
Quantitative synthesis of bis(cyclic carbonate)s by iron catalyst for non-isocyanate polyurethane synthesis†
Xingfeng
Sheng
ab,
Guanjie
Ren
ab,
Yusheng
Qin
*a,
Xuesi
Chen
a,
Xianhong
Wang
*a and
Fosong
Wang
a
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, People's Republic of China. E-mail: ysqin@ciac.jl.cn; xhwang@ciac.jl.cn; Fax: +86 431 85262252, +86 431 85689095; Tel: +86 431 85262252, +86 431 85262250
bUniversity of Chinese Academy of Sciences, Beijing 100039, People's Republic of China
First published on 8th September 2014
Abstract
Bis(cyclic carbonate)s were quantitatively prepared with high efficiency via the coupling reaction of carbon dioxide (CO2) with diglycidyl ethers by a [Fe(BPMCDAC)]/TBAB catalytic system, where glycol diglycidyl ether (1a) could be completely converted to the corresponding bis(cyclic carbonate) (2a) with a turnover number of 1000 at 100 °C and 3 MPa in 4 h. The obtained bis(cyclic carbonate) (2a) could be used to prepare hydroxyl-functional polyurethanes via reaction with diamines, which may be one alternative for obtaining conventional polyurethanes without the use of toxic phosgene or isocyanates. The number-average molecular weights of the obtained non-isocyanate polyurethanes (NIPUs) were up to 25.4–30.2 kg mol−1, and the polydispersity indexes (PDIs) were relatively narrow between 1.18 and 1.22. A typical NIPU showed a glass transition temperature of 9 °C and an initial degradation temperature (Td 5%) of 206 °C.
Introduction
Carbon dioxide (CO2) is regarded as the main reason for global warming;1 however, it is abundant, cheap and non-toxic, and its specific molecular structure makes it an important C1 feedstock in the industry. Employing CO2 as a raw material to prepare large scale chemicals may create a new way to a high value-added utilization of CO2. Among all the reported CO2 fixation routes, the synthesis of cyclic carbonates via the coupling reaction of CO2 with epoxides is one of the most attractive processes.2Because cyclic carbonates are widely used as green solvents, electrolytes, fuel additives and intermediates for the production of biodegradable polymers,3 numerous catalyst systems have been developed to promote this transformation.4 Prominent among these are salen complexes of cobalt,5 chromium,6 zinc7 and aluminium,8 which show high activities under mild conditions. However, most of the studies have focused on the coupling reaction of CO2 with mono-epoxides, whereas epoxides with two or more epoxy groups were rarely used in the literature. Lee and co-workers9 synthesized bis(cyclic carbonate)s via the addition reaction of carbon dioxide with diglycidyl ether derivatives in N-methyl-pyrrolidone (NMP) in the presence of a phase transfer catalyst, and the conversion of diepoxides was below 60% at 100 °C within 6 h. Endo et al.10 prepared bis(cyclic carbonate)s via the coupling reaction of CO2 with diepoxides in NMP at 100 °C under CO2 atmosphere. Furthermore, Wilkes11 found that epoxidized soybean oil could be effectively converted to carbonated soybean oil containing five-membered cyclic carbonates via reaction with CO2 employing tetrabutylammonium bromide (TBAB) as a catalyst at 110 °C. Mülhaupt and co-workers12 reported that TBAB could catalyze the coupling reaction of limonene dioxide, epoxidized linseed and soybean oil with CO2 at 120–140 °C, and the limonene dioxide could be completely converted to the corresponding bis(cyclic carbonate) at 140 °C and 3 MPa within 43 h. Recently, Zhang13 reported that bis(cyclic carbonate)s could be prepared via the coupling reaction of diepoxides with CO2 catalyzed by the zinc–cobalt double metal cyanide complex; conversion of 81% could be obtained at 120 °C and 5 MPa CO2 pressure within 9 h. Furthermore, Guillaume14 prepared carbonate-end functionalized PTMCs through a complex series of reactions with suitable solvents. However, according to the above literature, the complete conversion of diepoxide in the coupling reaction needed a suitable solvent, relatively long time and harsh reaction conditions. Therefore, though some investigations about the diepoxide/CO2 coupling reaction have been carried out, the convenient synthesis of bis(cyclic carbonate) via the diepoxide/CO2 coupling reaction with the complete conversion of diepoxide still remained a challenge, before this work.
Bis(cyclic carbonate)s can react with di- or poly-functional primary alkyl amines, leading to the formation of non-isocyanate polyurethanes (NIPUs), which is an alternative route for obtaining the polyurethanes without using toxic phosgene or isocyanates.10–15 In this process, the ratio of carbonate to amine group is a major factor influencing the property of NIPUs, whose accuracy is significantly affected by the conversion of diepoxides, and the ideal ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Thus, if the epoxy groups were not completely carbonated, the product was a mixture containing mono-cyclic carbonate, bis-cyclic carbonate and diepoxide, which are difficult to separate, resulting in the error of the carbonate/amine group ratio. Thus, the highly efficient and quantitative synthesis of bis(cyclic carbonate)s is extremely significant.
:1. Thus, if the epoxy groups were not completely carbonated, the product was a mixture containing mono-cyclic carbonate, bis-cyclic carbonate and diepoxide, which are difficult to separate, resulting in the error of the carbonate/amine group ratio. Thus, the highly efficient and quantitative synthesis of bis(cyclic carbonate)s is extremely significant.
In this work, we report that an iron complex (Fig. 1), [N,N′-bis-2-pyridinylmethyl-cyclohexane-1,2-diamine]iron(II) chloride [Fe(BPMCDAC)], shows high catalytic activity in combination with TBAB towards the coupling reaction of CO2 and diglycidyl ethers (Scheme 1), whose iron active site is an environmentally friendly alternative for toxic central metal like cobalt and chromium. Under solvent-free conditions, glycol diglycidyl ether (1a) could be completely converted to the corresponding bis(cyclic carbonate) (2a) with a turn over number of 1000 under relatively mild conditions (100 °C and 3 MPa) in 4 h, which could be conveniently purified by flash column chromatography using CH2Cl2 as the eluent. Furthermore, the obtained bis(cyclic carbonate) (2a) could react with 1,2-ethane diamine (EDA), 1,3-propane diamine (PDA) or 1,6-hexane diamine (HDA) to produce NIPUs (Scheme 2) in an accurate ratio of carbonate to amine group.
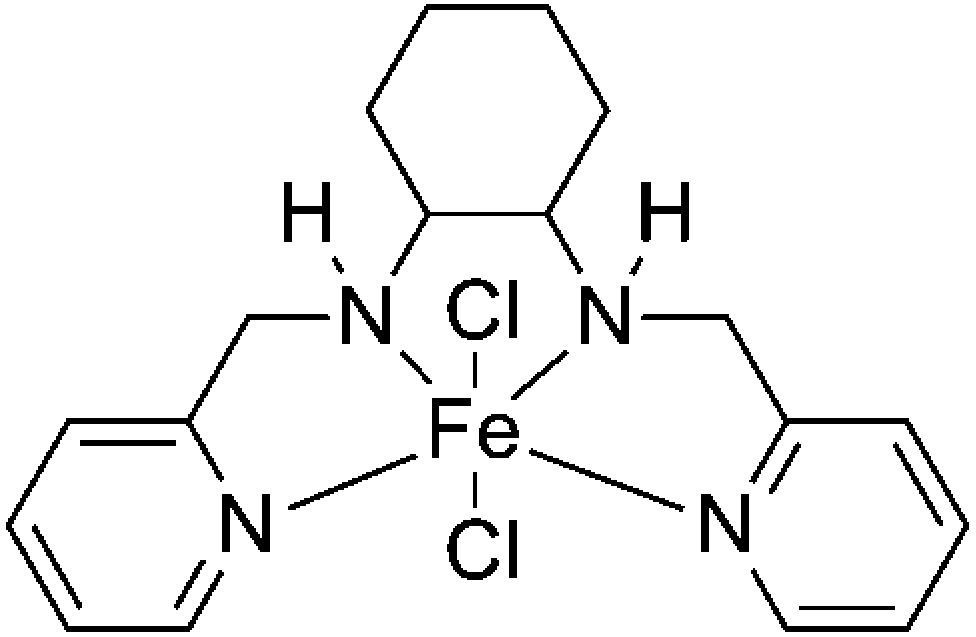 | ||
| Fig. 1 Structure of Fe(BPMCDAC). | ||
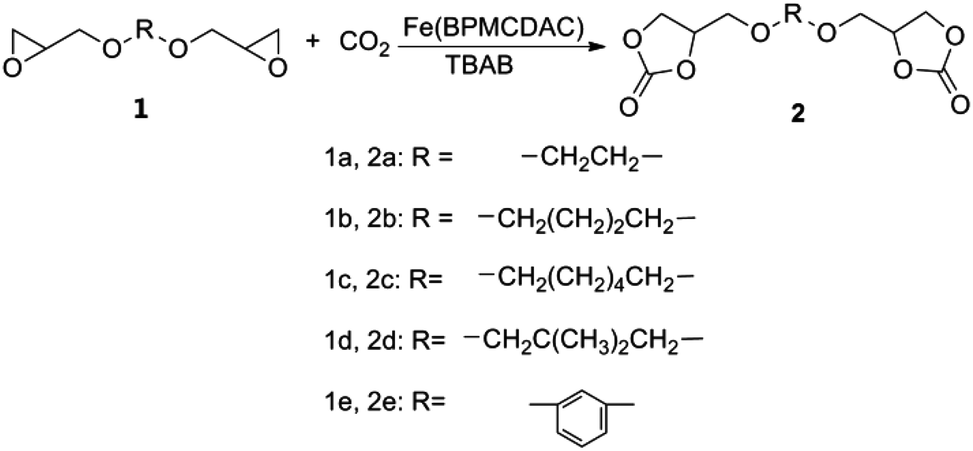 | ||
| Scheme 1 Coupling reaction of CO2 and diepoxides. | ||
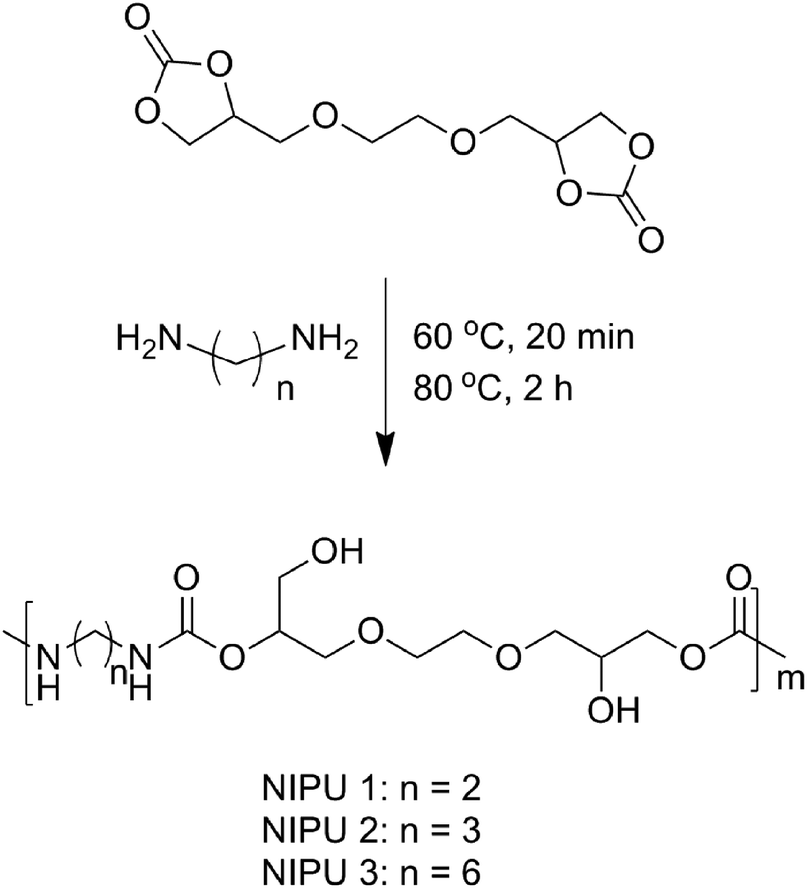 | ||
| Scheme 2 Synthesis of NIPUs from 2a and diamines. | ||
Experimental
Materials
Glycol diglycidyl ether (GDE) (1a) and other diglycidyl ether derivatives (1b–1e) were purchased from Ruipu Material Co. (China) and distilled over CaH2 under reduced pressure before use. [Fe(BPMCDAC)] was prepared following a literature procedure developed in this lab.16 Other reagents with analytical purity were used as received.Characterization
Synthesis of bis(cyclic carbonate)s
Diglycidyl ethers (1a–1e in Scheme 1), Fe(BPMCDAC) and TBAB were added to a stainless-steel autoclave with a magnetic stirrer in a glove box. CO2 was pressurized to this mixture, and the reaction was operated under determined conditions. After the reaction was completed, the autoclave was cooled to room temperature, and the CO2 pressure was released by opening the outlet valve. Then, the crude reaction mixture was obtained to calculate the conversion by 1H NMR spectroscopy in CDCl3. The catalysts were separated by flash column chromatography using CH2Cl2 as the eluent, and the obtained pure bis(cyclic carbonate) could be used for NIPU preparation.1a: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 3.72 (m, 2H), 3.62 (m, 4H), 3.36 (m, 2H), 3.10 (m, 2H), 2.72 (m, 2H), 2.54 (m, 2H).
1b: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 3.60 (m, 2H), 3.40 (m, 4H), 3.26 (m, 2H), 3.03 (m, 2H), 2.68 (m, 2H), 2.50 (m, 2H), 1.55 (m, 4H).
1c: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 3.61 (m, 2H), 3.41 (m, 4H), 3.26 (m, 2H), 3.03 (m, 2H), 2.68 (m, 2H), 2.48 (m, 2H), 1.48 (m, 4H), 1.26 (m, 4H).
1d: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 3.66 (m, 2H), 3.35 (m, 2H), 3.21 (m, 4H), 3.09 (m, 2H), 2.73 (m, 2H), 2.56 (m, 2H), 0.86 (m, 6H).
1e: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 7.17 (m, 1H), 6.51 (m, 3H), 4.21 (m, 2H), 3.91 (m, 2H), 3.34 (m, 2H), 2.88 (m, 2H), 2.74 (m, 2H).
2a: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 4.80 (m, 2H), 4.49 (m, 2H), 4.40 (m, 2H), 3.62–3.79 (m, 8H).
2b: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 4.76 (m, 2H), 4.43 (m, 2H), 4.30 (m, 2H), 3.44–3.63 (m, 8H), 1.55 (m, 4H).
2c: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 4.74 (m, 2H), 4.41 (m, 2H), 4.28 (m, 2H), 3.56 (m, 2H), 3.50 (m, 2H), 3.40 (m, 4H), 1.47 (m, 4H), 1.26 (m, 4H).
2d: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 4.78 (m, 2H), 4.42 (m, 4H), 3.62 (m, 2H), 3.52 (m, 2H), 3.18 (m, 4H), 0.86 (m, 6H).
2e: 1H-NMR (CDCl3, TMS, 300 MHz): δ (ppm) = 7.20 (m, 1H), 6.52 (m, 3H), 5.02 (m, 2H), 4.59 (m, 2H), 4.49 (m, 2H), 4.20 (m, 2H), 4.10 (m, 2H).
Synthesis of NIPUs
NIPUs could be synthesized through the polyaddition of bis(cyclic carbonate) (2a) and diamines (Scheme 2). 2a (60 mmol) and diamine (60 mmol) were added in a flask under N2 atmosphere. The mixture was stirred at 60 °C for 20 min, and then the temperature was raised to 80 °C and maintained for 2 h. All of the procedures were performed under N2 atmosphere.NIPU 1: 1H-NMR (d6-DMSO, TMS, 300 MHz): δ (ppm) = 7.12–6.75 (–NH), 4.65 (–OCH(CH2OH)CH2–), 3.36–3.91 (–OCH(CH2OH)CH2–, –CH2O–, –OCH2CH(OH)CH2–), 3.00 (–NHCH2). 13C-NMR (d6-DMSO, TMS, 75 MHz): δ (ppm) = 156.42, 156.06, 73.43, 72.33, 70.22, 69.79, 67.80, 65.69, 60.15, 40.22.
NIPU 2: 1H-NMR (d6-DMSO, TMS, 300 MHz): δ (ppm) = 7.09–6.72 (–NH), 4.66 (–OCH(CH2OH)CH2–), 3.36–4.05 (–OCH(CH2OH)CH2–, –CH2O–, –OCH2CH(OH)CH2–), 2.97 (–NHCH2), 1.50 (–CH2–). 13C-NMR (d6-DMSO, TMS, 75 MHz): δ (ppm) = 156.46, 156.12, 73.37, 72.39, 70.28, 69.85, 67.89, 65.67, 60.25, 38.03, 29.90.
NIPU 3: 1H-NMR (d6-DMSO, TMS, 300 MHz): δ (ppm) = 7.09–6.75 (–NH), 4.66 (–OCH(CH2OH)CH2–), 3.36–3.88 (–OCH(CH2OH)CH2–, –CH2O–, –OCH2CH(OH)CH2–), 2.93 (–NHCH2), 1.50–1.22 (–CH2–). 13C-NMR (d6-DMSO, TMS, 75 MHz): δ (ppm) = 156.33, 155.97, 73.14, 72.37, 70.20, 69.77, 67.84, 65.48, 60.19, 40.23, 29.43, 26.03.
Results and discussion
Reactions of carbon dioxide with glycol diglycidyl ether
Bis(cyclic carbonate)s were prepared by the cycloaddition reaction of diepoxides and CO2 catalyzed by Fe(BPMCDAC). Moreover, the selection of suitable co-catalysts was extensively investigated, and TBAB was found to be an effective co-catalyst in activating the iron catalytic system for the cycloaddition of epoxides and CO2 (in ESI Table S1†), which was consistent with our recent work.16 With TBAB as the co-catalyst, Fe(BPMCDAC) could effectively catalyze the coupling reaction of glycol diglycidyl ether (1a) and CO2 to generate the corresponding carbonate (2a). The data are listed in Table 1.| Entry | Temperature (°C) | Pressure (MPa) | Time (h) | Conversionb (%) | TONc | TOFd (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.1 mol% Fe(BPMCDAC), 0.1 mol% TBAB (relative to epoxy group). b Conversion was determined by 1H NMR spectroscopy. c Moles of epoxy group per mole of catalyst. d Moles of epoxy group produced per mole of catalyst per hour. | ||||||
| 1 | 100 | 3 | 1 | 73 | 730 | 730 |
| 2 | 100 | 3 | 2 | 91 | 910 | 455 |
| 3 | 100 | 3 | 3 | 96 | 960 | 320 |
| 4 | 100 | 3 | 4 | 100 | 1000 | 250 |
| 5 | 80 | 3 | 2 | 52 | 520 | 260 |
| 6 | 120 | 3 | 2 | 100 | 1000 | 500 |
| 7 | 100 | 1 | 2 | 91 | 910 | 455 |
| 8 | 100 | 2 | 2 | 90 | 900 | 450 |
The formation of the cyclic carbonate groups was quantitatively monitored via IR spectroscopy and 1H-NMR spectroscopy. Fig. 2 displays the IR spectra of diepoxide (1a) before and after the carbonation (2a). The strongest peak at 1795 cm−1 was ascribed to the carbonyl group of 2a. The 1H-NMR analysis was used to track the conversion of 1a to 2a at various times. Fig. 3 shows the 1H-NMR spectra of coupling products at 100 °C and 3 MPa (entries 1–4) using Fe(BPMCDAC)/TBAB as a catalyst. The signals between 2.54 and 3.10 ppm were ascribed to the methylene and methine protons of the epoxy group. By the decrease in the signal intensity at δ = 2.54–3.10 ppm, the conversion of 1a could be monitored. New signals at δ = 4.40–4.80 ppm were attributed to the methylene and methine protons of the cyclic carbonate group. The signals between 2.54 and 3.10 ppm completely disappeared at 100 °C and 3 MPa after 4 h, indicating that 1a could be completely converted to 2a under the applied reaction conditions, i.e., the quantitative synthesis of bis(cyclic carbonate) (2a) could be realized. This is extremely important for the preparation of NIPUs with high molecular weight.
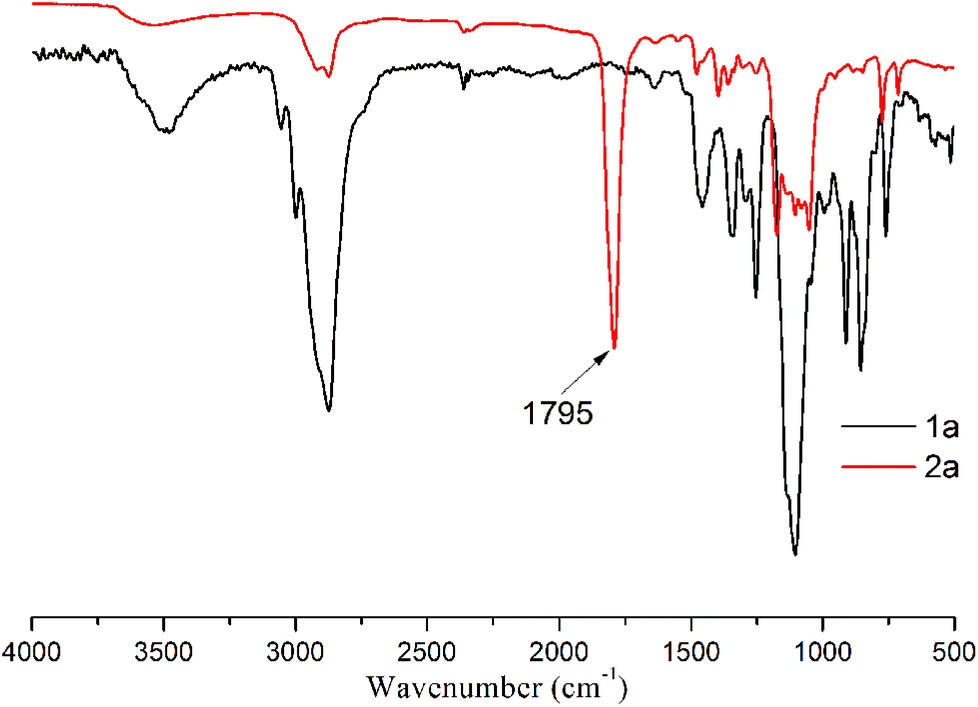 | ||
| Fig. 2 IR spectra of bis(cyclic carbonate) (2a) and diepoxide (1a). | ||
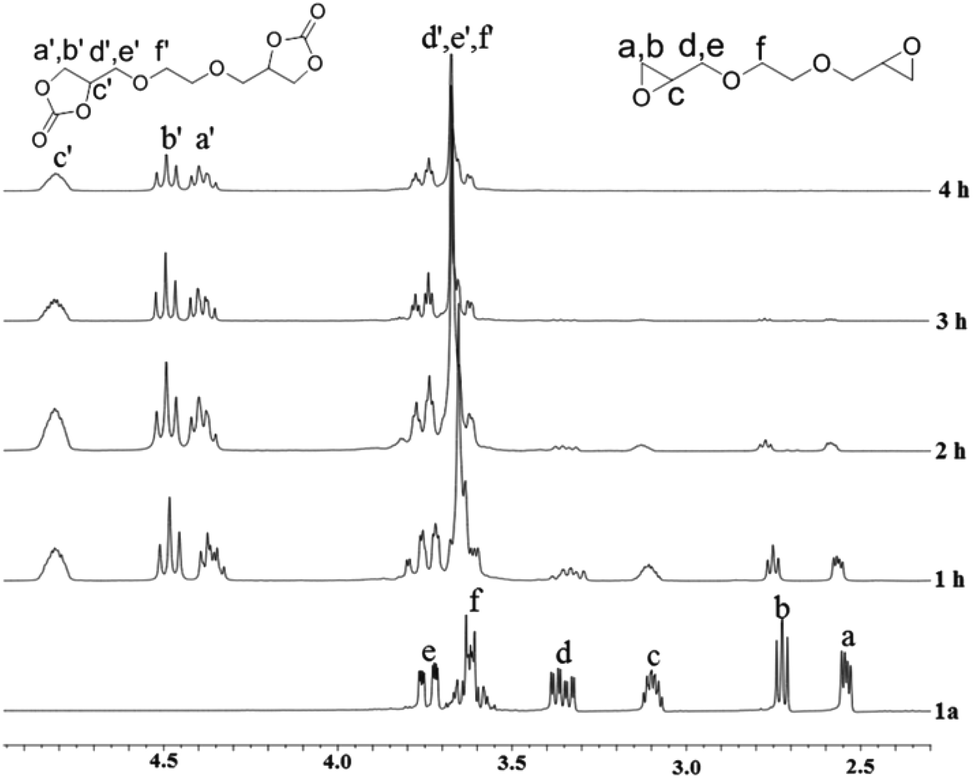 | ||
| Fig. 3 1H-NMR spectra of the coupling products at different times at 100 °C and 3 MPa using Fe(BPMCDAC)/TBAB as the catalyst. | ||
The reaction temperature is an important factor for the catalytic activity. As shown in Table 1, the catalytic activity significantly increased when the temperature increased from 80 to 120 °C. 1a was completely converted to the corresponding carbonate in 2 h at 120 °C (entry 6), whereas 4 h were required at 100 °C (entry 4). Higher temperature was beneficial for the conversion of quaternary ammonium salt into amine,17 the amine subsequently formed a carbamate salt with carbon dioxide; therefore, the activation of carbon dioxide was accelerated, leading to a faster coupling reaction.
The reactions with the iron catalyst were also conducted under different pressures. Using Fe(BPMCDAC) as the catalyst, the catalytic activities insignificantly varied when the CO2 pressure increased from 1 MPa to 3 MPa, indicating that the rate of the coupling reaction was independent of the CO2 pressure in this range.
Therefore, considering the catalytic performance, the easiness of operation, as well as economic efficiency, the optimal conditions for the coupling reaction of diepoxides/CO2 catalyzed by Fe(BPMCDAC) should be carried out at 100 °C and 3 MPa for 4 h in molar ratio [epoxy group/Fe(BPMCDAC)/TBAB] of 1000:1:1.
Furthermore, a reaction mechanism was proposed (Scheme 3). As reported in the literature,8c,17,18 coupling reaction of carbon dioxide with epoxides to generate corresponding cyclic carbonates requires activation, i.e., the Lewis acid activates the epoxide, whereas the Lewis base acts as a nucleophile to open the epoxide ring. In addition, the co-catalyst also plays an important role in the catalytic cycle. Based on the molecular structure of the complexes and the coupling reaction results, we propose a possible reaction pathway for the coupling reaction of diepoxides and carbon dioxide. It is possible that a chloride ligand, which is able to open the ring of epoxy group, dissociates from the iron centre forming a cationic iron species, which can coordinate and activate the epoxide.19 Furthermore, the co-catalyst (TBAB) has two important roles in the catalytic cycle:17a one is to provide a bromide for the ring-opening of the epoxide, and the other to form tributylamine that reacts with CO2 to form a carbamate salt for better CO2 activation. The activated CO2 can get inserted into the metal alkoxide bond to give a metal carbonate, which undergoes ring-closing to form the cyclic carbonate.
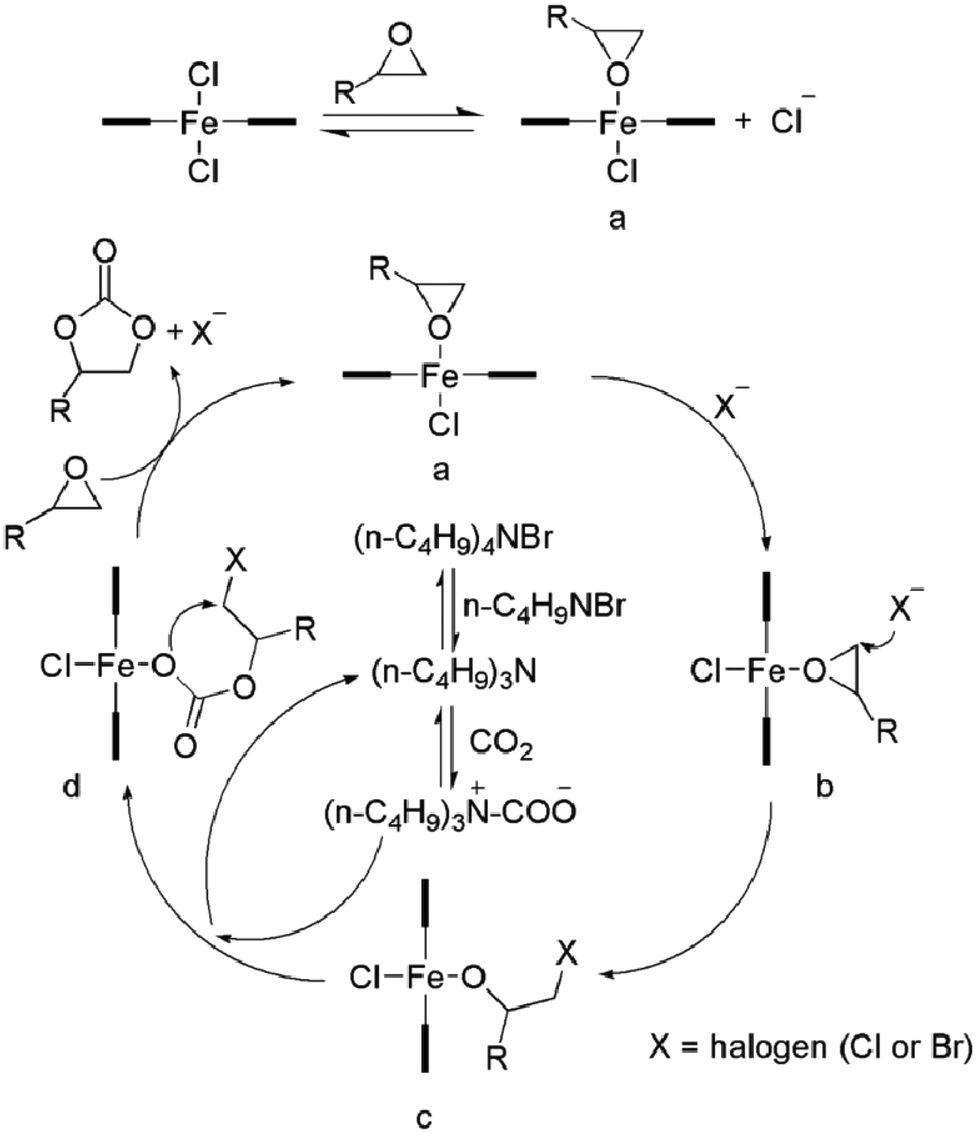 | ||
| Scheme 3 Proposed reaction mechanism for diepoxide/CO2 coupling. | ||
Reactions of carbon dioxide with a series of diglycidyl ether derivatives
To evaluate the versatility of Fe(BPMCDAC) catalyst for other diepoxides, the cycloaddition of CO2 with a variety of diepoxides (1b–1e) were investigated; the results are summarized in Table 2. In some cases it was necessary to use dichloromethane as the solvent during the conversion to avoid the high viscosity and/or solid nature of the reaction mixture.| Entry | Epoxide | Product | Time (h) | Conversion (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
|
a Reaction conditions: 0.1 mol% Fe(BPMCDAC), 0.1 mol% TBAB (relative to epoxy group), 3 MPa, 100 °C.
b Entry 2 and entry 4 from Table 1 were reproduced here for reference.
c Dichloromethane as the solvent, and the ratio of epoxide to solvent was 1:1 (v/v).
d Data from ref. 10c.
e Data from ref. 13.
f TON and TOF cannot be calculated due to heterogeneous catalysts.
|
||||||
| 1b | 1a | 2a | 2 | 91 | 910 | 455 |
| 2b | 1a | 2a | 4 | 100 | 1000 | 250 |
| 3 | 1b | 2b | 2 | 81 | 810 | 405 |
| 4 | 1b | 2b | 4 | 97 | 970 | 243 |
| 5 | 1c | 2c | 2 | 46 | 460 | 230 |
| 6 | 1c | 2c | 4 | 68 | 680 | 170 |
| 7 | 1d | 2d | 2 | 75 | 750 | 188 |
| 8 | 1d | 2d | 4 | 85 | 850 | 213 |
| 9 | 1e | 2e | 2 | 93 | 930 | 465 |
| 10 | 1e | 2e | 4 | 100 | 1000 | 250 |
| 11c | 1e | 2e | 2 | 39 | 390 | 195 |
| 12c | 1e | 2e | 4 | 55 | 550 | 138 |
| 13d | 1b | 2b | 72 | 74 | 132 | 1.8 |
| 14e | 1b | 2b | 9 | 82 | —f | —f |
The low loadings of catalyst (0.1 mol%) and co-catalyst (0.1 mol%) were generally effective at 100 °C and 3 MPa. All diepoxides can be effectively converted into the corresponding organic carbonates, and a conversion of over 50% was achieved, indicating the good versatility of this catalytic system. It should be noted that all the diepoxides could be completely converted into their corresponding organic carbonates within a sufficient reaction time. As anticipated, the conversions were time-dependent; they increased with longer reaction periods, while the turnover frequencies (TOFs) decreased. Furthermore, the catalyst system could not only catalyze the cycloaddition of the linear diepoxides and carbon dioxide, but could also tolerate the diepoxides that contain side chains or aromatic rings. Under the applied conditions, on prolonging the carbon chains, the conversions gradually decreased (entries 1, 3 and 5). A 91% conversion was obtained in 2 h for 1a, while it decreased to 81% for 1b, and only 46% conversion was observed for 1c. Moreover, a 75% conversion of 1d with a side chain was obtained in 2 h (entry 7), though it was much lower than that of 1a. Therefore, it can be assumed that the catalytic activity was affected by the aliphatic chain length in the monomer.9 As the aliphatic chain length increased; the addition of carbon dioxide was hindered because of chain folding or the fluidity of chains and the hindrance of methyl groups. The diepoxide 1e containing an aromatic ring had a relatively higher conversion; it reached 93% in 2 h, which was even higher than that for 1a (91%). The existence of an aromatic ring could increase the rigidity of chains and decrease the chain folding. Furthermore, phenoxy group could increase the Lewis acidity of the epoxy group to activate a diepoxide. Dichloromethane was used as the solvent because of the solid nature of the product (2e) at room temperature (entries 11 and 12). Unfortunately, the conversion of 1e significantly decreased compared to that without solvent because the solvent may reduce the diepoxide density leading to slow reaction rate. Furthermore, for diepoxides like 1b, Fe(BPMCDAC) exhibited great catalytic activity; a 97% conversion of 1b could be obtained in 4 h, which was considerably higher than the data mentioned in the literature (entries 4, 13 and 14).10c,13
Preparation of NIPUs
The obtained bis(cyclic carbonate)s could be applied to synthesize poly(hydroxyurethane)s via a polyaddition reaction with diamines. The polyaddition reactions of 2a with 1,2-ethane diamine (EDA), 1,3-propane diamine (PDA) and 1,6-hexane diamine (HDA) to produce NIPUs were carried out by a solvent-free and catalyst-free method, resulting in three polyurethanes with different backbones. The results are summarized in Table 3.| Entry | Diamine | NIPU | M n (kg mol−1) | PDIb (Mw/Mn) | T g (°C) | T d 5% (°C) | T d max (°C) |
|---|---|---|---|---|---|---|---|
|
a Reaction conditions: 2a:diamine (mol/mol) = 1:1, 60 °C for 20 min, then 80 °C for 2 h.
b Determined by GPC using dimethyl formamide (DMF) as the eluent.
c Measured by DSC.
d Measured by TGA.
|
|||||||
| 1 | EDA | NIPU 1 | 25.4 | 1.18 | 9 | 206 | 241 |
| 2 | PDA | NIPU 2 | 27.9 | 1.19 | 4 | 234 | 276 |
| 3 | HAD | NIPU 3 | 30.2 | 1.22 | −2 | 266 | 319 |
The polyadditon reactions of 2a and diamines were monitored by FT-IR spectroscopy. The FT-IR spectra in Fig. 4 shows a vibration peak at 1795 cm−1, which can be assigned to the carbonyl group of the cyclic carbonate. After polyaddition, the vibration peak at 1795 cm−1 disappeared, and new peaks appeared at 1700, 1540 and 3340 cm−1, which were typical for the carbonyl IR absorption of the urethane groups, N–H deformation of the urethane groups, and hydroxyl groups, respectively.
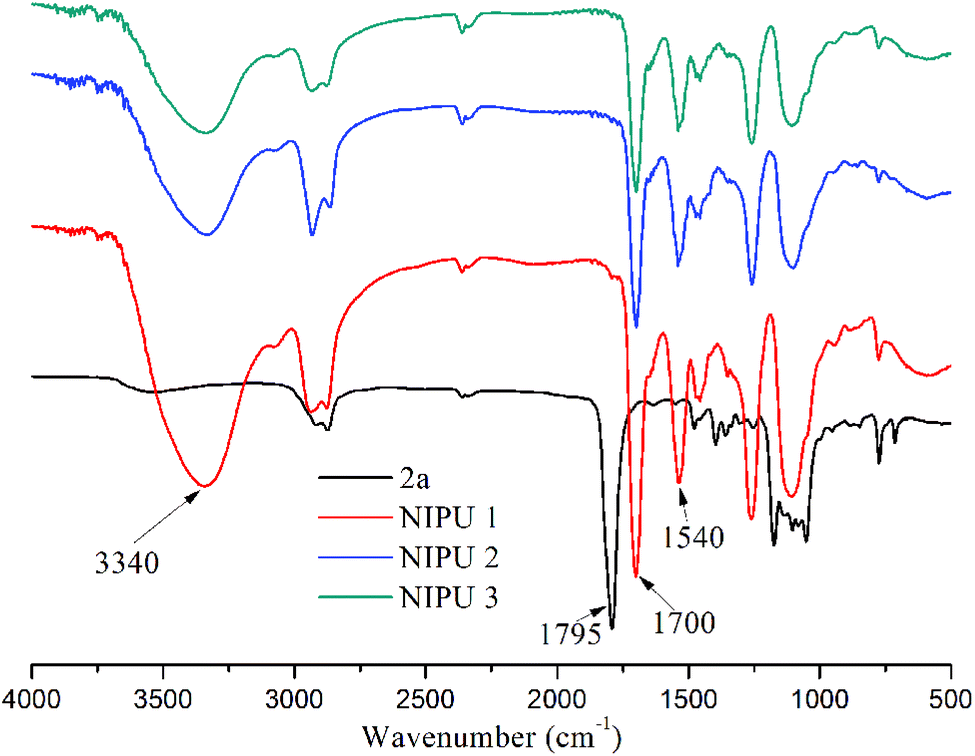 | ||
| Fig. 4 IR spectra of various NIPUs from diamine and 2a. | ||
As shown in Table 3, GPC analysis showed that the number-average molecular weights of the obtained NIPUs were between 25.4 and 30.2 kg mol−1, which were remarkable under solvent-free and catalyst-free conditions. Moreover, compared with the data in the literature,10,12a,13,15b the polydispersity indexes (PDIs) were relatively narrow between 1.18 and 1.22. Though the number-average molecular weights showed a slight decrease in comparison with the literature,14 the procedure to synthesize NIPUs was simple without the use of any solvent. Interestingly, the degrees of polymerization (DPs) of the resultant NIPUs were similar [m(NIPU 1) = 78.8, m(NIPU 2) = 82.9, m(NIPU 3) = 79.8] (Scheme 2), which were independent of the chain length of diamines.
The thermal properties of the NIPUs were characterized by DSC and TGA. The DSC thermograms are shown in Fig. S1,† and the evaluated data are presented in Table 3. The Tg values depend on the chemical structure of NIPUs. As expected, NIPU 1 had the highest Tg (9 °C) among the synthesized materials because of its smaller spacer length that limited the mobility of polymer chain segments for higher Tg.20 The thermal decomposition analysis under nitrogen of various NIPUs was investigated by TGA from ambient temperature to 500 °C. As shown in Fig. S2,† these NIPUs demonstrated initial degradation (Td = 5%) temperature between 206 and 266 °C, and maximum decomposition rate was between 241 and 319 °C. NIPU 3, which had the longest carbon chain, possessed the highest initial degradation temperature at 266 °C; its maximum rate of degradation occurred at relatively high temperature (319 °C), which was very interesting for these types of NIPUs, where the NIPU containing the longest spacer length showed the highest thermal stability.
Conclusions
Complete carbonation of diglycidyl ether derivatives with carbon dioxide was conveniently achieved by the Fe(BPMCDAC)/TBAB catalytic system under relatively mild conditions (100 °C and 3 MPa). Highly efficient and quantitative synthesis of 2a was realized with nearly 100% selectivity and 100% yield, with TON as high as 1000. The obtained bis(cyclic carbonate) (2a) could be used to prepare hydroxyl-functional green polyurethanes with diamines, which provided an alternative for conventional polyurethanes without toxic phosgene or isocyanates. Since iron is a green alternative to a toxic metal, and the synthesis of bis(cyclic carbonate)s and poly(hydroxyurethane)s does not use strongly toxic raw materials such as solvents, phosgene or isocyanates, these processes may exhibit great potentiality in industrial application.Acknowledgements
The authors thank financial support from the National Natural Science Foundation of China (grant no. 51321062, 51273197 and 21134002).Notes and references
- (a) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC; (b) M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 RSC.
- (a) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS; (b) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (c) W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A., 2009, 366, 2 CrossRef CAS PubMed; (d) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (e) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 RSC; (f) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS PubMed.
- (a) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259 CrossRef CAS; (b) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663 CrossRef CAS; (c) G. Rokicki and P. G. Parzuchowski, Polym. Sci.: Comprehensive Ref., 2012, 247 Search PubMed.
- (a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC; (b) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS PubMed; (c) J. M. Sun, S. I. Fujita, F. Y. Zhao, M. Hasegawa and M. Arai, J. Catal., 2005, 230, 398 CrossRef CAS PubMed; (d) H. S. Kim, J. J. Kim, H. Kim and H. G. Jang, J. Catal., 2003, 220, 44 CrossRef CAS; (e) H. Yasuda, L. N. He and T. Sakakura, J. Catal., 2002, 209, 547 CrossRef CAS; (f) Y. M. Shen, W. L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559 CrossRef CAS PubMed; (g) Y. Zhang, S. Yin, S. Luo and C. T. Au, Ind. Eng. Chem. Res., 2012, 51, 3951 CrossRef CAS; (h) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS PubMed.
- (a) R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622 RSC; (b) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC; (c) A. Sibaouih, P. Ryan, M. Leskela, B. Rieger and T. Repo, Appl. Catal., A., 2009, 365, 194 CrossRef CAS PubMed; (d) T. Roy, R. I. Kureshy, N.-u. H. Khan, S. H. R. Abdi and H. C. Bajaj, Catal. Sci. Technol., 2013, 3, 2661 RSC.
- (a) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS; (b) D. Adhikari, S. T. Nguyen and M.-H. Baik, Chem. Commun., 2014, 50, 2676 RSC; (c) S. Iksi, A. Aghmiz, R. Rivas, M. D. González, L. Cuesta-Aluja, J. Castilla, A. Orejón, F. El Guemmout and A. M. Masdeu-Bultó, J. Mol. Catal. A: Chem., 2014, 383, 143 CrossRef PubMed; (d) M.-H. Baik, D. Adhikari and S. Nguyen, Chem. Commun., 2014, 50, 2676 RSC; (e) Y. Niu, W. Zhang, H. Li, X. Chen, J. Sun, X. Zhuang and X. Jing, Polymer, 2009, 50, 441 CrossRef CAS PubMed.
- (a) Z. Wang, Z. Bu, T. Cao, T. Ren, L. Yang and W. Li, Polyhedron, 2012, 32, 86 CrossRef CAS PubMed; (b) D. Anselmo, V. Bocokic, A. Decortes, E. C. Escudero-Adan, J. Benet-Buchholz, J. N. H. Reek and A. W. Kleij, Polyhedron, 2012, 32, 49 CrossRef CAS PubMed; (c) C. Martin, C. J. Whiteoak, E. Martin, M. Martinez Belmonte, E. C. Escudero-Adan and A. W. Kleij, Catal. Sci. Technol., 2014, 4, 1615 RSC; (d) M. Taherimehr, A. Decortes, S. M. Al-Amsyar, W. Lueangchaichaweng, C. J. Whiteoak, E. C. Escudero-Adan, A. W. Kleij and P. P. Pescarmona, Catal. Sci. Technol., 2012, 2, 2231 RSC.
- (a) J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef; (b) J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577 RSC; (c) D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2029 CrossRef CAS; (d) S. Supasitmongkol and P. Styring, Catal. Sci. Technol., 2014, 4, 1622 RSC; (e) R. Luo, X. Zhou, S. Chen, Y. Li, L. Zhou and H. Ji, Green Chem., 2014, 16, 1496 RSC; (f) J. A. Castro-Osma, A. Lara-Sanchez, M. North, A. Otero and P. Villuendas, Catal. Sci. Technol., 2012, 2, 1021 RSC.
- M. R. Kim, S. R. Jeon, D. W. Park and J. K. Lee, J. Ind. Eng. Chem., 1998, 4, 122 CAS.
- (a) A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2375 CrossRef CAS; (b) B. Ochiai, S. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6613 CrossRef CAS; (c) N. Kihara and T. Endo, J. Polym. Sci., Part. A: Polym. Chem., 1993, 31, 2765 CrossRef CAS.
- B. Tamami, S. Sohn and G. L. Wilkes, J. Appl. Polym. Sci., 2004, 92, 883 CrossRef CAS.
- (a) M. Baehr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447 RSC; (b) M. Baehr and R. Muelhaupt, Green Chem., 2012, 14, 483 RSC.
- R.-j. Wei, X.-h. Zhang, B.-y. Du, Z.-q. Fan and G.-r. Qi, RSC Adv., 2013, 3, 17307 RSC.
- M. Helou, J.-F. Carpentier and S. M. Guillaume, Green Chem., 2011, 13, 266 RSC.
- (a) I. Javni, D. P. Hong and Z. S. Petrovic, J. Appl. Polym. Sci., 2013, 128, 566 CrossRef CAS; (b) A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205 RSC.
- X. Sheng, L. Qiao, Y. Qin, X. Wang and F. Wang, Polyhedron, 2014, 74, 129 CrossRef CAS PubMed.
- (a) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS PubMed; (b) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828 CrossRef CAS PubMed.
- X. B. Lu, B. Liang, Y. J. Zhang, Y. Z. Tian, Y. M. Wang, C. X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732 CrossRef CAS PubMed.
- J. E. Dengler, M. W. Lehenmeier, S. Klauss, C. E. Anderson, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2011, 336 CrossRef CAS.
- V. Besse, G. Foyer, R. Auvergne, S. Caillol and B. Boutevin, J. Polym. Sci., Part. A: Polym. Chem., 2013, 51, 3284 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental results, DSC and TGA thermograms of various NIPUs. See DOI: 10.1039/c4gc01294a |
| This journal is © The Royal Society of Chemistry 2015 |