
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquids and continuous flow processes: a good marriage to design sustainable processes
Eduardo
García-Verdugo
*a,
Belen
Altava
a,
M. Isabel
Burguete
a,
Pedro
Lozano
b and
S. V.
Luis
a
aDepartamento de Quíımica Inorgánica y Orgánica, Universidad Jaume I, Campus del Riu Sec, Avenida Sos Baynant s/n, E-12071, Castellón, Spain. E-mail: cepeda@uji.es; luiss@uji.es
bDepartamento de Bioquímica y Biología Molecular “B” e Inmunología, Facultad de Química, Universidad de Murcia, E-30.100, Murcia, Spain. E-mail: plozanor@um.es
First published on 18th March 2015
Abstract
In the last few years the use of Ionic Liquids (ILs) as alternative solvents for (bio)catalytic processes has increased substantially, and the benefits and different approaches reported to combine continuous flow systems and ILs are at the core of this overview. The synergy between both elements allowed us to highlight their great potential in manufacturing both bulk and fine chemicals by new and greener (bio)catalytic processes.
 Eduardo García-Verdugo | E. García-Verdugo was born in 1972 in Talavera de Reina, Spain. He obtained his chemistry degree at the University of Valencia (1995) and his PhD in chemistry at the University Jaume I (Spain) in 2001. Then, he worked as a researcher at Nottingham University (UK) under the supervision of M. Poliakoff (until 2004). He returned to Spain and worked as a Ramon y Cajal fellow at University Jaume I until 2009, and then he obtained a permanent position as Cientifico Titular of CSIC at ICP (Madrid) (2009–2010). In 2010, he moved to a permanent academic position at the University Jaume I. Currently, he is working on the integration of different so-called enabling techniques (catalysis, polymeric materials, continuous flow processes, microreactors, bio-catalysis, neoteric solvents (ILs, SCFs)) to develop more efficient and greener organic transformations. |
 Belen Altava | Belén Altava completed her degree in chemistry at the Universities of Valencia and Zaragoza and obtained her PhD in chemistry at the University Jaume I of Castellón in 1996. After postdoctoral stays at the University of Düsseldorf with Professor G. Wulff and at Novartis (Basel) with Dr H. U. Blaser she returned to Spain and obtained a permanent academic position at the University Jaume I in 2001. Her main areas of research are directly related to green and sustainable chemistry and involve the development of supported catalysts, asymmetric catalysis, the preparation and application of new task specific ionic liquids, in particular chiral ionic liquids, and the application of these compounds and other amino acid derived systems for selective recognition. |
 M. Isabel Burguete | M. Isabel Burguete graduated in chemistry at the University of Zaragoza in Spain and after a stay at the University of Pittsburgh (USA) under the supervision of Dr J. Rebek, she completed her PhD at the University of Valencia in 1989 under the direction of Dr F. Gaviña working on regulated crown ethers and convergent diacids. In 1989 she took an academic appointment at the University Jaume I in Castellón (Spain) where she is currently Professor of organic chemistry. Her main field of research is the development of new tools, in particular homogeneous and supported catalysis approaches, in green and sustainable chemistry. |
 Pedro Lozano | Pedro Lozano is a chemist born in 1961 in Ceutí, Spain. He graduated in Sciences (chemistry) at the University of Murcia in 1984. He received his PhD in Sciences (chemistry) from the University of Murcia in 1988. Between 1990 and 1991 he spent two years at the Centre de Bioingénierie Gilbert Durand, Toulouse (France) as a post-doctoral fellow. In 1993, he returned to the Faculty of Chemistry at the University of Murcia as Lecturer in Biochemistry and Molecular Biology, being finally promoted to Full Professor in 2004. During the period 1996–2014, he was Assistant-Dean of the Faculty of Chemistry and the coordinator of the Biochemistry Degree at the University of Murcia. Since 2014, Prof. Lozano has been the Dean of the Faculty of Chemistry at the University of Murcia. His research activity has always been related to enzyme technology for green chemistry, and his particular research interest is in the use of enzymes in ionic liquids and supercritical fluids. |
 S. V. Luis | Santiago V. Luis carried out his studies of chemistry at the University of Zaragoza in Spain and completed his PhD at the University of Valencia (Spain) in 1983 under the direction of Prof. F. Gaviña. After a postdoctoral stay in 1985 at the University of Pittsburgh (USA) under the supervision of Prof. J. Rebek, he obtained a permanent position at the University of Valencia (1987) and then a full professorship at the University Jaume I of Castellón in Spain (1995). His main areas of research involve supramolecular and biomimetic chemistry, particularly based on pseudopeptidic structures, and development of new tools from a sustainable and green chemistry perspective, with a special emphasis on catalysis and flow chemistry. Since the last decade he has been coordinating the Spanish MSc and PhD Interuniversity Programs in sustainable chemistry. |
1. Introduction
Over the last decade academia and industry have been working together in order to develop new greener alternative solvents.1 The main aim is to reduce significantly the important drawbacks and hazards associated with traditional solvents, replacing them with safer and more efficient alternatives. This can be achieved, ideally, through the design of new substances with specific and tailored properties. This is the case of ionic liquids (ILs), for which the performance can be tuned to be as good as or even better than that of conventional solvents for many applications.2 The application of other alternative reaction media including, amongst others, water, supercritical fluids, switchable solvents and fluorous solvents is also envisaged.3 In this regard, a variety of new chemical processes have been designed based on those alternative solvents in order to exploit their advantages and to implement their use in well-known chemical processes.An additional factor that can contribute to implement the sustainability of a chemical transformation is the substitution of batch processes by flow processes.4–8 Advantages associated with flow systems include the improvement in mass and heat transfer, with significant intensification of the process, making available systems working 24 h a day, 7 days a week (24/7), or their easier optimization through the adjustments of simple parameters such as flow, pressure or temperature. In addition, the scale-up of flow processes is generally more easily attainable than for batch processes via different approaches such as the scale-out or the number-up.4 Although the advantages of flow chemistry were soon realized by the bulk chemistry industry, the situation is quite different in the fine chemicals or pharmaceutical industries. Most industrial petrochemical processes, and many of those for the preparation of bulk chemicals, are carried out in flow, but batch processes still dominate in other industrial areas, and a high pressure is currently being exerted to introduce flow processes there. In the last few years, there has been a blossoming of new developments in the field of the application of flow processes in complex organic syntheses, especially those based on the application of microstructured devices.9
Here, we shall focus on recent advances in the use of ILs, working alone or together with other neoteric solvents (i.e. supercritical fluids), exploring their great potential to manufacture both bulk and fine chemicals by continuous flow systems. The combination of (bio)catalytic reactions in ILs with continuous flow processes can fulfil the needs and challenges of the chemical industry, achieving significant scientific and technological breakthroughs. Indeed, the examples here presented describe a series of techniques, methodologies and approaches that could allow the development of new and more efficient chemical processes, not only taking into account classical parameters such as yield, purity or selectivity, but also considering, for the overall equation, the environmental and hazard costs. Our goal in this review is to present some significant cases highlighting the main advantages and some of the shortcomings found for the combined use of ILs and continuous flow technologies.
2. Continuous flow processes for the synthesis and scale-up of ILs
A wide variety of ILs can be designed, taking into account their modular character, considering structural elements such as the nature of the cation, the nature of the anion, the ion substitution patterns, the introduction of either bio-renewable or chiral moieties, etc. Indeed, a wide range of structurally different ILs has been reported.10 Besides, ILs can be regarded as “design solvents” not only because their structure can be tailored on demand, but also because their physical and chemical properties can be tuned at the molecular level by selecting the right combination of structural elements. These unique opportunities offered by ILs have resulted in increasing their use as alternatives to classic organic solvents and their application in completely unexpected new fields.11In general, the synthesis of ILs is a simple process involving the quarternisation of an amine, imidazole, pyridine or phosphine, through an alkylation reaction, to form the corresponding cation. The variation in the nature of the nucleophile and the alkylating agent leads to ILs with different structures, typically having a halide as the counteranion. In the second step, a variety of anions can be introduced by anion metathesis (e.g., BF4−, PF6−, (CF3SO2)2N−). The most simple purification protocol for ILs is the distillation of an excess of alkylating agent prior to their use as solvents or reactants, according to their inherent nonvolatility. Although the performance often depends on the purity of ILs, especially for industrial applications, high purity ILs are essential to study in detail the fundamental science of this kind of new interesting material, which often involves a significant effort.12 Thus, tedious subsequent purification steps are usually required leading to high costs and limiting the potential for industrial application. In this context, the use of continuous flow systems and in particular those based on microstructured elements has shown significant potential for the preparation of ILs and the corresponding scale-up.13 In fact, systems containing micro-mixers and/or microreactors in the experimental set-up can provide very short diffusion pathways and large interfacial contact areas per unit volume (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 m2 m−3), which results in superior heat and mass transfer to conventional batch reactors. Thus, higher yields and selectivities, as well as improved product qualities, can be achieved by transferring the syntheses of ILs (especially those that are highly exothermic) from classical batch reactors to microstructured devices.14
000–50000 m2 m−3), which results in superior heat and mass transfer to conventional batch reactors. Thus, higher yields and selectivities, as well as improved product qualities, can be achieved by transferring the syntheses of ILs (especially those that are highly exothermic) from classical batch reactors to microstructured devices.14
In this context, Böwing et al. have described the continuous production of ethylmethyl imidazolium ethylsulfate ([EMIM][EtSO4]) by the alkylation of methyl imidazole.15 This compound can be considered both a bulk IL and a source of various other ILs, through a two-step synthesis approach. Thus, the first step involves the synthesis of a methylsulfate or ethylsulfate ionic liquid by direct alkylation. In the second step, this intermediate is transformed via a transesterification reaction, using different functionalized and nonfunctionalized alcohols, to the corresponding new alkylsulfate ILs.16 In this case, a simple tubular reactor (6 mm inner diameter and 4 m length) can produce up to 4 kg per day of [EMIM][EtSO4] without the need for a solvent, by simply pumping methylimidazole and ethylsulfate (Et2SO4) into a cooled reactor tube (303 K).15,17
The introduction of an experimental set-up of microstructured reactors (submillimetre range: 10–100 μm diameters) based on parallel microchannels instead of a simple tubular reactor can contribute to increase significantly the heat exchange capacity. Thus, Löwe et al. synthesised 1,3-dimethylimidazolium trifluoromethanesulfonate ([DMIM][OTf]) by alkylation of 1-methylimidazole (MIM) with methyltriflate (MeOTf), combining a microreactor and a heat pipe. The productivity of the system can reach up to 0.74 kg h−1.18 The combination of different types of mixers and reactor designs with different temperature profiles was assayed to adjust the overall performance of the process. Hence, a production rate up to 1.24 kg h−1 of 1-ethyl-3-methylimidazolium trifluoromethanesulfonate ([EMIM][OTf]) can be achieved.19,20 In a similar way, Waterkamp et al. have reported an approach to intensify the synthesis of 1-butyl-3-methylimidazolium bromide ([BMIM][Br]) by using a continuously operating micro-reactor system.21 The reactor set-up consists of a microstructured mixer of 450 μm channel width and a tubular reactor (inner diameter from 2 to 6 mm). Under the optimised conditions, production rates up to 9.3 kg [BMIM][Br] per day could be obtained. In this reactor system the strongly exothermic alkylation can be thermally controlled even at elevated temperatures, leading to high reaction rates in a solvent-free process. Thus, high purity (>99%) [BMIM][Br] was obtained even when the process was performed at moderate temperatures (85 °C). It is noteworthy that the process intensification involved more than a twenty-fold increase of the space–time yield (STY), as compared to the conventional synthesis in batch. By increasing the flow rate, the mass transfer of the reactant 1-methylimidazole from the product phase to the reaction phase could be enhanced. This can be achieved simply by means of higher flow velocities in the slug flow regime that is formed due to the generation of a two-phase liquid/liquid system. At high reaction temperatures (145 °C) an almost colorless ionic liquid could be produced by increasing the flow. In this case, a space–time yield of 1240 kg L−1 per day could be obtained.22
The remarkably versatile design of this continuous flow set-up has allowed the preparation of new imidazolium-type ILs bearing additional functionalities either in the cation or in the anion (Fig. 1).23 The continuous flow reactor set-up is based on the combination of HPLC pumps with a micromixer and a capillary residence tube. The family of ILs synthesized showed, according to the HPLC analysis, less than 1% of impurities (mostly unreacted starting materials). The good heat transfer properties of the continuous flow system allowed exploring windows of reaction conditions that should be unsafe under batch conditions. Indeed, the conventional batch synthesis of the considered imidazolium-type ILs bearing aromatic units takes place explosively at temperatures higher than 100 °C. Lower temperatures however involve the drawback of significantly slower kinetics. Thus, the continuous flow setup allows working safely at high temperatures without thermal overrun and subsequent loss of control over the reaction. Furthermore, temperatures and residence times can be individually chosen according to the substrates, without having to change the experimental setup and allowing the preparation of large product amounts with little experimental effort. In a typical experiment, using a single microstructured reactor, up to 200 mL of a high purity ionic liquid can be prepared. The employment of additional similar devices in parallel operation mode provides convenient access to multikilogram per hour scales without having to resort to a more complex system.
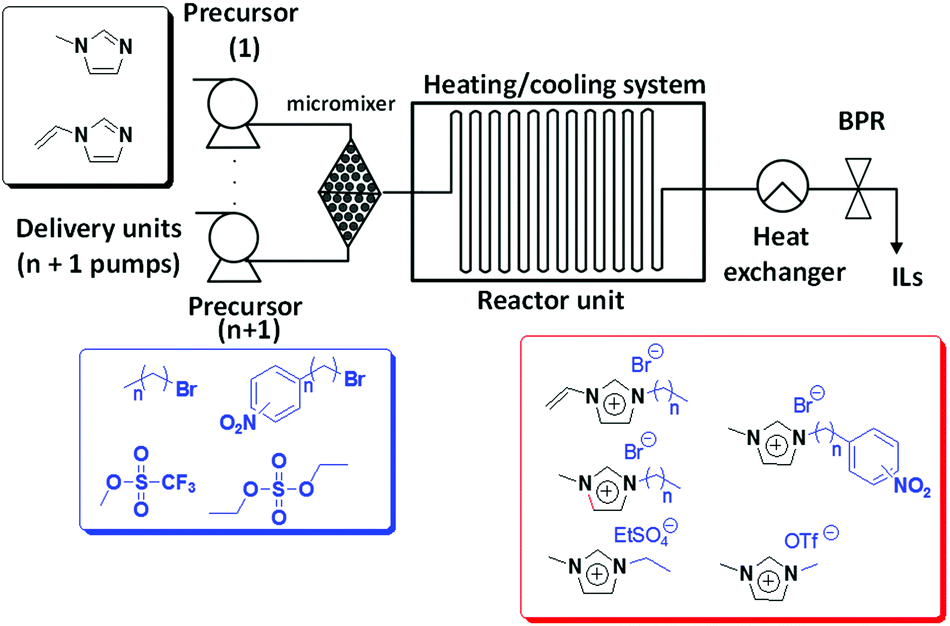 | ||
| Fig. 1 Schematic illustration of the experimental set-up applied for the continuous flow synthesis of different ILs.15–23 | ||
Water-soluble ionic liquids (1,3-dibutylimidazolium acetate [BBIM][OAc] and 1,3-dibutylimidazolium chloride [BBIM][Cl]) were synthesized by the modified Radziszewski reaction through a continuous procedure in a multipurpose microreaction device.24 Thus, [BBIM][Cl] can be obtained with up to 70% yield and with a STY of 0.12 kg L−1 h−1 and a capacity of 0.09 kg d−1. Using the same reaction, the corresponding parameters were adapted for the synthesis of 1,3-dibutylimidazolium acetate that was obtained with 91% yield (a STY of 0.35 kg L−1 h−1 and a capacity of 0.25 kg d−1). In comparison with the modified Radziszewski reaction in batch mode, the results clearly showed the potential of the microreaction technology in terms of process intensification. Indeed the reaction times were reduced by factors of 3 and 6 for chloride- and acetate-based ionic liquids.
Recently, Nokami, Itoh and co-workers have reported a simple microreactor system for the continuous flow synthesis of novel ionic liquids having a (2-methoxyethoxy)methyl or methoxymethyl substituent. Both N-bases and tributylphosphine ILs were efficiently prepared in the microreactor system. The authors claimed that the conversion rates for the synthesis of the ILs in the microreactor system are faster than those of the batch system due to the smaller diffusion distance in the tube reactor.25
The required IL-precursors can also be synthesised by continuous flow processes, especially when they bear specific structural features, as are certain functional groups or chiral structural elements. In this regard, a versatile and efficient method to synthesize tetrasubstituted imidazoles by means of a continuous flow process via a one-pot modified Debus–Radziszewski reaction and their subsequent transformation into the corresponding imidazolium ILs has recently been reported.26 This straightforward synthetic procedure allows for a fast and selective synthesis of tetrasubstituted imidazoles on a large scale (Fig. 2a). These substituted imidazoles can lead, through alkylation reactions, to new families of ILs.
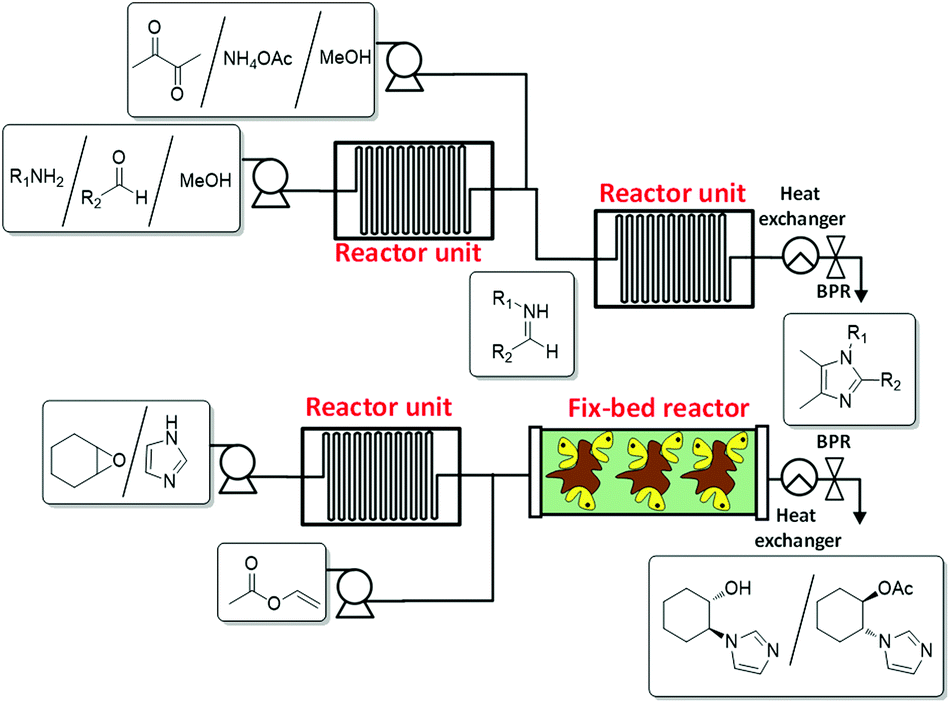 | ||
| Fig. 2 Continuous flow synthesis of IL precursors. (a) Substituted imidazoles (top).26 (b) Enantiopure imidazoles (bottom).27 | ||
The use of continuous flow systems has been demonstrated to be significantly more efficient than the corresponding batch processes for the synthesis of chiral imidazoles by the lipase-catalyzed kinetic resolution of racemic 2-(1H-imidazol-yl)cycloalkanols.27 The continuous flow biotransformations have allowed one to easily increase the production scale of these chiral imidazoles, which are adequate building blocks in the synthesis of chiral ionic liquids (Fig. 2b).28
In summary, the use of continuous flow systems facilitates the preparation of a wide variety of novel ionic liquids and/or their precursors by simple and precise control of the reaction parameters, avoiding time-consuming purification steps while meeting the strict demands for highly pure reaction products and simultaneously bearing the possibility to prepare “on demand” the desired compounds on a large scale. This will finally provide access to the rapid synthesis of tailor-made products for the steadily growing demand for ionic liquids.
3. Continuous flow processes using ILs as either solvents or catalysts
ILs have been intensely exploited as the homogeneous phase for the immobilization of different types of reagents or catalysts.29 The immobilization of a catalyst in the IL-phase can provide a series of advantages over the use of other supports, in particular solid supports: (i) first of all, no modification of the homogeneous catalyst is required for the immobilization, a factor that often affects the efficiency of solid-supported systems; (ii) the presence of counteranions can contribute to the activation of the catalyst; (iii) a large variety of available ILs can be used to fine-tune the overall efficiency of the catalytic system through straightforward modifications in the structure of the IL (anion and cation nature, aliphatic chains, the presence of additional functional groups…); (iv) finally, the IL phase can significantly contribute to the stabilization of the catalyst.30Furthermore, under certain experimental conditions, the tunable miscibility of ILs with other solvents may facilitate the separation of both the catalyst and the reaction medium from the products by means of liquid–liquid extraction. In terms of the potential industrial applications, this is a big advantage when considering the price of the metal catalysts, as the recovery of the catalytic IL-phase will facilitate recycling and reuse of these expensive catalysts (and that of the IL) for longer periods of time. Hence, it is feasible, in principle, to design a continuous flow system, in which a (bio)catalytic reaction is carried out in the homogeneous phase using an IL as the reaction medium. The experimental set-up should be completed by adding to the layout an efficient extraction/recycling device allowing for the recycling of the IL-phase containing the catalyst.
Indeed, based on these principles, Eastman Chemical Company developed in 1998 the first continuous flow industrial IL-based process using continuous stirred tank reactors.31 In this process, long-chain tetraalkylphosphonium salts are used as liquid phases for the isomerisation of 3,4-epoxybut-1-ene to 2,5-dihydrofuran, which is an important intermediate for the synthesis of higher added value products. This isomerisation requires a catalytic Lewis acid (a trialkyltin iodide) and an ionic liquid ([P88818][I]) as a Lewis base. The system operated with three continuous stirred-tank reactors, a wiped-film evaporator, a distillation train and a continuous, counter current, liquid–liquid extractor for recovering the IL-catalyst phase (Fig. 3). The plant was fully operative for eight years before being shut down as the market for the furan declined.
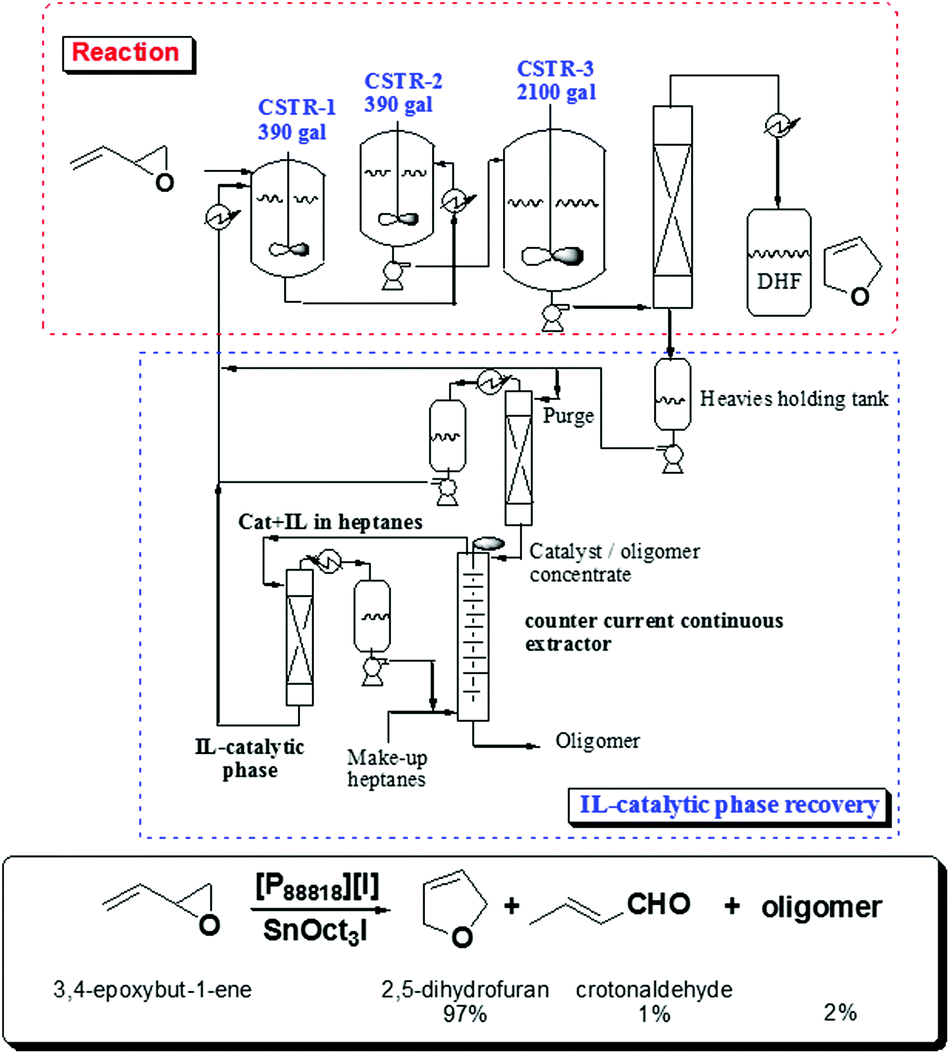 | ||
| Fig. 3 Continuous production of dihydrofuran by isomerisation of epoxybutadiene including the recycling of the catalytic IL-quaternary phosphonium phase.31 | ||
Liu et al. have developed a continuous microflow system for the synthesis of butyl cinnamate by a Mizoroki–Heck reaction catalysed by Pd–carbene species, which also allowed catalyst recovery, based on a similar approach combining reaction in ILs, product separation and recycling of the catalyst-IL phase.32 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide ([BMIM][NTf2]) was used as the homogeneous reaction medium due to its low viscosity. An automated continuous flow system was developed as shown in Fig. 4. The continuous flow reactor was designed in conjunction with two microextraction units. The first one allowed the separation of the product (butyl cinnamate) from the IL/catalyst phase by liquid/liquid extraction with hexane. In the second unit, water was used to extract from the IL-phase the salts (ammonium salts) formed during the reaction. Altogether, the system led to high yields of butyl cinnamate and efficient removal and recycling of the catalyst-IL phase, which could be used again in consecutive runs without any drop in the product yield (an overall yield of 80%, 115.3 g, 10 g h−1). The repeated use of the Pd catalyst supported in the ionic liquid allowed exploring a totally automated flow system with continuous catalyst recycling.
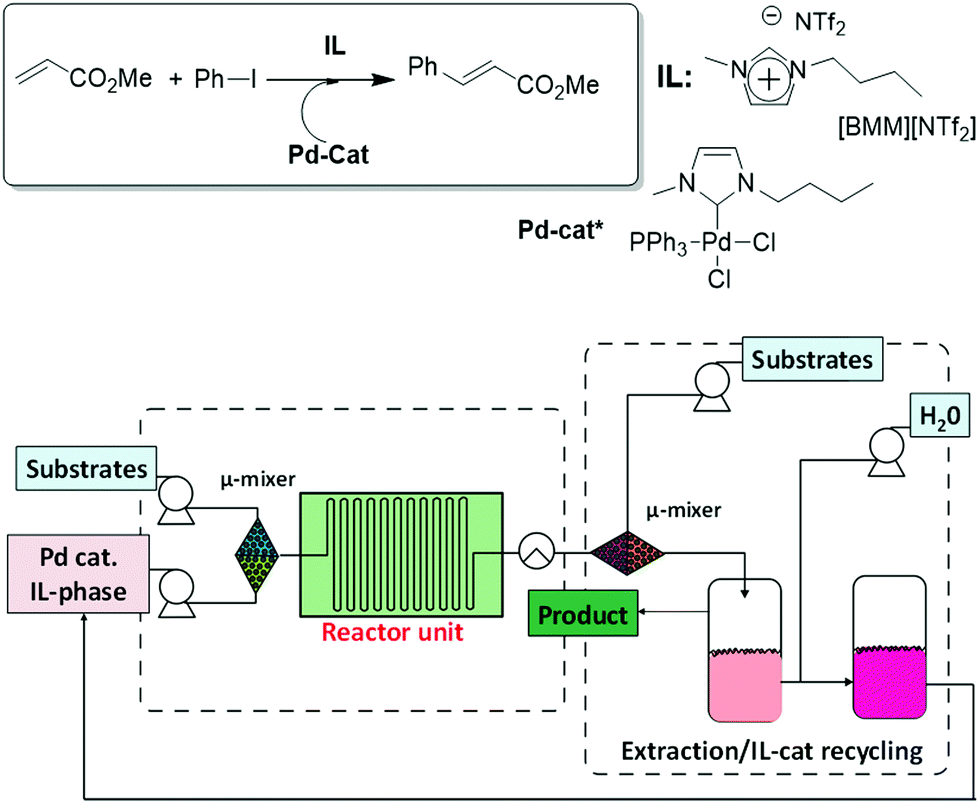 | ||
| Fig. 4 Schematic set-up for the automated microflow system (CP-CYTOS-labsystem), including a dual microextraction process, for the continuous Heck reaction.32 | ||
A similar set-up was used for the 100 g scale synthesis of a key matrix metalloproteinase inhibitor through a copper-free Sonogashira coupling reaction in [EMIM][NTf2] using a flow microreactor enabling continuous catalyst recycling.33
A different application of ILs to design continuous flow processes has been reported by Horii et al.34,35 They developed an electrosynthetic system for an anodic substitution reaction using a micro-flow reactor. This system enables nucleophilic reactions to overcome restraints such as the oxidation potential of nucleophiles and the stability of carbocations by the combined use of ILs as reaction media and a parallel laminar flow in the reactor. In this case, a stable liquid–liquid interface can be formed and mass transfer between input streams occurs only via diffusion (Fig. 5). The microflow system enables the precise control of the reactive intermediates formed and thereby facilitates highly selective reactions that are difficult to achieve in conventional reactors. The product yield was improved up to 91% when N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium bis(trifluoromethanesulfonyl)imide [DEME][NTf2] was used as the reaction medium in comparison with the 59% yield obtained in 2,2,2-trifluoroethanol (TFE), which is known as a stabilizing solvent for carbocations. These results suggest that ILs have an excellent stabilizing ability for carbocations, and therefore the carbocation generated from N-(methoxycarbonyl)pyrrolidine could react with allyltrimethylsilane before its decomposition.
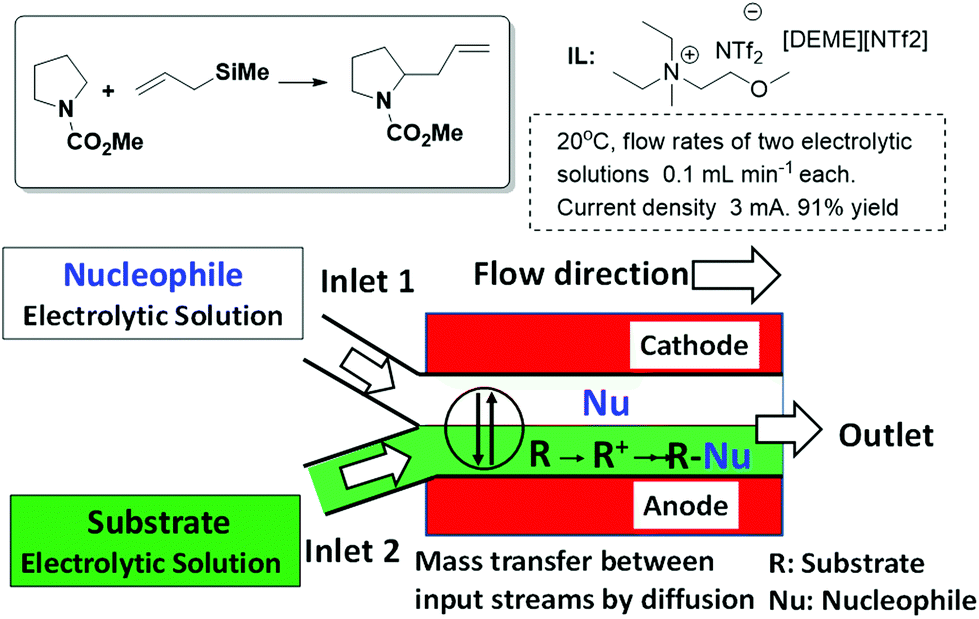 | ||
| Fig. 5 Schematic representation of the parallel laminar flow in the micro-flow reactor for an anodic substitution reaction.34,35 | ||
Another application of a continuous biphasic process is the well-known BASIL™ (Biphasic Acid Scavenging utilizing Ionic Liquids) process introduced by BASF in 2002.36 In this process, an IL (1-methyl-imidazolium chloride [HMIM][Cl]) is formed in situ by using 1-methylimidazole as the HCl scavenger in the production of diethylphenylphosphonite, which is used as a key raw material for the production of photoinitiators to cure coatings and printing inks by exposure to UV light. In the conventional process, tertiary amines were used to scavenge the HCl, but this led to thick, non-stirrable slurries, decreasing enormously the efficiency of the process.
The original process had to be carried out in batch mode in order to filter the product after the reaction, both the reaction and the isolation process being difficult to operate and providing low yields. The introduction of methyl imidazole as the acid scavenger affords 1-methyl-imidazolium chloride, which is a colourless mobile liquid that can be easily separated from the product (Fig. 6). Having eliminated the formation of any solid and having increased the reaction rate, development of new reactor concepts was possible. BASF was able to perform the same reaction formerly carried out in a large vessel in a small jet reactor having the size of a thumb. The productivity of the process was raised by a factor of 8 × 104 to 690000 kg m−3 h−1.
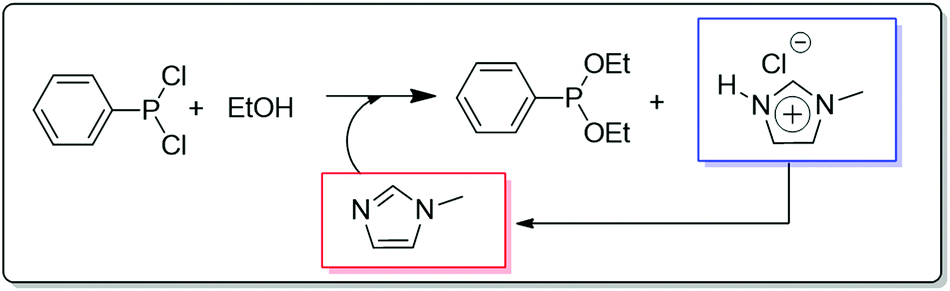 | ||
| Fig. 6 General features of the continuous flow BASF's BASIL process involving the acid scavenging to generate an IL that is continuously separated and regenerated.36 | ||
Another interesting development of an industrially relevant process is the dimerisation of alkenes, typically propene (Dimersol-G) and butenes (Dimersol-X), to the more valuable branched hexenes and octenes (Dimersol process) catalysed by Ni complexes. The use of 1,3-dialkylimidazolium chloroaluminates as solvents has been developed and pioneered at IFP. These ILs are good solvents for the butanes and also help in stabilising the Ni catalyst without even requiring an additional ligand. Under these conditions, the desired products are not soluble in the IL-phase. Thus, it is possible to establish a liquid–liquid biphasic system and the reaction can be performed in a continuous stirred tank-reactor (CSTR) followed by a phase separator. In the separator the upper organic phase containing the products escapes through an overflow for collection and processing, whilst the lower IL-phase containing the catalyst is recycled back to the reactor. This system dramatically decreases the Ni leaching when compared with the homogeneous Dimersol process in the absence of ILs.
Another example of a process facilitated by an IL has been recently reported by the joint efforts of the groups of Seddon and Kappe.37 In this case, tributylmethylammonium methylcarbonate ([Bu3NMe][MeCO2]) is employed as a catalytic base for the clean N-methylation of indole with dimethylcarbonate. [Me3NMe][MeCO2] can be generated in situ from the methylation of tributylamine with dimethylcarbonate. The reaction was performed under continuous flow conditions at high temperatures/high pressures (285 °C/150 bar) using a short (3 min) residence time and 2 mol% of the catalyst to efficiently methylate a variety of different amines, phenols, thiophenols and carboxylic acid substrates. The extremely short residence times, high versatility and selectivity have significant implications in the synthesis of a wide range of pharmaceutical intermediates. High product throughputs can be obtained via this scalable continuous flow protocol.
Biocatalytic processes can also be performed in continuous flow mode using ILs as the solvents. Indeed, [BMIM][Tf2N] has been used for the continuous synthesis of caffeic acid phenethyl ester using a packed bed microreactor based on a commercially available supported enzyme (Novozym 435). The system showed an excellent stability, with the efficient reuse of the biocatalyst for nine days without any decrease in activity.38 A similar pressure-driven miniaturized reactor packed with Novozym 435 beads has also been successfully applied for the synthesis of isoamyl acetate using [C7mmim][Tf2N] as the reaction medium.39 Further integration with a miniaturized continuous separator that exploited the high boiling point of ionic liquids as compared to those of products/substrates enabled, in principle, the isolation of the products and ionic liquid recycling (Fig. 7).40
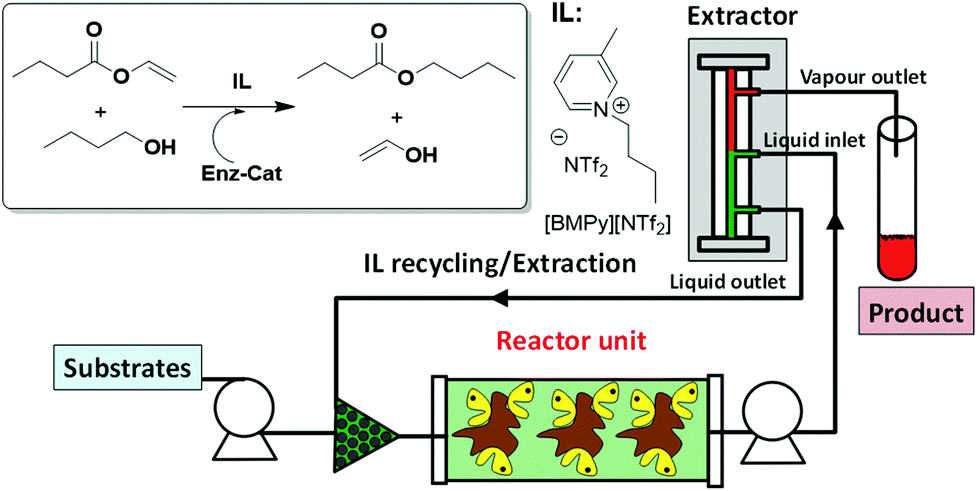 | ||
| Fig. 7 Continuous flow biocatalytic process in ILs integrating synthesis and isolation.40 | ||
Alternatively, reaction set-ups including membrane pervaporation systems have also been developed for carrying out the continuous biocatalytic synthesis of esters in ILs by an esterification reaction. The non-volatile character of ILs facilitates not only the separation of the ester but also their separation from the water produced during the esterification shifting the equilibrium reaction towards the production of the corresponding pure flavour or fragrances.41a By this approach, the immobilized CALB catalysed the synthesis of isoamyl acetate41b and ethyl acetate41c by esterification of acetic acid and the corresponding alcohol in a continuous stirred-tank reactor containing [Bmim][PF6] as the reaction medium. The coupling of two membrane units on the reactor allowed continuous removal of both the isoamyl acetate and water products by a double pervaporation system using hydrophobic and hydrophilic membranes, respectively, in continuous operation for 72 hours without any loss in the enzyme activity.41
Rahman et al. have reported a low pressure microflow system for continuous flow multiphase palladium catalyzed carbonylation reactions in ILs.42 The microflow system resulted in superior selectivity and higher yields in the carbonylative Sonogashira coupling and the amidation reactions of aryl iodides when compared to the related conventional batch systems (Fig. 8). The authors claimed that the superior efficiency of the microflow system to the batch reactor can be attributed to the occurrence of a plug flow. This flow regime might provide a large specific interfacial area between the CO and the liquid phase facilitating the diffusion of CO into the thin IL phase. This cannot be obtained by mechanical stirring, justifying the low efficiency of the batch reactions under similar conditions.
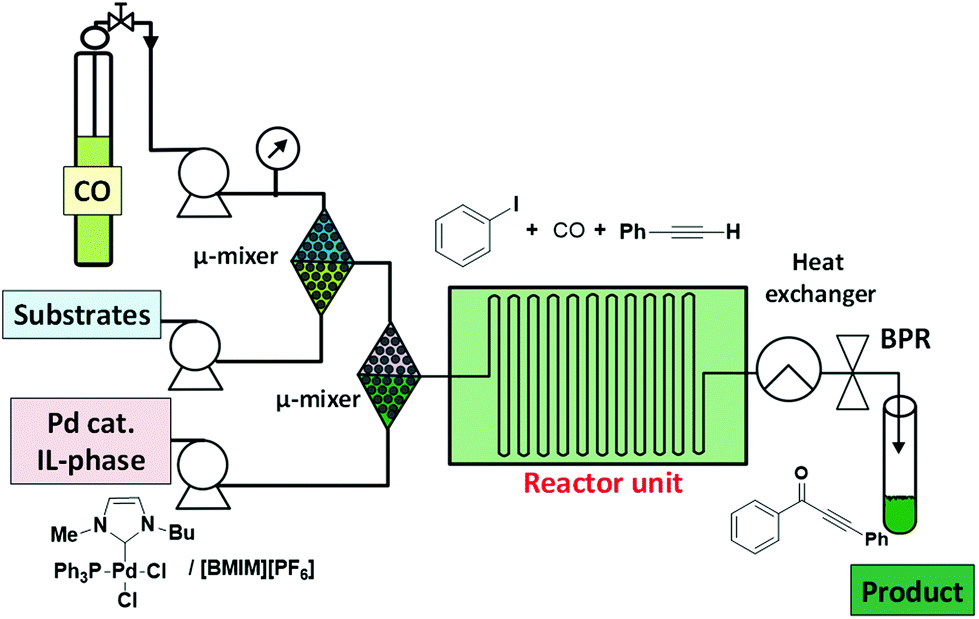 | ||
| Fig. 8 Pd-catalyzed carbonylative three-component coupling under continuous flow using ILs.42 | ||
Continuous flow set-ups can also be built on different types of biphasic systems derived from the IL phase and a supercritical fluid, in particular supercritical carbon dioxide (scCO2).30 These systems are based on the fact that scCO2 can show high solubility in some ionic liquids (up to 60 mol%), whilst the same ILs have no measurable solubility in CO2.43 In this way, it is relatively straightforward to develop continuous and semi-continuous IL/scCO2 set-ups. Generally, the IL-phase is used for the homogeneous immobilisation of the catalyst (metal complexes, enzymes, nanoparticles, etc.), while the scCO2 phase is intended to favour the delivery of substrates to the catalytic sites on the IL phase and to facilitate the extraction and separation of the final products.44Fig. 9 schematically shows the reactor set-ups used for biphasic continuous flow systems based on IL/scCO2. Very often, this combination allows optimisation of the efficiency of a given process leading to good yields, (enantio)selectivities and productivities by the fine tuning of the contact time of the scCO2/IL-phases using either the pressure or the flow rates.
 | ||
| Fig. 9 General set-up for biphasic continuous flow systems based on ILs/scCO2. | ||
In general, this excludes the need for other additional solvents and facilitates the isolation and separation of the products from the catalyst, being able to work 24/7, requiring less man power for operation and reducing the equipment size, maximizing at the same time the productivity. Therefore, they present great potential for the development of industrial processes. Although the examples reported here have been performed at the lab-scale their scale-up is feasible. Indeed, there is commercially available equipment designed for small-scale natural product extraction, which could be readily modified for this kind of purpose. As for any process involving scCO2, one of the limiting factors for the scale-up is the operating costs associated with the energy requirement for the compression cycles of CO2 between isolation and reaction.45
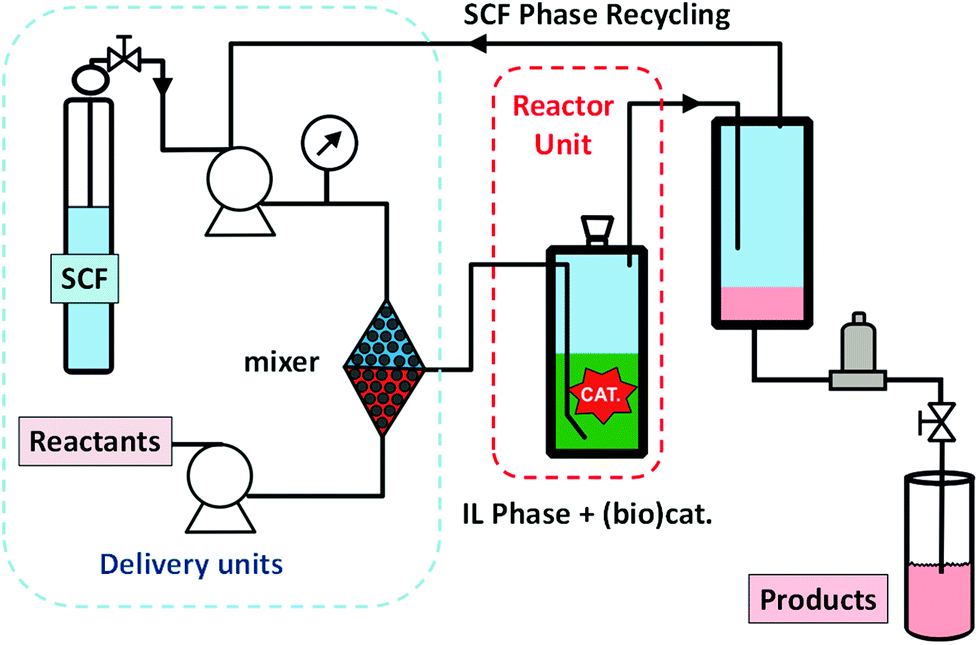
The pioneering work in this field was performed by Cole-Hamilton in the hydroformylation of long-chain olefins with rhodium complexes.46 The metal complex is dissolved in an IL and the reaction proceeded with high stability. Different catalysts and ILs have been assayed for the process (Fig. 10). After the optimisation of the catalyst, the IL and the flow conditions, excellent yields, with an improved TOF value of 517 h−1 and a long-term stability, over several weeks, could be achieved with almost negligible levels of rhodium leaching, down to 0.01 ppm.47,48 The hydroformylation of long chain alkenes can be achieved with selectivities to the desired linear aldehyde of 92%. The supercritical fluid–ionic liquid continuous flow system is based on the use of a xantphos-derived ligand attached to an imidazolium salt.49
 | ||
| Fig. 10 Continuous flow hydroformylation reaction in ILs/scCO2.46–49 | ||
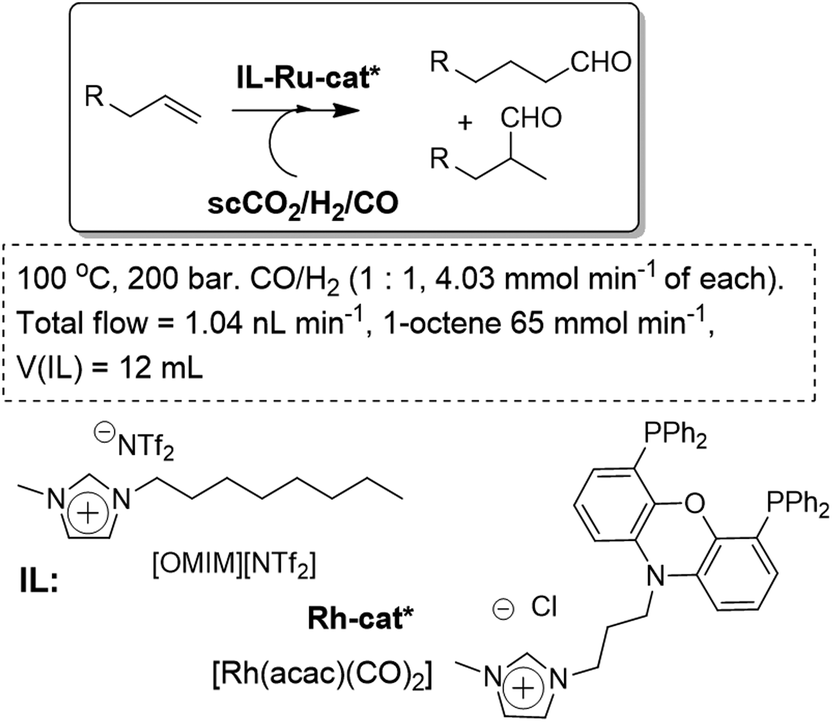
Leitner and co-workers have developed, based on a similar set-up, a simple system that allows for the synthesis of pure HCO2H in a single processing unit. The scCO2 phase is used as both the reactant feeding and extractive phase, resulting in the continuous removal of the product based on the negligible vapor pressure of the ruthenium catalyst in the IL phase ([EMIM][NTf2] (Fig. 11). Thus continuous and efficient separation and removal of pure HCO2H, free from any cross-contamination, is obtained. The adjustment of the right combination of the catalytic system in the IL-phase, together with the use of a supported base (QuadraPure-DMA), led to a stable system for continuous flow conditions. The initial activity was largely retained even after 200 h on-stream. In some cases the system was highly active, even surpassing the performance of similar ruthenium catalysts in conventional solvents.50
 | ||
| Fig. 11 Continuous flow hydrogenation of carbon dioxide in ILs/scCO2.50 | ||
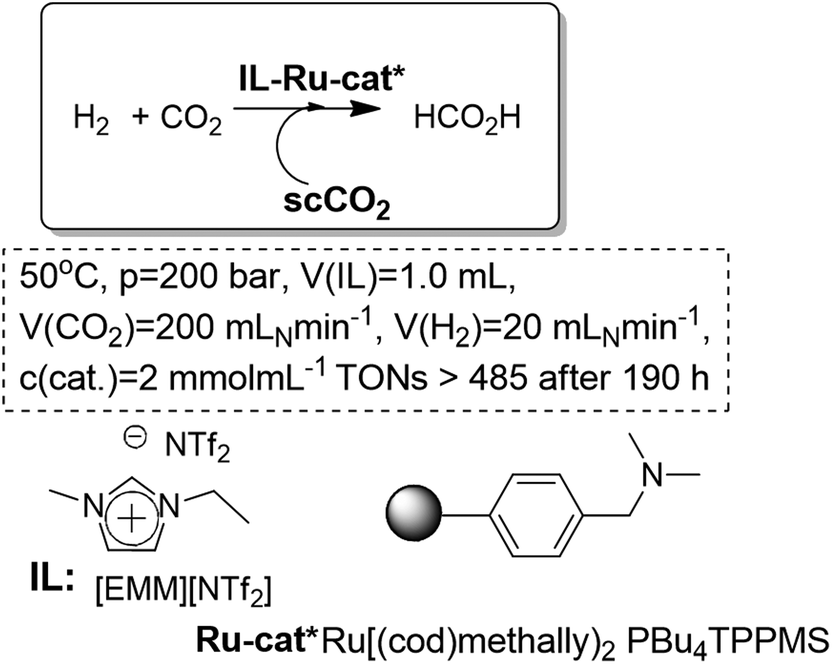
Similar biphasic systems can also be applied for the synthesis of chiral compounds. For instance, Leitner et al. have reported the enantioselective synthesis of N-(1-phenylethylidene)aniline by hydrogenation using a biphasic scCO2/[EMIM][NTf2] system. The combined use of IL-scCO2 reduces the required pressures of H2. Thus, under very mild conditions, a full conversion and a moderate enantioselectivity could be achieved (Fig. 12).51,52 Although the initial studies were performed under semicontinuous conditions, the easy isolation and recycling of the IL-catalytic phase by successive reaction–extraction cycles allowed the consecutive use of the system for at least seven cycles without any loss in conversion or enantioselectivity. The system was further improved to efficiently obtain a fully continuous process.53
 | ||
| Fig. 12 Continuous flow enantioselective hydrogenation of imines in ILs/scCO2.51–53 | ||
In a similar way, the continuous-flow asymmetric hydrogenation of methyl propionylacetate in a biphasic IL/scCO2 was reported. The synergic use of a chiral catalyst immobilised on an IL-phase with different acid additives led to very efficient catalytic systems showing a good conversion (>90%) and enantioselectivity (80–82% ee) with a single pass. The initial catalyst activity was retained at 91% after 100 h and at 69% after 150 h time-on-stream, whereas the enantioselectivity remained practically constant during the entire process. A total turnover number of ∼21000 and an averaged space–time yield of 149 g L−1 h−1 were obtained in a long-term experiment.54
The potential of this methodology has been extended for reactions where the catalyst involved is sensitive to air, changes in pressure, temperature, etc. For instance, the hydrovinylation of styrene using the Wilke's catalyst was carried out in an IL-scCO2 biphasic continuous flow system.55 The stability of the catalyst is improved (more than 60 h of continuous operation) in comparison with the recycling in the batch mode, where the catalyst was deactivated by the consecutive separation and reuse cycles.
Finally, the IL-scCO2 biphasic continuous flow systems can also be exploited to design biocatalytic processes. In general, the possibility of immobilization of biocatalysts onto IL-phases is a simple approach to enhance their stability. In this case, the biocatalyst is fixed (either dissolved or suspended) and activated by the IL phase, while the scCO2 becomes the mobile phase.56 Lozano and Leitner and Reetz were the pioneers in the development of such systems for the kinetic resolution of phenyl ethanol using CALB (Candida antarctica lipase B) as the biocatalyst (Fig. 13).57,58 The system could be further modified to fully explore the possibilities than supercritical fluids provide for the continuous separation of the two products formed based on their different solubilities in the scCO2 phase.59
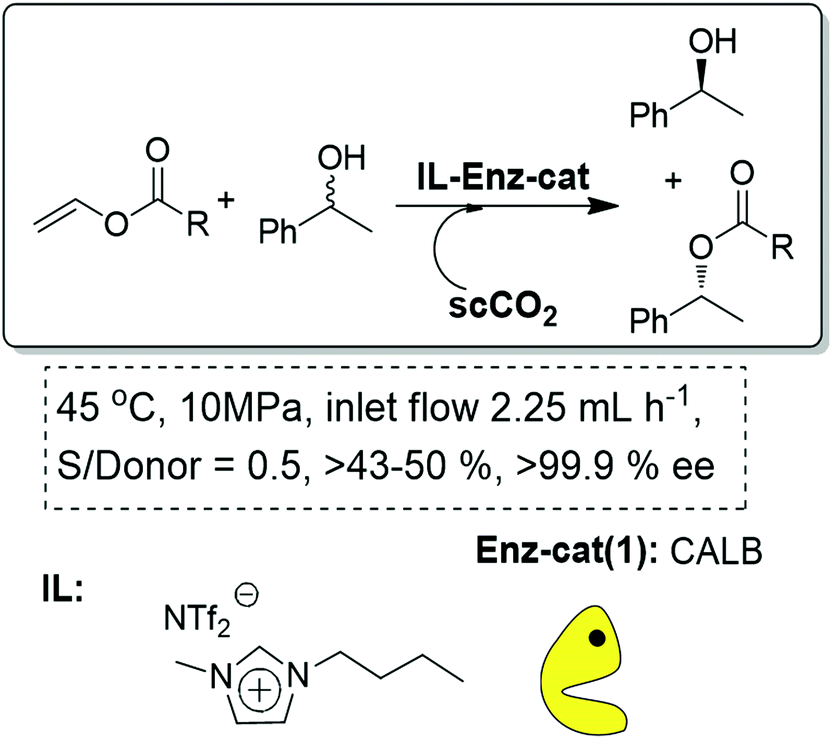 | ||
| Fig. 13 Continuous flow enzymatic resolution in ILs/scCO2.57–59 | ||
4. Supported ionic liquid phases for continuous flow processes
A step forward for the simple implementation of continuous flow processes based on ILs consists in the absorption of the ILs on a porous support material (Supported IL Phase: SILP).60 This strategy brings together the full potential of homogeneous biphasic IL-based systems with those provided by catalysts immobilised onto an insoluble support. The incorporation of the (bio)catalyst is then carried out by its adsorption on the surface modified with the IL-phase.61 The resulting continuous flow catalytic processes can use either a gas phase or a supercritical fluid phase as the mobile phase to deliver the reactants to the IL phase and to extract the products. The main limitation of this approach is the possible leaching of the IL-phase from the support. This phenomenon can be due to either certain solubility of the IL in the mobile phase or the products (which severely limits the use of liquid phases for delivery and extraction of reagents and products) or due to physical abrasion during extended exposition to the mobile phase. The type and morphology of the support is also a key parameter to achieve an efficient system. This type of set-up has been evaluated for the development of different industrial relevant processes under continuous flow conditions (Fig. 14).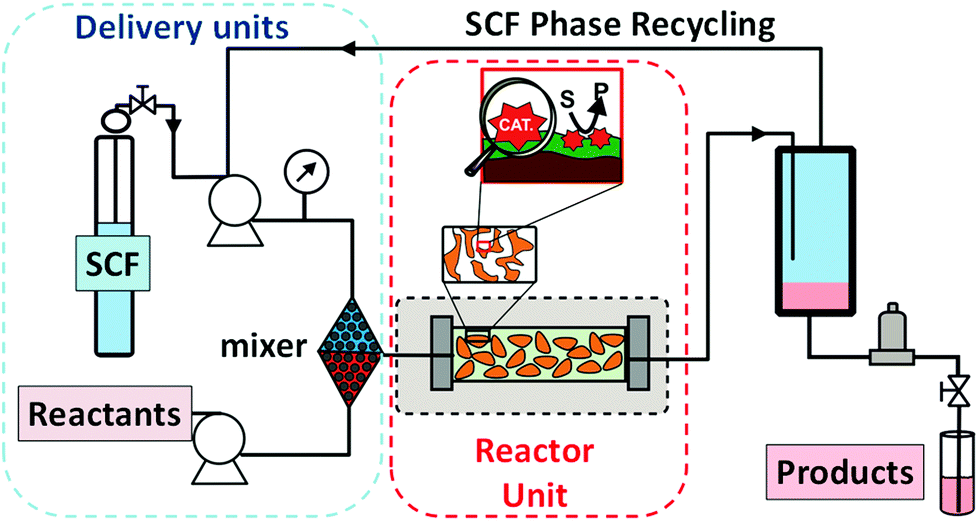 | ||
| Fig. 14 Continuous flow systems based on SILPs either using a gas-phase supply of the substrates (and removal of products) or with a scCO2 stream as the mobile phase. | ||
Different reports have described the application of this methodology for hydroformylation reactions in either scCO262 and/or in the gas phase.63 Thus, for the SILP/scCO2 system, higher rates than the batch bulk-IL/scCO2 system were obtained, with a TOF value of 800 h−1 and more than 3-fold enhancement of spatial-time yields. Furthermore, the system showed good stability maintaining the TOF values (500–800 h−1) for at least 40 h of continuous flow operation (TON > 20000) independently of the IL used to prepare the SILP.64
A similar system has been reported using as the gas phase a highly diluted, technical grade, C4 feed containing 1.5% 1-butene, 28.5% 2-butenes, and 70% of inert n-butane (Fig. 15). To obtain the desired product, n-pentanal, the Rh-biphephos sulfonated catalyst (Rh-sulfoxantphos) was immobilized in the SILP system to allow for consecutive isomerization/hydroformylation activity. The resulting SILP-catalyst system converted up to 81% of the reactive butenes, with a residence time of 155 s in the reactor. The n-pentanal selectivity was greater than 92% for more than 500 h time-on-stream in the continuous gas-phase reaction.65 The non-ionic Rh-diphosphite catalyst was also used to prepare a catalytic SILP system. The catalyst was found to be sensitive to hydrolysis. However, when a dried feed gas was used and an acid scavenger added to the immobilized ionic liquid, the catalyst stability could be extended to more than 30 days time-on-stream. During this operation time a total turnover number of more than 350000 was achieved. Under slightly harsher reaction conditions (120 °C, 25 bar total pressure) the performance of the Rh catalyst-SILP system was increased, with the same highly diluted feed, reaching a TOF of 3600 h−1 and a space–time yield of 850 kg of n-pentanal m3 h−1.66 Under these conditions, the selectivity towards n-pentanal always remained above 99%. It is worth mentioning that in an IL–organic biphasic operation, this non-ionic ligand would leach quickly into the product phase. A structural modification (e.g. attachment of ionic groups) would be necessary to immobilize it in the IL-phase. This is an important finding for the application of a much broader range of non-ionic ligand systems in future catalysis studies based on SILPs.
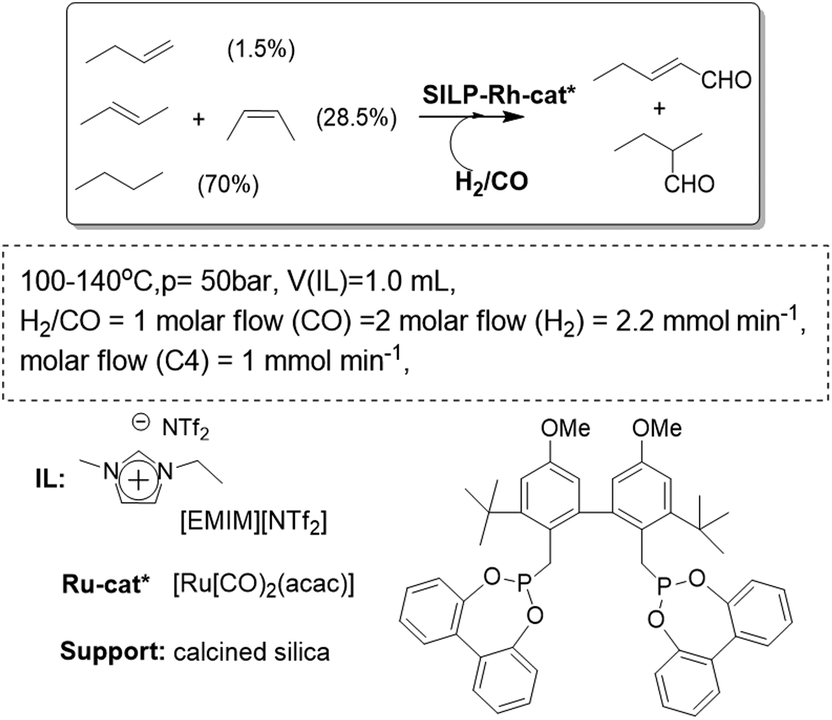 | ||
| Fig. 15 Continuous flow gas-phase hydroformylation in SILPs.65,66 | ||
A related approach has been developed for the synthesis of amines in a one-pot cascade reaction involving an alkene hydroformylation followed by a reductive amination. Both the nature of the IL and the characteristics of the support can be used to tune the efficiency and chemoselectivity between the desired amination/hydrogenation process and the undesired aldol reaction (Fig. 16).67 Thus, SILP-supported catalysts based on neutral oxides and porous carbon supports and ILs of low basicity and lipophilicity resulted in very selective hydroaminomethylation catalysis. The long-term stability of the developed systems is remarkably high, with an effective time-on-stream of more than 18 days, reaching a total turnover number of 115000.
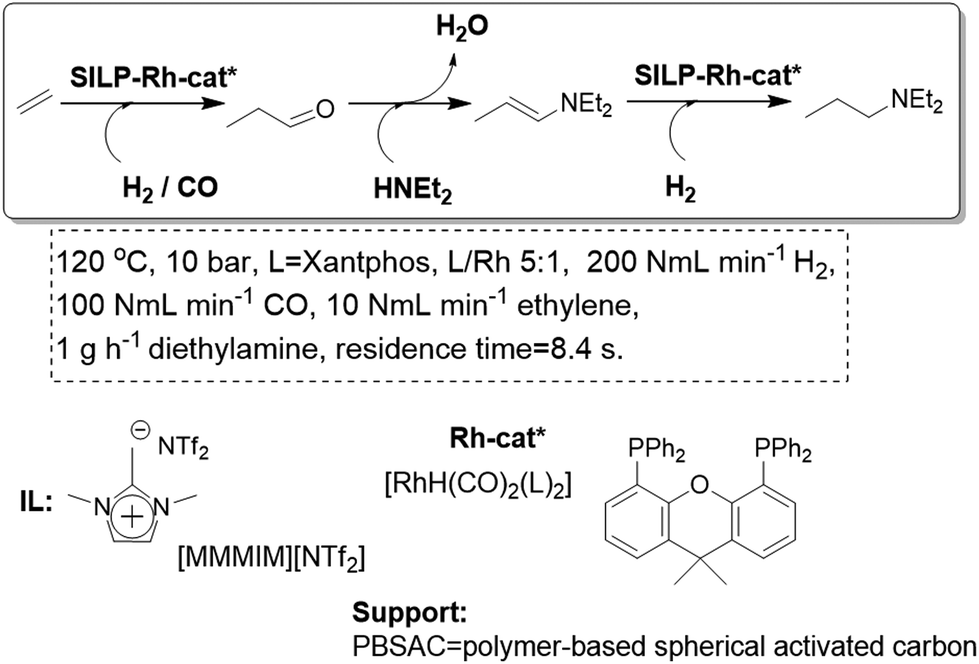 | ||
| Fig. 16 Continuous flow gas-phase hydroaminomethylation in SILPs.67 | ||
The continuous flow homogeneous alkene metathesis can also be performed using this methodology. The self-metathesis of oleochemicals is of considerable interest for the conversion of low value feedstocks into useful chemicals for petrochemistry and polymerization. The system involves a Ru homogeneous boomerang catalyst with an imidazolium tag on the reactive carbene. At a flow rate of 0.05 mL min−1 and a temperature of 50 °C, up to 64% conversion was obtained over the first 6 h. A slight deactivation was observed after 10 hour on a continuous flow stream (Fig. 17). In the most favorable cases, 6 g of substrate per hour can be converted to an equilibrium mixture of products in a 9 mL reactor. No solvent is present in the collected product and the only purification needed is the fractional distillation of the equilibrium mixture of products.68
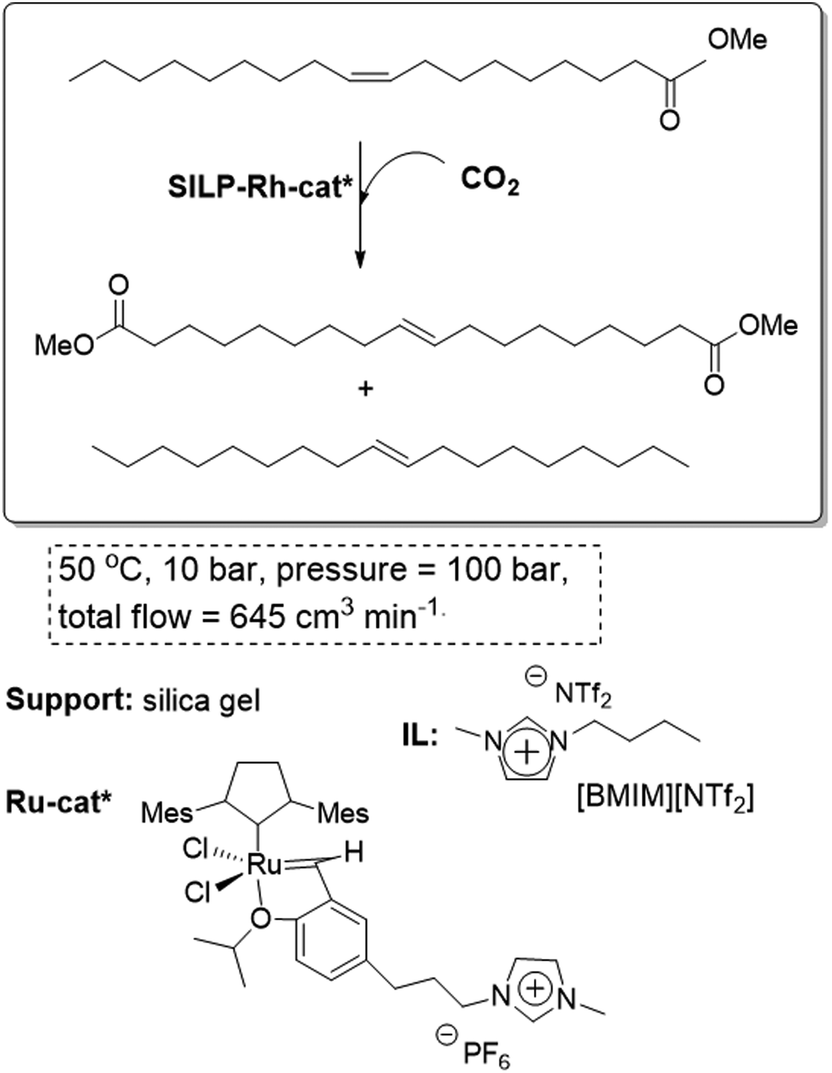 | ||
| Fig. 17 Continuous flow homogeneous alkene metathesis in SILPs.68 | ||
Ru-based SILP catalysts have also been applied for the gas phase asymmetric hydrogenation of α-keto esters.69 With Ru-BINAP, dissolved in the SILP 3-hydroxypropylpyridinium bis(trifluoromethylsulfonyl)imide on silica 100, a stable catalyst performance could be obtained for more than 50 h time-on-stream, although only a moderate enantioselectivity was reported.
The hydrogenation of methyl acetoacetate using dibromo[3-(2,5-(2R,5R)-dimethylphospholanyl-1)-4-di-o-tolylphosphino-2,5-dimethyl-thiophene]ruthenium as the catalyst in [EMIM][NTf2] immobilised on silica 30 rendered enantiomeric excesses in the range of 65–82% ee for more than 100 h time-on-stream of continuous operation.70
A highly enantioselective continuous transformation could be achieved, using a SILP-catalyst based on a chiral transition-metal complex and scCO2 as the mobile phase, for the asymmetric hydrogenation of dimethyl itaconate. A dehydroxylated silica was chosen as the support. The pretreated silica gel 100 (surface area = 300 m2 g−1, pore volume = 1 mL g−1) was coated with [EMIM][NTf2] saturated with QUINAPHOS-Rh as the catalyst (Fig. 18). Deposition of the IL solution onto the support caused the surface area to decrease to 130 m2 g−1 and the pore volume to decrease to 0.44 mL g−1. The full conversion of dimethyl itaconate with an excellent enantioselectivity of >99% ee for the desired alcohol could be achieved. The total TON reached a remarkable value of 115000 moles of substrate per mole of rhodium. After 65 h, 3 mg of catalyst in 0.4 mL of IL had converted over 50 g of substrate, corresponding to a high space–time yield of 0.3 kg h−1 L−1.71 Although good initial catalyst performance could be achieved with a variety of silica-based materials, the long-term stability was very sensitive to the surface properties of the support. The selection of hydrophobic, fluorinated materials (meso-scale) or the introduction of water scavengers into the processing unit (macro-scale) proved effective in retarding or preventing catalyst deactivation caused by water poisoning (molecular scale).72
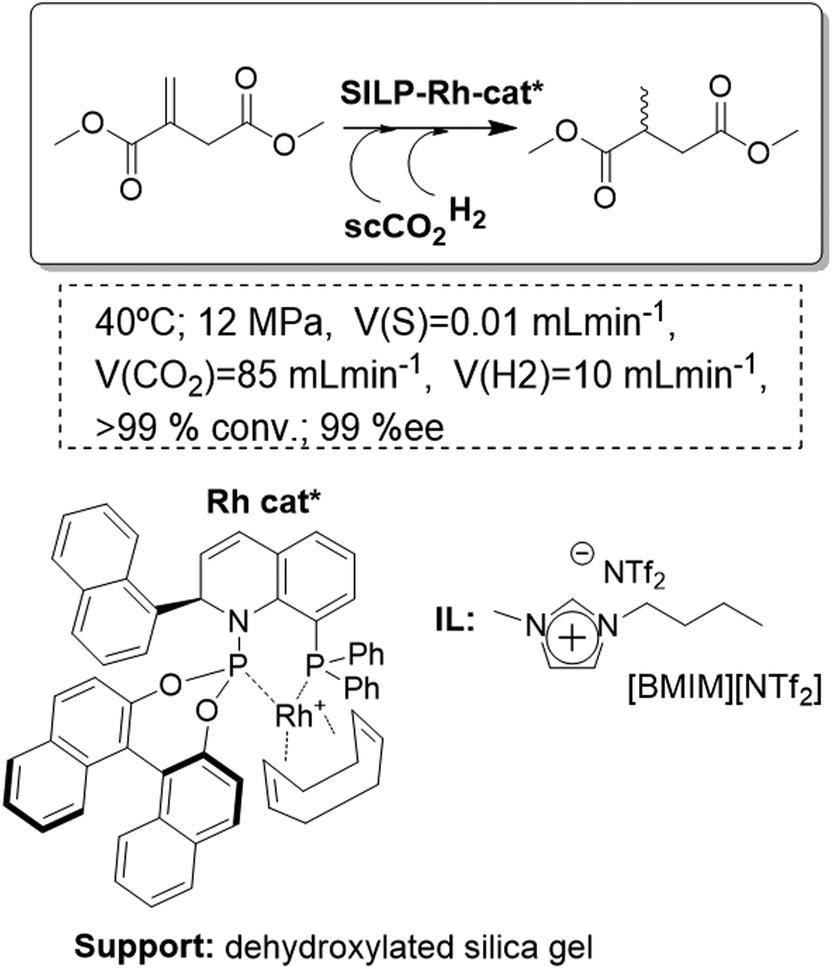 | ||
| Fig. 18 Continuous flow enantioselective hydrogenation in SILPs.71 | ||
PdNPs of 5 and 10 nm obtained in either [BMIM][PF6], or [BMIMOH][NTf2] have been supported on carbon nanofibers (CNF) anchored to sintered metal fibers (SMF) and applied for the selective acetylene hydrogenation under continuous-flow conditions. The lower solubility of ethylene as compared to acetylene in the IL results in a high selectivity to ethylene, up to 85% at 150 °C. The catalytic system also demonstrated high efficiency and long-term stability without any deactivation in ethylene-rich feed (2 vol% of acetylene, 40 vol% of ethylene, 10 vol% of H2 in Ar), and therefore the system shows promise for industrial application.73
The SILP methodology has been applied to screen transition metal complexes for their catalytic activity under very controlled steady-state conditions in the search for new water–gas shift (WGS) catalytic systems. Differing from classical batch experiments, this method provides a great amount of information on catalyst formation and deactivation phenomena. This makes the selection of the most promising candidates, and the structural motifs for the further development of even more effective WGS catalysts much more reliable. Using as a model feedstock H2O and CO, in a continuous screening reactor, an activity of 3 mol H2 mol−1 Ru h−1 was attained at 160 °C and 0.1 MPa after a prolonged induction period of more than 20 h. This moderate activity already exceeded that of a commercial copper-based catalyst under the same ultra-low temperature conditions (0.5 mol H2 mol−1 Cu h−1).74,75 The systematic development of a SILP-catalyst by variation of the ionic liquid, the support, the additive and the metal complex has shown a cooperative interplay of the influencing factors when combining the individually optimized factors in one system. In this way, a new SILP-WGS catalytic system of unprecedented activity, under extremely mild conditions, displaying a unique re-start behavior and robustness under typical storage conditions has been developed (Fig. 19).76
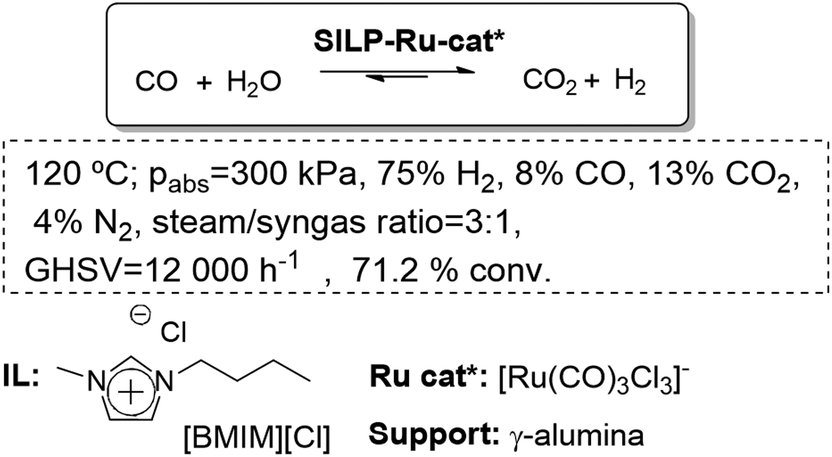 | ||
| Fig. 19 Continuous flow water–gas shift reaction in SILPs.76 | ||
Another interesting application of the SILP approach in the gas phase is the Friedel–Crafts isopropylation of toluene and cumene. It could be performed using an acidic SILP system, where the different Lewis acidity of the catalysts and the IL loading play a key role. The study showed that a moderate IL loading (α = 0.2) with a considerably high aromatic-to-propylene ratio (7:1 molar ratio) were the best conditions to suppress consecutive alkylation reactions that fill the catalyst pores with low-volatile heavies. Besides, the support had a beneficial effect on the selectivity to mono-alkylated products. The optimized acidic SILP system was able to effect the Friedel–Crafts alkylation for more than 210 h time-on-stream showing constantly high selectivity to the mono-alkylated product (>95%) and excellent selectivity to meta-cymene within the cymenes (up to 80%) (Fig. 20).77
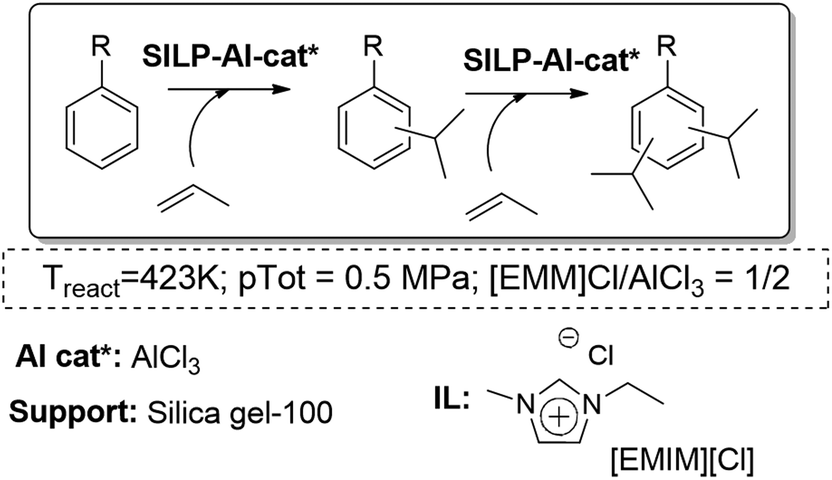 | ||
| Fig. 20 Continuous flow gas-phase isopropylation of toluene and cumene in SILPs.76,77 | ||
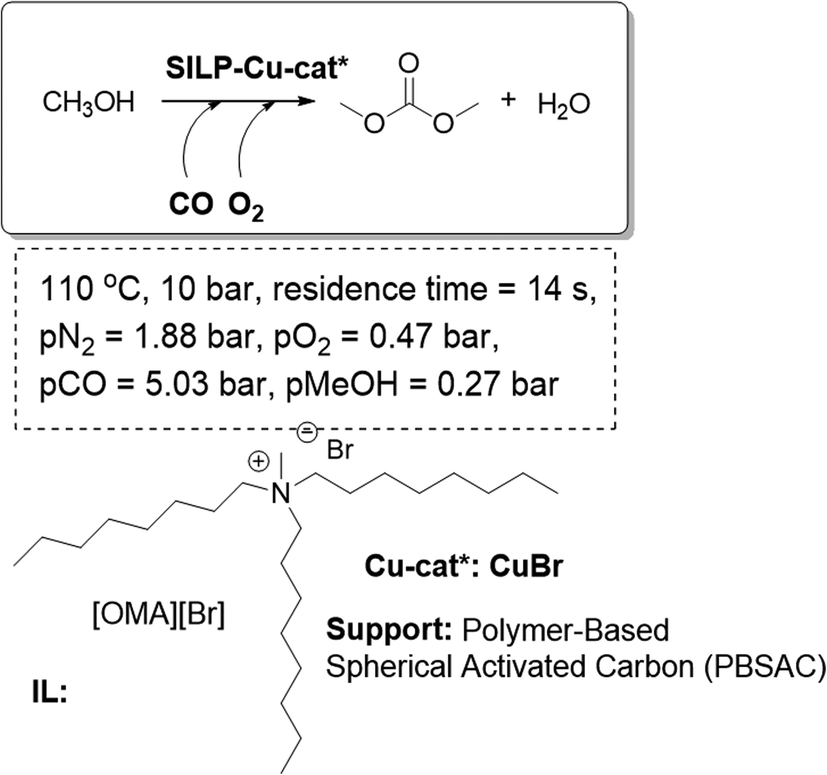
SILP-based systems have also been assayed for the continuous gas-phase methanol carbonylation to produce dimethyl carbonate (DMC), which is a potential alternative to phosgene in polycarbonate and isocyanate synthesis. Fixed-bed reactors based on silica-SILPs bearing a rhodium iodide Monsanto-type catalyst have been proven as good catalytic systems. The reaction proceeds with an excellent activity and selectivity towards acetyl products.78 Alternatively, a system based on CuBr as the catalyst on a Polymer-Based Spherical Activated Carbon (PBSAC) as the support and [OMA][Br] as the IL-phase yielded the best results (Fig. 21). An optimized SILP system was further developed including an increased Cu- and IL-loading and the addition of the ionic Lewis base [DMMAP][OTf]. This catalytic system, using the SILP concept, was tested over 50 h time-on-stream, achieving a total turnover number of more than 600, which is an unprecedented productivity in the gas-phase homogeneous oxycarbonylation.79 The process design requires a smaller reactor size than the existing technology, in order to obtain the same productivity, which makes the SILP carbonylation concept potentially interesting for technical applications.
 | ||
| Fig. 21 Continuous flow gas-phase oxycarbonylation of methanol for the synthesis of dimethyl carbonate in SILPs.76,79 | ||
Recently, nickel complexes were immobilized on Supported Ionic Liquid Phases (SILPs) and applied as catalysts for the tandem dimerization/isomerization of ethylene to 2-butene in a fluidized bed reactor. The better heat removal in the fluidized bed improved the catalyst stability and allowed for a more detailed investigation of the deactivation mechanism. Based on kinetic studies, a second order deactivation mechanism was proposed in which two nickel complexes dimerize if the supply of ethene is insufficient.80
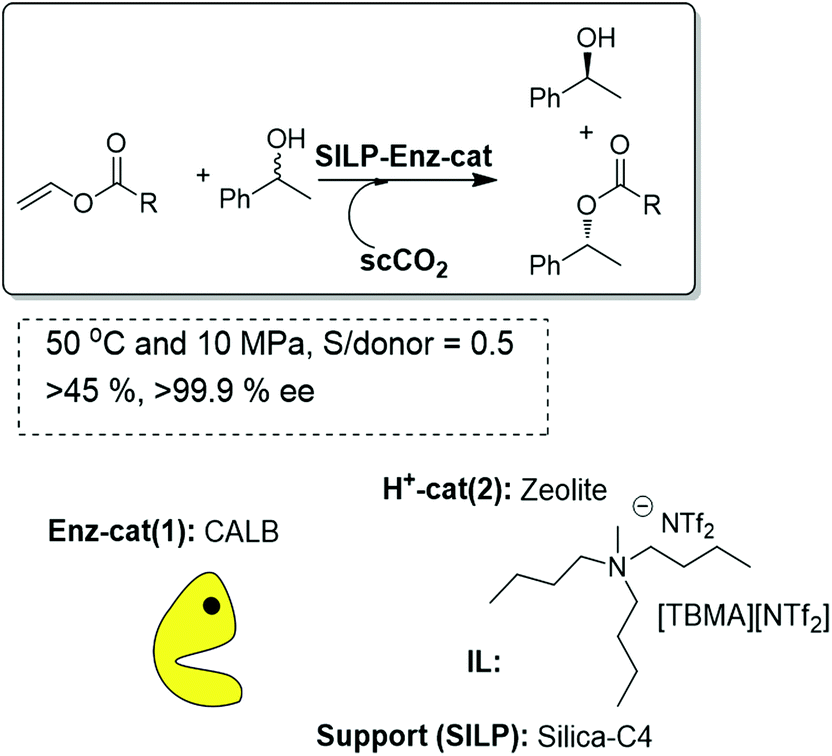
A completely different application of the SILP approach has been used for the design of enzymatic reactions. Lozano and co-workers have evaluated the immobilisation of free Candida antarctica lipase B (Novozyme 525L) by adsorption onto 12 different silica supports surface-modified with specific side chains (e.g. alkyl, amino, carboxylic, nitrile, etc.) for the kinetic resolution of 1-phenyl ethanol.81 Coating immobilized enzyme particles with ionic liquids (butyltrimethylammonium bistriflimide [BTMA][NTf2] or trioctylmethylammonium bistriflimide [OMA][NTf2]) resulted in an increase in activity (10 times) under batch conditions. It is worth mentioning that, in this case, the enzyme maintained an excellent activity and a reasonable stability even at 150 °C, which reveals the important stabilization associated with the combination of the biocatalyst with the IL phase. Very interestingly, the synthetic activity displayed by the CALB-C4-silica/[BTMA][NTf2] in the continuous flow scCO2 system was enhanced up to 35.1 U mg−1 protein, six-fold higher than that obtained in hexane/IL media (Fig. 22).
 | ||
| Fig. 22 Continuous flow enzymatic kinetic resolution in SILPs.76,81 | ||
As many commercial enzyme formulations contain the enzyme immobilized on different support materials (i.e. Novozyme 435 is a commercial product with CALB immobilized on an acrylic resin), a simple alternative for the preparation of the enzyme-SILP system is the direct coating of the commercial material with a thin layer of IL. Lozano et al. created a thin layer of the IL phase on the surface of the particles containing the enzyme and the resulting material was used in a packed bed reactor, using vinyl propionate as the acyl donor (50 °C, 15 MPa, substrate:donor ratio 0.5, conversion >35%, 99% ee for the product).82
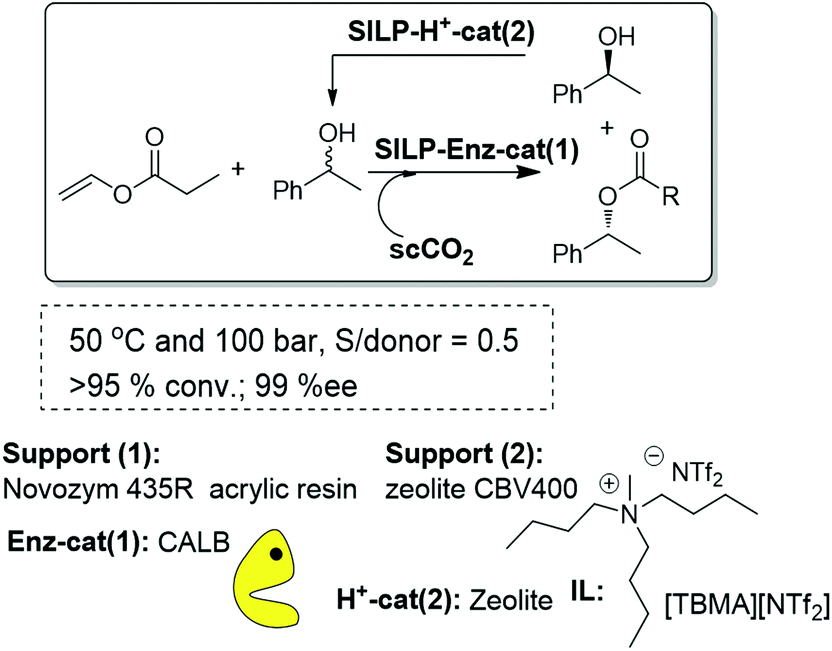
A logical step forward from the kinetic resolution process based on an enzyme-SILP system is the dynamic kinetic resolution (DKR). This can be achieved by introducing an additional chemocatalyst, either an acid or an organometallic catalyst, able to racemize the free alcohol present in solution. The key to success is the compatibility between the biocatalytic and chemocatalytic components, as the interaction between them leads, in general, to deactivation of the enzyme. In this regard, the use of immobilized systems coated with an IL thin film is also advantageous, as the immobilized IL-phase can greatly decrease the possibility of interparticle site–site interactions between two immobilized catalysts. Thus, the first attempts to develop a continuous DKR process of rac-1-phenylethanol combined in a single-pot the biocatalyst (Novozym 435) and the acid catalyst (a silica modified with benzenosulfonic acid groups, SCX) both coated and protected with an IL thin layer. Nevertheless, the direct mixture of the two components afforded a rapid deactivation of the enzyme, most likely because of the influence of the acidity originating at the SCX. To overcome this problem, a fixed bed tubular reactor was assembled, in this case with three different compartments. The SCX/IL was placed in the middle of a high-pressure cartridge, while the Novozym 435/IL mixture was placed at the two ends of the cartridge. The three different layers were separated by glass wool to avoid any physical contact. Using scCO2, at 40 °C and 100 MPa, containing the racemic substrate and the acyl donor as the flowing solvent, excellent results were obtained, with yields up to 75% and 91–98% ee for the R-ester.83 It is worth noting that the yield attained is the maximum that can be expected for a DKR with this configuration. A simpler and more effective approach for this purpose should be, however, the work with a single layer containing a mixture of the enzymatic and the acid catalyst. In order to avoid the interference of the acid catalysts with the enzyme, different acid catalysts of decreasing acidity have been assayed. Excellent results could be obtained, for this configuration (Fig. 23), using zeolites coated with an IL. The reactor was filled with a homogeneous mixture of both catalysts and finally assayed for the DKR of sec-alcohols under a flow of scCO2 at 50 °C and 10 MPa. Because of the lower acidity of the zeolites, no loss of enzymatic activity was observed. With this configuration, yields close to 100% could be obtained. The best results were observed for a multicatalytic reactor prepared by a combination of zeolite CBV400 and Novozym 435 both coated with [TBMA][NTf2]. In this case, the yield was 98% and the ee was 96%, and the system maintained the same activity and selectivity after continuous use for 14 days.84
 | ||
| Fig. 23 Continuous flow enzymatic dynamic kinetic resolution in SILPs.76,84 | ||
The biocatalytic synthesis of biodiesel can be improved by using ILs with cations bearing large alkyl side-chains (e.g. [C18mim][NTf2]), because they are able to dissolve the vegetable oil and methanol substrates in monophasic reaction media.85a The ILs also protect the enzyme against fast deactivation that results from direct contact with methanol, e.g. up to 1370 days half-life time at 60 °C.85b In this context, a continuous flow synthesis of biodiesel in scCO2 can be carried out by using a SILP based on immobilised lipase particles coated with a monolayer of the corresponding IL ([C18mim][NTf2]). Although the system can provide pure biofuel (95% yield at 18 MPa and 60 °C), it was not stable enough for long operation times. The biodiesel continuously produced during the operation was able to dissolve the protective IL shell around enzyme particles, producing a fast biocatalyst deactivation.85c These results underline some of the limitations of this approach and interest in developing approaches based on the permanent protection of the enzyme by ILs in scCO2 flow operation (e.g. by using covalently attached IL phases).
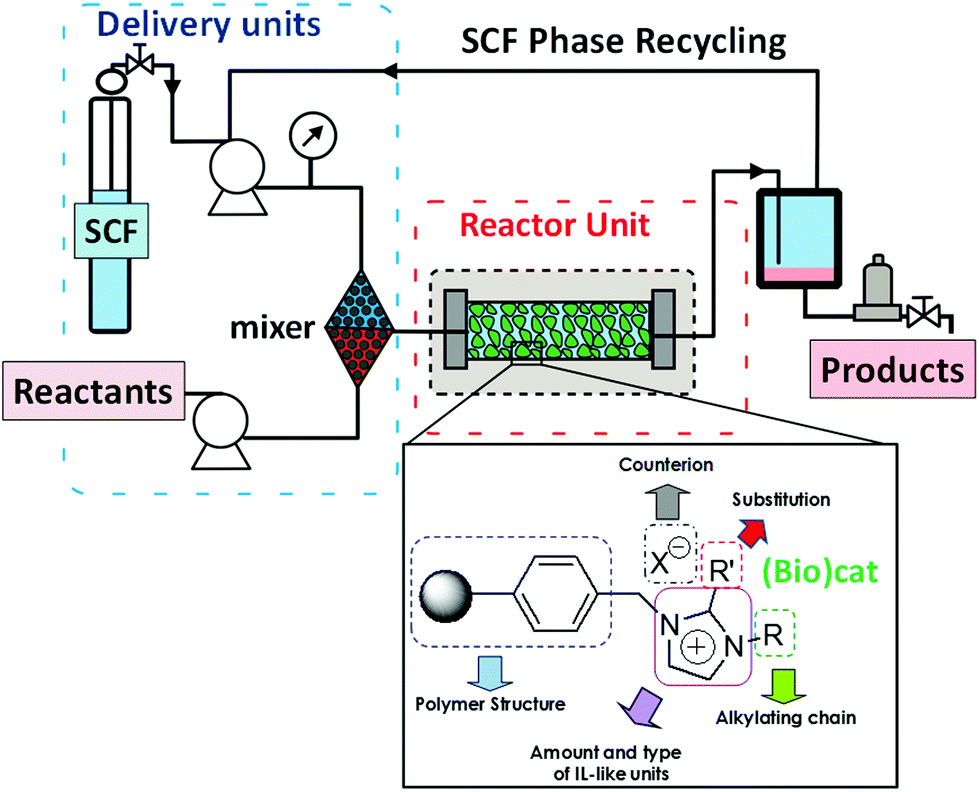
An alternative to the immobilization of the IL-phase by coating the surface of a support with a thin layer of the IL (SILPs) is to attach covalently structural fragments related to ILs to the functional groups present on the surface of the support. The resulting materials have been termed as Supported Ionic Liquid-Like Phases (SILLPs). Both inorganic (silica or other metal oxides) and organic (crosslinked polymers) supports have been used for the preparation of catalysts immobilized in SILLPs.86 Solid materials with multilayer IL structures can also be obtained by the polymerization of polymerizable task specific ILs either directly or in combination with a support.87,88 It is clear that in this case when the IL-like fragment is covalently attached to the support, the problem of leaching is totally eliminated. However, the transfer of the suitable essential properties of the corresponding ILs to the solid phase needs to be guaranteed, and this is not a simple matter. Fortunately, it has been demonstrated that, based on the proper selection of the different design elements involved, it is feasible to transfer the IL properties to the surface of the solid. Therefore, the resulting materials present the advantages associated with ILs but can overcome many of the drawbacks often associated with bulk ionic liquids, facilitating their technological applications.89,90 Indeed, their controlled solid nature enables them to be used to develop fixed-bed flow reactors for continuous flow processes (Fig. 24).
 | ||
| Fig. 24 Continuous flow systems based on Supported Ionic Liquid-like Phases (SILLPs) and using supercritical fluids (SCF) as the mobile phase.90 | ||
The incorporation of the catalyst to the SILLP is simply carried out by adsorption of the homogeneous catalyst as an adequate solution onto the modified surface of the support bearing IL-like units. The continuous catalytic reactions can be performed either in conventional solvents or alternatively, and moving towards greener methods, using different types of supercritical fluids or in solvent free processes.
The nature of the support provides an additional design vector, besides those related to the structure of the IL-like units, to tune the characteristics of the new materials, in particular in the case of polymeric supports. Thus, envisioning the different potential applications of SILLPs, a variety of polymeric supports can be obtained with different morphological, chemical and physical characteristics by controlled variation of their synthetic protocol and the existing functionalities. This modular design facilitates both the immobilization and the stabilization of the catalytic moieties.
Based on the fact that the polymeric SILLPs present at the molecular level the same fundamental properties of the bulk ILs,91 they can be used, for instance, for simple preparation and stabilisation of metal nanoparticles (MNPs) from different metal precursors, reproducing the analogous behaviour observed for bulk ILs.92 Thus, PdNPs supported onto monolithic SILLPs have been used as stable and active catalysts for the Heck reaction. As an alternative solvent to conventional organic solvents used for this reaction, hot pressurised ethanol was used (Fig. 25). Ethanol is not only benign, easily available and relatively cheap, but is also a solvent whose properties are easily adjustable with pressure and temperature. Supercritical ethanol provides a less corrosive solvent than supercritical water and with more accessible critical parameters (Tc = 516 K, Pc = 6.4 MPa). A stable performance on a continuous flow fixed bed reactor (ca. up to 91 bed-volumes) was observed for the Heck reaction in near critical EtOH (200 °C and 8 MPa). The presence of IL-like moieties in the polymer seems to help in stabilising the Pd(0), simultaneously reducing the leaching through a release and catch mechanism.93 The morphological and chemical properties of the support and the IL-like fragments, including the loading, in the polymeric ionic-liquid-like phase have been shown to play an active role in the catalytic cycle.94
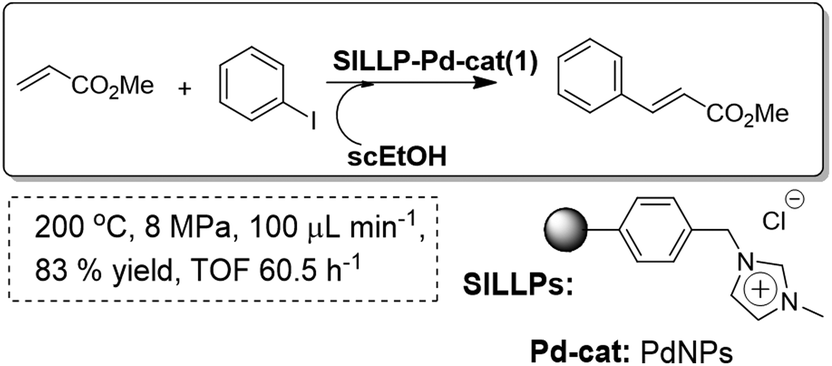 | ||
| Fig. 25 Continuous flow Heck reaction in SILLPs/scEtOH.93 | ||
A partly related approach has been the immobilization of a biocatalytic lipase onto a covalently Supported Ionic Liquid-Like Phase (SILLP).95 CALB supported on SILLPs was shown to be an efficient and very stable catalyst for the continuous flow synthesis of citronellyl propionate in scCO2. The presence of a high IL/enzyme ratio is reflected in high stabilization of the CALB in those systems. Thus, the process could be carried out at 80 °C with yields of 93% for more than 10 h without any appreciable deactivation of the enzyme (Fig. 26). The results substantially exceeded those obtained for packed-bed reactors with a supported silica-CALB-Si-4 catalyst under the same experimental conditions, highlighting the key role played by the covalently attached IL-like units. The SILLPs not only provide an adequate microenvironment for the lipase action, but also improve the mass-transfer phenomena of hydrophobic substrates and products from the scCO2 phase, leading to a highly selective and stable immobilised enzyme even at high temperatures and pressures.
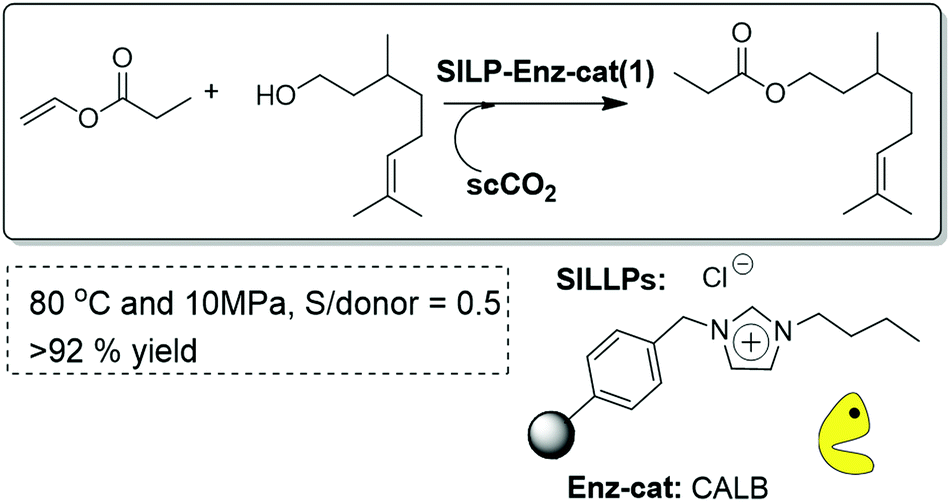 | ||
| Fig. 26 Continuous flow enzymatic synthesis of citronellyl propionate in SILLPs/scCO2.95 | ||
PS-DVB-derived SILLPs can also be prepared starting from commercial Merrifield resins in the form of beads. Both gel-type and macroporous resins containing covalently attached imidazolium subunits have shown to act efficiently for the immobilization of CALB, in particular for the polymers having a relatively high content in IL-like fragments. The macroporous resins are, however, more appropriate for being packed into tubular reactors and for the development of flow processes, according to their higher mechanical stability. In this way, CALB immobilised onto SILLPs bearing 1-decyl-2-methyimidazolium cations has been used as the biocatalyst for the continuous flow synthesis of biodiesel (methyl oleate) by the methanolysis of triolein in scCO2 (18 MPa, 45 °C).96 The system showed very good stability (85% mean yield of biodiesel after 45 cycles of 4–8 h) in scCO2. Again, the nature of the SILLPs regarding both the substitution pattern at the IL-like units and the type of polymeric support used were key factors to obtain an efficient supported biocatalyst. These elements determine the efficiency of the mass-transfer processes at the interfaces, either of the substrates to the active site or the products from the enzyme. The selection of an IL that contains a large alkyl chain in the cation results in a clear improvement of the efficiency for the biotransformation in monophasic liquid systems and the same takes place when the corresponding SILLPs are prepared. Thus, controlling the structure of the SILLPs, it is possible to develop efficient continuous processes for the transformation of vegetable oil into biodiesel (Fig. 27).
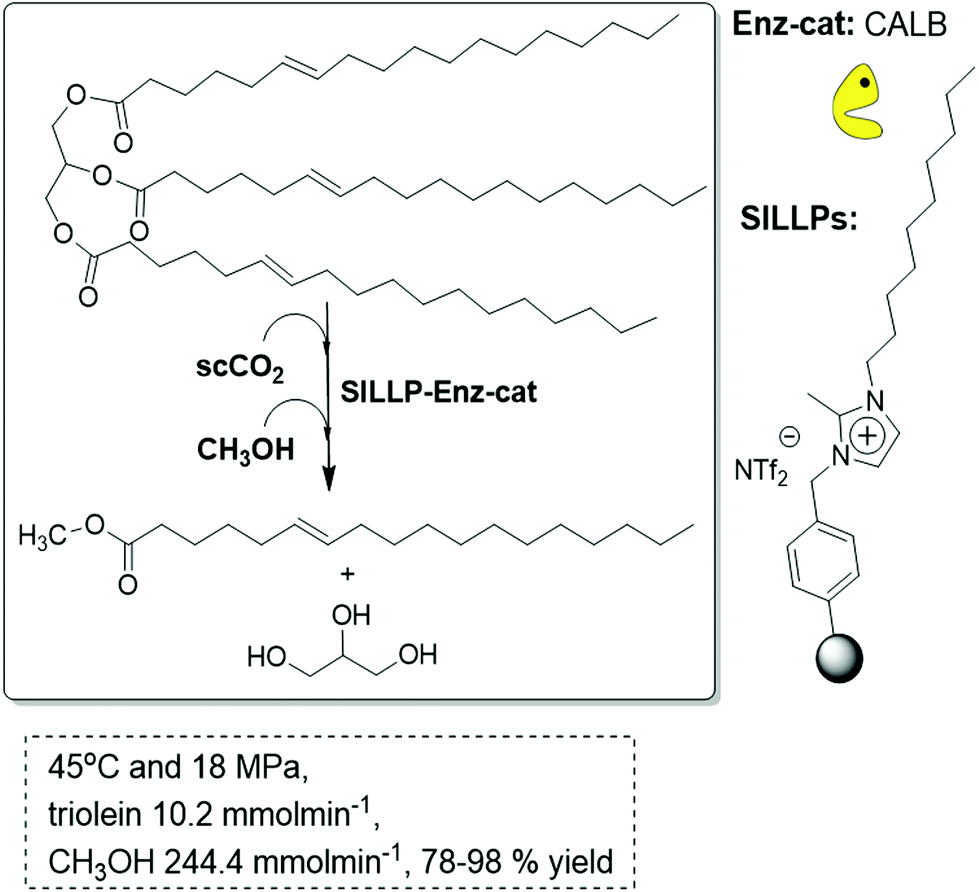 | ||
| Fig. 27 Continuous flow enzymatic biodiesel synthesis in SILLP-scCO2.96 | ||
The corresponding SILLP-based fixed-bed biocatalytic mini reactors were assayed for the continuous KR of sec-alcohols, using scCO2 at 50 °C and 10 MPa for feeding the substrates to the reactor. The results were excellent, as yields of 50% of the expected enantiopure ester were obtained. A high level of stability was also observed for the mini bioreactor, as the activity and selectivity was maintained without any significant change after 6 days of use under the above-mentioned conditions. It is remarkable that, in this case, resins with imidazolium fragments containing chloride as the counter anion showed to be more active, while the NTf2− anion had revealed to be the most appropriate for other enzymes supported either in SILPs or SILLPs.97
As was mentioned above, development of an efficient DKR allows raising the yields of the desired ester to 100% but requires the combination of the biocatalyst with an acidic or organometallic catalyst to produce the continuous racemization of the undesired enantiomer of the alcohol. Taking into account the former results discussed above, different zeolites (CP811C, CBV720 and CP811E) were studied as the acid catalysts for the racemization in combination with the CALB supported on SILLPs based on bead-type resins.
The complete isolation of the enzyme and the acid catalysts guarantees that no deactivation of the enzyme can be associated with the contact with the zeolite. The systems were maintained under continuous flow conditions using scCO2 at 50 °C and 10 MPa. A “one pot” single columnar minireactor was prepared using a mixture of the CALB-SILLP polymer with zeolite CP811E-150 (Fig. 28). Good results were obtained when the zeolite catalyst was coated with a small amount of an IL. This follows the same trend observed in the case of SILPs. Nevertheless, in contrast to the observations obtained in that case with the use of commercial immobilized CALB (Novozym 435), no additional coating with an IL of the biocatalyst was required for the stabilization of the enzyme. This clearly highlights how SILLPs are able to efficiently stabilize CALB against deactivation by scCO2 or in the presence of acidic catalysts.
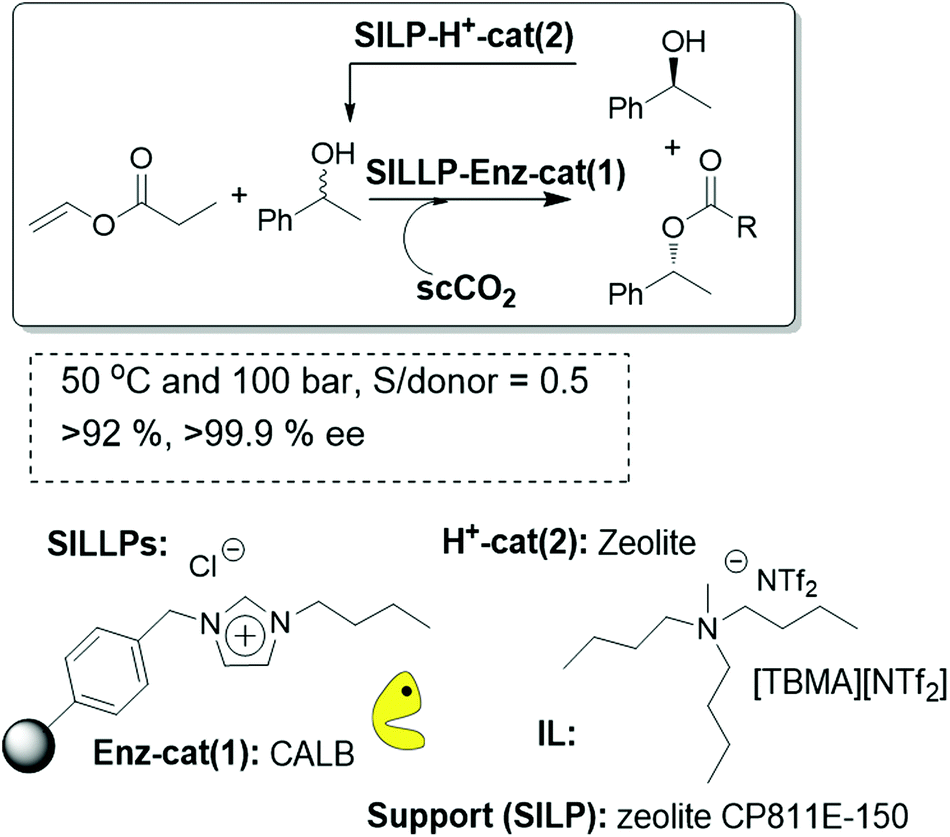 | ||
| Fig. 28 Continuous flow enzymatic dynamic kinetic resolution in SILLP-based systems.97 | ||
The combination in a single reactor of SILLPs with different functionalities can lead to new catalytic systems (catalytic cocktails) with improved properties towards their application for C–C coupling reactions. In this regard, it is important to understand that the chemical functionalities present in SILLPs can play different functions in the reaction medium. This can involve simultaneously (or consecutively) their actuation as supported reagents (i.e. supported bases or acids), precatalysts and scavengers of undesired reaction side-products.
The Heck reaction could also be conducted under continuous-flow conditions using a packed bed of a SILLP polymeric cocktail containing basic functionalities and PdNPs and employing supercritical carbon dioxide (scCO2) as the solvent (Fig. 29). This approach cannot be carried out with the use of soluble bases as this eventually leads to the clogging of the reactor. Finally, this kind of SILLP cocktail enables the combination of different reactions and purification steps in a single reactor, which represents a significant improvement in terms of process intensification and green chemistry. This is particularly true in the case of using scCO2 as this allows the direct production of pure crude materials not contaminated either by salts or by solvents.98
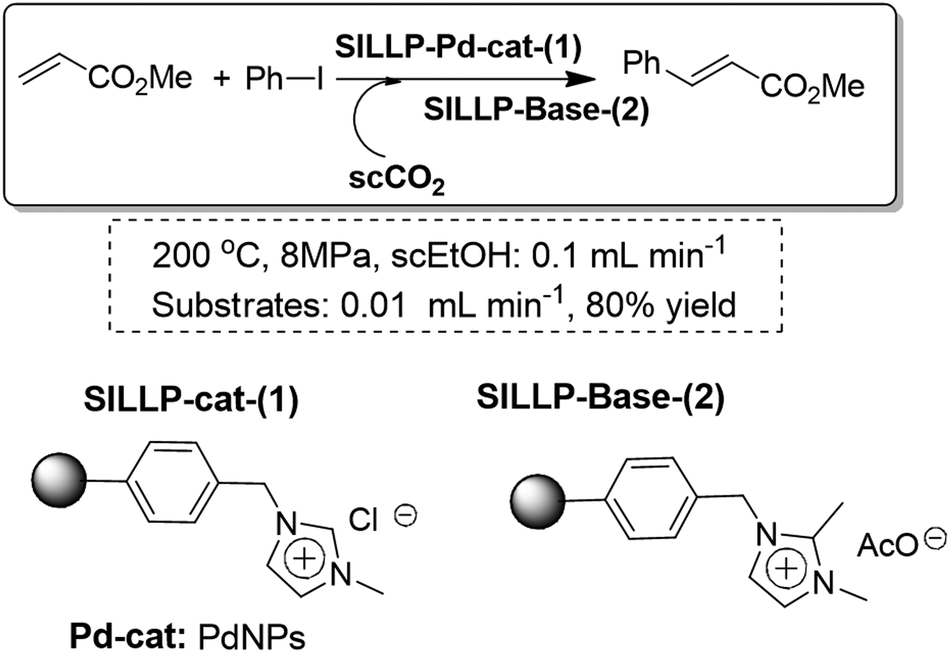 | ||
| Fig. 29 Continuous flow Heck reaction using a SILLP cocktail in scCO2.98 | ||
Supported ionic liquid-like phases (SILLPs) have also been efficiently applied as organocatalysts for the cyanosilylation of carbonyl compounds using trimethylsilyl cyanide under solvent-free conditions. They acted as efficient catalysts for a variety of carbonyl compounds including aromatic, aliphatic and α,β-unsaturated systems. Furthermore, the supported nature of the SILLPs allowed for the development of a continuous flow synthesis of cyanohydrin trimethylsilyl ethers (Fig. 30). The effect of the morphology of the polymer (gel-type or macroporous bead resins or, alternatively, a monolithic polymer) was also studied, which allowed the selection of the most suitable materials for the efficient continuous flow single pass synthesis of cyanohydrin trimethylsilyl ethers. The optimized system provided excellent productivities and high yields without observing any decrease in the activity of the catalyst with time. The results obtained showed a productivity of up to 45.1 g of cyanohydrin trimethylsilyl ether per g of SILLP used per hour. This process perfectly conforms to the features of green chemistry: no waste regarding side-products and unconverted reactants, metal-free, solvent-free, excellent catalytic activity, very low E factor and no requirement for separation.99
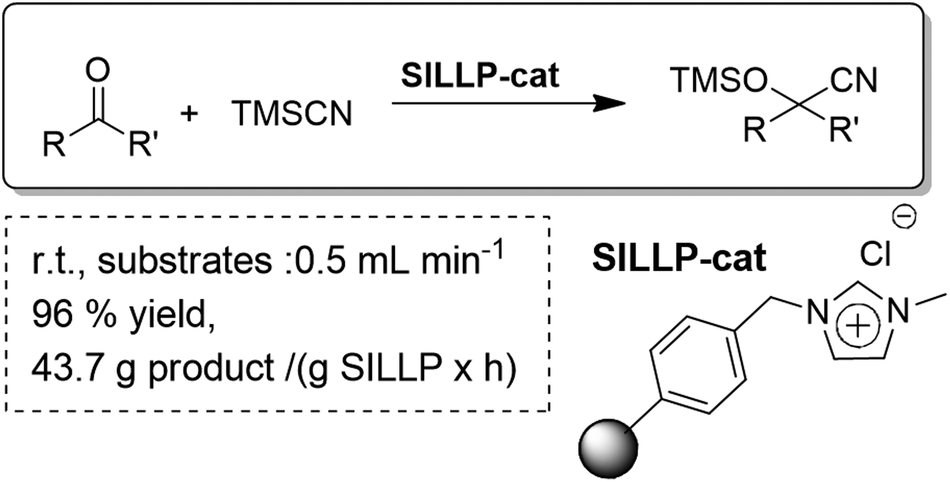 | ||
| Fig. 30 Continuous flow solvent-free cyanosilylation of carbonyl compounds catalysed by SILLPs.99 | ||
The exchange of the counter anion in the IL-fragments of SILLPs to introduce a basic anion such as OH− or AcO− led to the development of a basic catalyst for the continuous flow solvent-free Henry nitroaldol reaction (Fig. 31). A total conversion of the substrate into the product was achieved with residence times as short as 1–3 min, using a monolithic mini-reactor with a free-volume of ca. 500–800 μL.
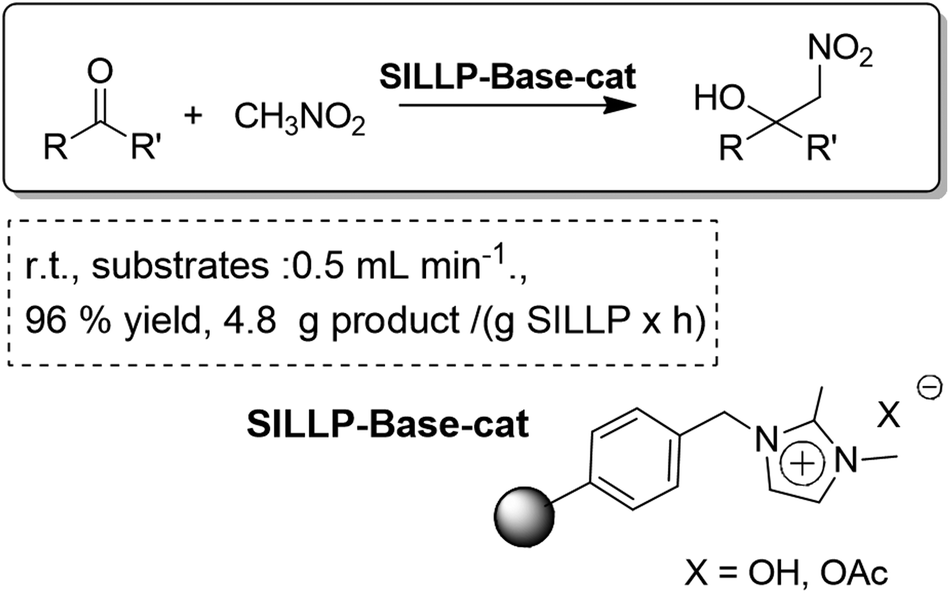 | ||
| Fig. 31 Continuous flow solvent-free Henry reaction catalysed by SILLPs.100 | ||
Indeed, if the TON values for the related batch and continuous flow processes are compared, higher values are obtained for the flow systems. The base-supported systems present a high throughput and are advantageous for the upscale of the process. Although the activity of the catalysts decays with time, the continuous flow set-up allows easy and efficient washing and recycling of the catalytic system affording complete recovery of the activity. Indeed, a set-up with parallel reactors would allow continuous production for long periods of time, by switching from one to the other when the catalytic activity decreases.100
5. New developments and future directions
Additional exciting possibilities of the marriage of ILs/capillary microfluidic devices are the generation of double and multiple emulsions, in a similar way to those of water-in-oil-in-water (W/O/W).101–104 Such two-phase systems based on the presence of an IL-phase enable for extraction/separation and continuous reaction. Thus, a biphasic system 1-butyl-3-methylpyridinium dicyanamide/n-heptane was found to be a highly efficient reaction medium for the biocatalytic synthesis of isoamyl acetate using acetic anhydride as the acyl donor and using 20 vol% of an aqueous CaLB solution.105 Under the optimized conditions, 48.4 g m−3 s−1 of isoamyl acetate were produced, which was almost three-fold better compared to the intensely mixed batch process. This was mainly a consequence of the efficient reaction–diffusion dynamics in the microchannel system, where the developed flow pattern involving intense emulsification provided a large interfacial area for the reaction and simultaneous product extraction to the desired phase.The combination of multi-phase microfluidics with magnetic fields extends these possibilities to the selective manipulation of fluids or nano/micro-particles in flow, through the interaction with these external magnetic fields.106,107 In this regard, a surfactant-free technique for the continuous generation of double emulsion droplets, composed of an organic solvent and a paramagnetic ionic liquid, has been recently reported.108 The specific magnetic properties of the IL allow, through the application of external magnetic fields, for selective and controllable manipulation of the individual emulsion droplets. Such systems might provide, even in a continuous flow set-up, a quick and simple method for the separation of a catalyst immobilized in the IL phase from the reaction mixture. The magnetic and catalytic behaviour of imidazole based magnetic ionic liquids was used to establish a so-called liquid fixed-bed (LFB) in a micro-/meso-structured reactor. As a proof of principle, the esterification of Ac2O with cyclohexanol was investigated by bubbling the generated regular microdroplets of the reaction mixture through the magnetically fixed ionic liquid catalyst. With residence times of approximately 1.4 s a yield of 78.5% of the target ester molecule was achieved.109
6. SWOT analysis of the process based on the combination of ILs and continuous flow processes
Once reviewed the state of the art and future trends in the field of ILs and continuous flow processes, it is important to properly assess the technologies and procedures based on their combination. We have applied a common tool used in strategic planning, viz. strength, weakness, opportunity, and threat (SWOT) analysis.110 SWOT analyses can highlight the advantages and disadvantages of their combined use. Table 1 summarises the main strengths, weaknesses, opportunities and threats based on the above reviewed examples.| Strengths | Weaknesses |
|---|---|
| Develop greener procedures | Full recovery of the ILs required |
| Synergy (better two than one) | Insoluble products (blocking) |
| Applicable for different types of catalysts | Scale-up |
| High quality IL synthesis | New engineering solutions needed |
| (Bio)catalysts more stable and reactive | IL leaching in SILPs |
| Opportunities | Threats |
|---|---|
| New process | Toxicity of ILs |
| New reactor design | IL stability for longer periods |
| Integration separation/reaction | Use and recovery (colouration/decomposition) |
| Intensification | Legislation (REACH) |
| Fast synthesis of new ILs | Cost issues related to ILs |
| New technologies/new IP | |
| Development of new materials (SILLPs) |
The strengths of this technology were highlighted in the previous sections with significant examples that demonstrated the synergies achieved by the combined use of ILs and flow processes. These achievements allow for the development of new greener processes for both the synthesis of high quality ILs (newly designed or already reported) and their application in (bio)catalytic processes. The IL phases generally contribute to the stabilisation of the catalyst and even, in some cases, to an enhancement of their catalytic activity. Thus, under a continuous flow stream, the productivity can significantly improve. In this way, new opportunities rise from the synergetic combination of these enabling tools. The greatest opportunities are the design of new processes based on new types of reactors integrating reaction and separation in a single unit. This will contribute to process intensification and it will require the joint effort of chemists and engineers. Regarding the synthesis of ILs, continuous flow synthesis offers a simple, fast and scalable approach for the synthesis of new ILs based on different building blocks. Furthermore, this combination provides exciting opportunities to generate valuable intellectual properties.
Regarding the weaknesses, recovery and reuse of ILs is a key parameter to be overcome when ILs are used. Although some of the methodologies reported in section 4 that are based on the immobilisation of ionic liquids (SILPs and SILLPs) can help in partially solving this problem, for the systems in which ILs are used as homogeneous solvents their recyclability would be the limiting step. Extraction of non-volatile products and distillation of volatile solutes/impurities from ILs are still the most common methods currently used to recycle and reuse ILs, however these approaches are energy and cost demanding. Alternative methods based on membrane processes such as filtration, pervaporation, reverse osmosis and electrodialysis have shown some promise for the recovery of ILs; however, the practical feasibility of these methods on a large scale needs to be proved.111 It seems evident that this is one of the weakest points of the methodology. Even when the ILs can be efficiently reused, there is still an uncertainty regarding the stability of the ILs under a prolonged use. Besides, some of the reported approaches may be difficult to scale-up or should be redesigned for industrial application.
The problems of blocking/clogging of the system related to the presence of insoluble products should also be mentioned as a possible disadvantage of the continuous flow systems. This is a major problem associated with the development of practical continuous flow systems and will also be problematic with the use of ILs under flow conditions.
Regarding possible industrial applications, it should be borne in mind that for any new industrial-scale application based on ionic liquids the implementation of the REACH (registration, evaluation, authorisation and restriction of chemicals) legislation is a threat, especially for new ILs, since they require a large initial financial investment for the registration and the gathering of the full physico-chemical characterization and eco-toxicity data required for their evaluation.
Finally cost issues related to ILs continue being an important threat for the generalization of the use of ILs and the possibility of scaling-up the corresponding applications up to the industrial level. This is particularly true in the case of flow applications involving the use of bulk ILs. The development of flow processes for the synthesis of ILs can make a significant contribution in this regard, but this approach still needs to be implemented and generalized. It is clear that the continuous effort towards the development and application of supported IL systems represents a positive answer to overcome this limitation.
7. Conclusions
A variety of technical approaches can be considered for the implementation of flow processes involving the use of ILs. In most cases, the ILs act as the catalytic phase, the IL acting directly as the catalyst or as the support for the immobilization of the corresponding catalyst (organometallic catalyst, catalytic MNP, organocatalyst or biocatalyst). From a practical point of view, both the full recovery of the catalyst and that of the IL used need to be always considered and this is improved, in general, with the use of continuous flow set-ups. Currently, two main approaches have been developed. The first one is associated with the use of biphasic systems involving a bulk IL, containing the catalytic system, and a second phase designed to deliver the substrates and extract the products from the IL phase. This has been the option initially selected for the development of industrial applications and has been often optimized through the use of microreactor technologies. The combination of IL-based systems with a gas phase or a supercritical fluid phase (particularly scCO2) is particularly well suited for the development of biphasic flow processes. A different alternative is the use of ILs supported on solid materials either by adsorption (SILPs) or by covalent attachment of IL-like fragments (SILLPs). This approach combines the advantages of the use of ILs as catalyst supports and the use of solid phase chemistry, well established for the development of flow chemistry processes.Most of the examples considered reveal that continuous flow processes provide significantly enhanced performances in comparison with the related batch processes. This is particularly true when they are carried out in combination with other key enabling techniques. The use of micro- or mini-reactor technologies, with different alternative reactor configurations, and the combination with supercritical fluids represents two important examples. Overall, the considered applications display, in general, important improvements in the space–time yield as well as excellent long-term stabilities for the corresponding catalytic systems.
New approaches are currently being developed that combine the use of ILs with flow chemistry. The incorporation of NP chemistry, the use of systems responsive to external stimuli (i.e. magnetic or electric fields) or the integration in a single process of the complex and often mutually incompatible multicatalytic systems represent some of the challenges to be achieved in the near future.
Acknowledgements
This work was partially supported by MINECO, Spain (Ref: CTQ2011-28903) and Generalitat Valenciana (PROMETEO 2012/020) and UJI (P1-1B 2013-37).Notes and references
- (a) P. Pollet, E. A. Davey, E. E. Ureña-Benavides, C. A. Ecker and C. L. Liotta, Green Chem., 2014, 16, 1034–1055 RSC; (b) P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC; (c) D. Reinhardt, F. Ilgen, D. Kralisch, B. König and G. Kreisel, Green Chem., 2008, 10, 1170 RSC.
- (a) P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) H. Zhao and S. V. Malhotra, Aldrichimica Acta, 2002, 35, 75–83 CrossRef CAS; (c) R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed; (d) J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS PubMed.
- (a) J. R. Vanderveen, J. Durelle and P. G. Jessop, Green Chem., 2014, 16, 1187–1189 RSC; (b) Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC; (c) C. Ruß and B. König, Green Chem., 2012, 14, 2969–2982 RSC; (d) J. Yang, J.-N. Tan and Y. Gu, Green Chem., 2012, 14, 3304–3317 RSC.
- S. V. Luis and E. Garcia-Verdugo, Chemical Reactions and Processes under Flow Conditions, RSC, Green Chemistry Series, 2009 Search PubMed.
- (a) L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC; (b) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC; (c) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (d) C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- (a) V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS; (b) C. Wiles and P. Watts, Chem. Commun., 2011, 47, 6512–6535 RSC.
- (a) U. Hintermair, G. Franciò and W. Leitner, Chem. Commun., 2011, 47, 3691–3701 RSC; (b) M. I. Burguete, E. García-Verdugo and S. V. Luis, Beilstein J. Org. Chem., 2011, 7, 1347–1359 CrossRef CAS PubMed.
- (a) D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS; (b) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- K. F. Jensen, B. J. Reizman and S. G. Newman, Lab Chip, 2014, 14, 3206–3212 RSC.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- (a) M. Smiglak, J. M. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. R. MacFarlane and R. D. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC; (b) G. Cevasco and C. Chiappe, Green Chem., 2014, 16, 2375–2385 RSC.
- (a) C. F. Poole, J. Chromatogr. A, 2004, 1037, 49–82 CrossRef CAS PubMed; (b) S. Ren, Y. Hou, W. Wu and W. Liu, J. Chem. Eng. Data, 2010, 55, 5074–5077 CrossRef CAS; (c) K. Takao and T. Tsubomura, J. Chem. Eng. Data, 2012, 57, 2497–2502 CrossRef CAS.
- M. N. Kashid, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2011, 66, 1480–1489 CrossRef CAS.
- L. R. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef PubMed.
- A. G. Böwing, A. Jess and P. Wasserscheid, Chem. Ing. Tech., 2005, 77, 1430–1439 CrossRef.
- S. Himmler, S. Hörmann, R. van Hal, P. S. Schulz and P. Wasserscheid, Green Chem., 2006, 8, 887–894 RSC.
- A. G. Böwing and A. Jess, Chem. Eng. Sci., 2007, 62, 1760–1769 CrossRef.
- H. Löwe, R. D. Axiente, D. Breuch and C. Hofmann, Chem. Eng. J., 2009, 155, 548–550 CrossRef.
- H. Löwe, R. D. Axiente, D. Breuch, T. Hang and C. Hofmann, Chem. Eng. Technol., 2010, 33, 1153–1158 CrossRef.
- H. Löwe, R. D. Axiente, D. Breuch, C. Hofmann, J. H. Petersen, R. Pommershein and A. Wang, Chem. Eng. J., 2010, 163, 429–437 CrossRef.
- D. A. Waterkamp, M. Heiland, M. Schlüter, J. C. Sauvageau, T. Beyersdorff and J. Thöming, Green Chem., 2007, 9, 1084–1090 RSC.
- D. A. Waterkamp, M. Engelbert and J. Thöming, Chem. Eng. Technol., 2009, 32, 1717–1723 CrossRef CAS.
- D. Wilms, J. Klos, A. F. M. Kilbinger, H. Löwe and H. Frey, Org. Process Res. Dev., 2009, 13, 961–964 CrossRef CAS.
- J. Zimmermann, B. Ondruschka and A. Stark, Org. Process Res. Dev., 2010, 14, 1102–1109 CrossRef CAS.
- T. Nokami, K. Matsumoto, T. Itoh, Y. Fukaya and T. Itoh, Org. Process Res. Dev., 2014, 18, 1367–1371 CrossRef CAS.
- (a) C. Maton, N. De Vos, B. I. Roman, E. Vanecht, N. R. Brooks, K. Binnemans, S. Schaltin, J. Fransaer and C. V. Stevens, ChemPhysChem, 2012, 13, 3146–3157 CrossRef CAS PubMed; (b) D. R. J. Acke, R. V. A. Orru and C. V. Stevens, QSAR Comb. Sci., 2006, 25, 474–483 CrossRef CAS.
- R. Porcar, V. Sans, N. Ríos-Lombardía, V. Gotor-Fernandez, V. Gotor, M. I. Burguete, E. García-Verdugo and S. V. Luis, ACS Catal., 2012, 2, 1976–1983 CrossRef CAS.
- N. Rios-Lombardia, E. Busto, V. Gotor-Fernandez, V. Gotor, R. Porcar, E. Garcia-Verdugo, S. V. Luis, I. Alfonso, S. Garcia-Granda and A. Menendez-Velazquez, Chem. – Eur. J., 2010, 16, 836–847 CrossRef CAS PubMed.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed.
- F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS PubMed.
- (a) G. W. Phillips, S. N. Falling, S. A. Godleski and J. R. Monnier, Continuous process for the manufacture of 2,5-dihydrofurans from γ,δ-epoxybutenes, US Pat., 5,315,019, 1994 Search PubMed; (b) S. N. Falling, S. A. Godleski and L. McGarry, Process for the separation of oligomeric materials from a catalyst mixture, US Pat., 5,238,889, 1993 Search PubMed.
- S. Liu, T. Fukuyama, M. Sato and I. Ryu, Org. Process Res. Dev., 2004, 8, 477–481 CrossRef CAS.
- T. Fukuyama, T. Rahman, Y. Sumino and I. Ryu, Synlett, 2012, 23, 2279–2283 CrossRef CAS.
- D. Horii, T. Fuchigami and M. Atobe, J. Am. Chem. Soc., 2007, 129, 11692–11693 CrossRef CAS PubMed.
- D. Horii, F. Amemiya, T. Fuchigami and M. Atobe, Chem. – Eur. J., 2008, 14, 10382–10387 CrossRef CAS PubMed.
- M. Maase and K. Massonne, in Ionic Liquids IIIB: Fundamentals, Progress, Challenges, and Opportunities – Transformations and Processes, ed. R. D. Rogers and K. R. Seddon, ACS Symp. Ser., American Chemical Society, Washington D.C., 2005, vol. 902, pp. 126–132 Search PubMed.
- T. N. Glasnov, J. D. Holbrey, C. O. Kappe, K. R. Seddon and T. Yan, Green Chem., 2012, 14, 3071–3076 RSC.
- J. Wang, S.-S. Gu, H.-S. Cui, X.-Y. Wu and F.-A. Wu, Bioresour. Technol., 2014, 158, 39–47 CrossRef CAS PubMed.
- M. Cvjetko, J. Vorkapić-Furač and P. Žnidaršič-Plazl, Process Biochem., 2012, 47, 1344–1350 CrossRef CAS.
- A. Pohar, I. Plazl and P. Žnidaršič-Plazl, Chem. Eng. J., 2012, 189–190, 376–382 CrossRef CAS.
- (a) Z. Findrik, G. Nemeth, D. Vasic-Racki, K. Belafi-Bako, Z. Csanadi and L. Gubicza, Process Biochem., 2012, 47, 1715–1722 CrossRef CAS; (b) K. Belafi-Bako, N. Dormo, O. Ulbert and L. Gubicza, Desalination, 2002, 149, 267–268 CrossRef CAS; (c) L. Gubicza, K. Bélafi-Bakó, E. Fehér and T. Fráter, Green Chem., 2008, 10, 1284–1287 RSC.
- M. T. Rahman, T. Fukuyama, N. Kamata, M. Sato and I. Ryu, Chem. Commun., 2006, 2236–2238 RSC.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- S. Keskin, D. Kayrak-Talay, U. Akman and Ö. J. Hortacsu, Supercrit. Fluids, 2007, 43, 150–180 CrossRef CAS.
- C. M. Gordon and W. Leitner, in Catalyst Separation, Recovery and Recycling - Chemistry & Process Design, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer, Heidleberg, Berlin, 2006, p. 215 Search PubMed.
- M. F. Sellin, P. B. Webb and D. J. Cole-Hamilton, Chem. Commun., 2001, 781–782 RSC.
- P. B. Webb, M. F. Sellin, T. E. Kunene, S. Williamson, A. M. Z. Slawin and D. J. Cole-Hamilton, J. Am. Chem. Soc., 2003, 125, 15577–15588 CrossRef CAS PubMed.
- P. B. Webb, T. E. Kunene and D. J. Cole-Hamilton, Green Chem., 2005, 7, 373–379 RSC.
- T. E. Kunene, P. B. Webb and D. J. Cole-Hamilton, Green Chem., 2011, 13, 1476–1148 RSC.
- S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS PubMed.
- S. Kainz, A. Brinkmann, W. Leitner and A. Pfaltz, J. Am. Chem. Soc., 1999, 121, 6421–6429 CrossRef CAS.
- M. Solinas, A. Pfaltz, P. G. Cozzi and W. Leitner, J. Am. Chem. Soc., 2004, 126, 16142–16147 CrossRef CAS PubMed.
- W. Leitner and M. Hölscher, Top. Organomet. Chem., 2008, 23, 91–108 CrossRef.
- J. Theuerkauf, G. Franciò and W. Leitner, Adv. Synth. Catal., 2013, 355, 209–219 CrossRef CAS.
- A. Bösmann, G. Franciò, E. Janssen, M. Solinas, W. Leitner and P. Wasserscheid, Angew. Chem., Int. Ed., 2001, 40, 2697–2699 CrossRef.
- P. Lozano, Green Chem., 2010, 12, 555–569 RSC.
- P. Lozano, T. De Diego, D. Carrié, M. Vaultier and J. L. Iborra, Chem. Commun., 2002, 692–693 RSC.
- M. T. Reetz, W. Wiesenhöfer, G. Franciò and W. Leitner, Chem. Commun., 2002, 992–993 RSC.
- M. T. Reetz, W. Wiesenhöfer, G. Franciò and W. Leitner, Adv. Synth. Catal., 2003, 345, 1221–1228 CrossRef CAS.
- (a) M. H. Valkenberg, C. de Castro and W. H. Holderich, Green Chem., 2002, 4, 88–93 RSC; (b) C. P. Mehnert, Chem. – Eur. J., 2004, 11, 50–56 CrossRef CAS PubMed; (c) A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Top. Catal., 2006, 40, 91–102 CrossRef CAS; (d) Y. Gua and G. Li, Adv. Synth. Catal., 2009, 351, 817–847 CrossRef; (e) C. Van Doorslaer, J. Wahlen, P. Mertens, K. D. Binnemans and D. De Vos, Dalton Trans., 2010, 39, 8377–8390 RSC; (f) B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- S. Werner, N. Szesni, M. Kaiser, M. Haumann and P. Wasserscheid, Chem. Eng. Technol., 2012, 35, 1962–1967 CrossRef CAS.
- U. Hintermair, G. Zhao, C. C. Santini, M. J. Muldoon and D. J. Cole-Hamilton, Chem. Commun., 2007, 1462–1464 RSC.
- M. Jakuttis, A. Schönweiz, S. Werner, R. Franke, K.-D. Wiese, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2011, 50, 4492–4495 CrossRef CAS PubMed.
- U. Hintermair, Z. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501–8510 RSC.
- M. Haumann, M. Jakuttis, R. Franke, A. Schçnweiz and P. Wasserscheid, ChemCatChem, 2011, 3, 1822–1827 CrossRef CAS.
- M. Jakuttis, A. Schnweiz, S. Werner, R. Franke, K.-D. Wiese, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2011, 50, 4492–4495 CrossRef CAS PubMed.
- M. J. Schneider, M. Lijewski, R. Woelfel, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2013, 52, 6996–6999 CrossRef CAS PubMed.
- R. Duque, E. Ochsner, H. Clavier, F. Caijo, S. P. Nolan, M. Mauduit and D. J. Cole-Hamilton, Green Chem., 2011, 13, 1187–1195 RSC.
- M. J. Schneider, M. Haumann and P. Wasserscheid, J. Mol. Catal. A: Chem., 2013, 376, 103–110 CrossRef CAS.
- E. Öchsner, M. J. Schneidera, C. Meyer, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2011, 399, 35–41 CrossRef.
- U. Hintermair, T. Hçfener, T. Pullmann, G. Franciò and W. Leitner, ChemCatChem, 2010, 2, 150–154 CrossRef CAS.
- U. Hintermair, G. Franciò and W. Leitner, Chem. – Eur. J., 2013, 19, 4538–4547 CrossRef CAS PubMed.
- M. Ruta, G. Laurenczy, P. J. Dyson and L. Kiwi-Minsker, J. Phys. Chem. C, 2008, 112, 17814–17819 CAS.
- S. Werner, N. Szesni, A. Bittermann, M. J. Schneider, P. Härter, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2010, 377, 70–75 CrossRef CAS.
- S. Werner, N. Szesni, R. W. Fischer, M. Haumann and P. Wasserscheid, Phys. Chem. Chem. Phys., 2009, 11, 10817–10819 RSC.
- S. Werner, N. Szesni, M. Kaiser, R. W. Fischer, M. Haumann and P. Wasserscheid, ChemCatChem, 2010, 2, 1399–1402 CrossRef CAS.
- J. Joni, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2010, 372, 8–15 CrossRef CAS.
- A. Riisager, B. Jørgensen, P. Wasserscheid and R. Fehrmann, Chem. Commun., 2006, 994–996 RSC.
- M. J. Schneider, M. Haumann, M. Stricker, J. Sundermeyer and P. Wasserscheid, J. Catal., 2014, 309, 71–78 CrossRef CAS.
- F. T. U. Kohler, K. Gärtner, V. Hager, M. Haumann, M. Sternberg, X. Wang, N. Szesni, K. Meyer and P. Wasserscheid, Catal. Sci. Technol., 2014, 4, 936–947 CAS.
- P. Lozano, T. De Diego, T. Sauer, M. Vaultier, S. Gmouh and J. L. Iborra, J. Supercrit. Fluids, 2007, 40, 93–100 CrossRef CAS.
- P. Lozano, T. De Diego, D. Carrie, M. Vaultier and J. L. Iborra, Biotechnol. Prog., 2003, 19, 380–382 CrossRef CAS PubMed.
- P. Lozano, T. De Diego, M. Larnicol, M. Vaultier and J. L. Iborra, Biotechnol. Prog., 2006, 28, 1559–1565 CAS.
- P. Lozano, T. De Diego, C. Mira, K. Montague, M. Vaultier and J. L. Iborra, Green Chem., 2009, 11, 538–542 RSC.
- (a) P. Lozano, J. M. Bernal, R. Piamtongkam, D. Fetzer and M. Vaultier, ChemSusChem, 2010, 3, 1359–1363 CrossRef CAS PubMed; (b) P. Lozano, J. M. Bernal, G. Sánchez-Gómez, G. López-López and M. Vaultier, Energy Environ. Sci., 2013, 6, 1328–1338 RSC; (c) P. Lozano, J. M. Bernal and M. Vaultier, Fuel, 2011, 90, 3461–3467 CrossRef CAS.
- P. Zhang, T. Wu and B. Han, Adv. Mater., 2014, 26, 6810–6827 CrossRef CAS PubMed.
- M. Gruttadauria, L. F. Liotta, A. M. P. Salvo, F. Giacalone, V. La Parola, C. Aprile and R. Noto, Adv. Synth. Catal., 2011, 353, 2119–2130 CrossRef CAS.
- P. Agrigento, S. M. Al-Amsyar, B. Sorée, M. Taherimehr, M. Gruttadauria, C. Aprile and P. P. Pescarmona, Catal. Sci. Technol., 2014, 4, 1598–1607 CAS.
- M. I. Burguete, E. García-Verdugo, N. Karbass, S. V. Luis, V. Sans and M. Sokolova, Pure Appl. Chem., 2009, 81, 1991–2000 CrossRef CAS.
- (a) M. I. Burguete, F. Galindo, E. García-Verdugo, N. Karbass and S. V. Luis, Chem. Commun., 2007, 3086–3088 RSC; (b) N. Karbass, M. Pawlak, M. I. Burguete, V. Compañ, E. García-Verdugo and S. V. Luis, Chem. – Eur. J., 2011, 17, 1894–1906 CrossRef PubMed.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS.
- M. I. Burguete, E. García-Verdugo, S. V. Luis and J. A. Restrepo, Phys. Chem. Chem. Phys., 2011, 13, 14831–14838 RSC.
- N. Karbass, V. Sans, E. García-Verdugo, M. I. Burguete and S. V. Luis, Chem. Commun., 2006, 3095–3097 RSC.
- M. I. Burguete, E. García-Verdugo, I. Garcia-Villar, F. Gelat, P. Licence, S. V. Luis and V. Sans, J. Catal., 2010, 269, 150–160 CrossRef CAS.
- P. Lozano, E. García-Verdugo, R. Piamtongkam, N. Karbass, T. De Diego, M. I. Burguete, S. V. Luis and J. L. Iborra, Adv. Synth. Catal., 2007, 349, 1077–1084 CrossRef CAS.
- P. Lozano, E. García-Verdugo, J. M. Bernal, D. F. Izquierdo, M. I. Burguete, G. Sanchez-Gómez and S. V. Luis, ChemSusChem, 2012, 5, 790–798 CrossRef CAS PubMed.
- P. Lozano, E. García-Verdugo, N. Karbass, K. Montague, T. De Diego, M. I. Burguete and S. V. Luis, Green Chem., 2010, 12, 1803–1810 RSC.
- V. Sans, F. Gelat, N. Karbass, M. I. Burguete, E. García-Verdugo and S. V. Luis, Adv. Synth. Catal., 2010, 352, 3013–3021 CrossRef CAS.
- S. Martín, R. Porcar, E. Peris, M. I. Burguete, E. García-Verdugo and S. V. Luis, Green Chem., 2014, 16, 1639–1647 RSC.
- M. I. Burguete, H. Erythropel, E. García-Verdugo, S. V. Luis and V. Sans, Green Chem., 2008, 10, 401–407 RSC.
- D. Tsaoulidis, V. Dore, P. Angeli, N. V. Plechkova and K. R. Seddon, Int. J. Multiphase Flow, 2013, 54, 1–10 CrossRef CAS.
- U. Novak, A. Pohar, I. Plazl and P. Žnidaršič-Plazl, Sep. Purif. Technol., 2012, 97, 172–178 CrossRef CAS.
- V. Dore, D. Tsaoulidis and P. Angeli, Chem. Eng. Sci., 2012, 80, 334–341 CrossRef CAS.
- F. Scheiff, A. Holbach and D. W. Agar, Chem. Eng. Technol., 2013, 36, 975–984 CrossRef CAS.
- A. Pohar, I. Plazl and P. Žnidaršič-Plazl, Lab Chip, 2009, 9, 3385–3390 RSC.
- M. Pamme, Lab Chip, 2006, 6, 174–178 RSC.
- N.-T. Nguyen, Microfluid. Nanofluid., 2012, 12, 1–16 CrossRef.
- V. Misuk, A. Mai, K. Giannopoulos, F. Alobaid, B. Epple and H. Loewe, Lab Chip, 2013, 13, 4542–4548 RSC.
- V. Misuk, D. Breuch and H. Löwe, Chem. Eng. J., 2011, 173, 536–540 CrossRef CAS.
- R. C. Appleby, Modern Business Administration, Pearson Education Ltd., 1994 Search PubMed.
- L. Mai, K. Ahn and Y.-M. Koo, Process Biochem., 2014, 49, 872–881 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |