
A biomass-derived safe medium to replace toxic dipolar solvents and access cleaner Heck coupling reactions†
Giacomo
Strappaveccia
a,
Ermal
Ismalaj
a,
Chiara
Petrucci
a,
Daniela
Lanari
b,
Assunta
Marrocchi
a,
Martin
Drees
c,
Antonio
Facchetti
*cd and
Luigi
Vaccaro
 *a
*a
aLaboratory of Green Synthetic Organic Chemistry, CEMIN – Dipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia, Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Fax: +39 075 5855560; Tel: +39 075 5855541
bDipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, Via del Liceo, 06123 Perugia, Italy
cPolyera Corporation, 8045 Lamon Avenue, Skokie, IL 60077, USA. E-mail: afacchetti@polyera.com
dDepartment of Chemistry and the Materials Research Center, Northwestern University, USA and and Center of Excellence for Advanced Materials Research (CEAMR), King Abdulaziz University, Jeddah, Saudi Arabia
First published on 4th September 2014
Abstract
γ-Valerolactone (GVL) is an excellent reaction medium, derived from biomasses, which can replace classic dipolar aprotic media such as DMF or NMP. In this contribution, we have investigated the use of GVL in the palladium-catalyzed Heck reaction and accessed the clean synthesis of several small molecules. Furthermore, we optimized a GVL-based protocol to synthesize a poly(phenylenevinylene) (PPV) derivative in high yields and purity, and with very low Pd-content. Finally, we demonstrated that GVL is superior to conventional dipolar media in terms of controlling palladium impurities, which may influence the performance of certain optoelectronic devices such as organic solar cells and field-effect transistors.
Introduction
Major challenges in Environmental Science relate to the evaluation and monitoring of the impact of chemical production. Thus, academic and industrial efforts are directed to the identification of novel routes towards the use of renewable raw materials and energy sources to maximize sustainability.1 Striking examples are given by the social challenges highlighted by the 2011 European Association for Chemical and Molecular Sciences (EuCheMS) roadmap,2 where the importance of designing new, efficient protocols for key synthetic methodologies employing catalysts based on exhaustible precious metals, such as palladium (Pd) catalyzed cross-coupling reactions, has been clearly highlighted. These synthetic methodologies have become fundamental for accessing several complex molecules including drugs, semiconducting materials for opto-electronic applications, and agrochemicals.2,3 Particularly, improved protocols for coupling reactions are required, aimed at minimizing waste volumes4,5 using, for instance, recoverable/recycling catalysts, which minimize metal and reagent consumption.The use of green alternative reaction media is also of key relevance, since organic solvents account for more than 80% of the waste production of a process. Cross-coupling reactions are commonly carried out in dipolar aprotic solvents such as dimethylformamide (DMF), N-methylpyrrolidin-2-one (NMP), and N,N-dimethylacetamide (DMA), thanks to their ability to solubilise several chemicals and promote a large number of reactions. However, they have significant environmental and safety issues due to their toxicity and problematic waste disposal originating from the aqueous/organic mixtures used in reaction work-up procedures. Such wastes are typically incinerated with consequent emission of toxic NOX.6 Indeed, DMF, NMP and DMA are among the first six chemicals of highest concern,7 and suitable replacements are therefore required.6,8–10 Furthermore during coupling reactions, dipolar aprotic solvents coordinate the catalytically active metal (e.g., Pd) causing its dissolution.11–15 Although this phenomenon (so-called leaching) may lead to enhanced catalytic activity, the final product is often metal-contaminated, which is not desirable.11–15 For instance, regulatory basis applies to the limit for palladium residues into drug chemicals, which must be kept at a very low level (<5 ppm).12,13 Moreover, it has been found that metal contamination of organic semiconductors may negatively affect their performance.11,14,15
Nowadays, the most recommended polar aprotic solvent is acetonitrile (MeCN),16 which is actually a relatively poor alternative medium because of large volatility and toxicity.17 In the search for safer organic solvents, special attention is directed towards bio-based solvents derived from biomass,18,19 since they hold the promise of low-cost, safety, biodegradability, and reduced toxicity. To this front, some of us have contributed to the development of synthetic protocols employing eco-compatible reaction media,20 solvent free conditions (SolFC),21 and supported organocatalysts,22 demonstrating that significant improvements in both chemical efficiency and process sustainability can be achieved. Thus, in this context, we directed our attention to the use of biomass-derived γ-valerolactone (GVL)18b,23–26 as a sustainable cross-coupling reaction medium. GVL27 is attractive because its polarity is comparable to that of classical polar aprotic solvents,28,29 and exhibits a high boiling point, high viscosity and a generally good stability to acidic and basic environments. Particularly, continuing our efforts for the sustainable and large scale synthesis of organic semiconductors,30 we here explore for the first time the use of GVL as a reaction medium31 in the Heck–Mirozoki coupling reaction.32 This reaction is widely employed to access several active pharmaceutical ingredients (API), materials and fine chemicals,33,34 and it is typically performed in DMF, NMP, DMA, or MeCN.33 Important to the scope of this study, a major issue in Heck–Mirozoki coupling is related to the gradual leaching of the catalytically active palladium35 into the final product. Although purification methods to remove palladium from APIs12,35,36 and from conjugated organic polymers37 are commonly used and have been reported, they are time/solvent-consuming and create a large amount of (contaminated) waste.
As a model of an organic semiconductor synthesized via the Heck–Mirozoki coupling, we employed a poly(p-phenylene)vinylene (PPV) derivative, specifically poly(2,5-dihexylphenylenedivinylene-alt-1,4-phenylenevinylene) 1 (Fig. 1). Furthermore the PPV family remains of prime importance for several opto-electronic applications including transistors, photovoltaic, and (bio)chemical sensor devices.38
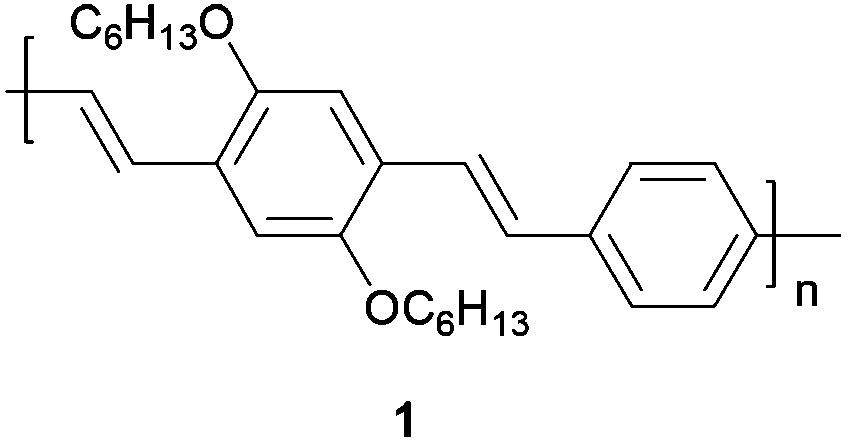 | ||
| Fig. 1 Poly(2,5-dihexylphenylenedivinylene-alt-1,4-phenylenevinylene). | ||
Our results demonstrate that GVL is an excellent medium to synthesize very pure PPVs in high yields and in a more sustainable way. It will be seen that GVL is superior to conventional dipolar NMP in terms of control of palladium impurities in PPV 1, and that it has influence on the material optoelectronic properties. These results are rationalized by the lower complexing ability of GVL in the Heck–Mirozoki cross-coupling procedure.
Results and discussion
In the following sections we first investigate whether GVL can be a solvent for the Heck–Mirozoki reactions by testing several protocol conditions and substrates. Second we compare vis-à-vis the properties of GVL with other polar aprotic solvents for a specific reaction, and then selected conditions are applied to the synthesis of PPV 1. Finally, we report the properties of the PPV derivative and its corresponding performance in two organic opto-electronic devices.Substrate scope and E-factor optimization
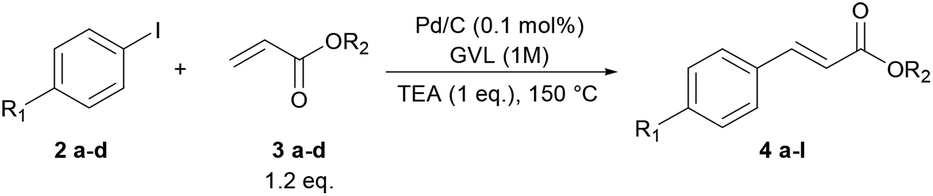
To verify the applicability of GVL as a reaction medium for the preparation of PPVs, we investigated first the Heck–Mirozoki coupling using iodobenzenes 2 and acrylic esters/acids 3 (Table 1) or styrenes 5 (Table 2) as models of monofunctional reagents. Furthermore, we decided to carry out these reactions by employing a supported catalyst. Indeed, heterogeneous palladium catalysts are one of the most useful tools to avoid the leaching of palladium into the product.39 In particular, we focused on the cheap, ligand-free palladium on activated charcoal (Pd/C), which has shown great potential in several Heck–Mirozoki reactions.40,41 We were pleased to find that using a low loading of Pd/C (0.1 mol%), only 1 equivalent of triethylamine (TEA) instead of over-stoichiometric amounts that are routinely employed, and high reactant concentrations (1 M) at 150 °C, a good reactivity of the substrates 2 in the reaction with alkenyl derivatives 3 or 5 was achieved. Thus, in short times (1–2 h) several types of cinnamate esters (4a–h), cinnamate acids (4i–l) (Table 1) and stilbenes (6a–h) (Table 2) were synthesized in good yield (80–90%). It should be noted that the use of 10, 20, or 40% water-GVL mixtures gave the same results. Therefore, the use of only GVL was preferred.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Yield (%) | Product | E-factor |
| a 1.5 eq. of acrylic acid was used. b This reaction has been also performed in 10, 20 and 40% GVL–water mixtures obtaining identical results. | ||||||
| 1 | –H | –Me | 1 | 82% | 4a | 25 |
| 2 | –OCH3 | –Me | 2 | 82% | 4b | 21 |
| 3 | –OCH3 | –Et | 2 | 90% | 4c | 18 |
| 4 | –COCH3 | –Me | 1.5 | 87% | 4d | 19 |
| 5 | –COCH3 | –Et | 1.5 | 85% | 4e | 18 |
| 6 | –NO2 | –Me | 1 | 84% | 4f | 19 |
| 7 | –NO2 | –Et | 1 | 86% | 4g | 18 |
| 8 | –NO2 | –nBu | 1 | 82% | 4h | 16 |
| 9a | –H | –H | 1 | 84% | 4i | 27 |
| 10a | –OCH3 | –H | 2 | 80% | 4j | 24 |
| 11a | –COCH3 | –H | 1.5 | 85% | 4k | 21 |
| 12a | –NO2 | –H | 1 | 83% | 4l | 21 |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Yield (%) (6a/6a′) | Product | E-factor |
| 1 | –H | –H | 2 | 83 (94/6) | 6a | 22 |
| 2 | –OCH3 | –H | 2.5 | 82 (95/5) | 6b | 18 |
| 3 | –COCH3 | –H | 1.5 | 86 (99/1) | 6c | 16 |
| 4 | –NO2 | –H | 1.5 | 84 (99/1) | 6d | 17 |
| 5 | –Me | –H | 2.5 | 83 (94/6) | 6e | 20 |
| 6 | –H | –Cl | 2 | 90 (92/8) | 6f | 16 |
| 7 | –OCH3 | –Cl | 2.5 | 90 (90/10) | 6g | 14 |
| 8 | –COCH3 | –Cl | 1.5 | 85 (97/3) | 6h | 14 |
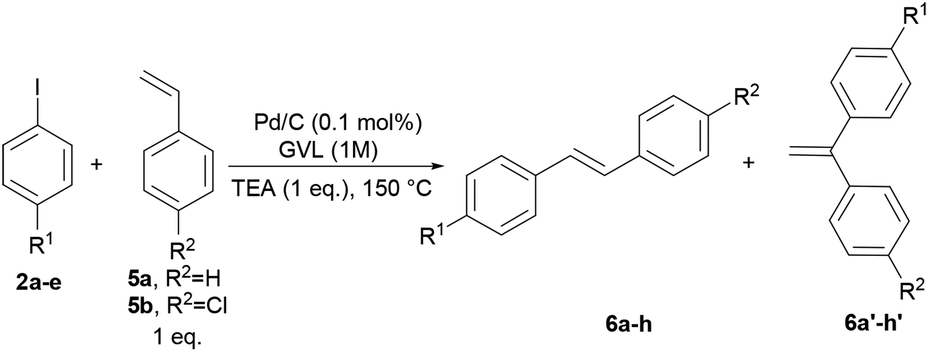
In the reaction of 2a–e with 5a–b, the formation of gem-substituted regioisomer 6a′–h′ was detected in traces,40a,b particularly when unsubstituted/electron-donating substituted iodobenzenes were used as substrates.
Next, to reduce the reactions’ environmental burden, we focused our attention on the work-up procedure optimization. The Pd/C catalyst was removed by a simple filtration of the reaction mixture. Notably, the pure product could be conveniently isolated by direct precipitation from a minimal amount of water, due to the high solubility in this medium of both GVL and triethylammonium iodide byproducts.
To monitor the amount of waste produced by the process we calculated the E-factor,42 which is defined as the kg of waste produced per kg of the desired product. For our reactions, we found that the optimized work-up protocol is very efficient for waste reduction. Indeed, the E-factors range from 14 to 27 (Tables 1 and 2), and are significantly smaller than those for conventional extraction work-up,38c where an average of ∼63 was obtained, thereby achieving a reduction of waste by 60% (see ESI for detailed work-up procedures and E-factor calculation).
Leaching of the palladium catalyst
To investigate the palladium leaching issue in details when employing GVL as the reaction medium, and in comparison with conventional dipolar aprotic media, we focused on the Heck–Mirozoki reaction between iodobenzene (2a) and methyl acrylate (3a) (Scheme 1). Thus, the model substrates showed a high reactivity in DMF and NMP (Table 3, entries 1 and 2), whereas other dipolar aprotic solvents, dimethyl sulfoxide (DMSO), MeCN and water, respectively, gave modest conversions (Table 3, entries 3–5). Similarly, a water–DMF mixture resulted in a conversion of 2a to 4a as high as ∼30% (Table 3, entry 6). GVL gave complete conversion at 130 °C but in a bit longer reaction time compared to DMF and NMP (Table 3, entry 7).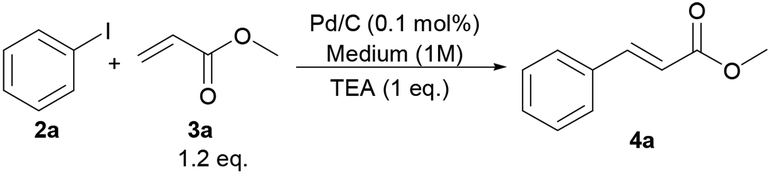 | ||
| Scheme 1 Synthetic protocol to 4a. | ||
| Entry | T (°C) | t (h) | Medium | Conversion (%) |
|---|---|---|---|---|
| 1 | 130 | 0.5 | DMF | 100 |
| 2 | 130 | 0.5 | NMP | 100 |
| 3 | 130 | 24 | DMSO | 75 |
| 4 | 70 | 24 | CH3CN | 36 |
| 5 | 100 | 24 | H2O | 33 |
| 6 | 100 | 24 | H2O–DMF (4/1) | 29 |
| 7 | 130 | 2 | GVL | 100 |
Next, we investigated the leaching of palladium in the solvent enabling the best reaction performance, and compared it with what was observed in GVL (Table 4).
Previous work showed that the palladium leaching was also influenced by the temperature,40 so we also carried out this model reaction at different temperatures in the selected media. Our results indicate that with GVL the leaching of palladium diminished with the increasing temperature. Indeed, inductively coupled plasma-atomic emission spectrometric (ICP-AES) measurements revealed a constant decrease of leached palladium in the products from 8.9 ppm for the reaction carried out at 130 °C (Table 4, entry 1), down to 3.9 ppm when heating at 200 °C (Table 4, entry 4).
By employing NMP and DMF as the reaction media, we found a much higher product contamination, reflecting a significant palladium leaching. Particularly, 4a through NMP contained 835 ppm and 279 ppm of palladium levels for a reaction occurring at 150 °C and 200 °C, respectively (Table 4, entries 5 and 6), which also confirmed the trend with temperature, whereas when DMF was used, the Pd level was found to be 50 ppm (150 °C) (Table 4, entry 7). Moreover, we demonstrated that with GVL the leaching of palladium was not substrate-dependent. Since comparable metal content was detected in product 6b when prepared under the same conditions (200 °C) as those for the synthesis of 4a (Table 4, entry 8).
All these findings confirm that GVL is the optimal medium to limit the leaching of palladium into the product. Thus, GVL is not only a safer and cleaner reaction medium, but it enables the formation of cleaner products thereby avoiding the need for further costly and tedious purification steps.
Heck–Mirozoki coupling copolymerization reaction
In light of these encouraging results, we aimed at the preparation of poly(2,5-dihexylphenylenedivinylene-alt-1,4-phenylenevinylene) 1 (Fig. 1, Scheme 2). To this end, the Heck–Mirozoki polymerization between 2,5-dihexyl-1,4-diiodobenzene (2f) and 1,4-divinylbenzene (5c) (Table 5) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-monomer molar ratio, 0.08 M concentration) was performed employing Pd/C as the catalyst (5 mol%), K2CO3 as the base (3 eq.), and GVL as the reaction medium (135 °C, 72 h). These conditions were found to lead to polymer 1 with Mn = 8.14 kDa and PDI of ∼2 (Table 5, entry 1) in 72% yield. Under similar conditions, but using NMP, polymer 1 was achieved with a higher molecular weight (Mn = 16.43 kDa, PDI = ∼2, Table 5, entry 2) and similar yields (70%). All polymer samples were purified through multiple Soxhlet extractions with acetone, hexane, and chloroform.
:1 co-monomer molar ratio, 0.08 M concentration) was performed employing Pd/C as the catalyst (5 mol%), K2CO3 as the base (3 eq.), and GVL as the reaction medium (135 °C, 72 h). These conditions were found to lead to polymer 1 with Mn = 8.14 kDa and PDI of ∼2 (Table 5, entry 1) in 72% yield. Under similar conditions, but using NMP, polymer 1 was achieved with a higher molecular weight (Mn = 16.43 kDa, PDI = ∼2, Table 5, entry 2) and similar yields (70%). All polymer samples were purified through multiple Soxhlet extractions with acetone, hexane, and chloroform.
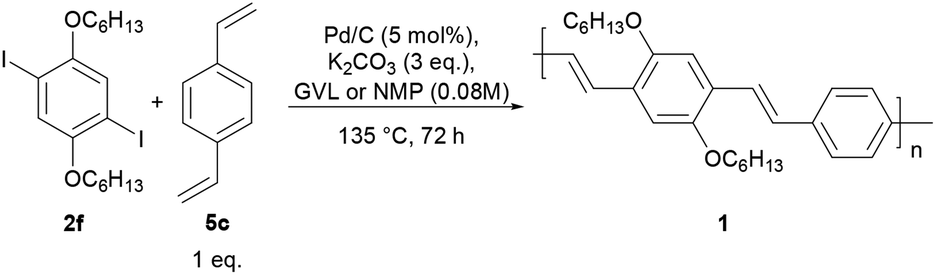 | ||
| Scheme 2 Synthesis of the PPV-based polymer 1. | ||
| Entry | Medium | M n (kDa) | M w (kDa) | PDI | Pd (ppm) |
|---|---|---|---|---|---|
| 1 | GVL | 8.14 | 16.27 | 1.99 | 6 |
| 2 | NMP | 16.43 | 36.98 | 1.99 | 860 |
However, the solvent type had a notable effect on the Pd residue in the polymer. Indeed, it was found that polymer 1 prepared employing NMP contained a Pd level of 860 ppm, whereas when using GVL the Pd level was reduced by two orders of magnitude (6 ppm), as accessed by atomic absorption spectroscopy (Table 5). Most importantly, this was achieved without any metal purification step. The lower complexing ability of GVL towards palladium relative to that of NMP during the Heck polymerization may account for a different catalyst activity, thereby resulting in lower molecular weight polymer chains, as well as for the negligible Pd impurity content.
Finally, the UV-Vis absorption spectrum of polymer 1 as a thin film synthesized with GVL is nearly identical to that of the polymer prepared via the Heck–Mirozoki cross-coupling in NMP (Fig. S1 ESI†), with the absorption maxima (λmax) located at 344 and 464 nm. The slightly stronger absorbance up to ∼450 nm for the NMP sample may be due to the greater molecular weight as this is known to promote interchain stacking and red-shift the optical absorption.43
Photovoltaic and transistor device performance
To demonstrate that GVL-based protocols can afford device-quality semiconductors and access the effect of the metal impurity on the device performance, bulk heterojunction (BHJ) solar cells and field-effect transistors were fabricated using different 1 samples. The BHJ devices used here have the following architecture: ITO/PEDOT:PSS/PPV:PC61BM/LiF/Al. The photoactive layer was spin-coated from chlorobenzene (7 mg mL−1, 1:1 = 1–PC61BM). Typical current–voltage (J–V) characteristics of the devices fabricated with PPV derivative samples under white light illumination (AM1.5G, 100 mW cm−2) are shown in Fig. S2 in ESI.†
A representative BHJ cell based on polymer 1via GVL featured Jsc = 2.4 mA cm−2, Voc = 0.92 V, and FF = 33.7%, leading to a PCE = 0.73% (Table 6, entry 1). These performances are typical of PPV derivatives.38 On the other hand, devices based on polymer 1 through NMP exhibited better performance characterized by a Voc of 0.91 V, Jsc = 3.8 mA cm2 and FF = 36.1%, giving a power conversion efficiency of 1.26% (Table 6, entry 2). To discriminate the influence of the polymer molecular weight from that of the metal content on the device performance,44 we prepared BHJ solar cells from polymer 1 through GVL with the addition of Pd using Pd(PPh3)4 to reach the contamination level of the polymer from the NMP-based protocol (∼800 ppm). Notably, both the relevant parameters Jsc = 1.6 mA cm−2 and Voc = 075 V obtained using the artificially contaminated polymer 1 through GVL in the BHJ active layer were lower (PCE = 0.39%, Table 6, entry 3) than those obtained from devices employing the clean counterpart (Table 6, entry 2). Thus, it is clear that the presence of palladium metal contaminant has a major adverse effect on the device performance, causing a ∼40% reduction in efficiency.
The detrimental effect of palladium impurities on the Jsc parameter can also be observed from the EQE spectra (Fig. S3 in ESI†) which show a large increase in current generation over the entire wavelength range for BHJ cell based on polymer 1 through GVL compared to those using the artificially Pd-contaminated counterpart.
Next, to investigate the influence of Pd impurities on in-plane charge-transport efficiency, also a critical parameter for organic photovoltaics,45 bottom-gate top-contact field-effect transistors (FETs) were also fabricated with films of polymer 1 spin-coated on Si/SiO2 substrates (6 mg ml−1 in chlorobenzene). The Si/SiO2 substrates were used without dielectric surface treatment. Vacuum deposited gold source/drain electrodes completed the OFET structure.
The transfer and output characteristics of transistors fabricated from polymer 1 samples are shown in Fig. S4 and S5 of the ESI.† The derived field effect transistor mobilities are modest, although consistent with those reported in the literature for PPV derivatives,38 with a maximum value of 1.21 × 10−3 cm2 V−1 s−1 (Ion/Ioff = 2 × 102) for devices based on the polymer prepared employing NMP as the solvent (Table 6, entry 2). For the batch achieved using GVL (Table 6, entry 1), a slightly lower FET mobility was achieved, however, the Ion/Ioff is larger. Considering the modest performance, the statistical FET transport characteristics are comparable. Furthermore, no significant effects of the palladium impurities on FET charge carrier mobility are observed (Table 6, entries 1 and 3). These results are in agreement with some general observations where OPV performance are more affected by metal impurities than FET charge transport.46
Experimental
Materials synthesis
The reagents 2,5-bis(hexyloxy)-1,4-diiodobenzene47 and 1,4-divinylbenzene48 were synthesized according to published procedures. All starting organic reagents and solvents were purchased from Aldrich, and used without further purification, unless otherwise noted. Tetrahydrofuran was distilled from sodium/benzophenone.General procedure for Heck–Mirozoki reaction and E-factor calculation (precipitation work-up)
In a screw capped vial equipped with a magnetic stirrer Pd/C 10 wt% (2.1 mg, 0.002 mmol, 0.1 mol%), GVL (2 mL), iodobenzene (1a) (416 mg, 0.228 mL, 2 mmol), triethylamine (202 mg, 0.279 mL, 2 mmol) and methyl acrylate (2a) (207 mg, 0.216 mL, 2.4 mmol) were consecutively added and the resulting mixture was left under stirring at 150 °C. After 1 hour the catalyst was filtered off, 3 mL of water were added and the mixture was cooled down to 0 °C; then the precipitate was filtered off and washed with 1 mL of cold water. Ultimately the obtained solid was dried under high vacuum. In this way 3a was obtained as a pale yellow solid (266 mg, 82% yield).49| E -factor = (4000 (H2O) + 2100 (GVL) + 416 + 202 + 207 + 2.1 – 266)/266 = 25 |
General procedure for Heck–Mirozoki reaction and E-factor calculation (extraction work-up)
In a screw capped vial equipped with a magnetic stirrer Pd/C 10 wt% (2.1 mg, 0.002 mmol, 0.1 mol%), GVL (2 mL), iodobenzene (1a) (416 mg, 0.228 mL, 2 mmol), triethylamine (202 mg, 0.279 mL, 2 mmol) and methyl acrylate (2a) (207 mg, 0.216 mL, 2.4 mmol) were consecutively added and the resulting mixture was left under stirring at 150 °C. After 1 hour the catalyst was filtered off, 6 mL of cyclopentyl methyl ether were added and the organic phase was washed with water (3 × 3 mL). Then the organic phase was dried under vacuum. Compound 3a was obtained as a pale yellow solid (266 mg, 82% yield).| E-factor = (9000 (H2O) + 2100 (GVL) + 5160 (cyclopentyl methyl ether) + 416 + 202 + 207 + 2.1 – 266)/266 = 63 |
Physical characterization
NMR spectra were recorded on a Bruker DRX-ADVANCE 400 MHz (1H at 400 MHz and 13C at 100.6 MHz) spectrometer in CDCl3 using TMS as the internal standard.The molecular weights of the polymer samples were determined on an Agilent 1100 series HPLC, equipped with Phenomenex Phenogel columns, and polystyrene (Mp 400–2000000) was used as the calibration standard.
The amount of residual palladium in the polymer samples was determined by ICP-AES (Inductively Coupled Plasma-Atomic Emission Spectrometry). The samples (∼25 mg) were dissolved in aqua regia at r.t., diluted to 5 mL with deionized water, filtered and subjected to analysis.
UV-visible spectroscopy was performed on a Varian Cary 5000 UV-vis-NIR spectrophotometer.
Organic field effect transistors (OFETs) were fabricated on p+-Si/SiO2 (300 nm thermal oxide). Top contact gold (500 Å) were deposited by evaporating gold (pressure <10−5 Torr); channel dimensions were 100 μm (L) by 1.0 mm (W). The capacitance of the insulator is 10.5 nF cm−2 for 300 nm SiO2. Device measurements were carried out at room temperature in a customized probe station under nitrogen. OFET characterizations were performed with a Keithley 6430 subfemtoamp (drain) and a Keithley 2400 (gate) source meter, operated by a locally written Labview program.
OPV characterization was performed on a Spectra-Nova Class A Solar Simulator with AM1.5G light (100 mW cm−2) from a Xe arc lamp. The light source was calibrated with a NREL-certified Si diode equipped with a KG3 filter to bring spectral mismatch to unity. Current versus potential (J–V) measurements were recorded with a Keithley 2400 digital source meter. Power conversion efficiencies were calculated from the following equation: PCE = (JscVocFF)/P0, where Jsc (mA cm−2) is the short circuit current, Voc (V) the open circuit voltage, FF the fill factor, and P0 the power of the incident light source (mW cm−2).
External quantum efficiency (EQE) was measured using an Oriel model QE-PV-SI (Newport instruments) equipped with a NIST (National Institute of Standards and Technology)-certified Si-diode and a Merlin lock-in amplifier and optical chopper. Monochromatic light was generated from a 300 W Xe arc lamp.
Conclusions
In conclusion, in this contribution we have shown that GVL is an excellent reaction medium, derived from biomasses, which can replace classic dipolar aprotic media such as DMF or NMP. The palladium-catalyzed Heck reaction has been investigated as a representative and widely important process for obtaining chemicals used in several applied fields. Specific attention in this study has been devoted to the critical issue of palladium leaching, which commonly occurs in Heck reactions, leading to a high metal impurity content in the final products for both small and macro-molecules.We have used GVL for the clean preparation of several small molecules (4a–l, and 6a–h) and also for the synthesis of a very pure PPV semiconductor (1) in high yields and with a very low Pd-content. We have proved that GVL is superior to conventional dipolar media in terms of controlling palladium impurities. These results are rationalized by the lower complexing ability of GVL in the Heck–Mirozoki cross-coupling procedure. Furthermore, OPV and FET data demonstrate that device-quality PPV 1 can be achieved without extensive purification processes, complementing recent synthetic efforts on semiconducting materials without the use of toxic Sn metal.30c,50 These results are extremely encouraging and prove that GVL, as a biomass derived solvent, is a promising medium, and its use in strategic processes may significantly reduce the environmental impact and energy cost of the chemical industry.
Acknowledgements
This research has been developed and partially financed within the National project “BIT3G” – Italian Green Chemistry Cluster. We gratefully acknowledge the Università degli Studi di Perugia, and the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) for the financial support within the program “Firb-Futuro in Ricerca” ref. N. RBFR08JKHI and RBFR08J78Q. We also thank the “Fondazione Cassa di Risparmio di Terni e Narni” for financial support.References
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederik Jr., J. P. Hallet, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- European Association for Chemical and Molecular Sciences EuCheMS, Chemistry-Developing solutions in a changing world, 2011 Search PubMed.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053–2066 CAS.
- (a) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC; (b) C. Seyler, C. Capello, S. Hellweg, C. Bruder, D. Bayne, A. Huwiler and K. Hungerbühler, Ind. Eng. Chem. Res., 2006, 45, 7700–7709 CrossRef CAS; (c) C. Jiménez-González, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess., 2004, 9, 114–121 CrossRef.
- (a) E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038–1049 RSC; (b) A. D. Curzons, D. J. C. Constable, D. N. Mortimera and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- D. J. C. Constable, C. Jiménez-González and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 CrossRef.
- (a) T. Laird, Org. Process Res. Dev., 2012, 16, 1–2 CrossRef CAS; (b) G. M. Kemeling, ChemSusChem, 2012, 5, 2291–2292 CrossRef CAS PubMed.
- M. P. Nikiforov, B. Lai, W. Chen, S. Chen, R. D. Schaller, J. Strzalka, J. Maserb and S. B. Darling, Energy Environ. Sci., 2013, 6, 1513–1520 CAS.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
- Note for Guidance on Specification Limits for Residues of Metal Catalysts, The European Agency for the Evaluation of Medicinal Products, Evaluation of Medicines for Human Use; London, 17 December 2002; http//www.emea.eu.int.
- For a recent example see: J. Kuwabara, T. Yasuda, S. J. Choi, W. Lu, K. Yamazaki, S. Kagaya, L. Han and T. Kanbara, Adv. Funct. Mater., 2014, 24, 3226–3233 CrossRef CAS.
- (a) A. Saeki, M. Tsuji and S. Seki, Adv. Energy Mater., 2011, 1, 661–669 CrossRef CAS; (b) P. A. Troshin, D. K. Susarova, Y. L. Moskvin, I. E. Kuznetsov, S. A. Ponomarenko, E. N. Myshkovskaya, K. A. Zakharcheva, A. A. Balakai, S. D. Babenko and V. F. Razumov, Adv. Funct. Mater., 2010, 20, 4351–4357 CrossRef CAS.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- (a) Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC; (b) I. T. Horváth, Green Chem., 2008, 10, 1024–1028 RSC.
- (a) C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed; (b) J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC; (c) D. R. Dodds and R. A. Gross, Science, 2007, 318, 1250–1251 CrossRef CAS PubMed; (d) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- (a) T. Angelini, D. Lanari, R. Maggi, F. Pizzo, G. Sartori and L. Vaccaro, Adv. Synth. Catal., 2012, 354, 908–916 CrossRef CAS; (b) D. Lanari, O. Piermatti, F. Pizzo and L. Vaccaro, Synthesis, 2012, 2181–2184 Search PubMed; (c) S. Bonollo, D. Lanari, F. Pizzo and L. Vaccaro, Org. Lett., 2011, 13, 2150–2152 CrossRef CAS PubMed; (d) S. Bonollo, D. Lanari and L. Vaccaro, Eur. J. Org. Chem., 2011, 2587–2598 CrossRef CAS.
- (a) D. Lanari, R. Ballini, A. Palmieri, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2011, 2874–2884 CrossRef CAS; (b) F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Green Chem., 2010, 12, 1301–1305 RSC; (c) A. Zvagulis, S. Bonollo, D. Lanari, F. Pizzo and L. Vaccaro, Adv. Synth. Catal., 2010, 352, 2489–2496 CrossRef CAS.
- (a) M. Alonzi, M. P. Bracciale, A. Broggi, D. Lanari, A. Marrocchi, M. L. Santarelli and L. Vaccaro, J. Catal., 2014, 309, 260–267 CrossRef CAS PubMed; (b) S. Bonollo, D. Lanari, J. M. Longo and L. Vaccaro, Green Chem., 2012, 14, 164–169 RSC; (c) S. Bonollo, D. Lanari, T. Angelini, F. Pizzo, A. Marrocchi and L. Vaccaro, J. Catal., 2012, 285, 216–222 CrossRef CAS PubMed.
- (a) E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed; (b) L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS PubMed.
- (a) D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Catal. Sci. Technol., 2013, 3, 927–931 RSC; (b) P. Azadi, R. Carrasquillo-Flores, Y. J. Pagán-Torres, E. I. Gürbüz, R. Farnood and J. A. Dumesic, Green Chem., 2012, 14, 1573–1576 RSC; (c) Z. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- (a) D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081 CrossRef CAS PubMed; (b) J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed; (c) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC; (d) I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- (a) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC; (b) J.-P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488–3490 RSC.
- GVL dielectric constant at 25 °C is 36.47; S. Aparicio and R. Alcalde, Phys. Chem. Chem. Phys., 2009, 11, 6455–6467 RSC.
- Dielectric constant (25 °C) for: acetonitrile = 37.5, DMF = 36.7, NMP = 32.0, DMA = 37.8. B. S. Furniss, A. J. Hannaford, V. Rogers, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman, London and New York, 4th edn, 1978 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- (a) E. Bartollini, M. Seri, S. Tortorella, A. Facchetti, T. J. Marks, A. Marrocchi and L. Vaccaro, RSC Adv., 2013, 3, 9288–9295 RSC; (b) M. Alonzi, D. Lanari, A. Marrocchi, C. Petrucci and L. Vaccaro, RSC Adv., 2013, 3, 23909–23923 RSC; (c) A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- (a) J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85 RSC; (b) Z.-Q. Duan and F. Hu, Green Chem., 2012, 14, 1581–1583 RSC; (c) S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 RSC; (d) L. Qi and I. T. Horváth, ACS Catal., 2012, 2, 2247–2249 CrossRef CAS; (e) D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992 RSC.
- (a) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed; (c) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (b) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed; (c) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed; (d) V. Farina, Adv. Synth. Catal., 2004, 346, 1553–1582 CrossRef CAS; (e) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed.
- (a) Y. Lim, Y.-S. Park, Y. Kang, D. Y. Jang, J. H. Kim, J.-J. Kim, A. Sellinger and D. Y. Yoon, J. Am. Chem. Soc., 2011, 133, 1375–1382 CrossRef CAS PubMed; (b) C. Torborga and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef; (c) Y. Lee, Y. Liang and L. Yu, Synlett, 2006, 2879–2893 CAS.
- (a) D. Barbaras, J. Brozio, I. Johannsen and T. Allmendinger, Org. Process Res. Dev., 2009, 13, 1068–1079 CrossRef CAS; (b) M. J. Girgis, L. E. Kuczynski, S. M. Berberena, C. A. Boyd, P. L. Kubinski, M. L. Scherholz, D. E. Drinkwater, X. Shen, S. Babiak and B. G. Lefebvre, Org. Process Res. Dev., 2008, 12, 1209–1217 CrossRef CAS.
- (a) C. J. Welch, J. Albaneze-Walker, W. R. Leonard, M. Biba, J. DaSilva, D. Henderson, B. Laing, D. J. Mathre, S. Spencer, X. Bu and T. Wang, Org. Process Res. Dev., 2005, 9, 198–205 CrossRef CAS; (b) V. W. Rosso, D. A. Lust, P. J. Bernot, J. A. Grosso, S. P. Modi, A. Rusowicz, T. C. Sedergran, J. H. Simpson, S. K. Srivastava, M. J. Humora and N. G. Anderson, Org. Process Res. Dev., 1997, 1, 311–314 CrossRef CAS.
- (a) K. T. Nielsen, H. Spanggaard and F. C. Krebs, Macromolecules, 2005, 38, 1180–1189 CrossRef CAS; (b) K. T. Nielsen, K. Bechgaard and F. C. Krebs, Macromolecules, 2005, 38, 658–659 CrossRef CAS; (c) K. T. Nielsen, K. Bechgaard and F. C. Krebs, Synthesis, 2006, 1639–1644 CrossRef CAS PubMed.
- (a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2011, 112, 2208–2267 CrossRef PubMed; (b) A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC; (c) A. C. Grimsdale and A. B. Holmes, in Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, 3rd edn, 2007 Search PubMed.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Béland and P. D. Carà, ChemCatChem, 2012, 4, 432–445 CrossRef CAS.
- (a) R. G. Heidenreich, J. G. E. Krauter, J. Pietsch and K. Köhler, J. Mol. Catal. A: Chem., 2002, 183, 499–509 CrossRef; (b) K. Köhler, R. G. Heidenreich, J. G. E. Krauter and J. Pietsch, Chem. – Eur. J., 2002, 8, 622–631 CrossRef; (c) F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem. – Eur. J., 2000, 6, 843–848 CrossRef CAS.
- S.-Y. Liu, H.-Y. Li, M.-M. Shi, H. Jiang, X.-L. Hu, W.-Q. Li, L. Fu and H.-Z. Chen, Macromolecules, 2012, 45, 9004–9009 CrossRef CAS.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- R. J. Kline and M. D. McGehee, Macromolecules, 2005, 38, 3312–3319 CrossRef CAS.
- C. J. Brabec, M. Heeney, I. McCulloch and J. Nelson, Chem. Soc. Rev., 2011, 40, 1185–1199 RSC.
- (a) G. Y. Sang, Y. P. Zou and Y. F. Li, J. Phys. Chem. C, 2008, 112, 12058–12064 CrossRef CAS; (b) M. Morana, P. Koers, C. Waldauf, M. Koppe, D. Muehlbacher, P. Denk, M. Scharber, D. Waller and C. Brabec, Adv. Funct. Mater., 2007, 17, 3274–3283 CrossRef CAS.
- (a) P. Sonar, A. C. Grimsdale, M. Heeney, M. Shkunov, I. McCulloch and K. Muellen, Synth. Met., 2007, 157(21), 872–875 CrossRef CAS PubMed; (b) N. Bjoerklund, J.-O. Lill, J. Rajander, R. Oesterbacka, S. Tierney, M. Heeney, I. McCulloch and M. Coelle, Org. Electron., 2009, 10, 215–221 CrossRef CAS PubMed; (c) M. P. Nikiforov, B. Lai, W. Chen, S. Chen, R. D. Schaller, J. Strzalk, J. Maserb and S. B. Darling, Energy Environ. Sci., 2013, 6, 1513–1520 RSC; (d) P. A. Troshin, D. K. Susarova, Y. L. Moskvin, I. E. Kuznetsov, S. A. Ponomarenko, E. N. Myshkovskaya, K. A. Zacharcheva, A. A. Balakai, S. D. Babenko and V. F. Razumov, Adv. Funct. Mater., 2010, 20, 4351–4357 CrossRef CAS; (e) W. L. Leong, G. C. Welch, L. G. Kaake, C. J. Takacs, Y. Sun, G. C. Bazan and A. J. Heeger, Chem. Sci., 2012, 3, 2103–2109 RSC; (f) A. Saeki, M. Tsuji and S. Seki, Adv. Energy Mater., 2011, 1, 661–669 CrossRef CAS.
- P. Singh, U. Saha and T. H. Goswami, Synth. Met., 2012, 162, 1240–1254 CrossRef PubMed.
- T. Ohishi, T. Masukawa, S. Fujii, A. Yokoyama and T. Yokozawa, Macromolecules, 2010, 43, 3206–3214 CrossRef CAS.
- Z. Zhang, Z. Zha, C. Gan, C. Pan, Y. Zhou, Z. Wang and M. M. Zhou, J. Org. Chem., 2006, 71, 4339–4342 CrossRef CAS PubMed.
- (a) V. Senkovskyy, R. Tkachov, H. Komber, M. Sommer, M. Heuken, B. Voit, W. T. S. Huck, V. Kataev, A. Petr and A. Kiriy, J. Am. Chem. Soc., 2011, 131, 19966 CrossRef PubMed; (b) V. Senkovskyy, R. Tkachov, H. Komber, A. John, J.-U. Sommer and A. Kiriy, Macromolecules, 2012, 5, 7770 CrossRef; (c) R. Tkachov, Y. Karpov, V. Senkovskyy, I. Raguzin, J. Zessin, A. Lederer, M. Stamm, B. Voit, T. Beryozkina, V. Bakulev, A. Facchetti and A. Kiriy, Macromolecules, 2014, 47, 3845–3851 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: OFET/OPV fabrication details, synthetic procedures/spectroscopic data/E-factor calculation, synthesis optimization screening. See DOI: 10.1039/C4GC01677G |
| This journal is © The Royal Society of Chemistry 2015 |