precursors in the prototypical hydrogenation of methyl α-N-acetamido acrylate (4a, Scheme 3) in methanol under standard conditions (4 bar of H2, room temperature). Thus, we observed full conversion and an excellent enantioselectivity using complex 3a (99% ee; entry 1, Table 1). This catalyst showed lower activity, while maintaining constantly high enantioselectivity upon recycling, as 87% (99% ee) and 78% (99% ee) conversions were obtained in the second and third cycles, respectively (entries 2 and 3). Moreover, the same catalyst exhibited good activity although somewhat lower enantioselectivity at a lower catalyst loading (entry 4).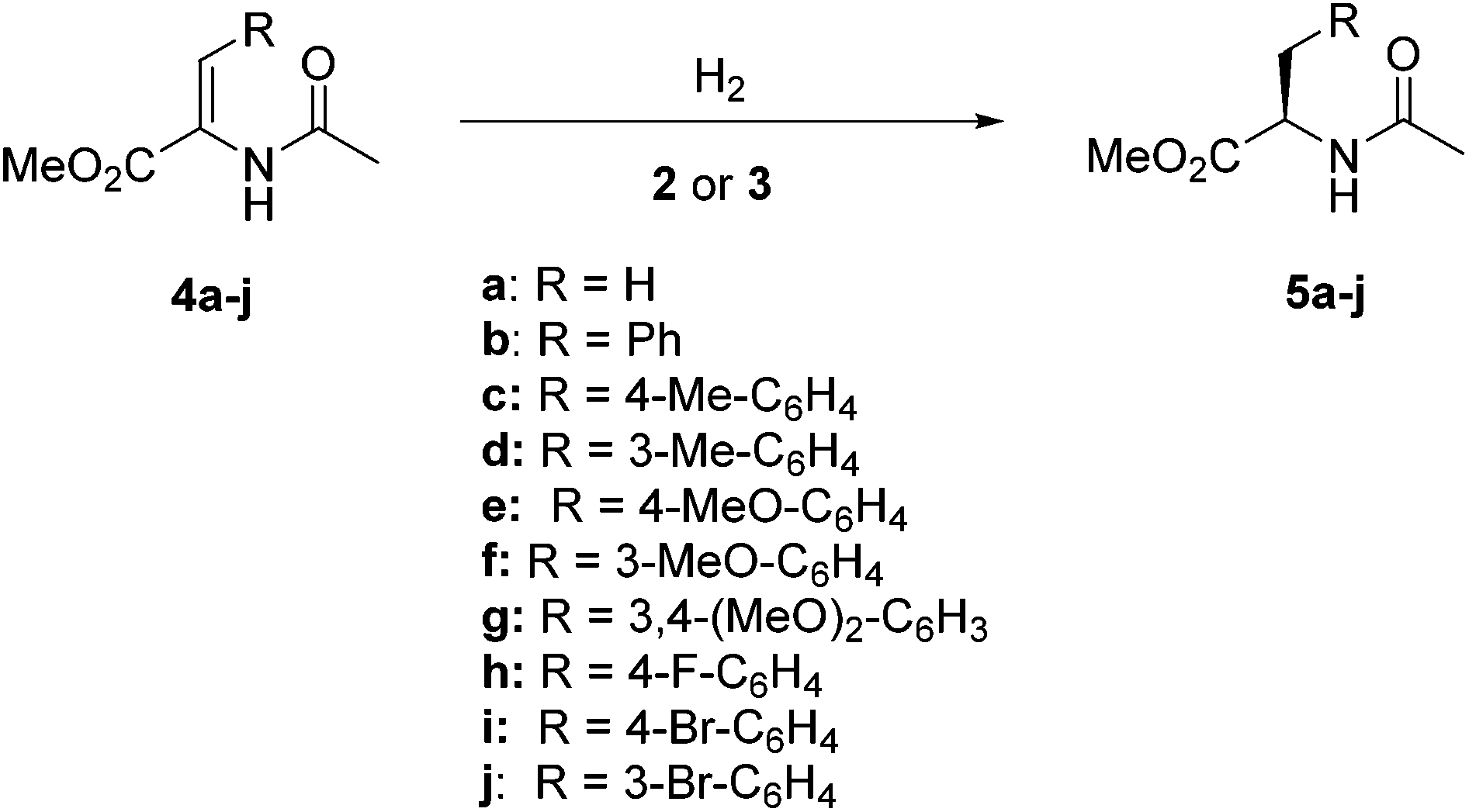 | ||
| Scheme 3 Hydrogenation of enamides. | ||
| Entry | Cat. | S/C | Timeb | % Conv. | % ee (Conf.) |
|---|---|---|---|---|---|
| a Reactions were performed using the suitable amounts of 4a and the resin supported complex under 4 bar H2 at room temperature in MeOH (3 mL), unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess was analyzed by chiral GC. Product configuration was assigned by comparison with the literature data. b Reaction time in hours. In recycling experiments, the number of the cycle is stated in brackets. c Reaction performed in 1 mL of MeOH. | |||||
| 1 | 3a | 80 | 2 (1st cycle) | 95 | 99 (R) |
| 2 | 80 | 2 (2nd cycle) | 87 | 99 (R) | |
| 3 | 80 | 3 (3rd cycle) | 78 | 99 (R) | |
| 4 | 3a | 320 | 14 | >99 | 94 (R) |
| 5 | 3b | 70 | 2 | 43 | 35 (R) |
| 6 | 3c | 70 | 2 | 97 | 48 (R) |
| 7 | 2d | 60 | 2 (1st cycle) | >99 | 80 (R) |
| 8 | 60 | 2 (2nd cycle) | >99 | 73 (R) | |
| 9 | 60 | 2 (3rd cycle) | >99 | 60 (R) | |
| 10 | 2d | 600 | 16 | >99 | 66 (R) |
| 11c | 3g | 100 | 14 | >99 | 83 (R) |
For comparison, we tested catalyst precursors bearing PEt2 (3b) and PCy2 (3c) groups, although they were significantly less enantioselective than 3a (entries 5, 6). On the other hand, binaphthyl based complex 2d provided 80% ee and a good activity (entry 7). Upon recycling, activity was maintained while a severe drop in enantioselectivity was observed in the second and third cycles (entries 8 and 9). This catalyst provided full conversion and a significant decrease in enantioselectivity at a lower catalyst loading (66% ee, S/C = 600, entry 10). These results seem to indicate that a slight degradation of the catalyst generated from 2d in methanol occurs, leading to a species featured by a less effective asymmetric transfer. Finally, a good activity and a moderate enantioselectivity were obtained with ethane bridged complex 3g (83% ee, entry 11).
After initial experiments, we turned our attention to the hydrogenation of methyl α-acetamido cinnamate (4b) as a representative substrate for the synthesis of important β-aryl-α-amino-acid derivatives.25 Initially, low conversion (40%) and good enantioselectivity (94% ee, entry 1, Table 2) were obtained with complex 3a. In contrast, catalyst precursor 2d showed full conversion, but rather low enantioselectivity (43% ee, entry 2). Moreover, this catalyst provided high conversion (84%) but an even lower enantioselectivity in a reaction performed at S/C = 600 (entry 3). Remarkably, no catalytic activity of the supernatant solution was observed when it was transferred to another reactor under hydrogen.26 It should be noticed, in addition, that a significant Rh leaching has been observed in reactions performed in MeOH (see the ESI† for details). Thus, for reactions using 2d, 15 and 18% were observed in the hydrogenations of 4a (entry 7, Table 1) and 4b (entry 2, Table 2), respectively. Moreover, in reactions with 3a a moderate leaching was also observed. Thus, in the hydrogenation of 4a (entry 1, Table 1) and 4b (entry 1, Table 2), Rh leaching values of 12 and 9% were observed, respectively. These values are higher than those reported for supported catalysts based on diphosphine and diphosphinite ligands21 and may be connected to some degradation of the phosphite fragment in methanol under hydrogenation conditions.
| Entry | Cat. | S/C | Timeb | % Conv. | % ee (Conf.) |
|---|---|---|---|---|---|
| a Reactions were performed using the suitable amounts of 4b and the resin supported complex under 4 bar H2 at room temperature in MeOH (1 mL). Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral HPLC. Product configuration was assigned by comparison with the literature data. b Reaction time in hours. | |||||
| 1 | 3a | 70 | 14 | 40 | 94 (R) |
| 2 | 2d | 60 | 14 | >99 | 43 (R) |
| 3 | 2d | 600 | 16 | 84 | 33 (R) |
| 4 | 3e | 50 | 14 | >99 | 84 (R) |
Study of the model hydrogenation of 4a in water
The practical application of the present supported catalysts in methanol is strongly limited by the high Rh leaching observed and prompted us to find a more robust catalytic system. In this context, the study by Sheldon and coworkers highlighted above also reported a high metal leaching in hydrogenations of 4a in methanol with a Rh-phosphoramidite catalyst supported on Nafion. Moreover, this catalyst provided a significantly lower leaching and a better enantioselectivity in water.19g Following this precedent, we examined the behavior of catalyst precursors 2–3 in water. Initially, hydrogenation of water soluble 4a with catalyst precursor 2d at S/C = 60 showed full conversion and 84% ee (entry 1, Table 3). It is worth noting that this catalyst exhibited a good performance on recycling as complete reactions and constant enantioselectivities were obtained in the second and third cycles (entries 2 and 3). These observations contrast with the decrease in enantioselectivity observed in recycling experiments in MeOH and therefore suggest a higher stability in water of catalyst generated from 2d. Moreover, this catalyst exhibited enough activity to finish in 24 h a reaction performed at S/C = 600, with a moderate enantioselectivity (84% ee, entry 4). Moreover, the reaction was also complete at S/C = 1800, but a significant decrease in enantioselectivity was observed (77% ee; entry 5). These results clearly show a better performance of 2d in water than in MeOH. On the other hand, 3a showed an outstanding enantioselectivity in the hydrogenation of 4a. Thus, for a reaction performed at S/C = 160, 91% conversion and 99% ee were obtained (entry 6). A good indication of the beneficial effect of supporting the catalyst is demonstrated by the relatively poor performance of [Rh(NBD)(1a)](BF4) in the hydrogenation of 4a (S/C = 100, 4 bar H2, room temperature), which after 24 h showed 90% conversion and 65% ee. In addition, a satisfactory behavior upon recycling was observed for 3a, affording high conversion and enantioselectivity (91–96% ee, entries 7–9). Interestingly, a lower enantioselectivity was observed in the first cycle. This phenomenon has already been reported and appears to be related to a lower enantioselectivity in the initial step of the reaction or during the activation process.21a Moreover, a reaction performed at S/C = 2300 showed 80% conversion and 99% ee after 24 h, while full conversion and 99% ee were observed after 72 h (entry 10). On the other hand, as observed in MeOH reactions, the substitution of the PPh2 group by a PCy2 fragment has a detrimental effect. Thus, catalyst precursor 3c provided 88% conversion and a low enantioselectivity (47% ee, entry 11), well below results provided by 3a.| Entry | Cat. | S/C | Timeb | % Conv. | % ee (Conf.) |
|---|---|---|---|---|---|
| a Reactions were performed at room temperature in water (3 mL) using the resin supported catalyst and the suitable amount of 4a under 4 bar H2 unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral GC. b Reaction time in hours. In recycling experiments, the number of the cycle is stated in brackets. c Reaction performed in 1 mL of water. | |||||
| 1 | 2d | 60 | 2 (1st cycle) | >99 | 84 (R) |
| 2 | 60 | 2 (2nd cycle) | >99 | 83 (R) | |
| 3 | 60 | 2 (3rd cycle) | >99 | 83 (R) | |
| 4 | 2d | 600 | 16 | >99 | 84 (R) |
| 5 | 2d | 1800 | 24 | >99 | 77 (R) |
| 6 | 3a | 160 | 2 | 91 | 99 (R) |
| 7 | 3a | 80 | 2 (1st cycle) | >99 | 91 (R) |
| 8 | 3 (2nd cycle) | >99 | 95 (R) | ||
| 9 | 4 (3rd cycle) | >99 | 96 (R) | ||
| 10 | 3a | 2300 | 24 | 80 | 99 (R) |
| 72 | >99 | 99 (R) | |||
| 11 | 3c | 70 | 2.5 | 88 | 47 (R) |
| 12c | 3e | 100 | 24 | >99 | 97 (R) |
| 13c | 3e | 500 | 24 | >99 | 96 (R) |
| 14c | 3e | 1500 | 24 | >99 | 93 (R) |
| 15 | 3e | 50 | 2 (1st cycle) | 98 | 89 (R) |
| 16 | 50 | 2 (2nd cycle) | >99 | 94 (R) | |
| 17 | 50 | 2 (3rd cycle) | >99 | 95 (R) | |
| 18c | 3f | 1500 | 19 | >99 | 88 (R) |
| 19c | 3g | 70 | 24 | >99 | 89 (R) |
Following the dissimilar performance of 2d and 3a, we rationalized that the use of a phosphite fragment with intermediate steric properties could provide a better catalyst. Complex 3e, characterized by a binaphthyl fragment with methyl substituents at positions 3 and 3′, was accordingly prepared and tested. We were delighted to observe that the corresponding catalyst showed both good activity and enantioselectivity. Thus, it finished in 24 h a reaction performed at S/C = 100 with 97% ee (entry 12). Moreover, this catalyst exhibited enough activity to complete reactions performed at S/C ratios of 500 and 1500 with 96 and 93% ee (entries 13 and 14), respectively. Most remarkably, very low Rh leaching (lower than 0.8%) was observed in both reactions. In addition, this catalyst maintained a good performance upon recycling. Thus, full conversion was obtained in three cycles while the enantioselectivity observed in the first cycle (89% ee, entry 15) improved up to 94 and 95% ee in the second and third cycles, respectively (entries 16 and 17). It is also interesting to note that 3e displayed a significantly better performance in water than in MeOH (entry 4, Table 2). On the other hand, complex 3f, characterized by a H8-binaphthyl fragment, also exhibited a high reactivity and was able to complete a reaction at S/C = 1500. However, enantioselectivity for this reaction (88% ee, entry 18) was slightly lower than that provided by 3e. Finally, complex 3g, possessing an ethane bridged ligand, also provided full conversion and a good enantioselectivity (89% ee, entry 19). It did not improve, however, the value provided by the corresponding benzene bridged complex 3a.
Hydrogenations of β-aryl-dehydroaminoacids in water
The satisfactory results obtained with 4a committed us to study the more challenging hydrogenation of 4b in water. This reaction is hampered by the low solubility of this compound in water (7.3 × 10−3 mol L−1 at 25 °C),27 as substrate amounts typically used in the present hydrogenations (0.06–0.3 mol L−1, for S/C = 100–500) are well above this solubility value. As an alternative to the substrate slurry, we have initially tested a mixture of water and environmentally friendly 2-Me-thf (9:1 v/v, reaction conditions A), which is able to dissolve the required amount of 4b. Hydrogenation of the resulting solution with catalyst precursor 2d provided full conversion for a reaction performed at S/C = 120 with 88% ee (entry 1, Table 4). On the other hand, precatalyst 3a showed a very slow reaction under these conditions, with a conversion lower than 10% in 14 h (entry 2). In contrast, 3e provided full conversion and a remarkable 96% ee (entry 3). Moreover, an increase in the reaction temperature up to 40 °C only caused a small decrease in enantioselectivity (94% ee, entry 4).
| Entry | Cond.b | Cat. | S/C | Time | % Conv. | % ee (Conf.) |
|---|---|---|---|---|---|---|
|
a Reactions were performed at room temperature using the resin supported catalyst and the suitable amount of 4b in the corresponding solvent (2 mL) under 4 bar H2 unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral HPLC.
b Conditions A: reactions performed in H2O/2-Me-thf (9:1 v/v); conditions B: Triton-X100 added, % mol relative to the substrate in brackets.
c Reaction at 40 °C.
|
||||||
| 1 | A | 2d | 120 | 14 | >99 | 88 (R) |
| 2 | A | 3a | 160 | 14 | 10 | n.d. |
| 3 | A | 3e | 100 | 18 | >99 | 96 (R) |
| 4c | A | 3e | 100 | 18 | >99 | 94 (R) |
| 5 | B (10%) | 3a | 160 | 40 | 30 | 88 (R) |
| 6 | B (10%) | 3e | 100 | 18 | >99 | 96 (R) |
| 7 | B (10%) | 3e | 200 | 18 | >99 | 97 (R) |
| 8 | B (5%) | 3e | 200 | 18 | >99 | 96 (R) |
| 9c | B (5%) | 3e | 200 | 18 | >99 | 95 (R) |
| 10c | B (2.5%) | 3e | 500 | 24 | >99 | 93 (R) |
As an additional approach to facilitate the hydrogenation of 4b in water, we have considered the addition of a surfactant to disperse the substrate in the reaction media. In order not to interfere in the electrostatic catalyst immobilization, a polar surfactant instead of an ionic one was preferred. Thus, a set of reactions were performed by adding Triton-X100 to the reaction medium (reaction conditions B).28 Initially, a reaction under these conditions using catalyst precursor 3a and 10 mol% of surfactant relative to the substrate showed a very slow reaction and a low conversion was observed after a prolonged reaction time (30%, entry 5). Alternatively, the use of complex 3e under these reaction conditions led to complete conversion and a high enantioselectivity (96% ee, entry 6). Moreover, a similar performance was observed at an S/C ratio of 200 (97% ee, entry 7). In addition, a decrease by half of the amount of surfactant did not affect catalyst performance appreciably (entry 8). Moreover, an increase in temperature up to 40 °C only produces a small decrease in enantioselectivity (95% ee, entry 9). A further reduction of the amount of surfactant to 2.5 mol % and of the catalyst loading (S/C = 500) also showed full conversion, while enantioselectivity was reduced slightly (93% ee, entry 10).
Considering the high water content of the swollen resin, results obtained in the hydrogenation of 4b with catalyst precursor 3e under conditions A and B indicate that the supported catalyst is able to provide good catalyst activity and enantioselectivity for this substrate in an essentially aqueous reaction medium. Accordingly, we rationalized that if the substrate has some solubility in water, an effective hydrogenation of this substrate could then be effected in neat water. Thus, we next explored the hydrogenation of a slurry of 4b.6 Initially, complex 3a only provided low conversion at room temperature (8%), although the corresponding catalyst afforded very high enantioselectivity (98% ee, entry 1, Table 5). In order to increase conversion, a reaction at 40 °C was also explored. This provided a moderate conversion (44%, entry 2) and a high enantioselectivity (98% ee). An alternative experiment performed at 40 °C under 20 bar H2 showed that the reaction was complete, although a decrease in enantioselectivity was observed (92% ee, entry 3). In contrast, catalysts formed from 2d and 3e were more active and they completed the reaction in 14 h at room temperature under 4 bar H2 (entries 4, 5). It is worth noting that 3e provided a remarkable enantioselectivity of 97% ee. Also it is pertinent to note that an increase in the reaction temperature up to 40 °C had a negligible effect on enantioselectivity (entry 6). Under these conditions, complex 3f also provided a high catalyst activity and a good enantioselectivity (91% ee, entry 7), while 3h offered high conversion (90%) and an outstanding enantioselectivity (97% ee, entry 8).
| Entry | Cat. | S/C | Time | % Conv. | % ee |
|---|---|---|---|---|---|
| a Reactions were performed at room temperature using the resin supported catalyst and a slurry of 4b in water (1 mL) under 4 bar H2 unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral HPLC. b Reaction at 40 °C. c Reaction at 20 bar H2. d Slurry in 6 mL of water. e Reaction at 50 °C. | |||||
| 1 | 3a | 160 | 14 | 8 | 98 (R) |
| 2b | 3a | 160 | 24 | 44 | 98 (R) |
| 3b,c | 3a | 160 | 24 | >99 | 92 (R) |
| 4 | 2d | 120 | 14 | >99 | 89 (R) |
| 5 | 3e | 100 | 14 | >99 | 97 (R) |
| 6b | 3e | 100 | 24 | >99 | 96 (R) |
| 7b | 3f | 100 | 24 | >99 | 91 (R) |
| 8b | 3h | 130 | 24 | 90 | 97 (R) |
| 9d | 3e | 500 | 24 | 75 | 97 (R) |
| 10b,d | 3e | 500 | 24 | 93 | 96 (R) |
| 11d,e | 3e | 500 | 24 | >99 | 95 (R) |
Among these catalysts, 3e displayed a suitable performance. Initially, a reaction performed at room temperature and an S/C ratio of 500 showed high enantioselectivity (97% ee), although the conversion was only moderate (75%, entry 9). However, raising the reaction temperature to 40 °C significantly increased conversion to 93%, without any important effect on enantioselectivity (entry 10). Finally, full conversion and a remarkable enantioselectivity of 95% ee were obtained at 50 °C (entry 11).
To the best of our knowledge, the results presented in Table 5 provide the highest enantioselectivities reported to date in the hydrogenation of 4b in neat water. Most interestingly, apart from the practical advantages associated with catalyst heterogenization, the support also has a remarkable effect on catalyst performance. Thus, the hydrogenation of 4b in H2O using [Rh(NBD)(1e)](BF4) (S/C = 200, 4 bar H2, 40 °C) showed 40% conversion and 32% ee after 24 h. These values indicate lower catalyst activity and enantioselectivity than those provided by the corresponding supported catalyst 3e.
An analysis of the Rh leaching in hydrogenations of 4b in water with complex 3e indicates generally low values. Thus, for type A reaction conditions, the Rh concentration was lower than 0.2 ppm, which corresponds to a leaching lower than 0.9% (entry 3, Table 4). This value was however increased up to 2.4% in the reaction performed at 40 °C (entry 4). On the other hand, the use of 10 mol% surfactant under conditions of type B also showed values lower than 0.9% for reactions performed at room temperature (entries 6 and 7). In contrast, the metal leaching significantly increased up to 2.0% for the reaction heated at 40 °C (entry 9). Most remarkably, reactions performed in neat water at 40 °C exhibited a very low metal leaching. Thus, values lower than 0.5% were observed for reactions performed at S/C ratios of 100 and 500 (entries 6 and 10, Table 5). Likewise, the Rh leaching also remained lower than 0.5% for the reaction performed at 50 °C (entry 11).
It is also worth commenting that a comparison of results of hydrogenations of 4a and 4b indicates that both the nature of phosphine and phosphite fragments have a strong impact on the catalytic hydrogenation. On the one hand, catalysts bearing a PPh2 fragment provided better results than those bearing dialkylphosphino groups. On the other hand, substitution of the phosphite biphenyl fragment is required for the attainment of high enantioselectivity. Moreover, the size of these substituents also has an impact on catalyst activity. Thus, t-Bu substituted 3a provided the best enantioselectivities, but it displayed a lower catalyst activity. In contrast, less sterically hindered 2d and 3f provided higher catalyst activity but lower enantioselectivities. Among them, complex 3e, which possesses phosphite steric properties between the two prior limiting cases, compromises both suitable catalyst activity and good enantioselectivity.
Having achieved good results in the hydrogenation of representative substrate 4b in water, we investigated the asymmetric hydrogenation of related substrates 4c–4j. However, to broaden the scope of the present catalytic system is not trivial, as the introduction of aryl substituents further reduces substrate solubility in water, ranging between 1.4 × 10−3 mol L−1 (4j) and 3.9 × 10−3 mol L−1 (4g) at 25 °C.27 Initially, hydrogenation of the p-Me enamide 4c using precatalyst 3e at 40 °C provided full conversion and 94% ee (entry 1, Table 6), although the catalyst was not reactive enough to complete a reaction at S/C = 250 (entry 2). On the other hand, the corresponding m-Me substrate 4d produced a complete reaction with 96% ee (entry 3). In addition, the p-MeO substrate 4e provided full conversion and 95% ee (entry 4). An increase to S/C = 250 also showed a complete reaction, while the enantioselectivity decreased down to 90% ee (entry 5). On the other hand the m-MeO enamide 4f also yielded a complete reaction and 97% ee under our standard conditions (entry 6). In contrast to the above results, dimethoxy substrate 4g was less reactive and only 60% conversion was observed, although it provided a high enantioselectivity (96% ee, entry 7). However, an increase in reaction temperature up to 50 °C yielded full conversion without erosion on enantioselectivity (97% ee, entry 8). Moreover, p-F substrate 4h also provided good results, with full conversion and 96% ee under our standard conditions (entry 9). It is also pertinent to comment on the visual changes produced by the reaction. Thus, hydrogenations providing low conversion clearly show the presence of the remaining solid substrate (Fig. 1a). In reactions with high conversion, disappearance of the suspended solid is observed and product 5 can generate a new oily phase over the aqueous one (Fig. 1b and c), or apparently be dissolved in the aquatic phase (Fig. 1d and e).
 | ||
| Fig. 1 Photographs of vials corresponding to hydrogenations of diverse substrates 4 performed with complex 3e (4 bar H2, 40 °C, 24 h). (a) 4d (S/C = 250; 40% conv.); (b) 4d (S/C = 100; >99% conv.); (c) 4e (S/C = 250; >99% conv.); (d) 4b (S/C = 100; >99% conv.); (e) 4a (S/C = 100; >99% conv.). | ||
| Entry | Subs. | Cat. | S/C | Timeb (h) | % Conv. | % ee |
|---|---|---|---|---|---|---|
|
a Reactions were performed at 40 °C using the resin supported catalyst and a slurry of substrate in water (1 mL) under 4 bar H2 unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral HPLC.
b In recycling experiments, the number of the cycle in brackets.
c Reaction at 50 °C.
d Mixture of 5i:5a products in a 50:50 ratio obtained.
e Mixture of 5j:5a products in an 80:20 ratio obtained.
f Mixture of 5i:5a products in an 80:20 ratio obtained.
g Mixture of 5j:5a products in an 85:15 ratio obtained.
h Slurry in 6 mL of water.
|
||||||
| 1 | 4c | 3e | 100 | 24 | >99 | 94 |
| 2 | 4c | 3e | 250 | 24 | 40 | 94 |
| 3 | 4d | 3e | 100 | 24 | >99 | 96 |
| 4 | 4e | 3e | 100 | 24 | >99 | 95 |
| 5 | 4e | 3e | 250 | 24 | >99 | 90 |
| 6 | 4f | 3e | 100 | 24 | >99 | 97 |
| 7 | 4g | 3e | 100 | 24 | 60 | 96 |
| 8c | 4g | 3e | 100 | 24 | >99 | 97 |
| 9 | 4h | 3e | 100 | 24 | >99 | 96 |
| 10d | 4i | 3e | 100 | 24 | 80 | 87 |
| 11e | 4j | 3e | 100 | 24 | 75 | 88 |
| 12f | 4i | 3f | 100 | 24 | 85 | 85 |
| 13g | 4j | 3f | 100 | 24 | 95 | 84 |
| 14 | 4c | 3h | 130 | 24 | 50 | 96 |
| 15 | 4d | 3h | 130 | 24 | 90 | 95 |
| 16 | 4e | 3h | 130 | 24 | 55 | 96 |
| 17 | 4f | 3h | 130 | 24 | 65 | 96 |
| 18 | 4g | 3h | 130 | 24 | 15 | 86 |
| 19 | 4h | 3h | 130 | 24 | 80 | 94 |
| 20 | 4i | 3h | 130 | 24 | 25 | 98 |
| 21 | 4j | 3h | 130 | 24 | 50 | 94 |
| 22h | 4f | 3e | 50 | 12 (1st cycle) | >99 | 97 |
| 23h | 50 | 12 (2nd cycle) | >99 | 97 | ||
| 24h | 50 | 12 (3rd cycle) | 60 | 86 | ||
| 25h | 50 | 12 (4th cycle) | 5 | n.d. | ||
In contrast with the previous reactions, p-Br substrate 4i showed a different reactivity than previous enamides. Thus, a hydrogenation performed under our standard conditions afforded a product mixture (80% conv.) composed of desired product 5i and debrominated product 5b in a 50:50 ratio (entry 10). On the other hand, m-Br substrate 4j showed a cleaner reaction towards the desired hydrogenated product 5j, although it also showed an appreciable formation of 5b in the reaction. Thus, the 5j:5b ratio was 80:20 (75% conversion, entry 11). Pursuing more selective reactions with Br substituted substrates, complex 3f was also tested. The corresponding catalyst afforded higher conversion and selectivity, providing 80% of 5i and 85% of 5j, respectively, in the corresponding products. Moreover, enantioselectivities were only slightly lower than those provided by 3e and values of 85 and 84% ee were obtained for 5i and 5j, respectively (entries 12 and 13).
In order to provide additional information about the unexpected debromination of 4i some additional experiments were also performed. We first examined the behavior of 4i under standard homogeneous hydrogenation conditions. Thus, complete conversion of 4i into 5i (99% ee, S/C = 100, 24 h) was observed in a reaction performed with unsupported [Rh(NBD)(1e)](BF4) in CH2Cl2. Moreover, formation of 5a was not observed in the hydrogenation of 4i in MeOH using complex 3e (>99% conversion, 59% ee, 14 h). It seems therefore that the interplay of water and the supported catalyst is required for debromination. Moreover, we have observed that 5i can undergo the debromination reaction under hydrogenation conditions. Thus, when a solution of 5i was exposed to 4 bar H2 in the presence of 3e, a mixture of 5a and 5i in a 15:85 ratio was observed after 14 h at 40 °C. As expected, this reaction was not observed when 3e was substituted by the complex-free Li-resin. It is worth noting that along with the debromination we have observed a significant Li leaching of the resin, while Rh leaching remained lower than 0.5%. Thus, for hydrogenations of 4i and 4j (0.06 mmol of substrate in 1 mL of water) using complex 3e (entries 10, 12), values of [Li] = 3.9 × 10−3 M (22% Li leaching) and [Li] = 3.6 × 10−3 M (20% Li leaching) were obtained, respectively. As a reference value, [Li] = 1.1 × 10−3 M (6% Li leaching) was observed after the hydrogenation of 4b under identical reaction conditions. In contrast, in the hydrogenation of 4i with 3e in MeOH, a negligible Li leaching was observed (0.1%). We speculated that this leaching could be favored by the formation of HBr under hydrogenation conditions,29,30 which should result in the generation of 5b from 5i (or of 4b from 4i).31 Thus, the presence of an acid in the solution may produce some Li/H exchange in the resin, due to the low selectivity coefficient of Li cations on polystyrene sulphonated resin.32 According to this, treatment of Li-resin (5.0 mg) with solutions of HBr (1.0 mL) at 40 °C for 24 h showed a higher Li concentration when the concentration of the acid was raised. Thus, when the latter grew from 0.006 M to 0.058 M, an increase in [Li] from 2.2 × 10−3 M to 7.3 × 10−3 M was observed.
Besides these experiments, due to the high enantioselectivity displayed in the hydrogenation of 4b, we were also interested in examining the performance of complex 3h in the hydrogenation of substrates 4c–4j. Then, a set of reactions under standard conditions were performed (entries 14–21). The enantioselectivities observed were generally high, between 94 and 98% ee, with the exception of dimethoxy substituted substrate 4g, for which a very low conversion and a value of 86% ee were obtained (entry 18). Despite the high enantioselectivities, the catalytic activity shown by complex 3h was however lower than that provided by 3e.
Finally we attempted to perform catalyst recycling in water with complex 3e. In this regard it should be recalled that due to their low solubility, solid substrates 4b–4j cannot be transferred as water solutions at the required concentration for the hydrogenations. Moreover, the present hydrogenation catalysts are very reactive towards oxygen; therefore recycling operations should be made under an inert atmosphere or preferably under hydrogen. Upon these considerations we devised a simple procedure to add solid substrates to the reactor inside a piece of Teflon tubing (see the ESI†). To test this procedure, we examined the hydrogenation of 4f. Results obtained indicated full conversion and 97% ee in the first and second cycles (entries 22, 23), while, unfortunately, a rapid degradation in catalyst performance was observed in the third and fourth cycles (entries 24, 25). It is worth noting that the Rh concentration in solution remained lower than 0.2 ppm (Rh leaching lower than 2%) in all cycles. We therefore speculated that the loss of catalyst performance observed may be associated with the introduction of oxygen traces upon recycling. Therefore, an improved reactor design is required to implement an efficient catalyst recycling of the present catalysts in water.
Mechanistic studies
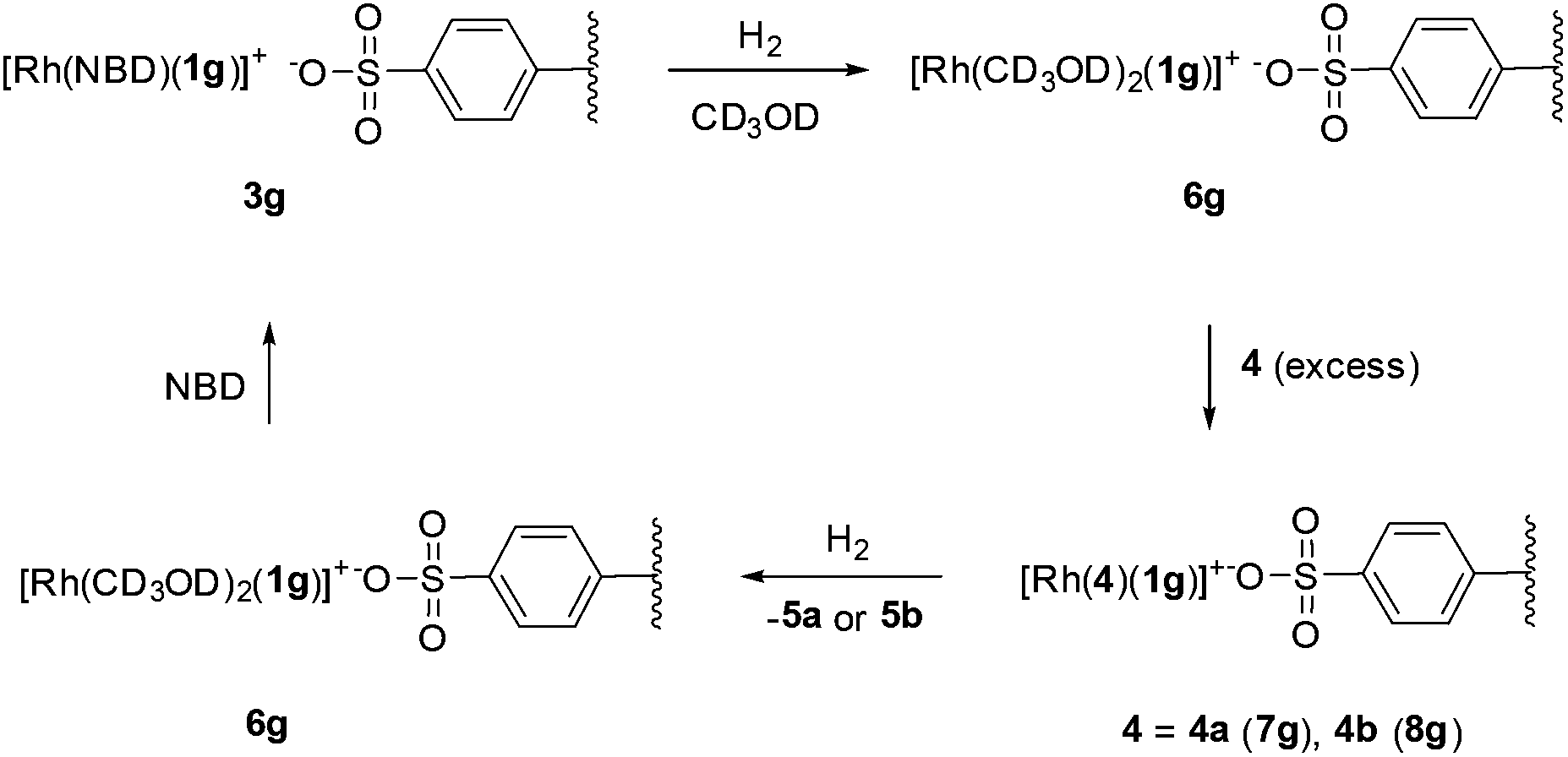
In order to provide additional information, we have monitored by gel phase 31P{1H} NMR the progress of a selected hydrogenation.33 Initially, we observed that complex 3g provided a spectrum of satisfactory quality in CD3OD. In this spectrum the typical pattern for a phosphine–phosphite ligand coordinated to a rhodium atom is observed (Fig. 2a). The phosphite group appears as a doublet of doublets centered at 123.6 ppm (1JRhP = 259 Hz, 2JPP = 69 Hz), while for the phosphine, the corresponding signal appears as a doublet of doublets centered at 7.4 ppm (1JRhP = 147 Hz). It is worth noting that a spectrum of the methanol supernatant only showed low intense singlets assigned to phosphorus impurities after standing 3 days at room temperature, indicating the stability of the catalyst precursor under these conditions. As expected, these data closely match the data for the corresponding soluble [Rh(NBD)(1g)](BF4) in CD3OD [δPO = 123.1 (dd, J(P, Rh) = 257 Hz, J(P, P) = 68 Hz, PO), δPC = 8.7 (dd, J(P, Rh) = 147 Hz].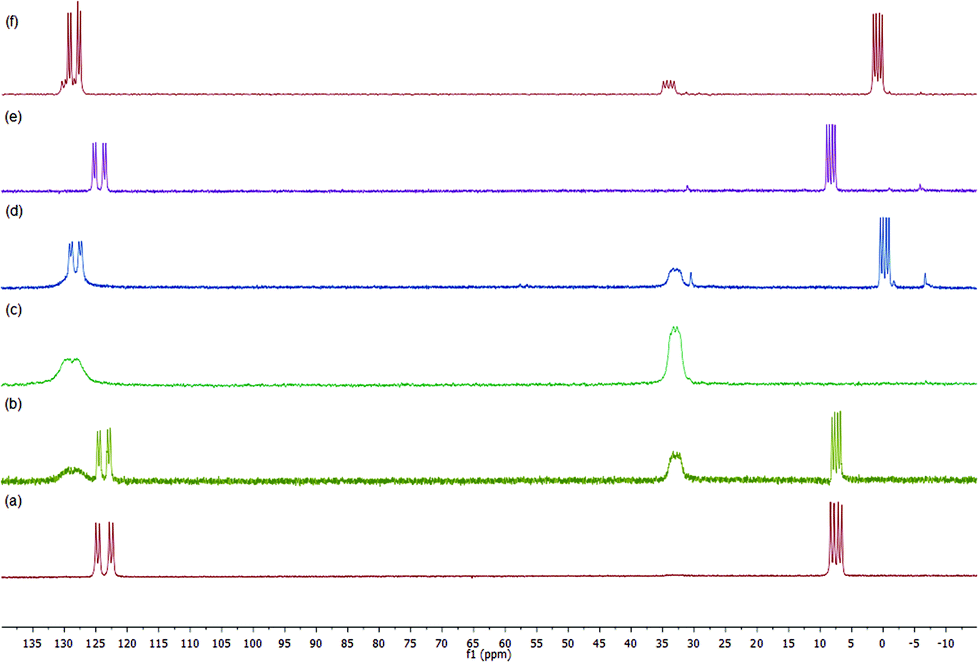 | ||
| Fig. 2 Gel-phase 31P{1H} NMR spectra in CD3OD of the following samples: (a) complex 3g; (b) after pressurizing with 4 bar H2; (c) 0.5 h later; (d) after addition of 12 equivalents of 4b; (e) after addition of 12 equivalents of NBD; (f) 31P{1H} NMR spectrum of the sample obtained after hydrogenation of [Rh(NBD)(1g)](BF4) in CD3OD followed by the addition of 20 equivalents of 4b. | ||
When the sample of 3g was pressurized with 4 bar of H2, a fast reaction was detected and immediate disappearance of 3g was observed along with the formation of new species 6g (Scheme 4). The latter is characterized in the 31P{1H} NMR by two multiplets centred at 127.7 and 33.0 ppm (Fig. 2b), corresponding to phosphite and phosphine groups, respectively (Fig. 2c). Remarkably, no signals were detected in the hydride region of the 1H NMR experiment. By comparison with the spectrum obtained after hydrogenation of [Rh(NBD)(1f)](BF4) in CD3OD, 6g can be assigned to the supported methanol solvate. As a next step we investigated the formation of complexes of enamides 4. Thus, addition of 3 equivalents of 4b to 6g showed a mixture of the latter and new species 8g in ca. 3:1 ratio.34 A further addition of 4b up to 12 equivalents increased the amount of 8g and a 0.7:1 6g:8g ratio was then observed (Fig. 2d). Complex 8g is characterized by a doublet of doublets in the coordinated phosphine region centered at 0.3 ppm (1JRhP = 152 Hz, 2JPP = 70 Hz), while in the phosphite region signals corresponding to 6g and 8g overlap in a broad doublet of doublets centred at 128.2 ppm (1JRhP = 246 Hz, 2JPP = 70 Hz). Data for 8g are in good accord with those of soluble enamide adducts prepared in our laboratory.23c For further comparison, a sample of [Rh(CD3OD)2(1g)](BF4) was treated with 20 equivalents of 4b. This showed a mixture of the deuteromethanol adduct and [Rh(4b)(1g)](BF4) in a 1:3 ratio. The latter species was characterized by phosphine and phosphite signals at δ = 0.8 (1JRhP = 151 Hz, 2JPP = 70 Hz) and δ = 128.4 (1JRhP = 243 Hz, 2JPP = 70 Hz), respectively (Fig. 2f). Thus, an analogous behavior is observed for the immobilized and the homogeneous systems. On the other hand, an addition of 3 equiv. of 4a to 6g showed nearly full displacement of CD3OD by the enamide. A subsequent increase of the amount of 4a up to 12 equiv. showed full conversion into the corresponding enamide adduct 7g. This compound shows rather similar data to 8g. Thus, 7g is characterized by two doublet of doublets centered at δ = 2.3 (1JRhP = 152 Hz, 2JPP = 70 Hz) and a broad doublet of doublets at δ = 127.5 with 1JRhP and 2JPP roughly in the range of 230 and 70 Hz, respectively. Moreover, exposing the latter sample to 4 bar H2 only showed the presence of the supported deuteromethanol adduct 6g, while no intermediates in the catalytic cycle were observed. In connection with this, Reek and coworkers reported a similar observation in studies with Rh complexes bearing Indolphos ligands, which possess similar electronic properties to P–OP.35 As a final remark, the high coordinating ability of NBD in the present system should be stressed. Thus, addition of an excess of NBD (12 equivalents) to the 6g:8g mixture cleanly regenerated 3g (Fig. 2e). A similar observation was observed in the case of 7g.
 | ||
| Scheme 4 | ||
The NMR study described above indicates a close similarity between the homogeneous and the resin immobilized catalytic hydrogenation systems. It should be recalled, in connection with this, that a product labelling at position α in the hydrogenation of 4a and 4b in D2O using water soluble complexes has been described in the literature.10b,d This selective labeling has been proposed to result from an exchange between the hydrido-alkyl intermediate and D2O, leading to the corresponding Rh deuteride (Scheme 5).
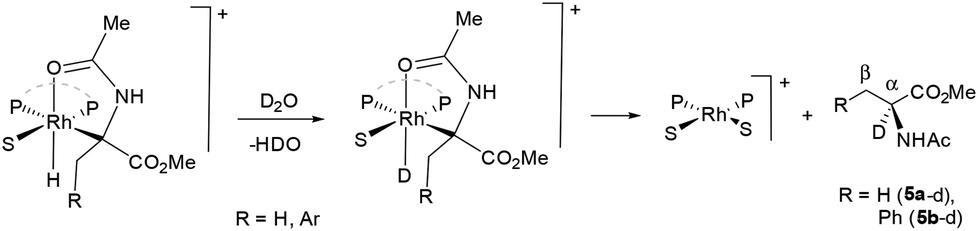 | ||
| Scheme 5 Proposed formation of deuterated 5a–d and 5b–d. | ||
With the intention of investigating whether such effect operates in the present system, we have performed some hydrogenations of 4b in D2O. Thus, the reaction with complex 2d exhibited a deuterium labelling of 67% at position α (entry 1, Table 7), while 9% D was observed at position β. On the other hand, the reaction performed with complex 3e showed 40% D at position α, while no labelling was observed at position β (entry 3). Finally, a smaller labelling (5%) was observed in the reaction performed with complex 3a (entry 2). Likewise, in hydrogenations of 4a, a low labelling (4%, entry 4) was observed in the reaction using 3a, while the corresponding hydrogenation using 2d as a catalyst precursor showed a significantly higher value of deuterium incorporation at position α (75%, entry 5).
| Entry | Subst. | Cat. | S/C | % D (α) | % Conv. | % eeb |
|---|---|---|---|---|---|---|
| a Conditions: reactions performed at 40 °C in D2O for 24 h unless otherwise stated. Conversion was determined by 1H NMR. Enantiomeric excess analyzed by chiral GC or HPLC. b Enantioselectivity of the corresponding reaction in H2O in brackets. c Reaction performed at room temperature. | ||||||
| 1 | 4b | 2d | 120 | 67 | 95 | 85 (83) |
| 2 | 4b | 3a | 160 | 5 | 40 | 97 (98) |
| 3 | 4b | 3e | 100 | 40 | >99 | 96 (97) |
| 4c | 4a | 3a | 500 | 4 | 80 | 99 (99) |
| 5c | 4a | 2d | 500 | 75 | >99 | 83 (84) |
These results outline a similar behavior to that reported for water soluble catalysts. Moreover, in good accord with the proposed labelling mechanism involving exchange between the alkyl-hydride intermediate and water (i.e. after the generation of the stereogenic center), the enantioselectivities obtained in these reactions are very close to those obtained in H2O. Finally, a dependence of labelling with the steric properties of the ligand, being the labelling higher with less sterically encumbered ligands, has been observed. In this regard, a slower reaction with D2O due to steric hindrance caused by the t-Bu substituents may tentatively be proposed to explain the results.
Conclusions
In this contribution we present a convenient immobilization of rhodium phosphine–phosphite catalysts on an ionic resin. The procedure is simple, as it does not require chemical modification of the readily accessible cationic rhodium complexes and provides the supported complexes in moderate to high yield. The corresponding catalysts show a good performance in the hydrogenation of 4a in MeOH, with enantioselectivities up to 99% ee. On the other hand, no catalyst was found to provide both high activity and enantioselectivity in the hydrogenation of 4b in MeOH. Moreover, a high metal leaching was observed in these reactions. In contrast, a suitable catalyst (3e) for the hydrogenation of 4b in water, providing good activity and an enantioselectivity up to 97% ee, was found following a rational approach. For this substrate, extensively studied in the literature, this is the highest value obtained in neat water. This catalyst has a broader scope and provides both good activity and enantioselectivity, between 87 and 97% ee, in the hydrogenation of several β-aryl dehydroaminoacids (4c–4j). Thus, besides the general advantage of an easier catalyst separation provided by immobilization, the high swelling of the resin supporting the catalyst enables to perform efficient hydrogenations in water in the present case. Moreover, the hydrophilic support significantly enhances the performance of the catalyst in comparison with that observed by the corresponding tetrafluoroborates in water. On the other hand, in the hydrogenation of brominated substrates 4i and 4j an unexpected formation of debrominated 5b was observed as a side reaction. This phenomenon seems associated with the use of both water and the Li-resin. In addition, NMR studies show a similar behavior of the supported catalysts to the homogeneous one. Likewise, a similar deuterium labelling to that reported in the literature for homogeneous hydrogenation of 4a in water has also been observed in the present system.Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme through the Marie Curie Initial Training Network NANO-HOST (grant agreement no. 215193), Junta de Andalucía (2009/FQM-4832) and CSIC (grant 201480E031). We thank Carmen Moreno-Marrodán and Francesca Liguori for technical assistance. We also gratefully acknowledge the Microanalysis and Mass Spectrometry Services of Universidad de Sevilla (CITIUS) for the analytical and mass spectrometry determinations, respectively. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2010, 111, 1713 CrossRef PubMed; (c) D. J. Ager, A. H. M. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340 RSC.
- For some recent examples, see: (a) T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754 CrossRef CAS PubMed; (b) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235 CrossRef CAS PubMed; (c) K. Dong, Y. Li, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 14191 CrossRef CAS PubMed; (d) W. Chen, F. Spindler, B. Pugin and U. Nettekoven, Angew. Chem., Int. Ed., 2013, 52, 8652 CrossRef CAS PubMed; (e) J. Jiang, Y. Wang and X. Zhang, ACS Catal., 2014, 4, 1570 CrossRef CAS.
- (a) H.-U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005, 231, 1 CrossRef CAS PubMed; (b) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed; (c) C. S. Shultz and S. W. Krska, Acc. Chem. Res., 2007, 40, 1320 CrossRef CAS PubMed; (d) P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728 RSC.
- (a) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC; (b) R. A. Sheldon, Chem. Commun., 2008, 3352 RSC; (c) B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952 CrossRef CAS PubMed; (d) R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1479 CrossRef CAS.
- For reviews on this topic, see: (a) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef PubMed; (b) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed; (c) E. Wiebus and B. Cornils, inOrganic Reactions in Water, Blackwell Publishing Ltd, 2007, pp. 366–397 Search PubMed; (d) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (e) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- For examples of slurry hydrogenations in water, see: (a) I. Tóth, B. Hanson and M. Davis, Catal. Lett., 1990, 5, 183 CrossRef; (b) T. Malmström and C. Andersson, J. Mol. Catal. A: Chem., 1999, 139, 259 CrossRef.
- (a) I. Tóth, B. E. Hanson and M. E. Davis, Tetrahedron: Asymmetry, 1990, 1, 913 CrossRef; (b) J. Holz, D. Heller, R. Stürmer and A. Börner, Tetrahedron Lett., 1999, 40, 7059 CrossRef CAS; (c) T. V. RajanBabu, Y.-Y. Yan and S. Shin, J. Am. Chem. Soc., 2001, 123, 10207 CrossRef CAS PubMed.
- (a) R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS; (b) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed.
- For an olefin hydrogenation study considering hydrophobic effects, see: J. Bakos, I. Tóth, B. Heil, G. Szalontai, L. Párkányi and V. Fülöp, J. Organomet. Chem., 1989, 370, 263 CrossRef CAS.
- (a) Y. Amrani, L. Lecomte, D. Sinou, J. Bakos, I. Toth and B. Heil, Organometallics, 1989, 542 CrossRef CAS; (b) M. Laghmari and D. Sinou, J. Mol. Catal., 1991, 66, L15 CrossRef CAS; (c) F. Joo, P. Csiba and A. Benyei, J. Chem. Soc., Chem. Commun., 1993, 1602 RSC; (d) J. Bakos, R. Karaivanov, M. Laghmari and D. Sinou, Organometallics, 1994, 13, 2951 CrossRef CAS; (e) A. Bakac, Dalton Trans., 2006, 1589 RSC; (f) X. Wu and J. Xiao, in Metal Catalyzed Reactions in Water, Wiley-VCH, 2013, pp. 173–242 Search PubMed.
- (a) J. M. Brown and P. A. Chaloner, J. Am. Chem. Soc., 1980, 102, 3040 CrossRef CAS; (b) C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746 CrossRef CAS; (c) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (d) S. Feldgus and C. R. Landis, J. Am. Chem. Soc., 2000, 122, 12714 CrossRef CAS.
- (a) D. Sinou, Adv. Synth. Catal., 2002, 344, 221 CrossRef CAS; (b) T. Dwars and G. Oehme, Adv. Synth. Catal., 2002, 344, 239 CrossRef CAS.
- For a review on this topic, see: K. H. Shaughnessy, Chem. Rev., 2009, 109, 643 CrossRef CAS PubMed.
- (a) T. Malmstrom and C. Andersson, Chem. Commun., 1996, 1135 RSC; (b) M. T. Zarka, O. Nuyken and R. Weberskirch, Chem. – Eur. J., 2003, 9, 3228 CrossRef CAS PubMed; (c) B. Pugin and H.-U. Blaser, Adv. Synth. Catal., 2006, 348, 1743 CrossRef CAS PubMed.
- For representative examples, see: (a) L. Lecomte, D. Sinou, J. Bakos, I. Tóth and B. Heil, J. Organomet. Chem., 1989, 370, 277 CrossRef CAS; (b) Ref. 7a; ; (c) G. Oehme, E. Paetzold and R. Selke, J. Mol. Catal., 1992, 71, L1 CrossRef CAS; (d) S. Shin and T. V. RajanBabu, Org. Lett., 1999, 1, 1229 CrossRef CAS; (e) F. Robert, G. Oehme, I. Grassert and D. Sinou, J. Mol. Catal. A: Chem., 2000, 156, 127 CrossRef CAS.
- (a) Y. Amrani, L. Lecomte, D. Sinou, J. Bakos, I. Toth and B. Heil, Organometallics, 1989, 8, 542 CrossRef; (b) H. Ding, B. E. Hanson and J. Bakos, Angew. Chem., Int. Ed. Engl., 1995, 34, 1645 CrossRef CAS PubMed; (c) K. Yonehara, T. Hashizume, K. Mori, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 5593 CrossRef CAS PubMed.
- (a) G. Oehme, E. Paetzold and R. Selke, J. Mol. Catal., 1992, 71, L1 CrossRef CAS; (b) I. Grassert, E. Paetzold and G. Oehme, Tetrahedron, 1993, 49, 6605 CrossRef CAS; (c) A. Kumar, G. Oehme, J. P. Roque, M. Schwarze and R. Selke, Angew. Chem., Int. Ed. Engl., 1994, 33, 2197 CrossRef PubMed; (d) K. Yonehara, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 9381 CrossRef CAS; (e) F. Robert, G. Oehme, I. Grassert and D. Sinou, J. Mol. Catal. A: Chem., 2000, 156, 127 CrossRef CAS; (f) R. Hoen, S. Leleu, P. N. M. Botman, V. A. M. Appelman, B. L. Feringa, H. Hiemstra, A. J. Minnaard and J. H. van Maarseveen, Org. Biomol. Chem., 2006, 4, 613 RSC.
- For reviews on this topic, see: (a) Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS PubMed; (b) P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC; (c) J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456 CrossRef CAS PubMed; (d) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418 CrossRef CAS PubMed; (e) A. E. C. Collis and I. T. Horvath, Catal. Sci. Technol., 2011, 1, 912 RSC.
- (a) I. Tóth, B. E. Hanson and M. E. Davis, J. Organomet. Chem., 1990, 397, 109 CrossRef; (b) R. Augustine, S. Tanielyan, S. Anderson and H. Yang, Chem. Commun., 1999, 1257 RSC; (c) A. Wolfson, S. Janssens, I. Vankelecom, S. Geresh, M. Gottlieb and M. Herskowitz, Chem. Commun., 2002, 388 RSC; (d) C. Simons, U. Hanefeld, I. W. C. E. Arends, R. A. Sheldon and T. Maschmeyer, Chem. – Eur. J., 2004, 10, 5829 CrossRef CAS PubMed; (e) X. Wang and K. Ding, J. Am. Chem. Soc., 2004, 126, 10524 CrossRef CAS PubMed; (f) W. P. Hems, P. McMorn, S. Riddel, S. Watson, F. E. Hancock and G. J. Hutchings, Org. Biomol. Chem., 2005, 3, 1547 RSC; (g) C. Simons, U. Hanefeld, I. W. C. E. Arends, T. Maschmeyer and R. A. Sheldon, J. Catal., 2006, 239, 212 CrossRef CAS PubMed; (h) L. Shi, X. Wang, C. A. Sandoval, Z. Wang, H. Li, J. Wu, L. Yu and K. Ding, Chem. – Eur. J., 2009, 15, 9855 CrossRef CAS PubMed; (i) S. Balogh, G. Farkas, J. Madarasz, A. Szollosy, J. Kovacs, F. Darvas, L. Urge and J. Bakos, Green Chem., 2012, 14, 1146 RSC.
- (a) P. Barbaro, Chem. – Eur. J., 2006, 12, 5666 CrossRef CAS PubMed; (b) J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2008, 109, 360 CrossRef PubMed; (c) P. Barbaro and F. Liguori, Chem. Rev., 2008, 109, 515 CrossRef PubMed.
- (a) R. Selke, J. Mol. Catal., 1986, 37, 227 CrossRef CAS; (b) R. Selke, K. Häupke and H. W. Krause, J. Mol. Catal., 1989, 56, 315 CrossRef CAS; (c) H. N. Flach, I. Grassert and G. Oehme, Macromol. Chem. Phys., 1994, 195, 3289 CrossRef PubMed; (d) P. Barbaro, C. Bianchini, G. Giambastiani, W. Oberhauser, L. M. Bonzi, F. Rossi and V. Dal Santo, Dalton Trans., 2004, 1783 RSC.
- P. Barbaro, L. Gonsalvi, A. Guerriero and F. Liguori, Green Chem., 2012, 14, 3211 RSC.
- For representative studies of Rh phosphine–phosphite catalysts in homogeneous hydrogenation, see: (a) S. Deerenberg, O. Pàmies, M. Diéguez, C. Claver, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Org. Chem., 2001, 66, 7626 CrossRef CAS PubMed; (b) O. Pàmies, M. Diéguez, G. Net, A. Ruiz and C. Claver, J. Org. Chem., 2001, 66, 8364 CrossRef PubMed; (c) A. Suarez, M. A. Mendez-Rojas and A. Pizzano, Organometallics, 2002, 21, 4611 CrossRef CAS; (d) Y. Yan, Y. Chi and X. Zhang, Tetrahedron: Asymmetry, 2004, 15, 2173 CrossRef CAS PubMed; (e) X. Jia, X. Li, W. S. Lam, S. H. L. Kok, L. Xu, G. Lu, C.-H. Yeung and A. S. C. Chan, Tetrahedron: Asymmetry, 2004, 15, 2273 CrossRef CAS PubMed; (f) H. Fernández-Pérez, M. A. Pericàs and A. Vidal-Ferran, Adv. Synth. Catal., 2008, 350, 1984 CrossRef PubMed; (g) T. Robert, Z. Abiri, A. J. Sandee, H.-G. Schmalz and J. N. H. Reek, Tetrahedron: Asymmetry, 2010, 21, 2671 CrossRef CAS PubMed; (h) G. Farkas, S. Balogh, J. Madarasz, A. Szollosy, F. Darvas, L. Urge, M. Gouygou and J. Bakos, Dalton Trans., 2012, 41, 9493 RSC.
- (a) M. Rubio, S. Vargas, A. Suárez, E. Alvarez and A. Pizzano, Chem. – Eur. J., 2007, 13, 1821 CrossRef CAS PubMed; (b) S. Vargas, A. Suárez, E. Alvarez and A. Pizzano, Chem. – Eur. J., 2008, 14, 9856 CrossRef CAS PubMed; (c) P. Etayo, J. L. Núñez-Rico and A. Vidal-Ferran, Organometallics, 2011, 30, 6718 CrossRef CAS; (d) M. Á. Chávez, S. Vargas, A. Suárez, E. Álvarez and A. Pizzano, Adv. Synth. Catal., 2011, 353, 2775 CrossRef PubMed; (e) P. Etayo, J. L. Núñez-Rico, H. Fernández-Pérez and A. Vidal-Ferran, Chem. – Eur. J., 2011, 17, 13978 CrossRef CAS PubMed; (f) J. L. Núñez-Rico, P. Etayo, H. Fernández-Pérez and A. Vidal-Ferran, Adv. Synth. Catal., 2012, 354, 3025 CrossRef PubMed; (g) P. Kleman, P. J. González-Liste, S. E. García-Garrido, V. Cadierno and A. Pizzano, ACS Catal., 2014, 4, 4398 CrossRef CAS.
- C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed.
- (a) J. E. Hamlin, K. Hirai, A. Millan and P. M. Maitlis, J. Mol. Catal., 1980, 7, 543 CAS; (b) J. P. Collman, K. M. Kosydar, M. Bressan, W. Lamanna and T. Garrett, J. Am. Chem. Soc., 1984, 106, 2569 CrossRef CAS.
- Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2015 ACD/Labs). For a review covering organic reactions in water, including the use of calculated solubility values, see ref. 8b.
- Triton-X100: polyethylene glycol p-(1,1,3,3-tetramethylbutyl)-phenyl ether. For examples of hydrogenations using this surfactant, see: (a) G. Oehme, E. Paetzold and R. Selke, J. Mol. Catal., 1992, 71, L1 CrossRef CAS; (b) M. Schwarze, J. S. Milano-Brusco, V. Strempel, T. Hamerla, S. Wille, C. Fischer, W. Baumann, W. Arlt and R. Schomacker, RSC Adv., 2011, 1, 474 RSC.
- (a) T. Okamoto and S. Oka, Bull. Chem. Soc. Jpn., 1981, 54, 1265 CrossRef CAS; (b) J. Chen, Y. Zhang, L. Yang, X. Zhang, J. Liu, L. Li and H. Zhang, Tetrahedron, 2007, 63, 4266 CrossRef CAS PubMed.
- For related dehalogenation reactions, see: (a) K.-i. Fujita, M. Owaki and R. Yamaguchi, Chem. Commun., 2002, 2964 RSC; (b) M. L. Buil, M. Esteruelas, S. Niembro, L. Orzechowski, C. Pelayo and A. Vallribera, Organometallics, 2010, 29, 4375 CrossRef CAS.
- As it is not expected that the presence of the olefin bond in 4i will significantly affect the reactivity of the C–Br bond it is reasonable to expect as well the debromination of 4i to give 4b.
- O. D. Bonner, J. Phys. Chem., 1954, 58, 318 CrossRef CAS.
- (a) C. R. Johnson and B. Zhang, Tetrahedron Lett., 1995, 36, 9253 CrossRef CAS; (b) B. H. G. Swennenhuis, R. Chen, P. W. N. M. van Leeuwen, J. G. de Vries and P. C. J. Kamer, Eur. J. Org. Chem., 2009, 5796 CrossRef CAS PubMed.
- For related spectroscopic studies on similar enamide adducts, see: (a) C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746 CrossRef CAS; (b) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (c) E. Alberico, W. Baumann, J. G. de Vries, H.-J. Drexler, S. Gladiali, D. Heller, H. J. W. Henderickx and L. Lefort, Chem. – Eur. J., 2011, 17, 12683 CrossRef CAS PubMed.
- J. Wassenaar, M. Kuil, M. Lutz, A. L. Spek and J. N. H. Reek, Chem. – Eur. J., 2010, 16, 6509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of ligands and Rh complexes, representative procedures for asymmetric hydrogenation, NMR spectra and chromatograms. See DOI: 10.1039/c5gc00485c |
| This journal is © The Royal Society of Chemistry 2015 |