
Reductive functionalization of CO2 with amines: an entry to formamide, formamidine and methylamine derivatives
Anis
Tlili
,
Enguerrand
Blondiaux
,
Xavier
Frogneux
and
Thibault
Cantat
*
CEA, IRAMIS, SIS2M, CNRS UMR 3299, 91191 Gif-sur-Yvette, France. E-mail: thibault.cantat@cea.fr; Fax: +33 1 6908 6640; Web: http://iramis.cea.fr/Pisp/thibault.cantat/index.html
First published on 23rd October 2014
Abstract
CO2 utilization for the production of C1-containing molecules is a desirable route to value-added chemicals. In this perspective, we summarize the recent results devoted to the formation of nitrogen compounds obtained by reductive functionalization of CO2 in the presence of amines. Using mild reductants, such as molecular hydrogen, hydrosilanes and hydroboranes, novel catalytic reactions have been designed in the last few years to facilitate the reductive functionalization of CO2 to formamide, formamidine and methylamine derivatives. While early efforts were devoted to the formylation of N–H bonds, efficient organic and metal catalysts have been developed lately to promote the complete deoxygenation of CO2 to benzimidazoles, quinazolinones, formamidines and methylamines. Finally, the opportunities and challenges facing the practical use of CO2 in the production of nitrogen-containing molecules are discussed.
1. Introduction
While the CO2 concentration in the atmosphere has exceeded the symbolic 400 ppm level this year, the search for technologies to reduce CO2 emissions is becoming urgent.1 A myriad of solutions have been proposed and are currently explored, either at the research or technological stages. CO2 Capture and Storage (CCS) technologies have the potential to mitigate meaningful volumes of CO2 and a few demonstration plants are slowly emerging.2 It is foreseeable, however, that this technology will suffer from the lack of added-value able to compensate for the energetic and economic cost of CO2 capture and deposition and the monitoring of the storage sites. The 30 Gt-CO2 emitted annually primarily derives from the mineralization of carbon fossil resources, i.e. hydrocarbons, coal and gas.3 In fact, utilization of these feedstocks in the fuel sector represents 80% of the world energy portfolio and their transformation in the chemical industry accounts for the production of 95% of organic chemical commodities.3 In this context, CO2 transformation into fuels and chemicals is a desirable alternative to CO2 storage for cutting the emissions of this greenhouse gas, while creating added value able to compensate for the costs of its capture.Yet, the chemical recycling of CO2 faces two energetic challenges. First, the thermodynamic stability of CO2 imposes an input of energy to convert CO2 into fuels and chemicals. The second challenge is kinetic in nature: catalysts are required to ensure that the activation barriers remain as low as possible along the chemical transformation pathways so that the overall carbon balance for CO2 utilization is not hampered by thermal loading needed to overcome high energy transition states. Obviously, these energetic considerations are associated with strong constraints on the resources utilized for CO2 conversion as the energy input must be carbon-free and rare or toxic metal catalysts must be avoided.
As a result of these limitations, only a handful of processes utilizing CO2 as a C1 building block have been industrialized to date, namely the Bosch–Meiser process for the production of urea from CO2 and ammonia, the Kolbe–Schmitt synthesis of salicylic acid (from CO2 and phenol) and the transformation of CO2 into carbonates.4 In total, they represent more than 70% of the total utilization of CO2 and less than 1% of the current anthropogenic CO2 emissions (Fig. 1).3 It is worth noting that, in these processes, CO2 is only functionalized: new C–O and C–N bonds are formed with no formal reduction of the CO2 carbon atom. In this context, a research field is expanding quickly to provide innovative transformations to produce organic ureas, urethanes, isocyanates and N-heterocycles featuring a CIV center, by CO2 functionalization.5
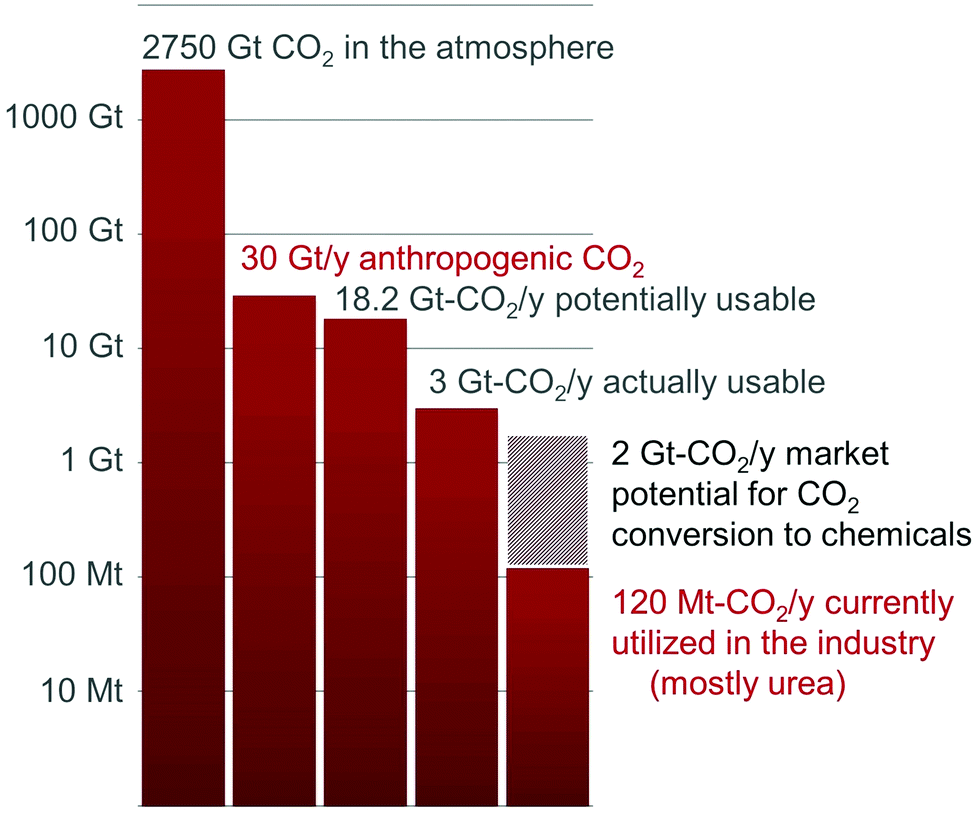 | ||
| Fig. 1 Current and potential market volumes for the conversion of CO2 to chemicals. | ||
In contrast, CO2reduction to formic acid, methanol, methane or light alkanes is attracting increased attention because of the potential utilization of these molecules as fuels or hydrogen carriers.2a,6 CO2 reduction to fuels has the capacity to reduce significantly CO2 emissions by recycling about 95% of the 30 Gt-CO2 released every year by human activities.3 Nonetheless, the low market value of these compounds is set by the petrochemical industry and their competitive production from CO2 remains an unsolved challenge. Indeed, these molecules are energy intensive and their production from CO2 requires a large amount of “carbon-free” hydrogen, whose cost exceeds natural gas reforming.7 Remarkably, a Mt per year pilot-plant is operated by Carbon Recycling International (CRI) that takes advantage of renewable geothermal energy in Iceland to produce methanol from CO2.7,8
On the other hand, CO2 conversion to chemicals might offer niche applications in the short term, because the end-products possess an added-value able to balance the cost of CO2 capture and transformation. In this regard, CO2 is an attractive carbon feedstock in synthesis, as it is non-toxic, cost-efficient, secured and well-distributed. It should be pointed out that this application will not reduce significantly our carbon footprint, as it scales to a maximum of 7% of the anthropogenic CO2 emissions and 11% of the 18.2 Gt-CO2 per year produced at point sources (Fig. 1).3 However, this field could offer viable technologies for the recycling of CO2 and increase at the same time our knowledge on CO2 reduction to foster emerging industrial applications.7 Notably, the scope of chemicals directly available from CO2 remains very narrow, compared to current available petrochemicals. Even at the laboratory scale, the majority of the basic organic functional groups, such as ketones, amides and esters, are presently unavailable from carbon dioxide.5 Indeed, these functionalities feature a carbon atom that is both functionalized and reduced. As a result of these limitations, considerable efforts have been devoted recently to promote the reductive functionalization of CO2 and widen the spectrum of chemicals accessible from CO2. For example, Willauer et al. have recently investigated the formation of light olefins from CO2/H2 over heterogeneous catalysts.6n, 9 The utilization of CO2 as a carboxylation reagent for the formation of C–C bonds is also a hot topic which has been the subject of excellent reviews.10
Herein, we review the results obtained in CO2 conversion using amines as functionalizing reagents, in the presence of mild reductants having a redox potential close to the CO2/CH3OH couple (e.g. H2, hydrosilanes and hydroboranes). Initiated in the 1970s, this field has witnessed a rapid development over the past five years with the design of novel processes able to transform CO2 and amines into formamide, formamidine and methylamine derivatives.
2. Formylation of N–H bonds with CO2
The formylation of amines with CO2 is an important transformation since the desired formamide products are versatile chemicals and key building blocks. Among them, N,N-dimethylformamide (DMF) is a widely utilized polar solvent as well as an important chemical reagent utilized in a multitude of applications in organic synthesis.11 It is produced industrially by the carbonylation of dimethylamine (DMA) in the presence of methanol.12 An alternative synthesis route based on the use of CO2 instead of the toxic carbon monoxide and starting with dimethylamine in the presence of a reductant has attracted several research groups for a long time. Thereby especially homogeneous or heterogeneous transition metal catalysts have been proven to have high potential. The most efficient and promising systems are overviewed thereafter.The successive reduction of CO2 with hydrogen can afford formic acid, formaldehyde, methanol and methane. Although the formation of methanol from CO2/H2 is thermodynamically favourable, the first reduction of CO2 to formic acid is uphill. As such, the addition of a base is usually needed to facilitate CO2 reduction to a formate derivative. Using a secondary amine, such as DMA, as a base for the hydrogenation of CO2 led to the formation of the corresponding formamide by condensation of the amine to the formate and paved the way to the catalytic formylation of amines with CO2/H2 mixtures. The first catalytic examples reported in the literature were based on the use of the stable heterogeneous RANEY® Nickel as a catalyst in 1935.13 Some amines including secondary amines (piperidine, phenylethylamine) as well as primary amines (n-amylamine) were carbonylated to their corresponding formamides under quite drastic reaction conditions (60 bar of CO2, up to 160 bar of H2 and temperatures up to 250 °C) with low to moderate yields. The first homogeneous catalyst was reported by Haynes et al. in 1970.14 Several well-defined complexes were tested for the synthesis of DMF, the most active being (dppe)2CoH, (PPh3)2(CO)IrCl and (PPh3)3CuCl (dppe: 1,2-bis(diphenylphosphino)ethane) with TONs (TurnOver Numbers) reaching 1000. The reactions were performed at 125 °C, with an equimolar mixture of H2 and CO2, at a total pressure of 54 bar (Table 1, entries 1 and 2). It is noteworthy that different secondary as well as primary amines could be formylated also under similar reaction conditions in the presence of Rh or Pt based catalysts.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | CO2 (bar) | H2 (bar) | T (°C) | TON | Ref. |
| a Starting from dimethylammonium dimethylcarbamate. | ||||||
| 1 | (PPh3)3CuCl | 27 | 27 | 125 | 900 | 9 |
| 2 | (PPh3)2(CO)IrCl | 27 | 27 | 125 | 1200 | 9 |
| 3 | PdCl2 | 40 | 80 | 170 | 34 | 10 |
| 4 | ([Pt2(μ-dppm)3]) | 12 | 94 | 75 | 1460 | 12 |
| 5 | RuCl2[P(CH3)3]4 | 130 | 80 | 100 | 370![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
14 |
| 6 | RuCl2(dppe)2 | 130 | 85 | 100 | 740000 |
16 |
| 7 | [Mo] (1) | 25 | 35 | 110 | 115 | 19 |
| 8a | Cu/ZnO | 60 | 60 | 140 | 70 | 20 |
| 9 | Fe(BF4)2·6H2O/PP3 | 30 | 60 | 100 | 727 | 6i |
| 10 | Co(BF4)2·6H2O/PP3 | 30 | 60 | 100 | 1300 | 21 |
| 11 | Fe(BF4)2·6H2O/PPAr3 | 30 | 70 | 100 | 5100 | 22 |
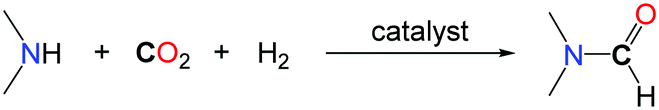
Afterwards, Kudo et al. demonstrated that simple PdCl2 salt is an effective catalyst, with a catalyst loading of about 3 mol%, for the synthesis of DMF.15 The reactions were performed in the presence of 40 bar of CO2, 80 bar of H2 and potassium bicarbonate as the base. The best result was obtained when the reaction mixture was heated at 170 °C (Table 1, entry 3).
Inspired by the system of Kudo et al. Morimoto et al. developed a new palladium based catalyst for the synthesis of diethylformamide.16 An optimized ratio of sodium formate and 2-methoxyethanol (0.05 mmol mL–1) in the presence of a catalytic amount of PdCl2(MeCN)2 (2.5 mol%) under an atmospheric pressure of CO2 were sufficient to obtain diethylformamide in good yields, without the need for hydrogen (Scheme 1). It is unclear however whether CO2 is the source of the formyl group in this transformation, as the reaction was undertaken in the presence of a formate reagent which can either act as a formyl donor or a reductant for CO2. Interestingly, the authors demonstrated that tetraethylurea could also be obtained with very high activity and selectivity in the presence of a well-defined ratio of PPh3/MeCN/CCl4, in the absence of any reductant.
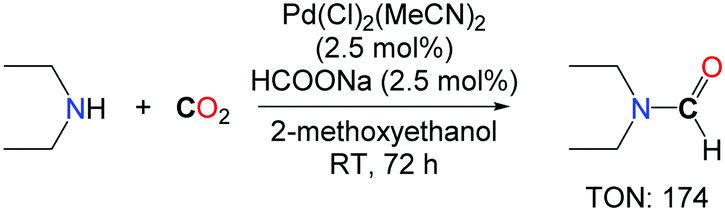 | ||
| Scheme 1 Pd-promoted synthesis of diethylformamide. | ||
A series of transition metals have also been tested by the group of Vaska.17 The most active catalyst for the synthesis of DMF starting from DMA and carbon dioxide (10–12 bar) in the presence of molecular hydrogen (67–94 bar) turned out to be a platinum based catalyst ([Pt2(μ-dppm)3]) (dppm: bis(diphenylphosphino)methane), working in toluene at 75 °C (Table 1, entry 4). Importantly, the authors demonstrated that the synthesis of DMF could be achieved under very mild reaction conditions and 1 bar of each of the three gas reactants was successfully transformed at room temperature. Moreover, they demonstrated that in the presence of [Pt2(μ-dppm)3]) the formation of DMF is readily reversible by heating DMF at 150 °C under low pressure of H2 (1 bar).
The use of NH3 as a reactive substrate presents a further challenge as three N–H bonds are potentially active in the formylation with CO2 and H2. The iridium complex [IrCl(CO)(PPh3)2] proved to be an effective catalyst for the synthesis of N-formylamine (FA) from NH3, CO2 and H2, in toluene or methanol solutions (Scheme 2).18 A TON of 1145 could be reached after 10 days. Yet, the source of the methyl groups in the methylated by-products (MMF: monomethylformamide, and DMF) was not established undoubtedly and could either originate from the hydrogenation of CO2 or the MeOH solvent.
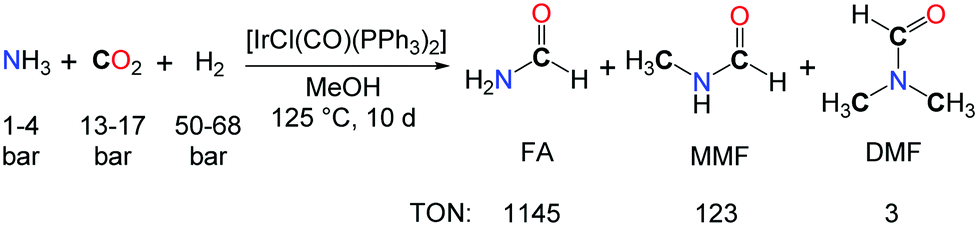 | ||
| Scheme 2 Formylation of ammonia promoted by [IrCl(CO)(PPh3)2]. | ||
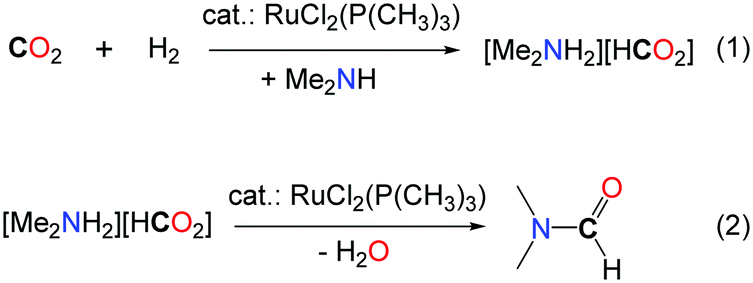
An elegant procedure has been developed by Noyori in 1994 for the synthesis of DMF with TONs up to 370000 and with overall rates up to 10000 h–1.19 The key of success of this system is the use of a well-defined homogeneous ruthenium catalyst, namely (RuCl2[P(CH3)3]4), with 80 bar H2, in supercritical CO2 (scCO2, 130 bar). scCO2 acts both as the reaction medium and the reactant and increases the solubility of the organic and gaseous reactants. It is noteworthy that the same results were obtained when the reactions were carried out with dimethylamine or the liquid dimethylammonium dimethylcarbamate as the source of DMA (Table 1, entry 5). The authors proposed that the formation of DMF proceeds via two steps. First, the ruthenium catalyst facilitates the hydrogenation of CO2 and the amine stabilizes the corresponding ammonium formate salt (Scheme 3, eqn (1)). In the second step, the catalyst promotes the condensation of the amine to the formate, to yield DMF (Scheme 3, eqn (2)). The same group demonstrated later on that the developed catalytic system could also be extended to the use of diethylamine and di-n-propylamine leading to the corresponding amides, albeit with a lower activity (TONs of 820 and 260, respectively).20 Moreover, when bulky dialkylamines were employed, only the corresponding ammonium formate salts were formed. It is likely that the condensation step is controlled both by the steric hindrance of the amine reagent and the solubility of the formate salt.
 | ||
| Scheme 3 Ru-catalyzed synthesis of DMF from DMA, using scCO2 and H2. | ||
Inspired by the discovery of the group of Noyori, the group of Baiker reported a more efficient protocol for DMF synthesis.21 A series of ruthenium complexes of the formula [RuCl2L2] have been prepared (L: dppm, dppe or dppp (1,3-bis(diphenylphosphino)propane)). The best catalyst system turned out to be [RuCl2(dppe)2] with very high TOFs (TurnOver Frequencies), up to 360000 h–1 (Table 1, entry 6). The experiments were performed at 100 °C in the presence of 130 bar of CO2 and 85 bar of H2. Under these conditions, 530 kg of DMF could be produced within 2 hours. In continuity with this promising result, the group of Baiker focused on the immobilisation of ruthenium/phosphorus catalysts.22 The synthesis of mesoporous ruthenium silica hydride aerogel has been achieved using the sol–gel process. The obtained materials showed high catalytic activities in DMF synthesis, leading to turnover frequencies up to 18400 h–1.
After this work, the group of Tumas and Baker proposed a new alternative based on a biphasic system.23 The association of scCO2 with 1-butyl-3-methylimidazolium hexafluorophosphate as the ionic liquid allowed for the hydrogenation of CO2 and the trapping of the formate ion by amines, catalyzed by RuCl2(dppe)2, to yield the corresponding formamides under 55 bar of H2 and 221 bar of scCO2 at 80 °C. Excellent selectivity has been observed with di-n-propylamine reaching a TON of 110.
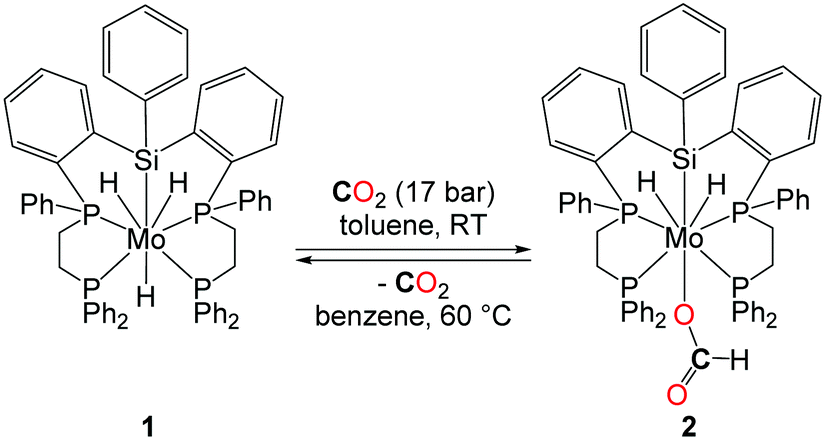
In 2001, the group of Ito reported the application of a new class of molybdenum-based catalysts in DMF synthesis (Table 1, entry 7).24 Even though the catalytic productivity of this well-defined molybdenum–silyl/phosphorus catalyst with a TON of 115 (under CO2/H2: 25/35 bar) is lower than those of other noble metals reported, the introduction of a noble metal-free system is highly desirable for such transformations. It is worth noting here that the molybdenum complex 1, depicted in Scheme 4, reacts under 17 bar of CO2 at room temperature yielding the formate complex 2. The insertion of CO2 into the Mo–H bond turned out to be reversible and 2 decarboxylates at 60 °C in benzene. Furthermore, 2 proved to be active in the catalytic formation of DMF, suggesting that it could be a potential intermediate in the catalytic reaction pathway.
 | ||
| Scheme 4 Reversible insertion of CO2 in the Mo–H bond of complex 1. | ||
More recently, the group of Han disclosed a novel heterogeneous noble metal-free system.25 A mixture of Cu–ZnO (3:2) allows for the synthesis of DMF starting from dimethylammonium dimethylcarbamate with a very good yield of 97% (Table 1, entry 8). The reactions were undertaken at 140 °C for 6 h under 120 bar of an equimolar mixture of CO2/H2.
Exploring the hydrogenation of CO2 with noble metal-free catalysts, the group of Beller, Laurenczy and co-workers described an efficient iron-based system.6i The active iron catalyst is generated in-situ from Fe(BF4)2·6H2O and PP3 (tris[2-(diphenylphosphino)ethyl]phosphine) as a ligand. DMF was obtained in a very good yield (75%) with a TON of 727, after 20 h at 100 °C, in the presence of 60 bar of molecular hydrogen and 30 bar of CO2 (Table 1, entry 9). Following this work, a novel cobalt catalyst (Co(BF4)2·6H2O/PP3) was shown to exhibit an improved catalytic activity with a TON of 1308 for the production of DMF, using similar conditions (Table 1, entry 10).26 In continuity, they presented later on a more efficient iron-based catalyst, with a new tetradentate ligand (PPAr3: tris(2-(diphenylphosphino)phenyl)phosphine) able to promote successfully the hydrogenation of CO2 with H2 (Table 1, entry 11). DMF as well as diethylformamide could be obtained with good to moderate yields and with TONs respectively of 5104 and 2114.27
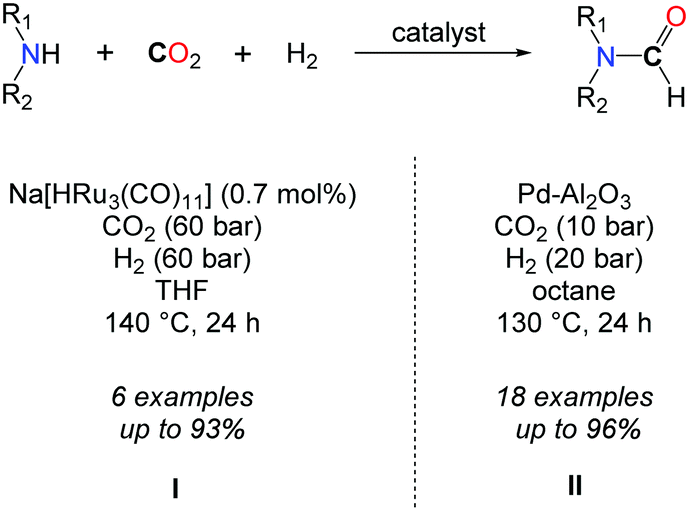
In parallel to the search for highly active catalysts in the formylation of amines with CO2/H2, efforts were devoted to enlarging the scope of active amines in this transformation. In this respect, Süss-Fink et al. showed, in 1989, that the ruthenium clusters, e.g. Ru3(CO)12, [HRu3(CO)11]− and [H3Ru4(CO)12]− (0.7 mol%), were active under 60 bar of CO2 and 60 bar of H2 in THF at 140 °C to convert piperidine, 3-methylpiperidine, pyrrolidine, heptamethylene imine, morpholine and piperazine to their formamides (Scheme 5, I).28 Even though good selectivities as well as good yields could be obtained, lower TONs (down to 43) of the catalyst were observed. With a similar scope of amines, increased activity was noticed with [RuCl2(dppe)2], in the presence of supercritical CO2, with TONs ranging from 850 to 210000, depending on the substrate and reaction conditions.29
 | ||
| Scheme 5 Catalyzed formylation of amines using CO2 and H2. | ||
The low basicity of aromatic amines significantly hampers the rates for the hydrogenation of CO2 and, in turn, the formylation of anilines with CO2 and H2 was found problematic. Interestingly, this limitation was circumvented by the group of Jessop with the use of extra bases.30 Indeed, conversion of PhNH2 to formanilide was achieved in 85% yield, using Noyori's system (RuCl2(P(CH3)3)4) at 100 °C for 10 h, in the presence of 2 molar equivalents of DBU (1,8-diazabicyclo-[5.4.0]undec-7-ene) as the additional base (Scheme 6).
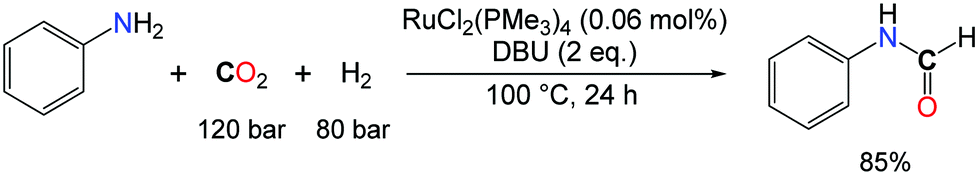 | ||
| Scheme 6 Ru-promoted synthesis of formanilide from aniline. | ||
To the best of our knowledge, only a heterogeneous palladium based catalyst, recently developed by the group of Shi, allows for the formylation of both aromatic and aliphatic secondary amines using CO2 in conjunction with H2 at quite low pressures (CO2/H2: 10/20 bar) (Scheme 5, II).31 A key feature of this system is the design of discrete palladium species supported on aluminium oxide. A variety of secondary aliphatic amines were converted into their corresponding formamide derivatives in very good to excellent yields. The authors demonstrated that the system is reusable and consistently good yields could be obtained after three runs. Nevertheless, it is not general since anilines and primary amines were very poorly converted to the desired products under the reaction conditions.
The search for alternative reductants able to promote the formylation of N–H bonds with CO2 was motivated by the narrow scope encountered with H2. In this respect, the group of Tanaka proposed a novel approach for the synthesis of DMF based on the use of the discrete ruthenium complex ([Ru(bpy)2(CO)2]2+) via the electrochemical reaction of CO2 with a mixture of dimethylamine and dimethylamine hydrochloride (Me2NH·HCl) with an efficiency of 21% with respect to DMF (Scheme 7).32
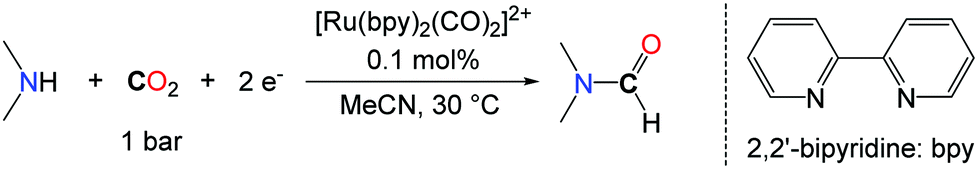 | ||
| Scheme 7 Electrochemical synthesis of DMF. | ||
The key of success of this system is the reactivity of DMA with the ruthenium carbonyl complex that leads to the formation of a carbamoyl complex ([Ru(bpy)2(CO)C(O)NMe2]+), which undergoes a two electron reduction to yield DMF.
The development of more general methodologies is of high interest to establish the synthetic utility of CO2 as a C1 building block in the formylation of amines. Hydrosilanes (R3Si–H) and hydroboranes (R2B–H) are excellent candidates for the reduction of CO2 because the slightly polar and weaker Si–H and B–H bonds (bond dissociation energy (BDE) 92 kcal mol–1 in SiH4 and 99 kcal mol–1 in BH3) are easier to activate than the strong non-polar H–H bond (BDE 104 kcal mol–1). These reductants could in principle enlarge the spectrum of active substrates. In this context, in 2011, our group reported a novel catalytic reaction to promote the formylation of N–H bonds, using CO2 as a carbon source and hydrosilanes as reductants.33
Interestingly, the kinetic advantage associated with the use of a hydrosilane reductant allows for the use of a metal-free system in the reduction of CO2. In fact, TBD (1,5,7-triazabicyclo[7.4.0]dec-5-ene) (5.0 mol%) is able to convert secondary amines with good to excellent yields when the reactions are undertaken in THF at 100 °C, with PhSiH3 (Scheme 8, I). Notably, the increased polarity of the solvent has a positive influence on the rate of the reaction and the best results were obtained in the absence of the solvent, where the polarity of the medium is augmented by the formation of the formamide product. While DMF was obtained in 80% yield from the carbamate [Me2NCO2][H2NMe2], the catalyst also enables the formylation of morpholine and diethylamine. Yet, primary amines such as n-hexylamine proved to be less reactive and aromatic primary amines are reluctant to formylation.
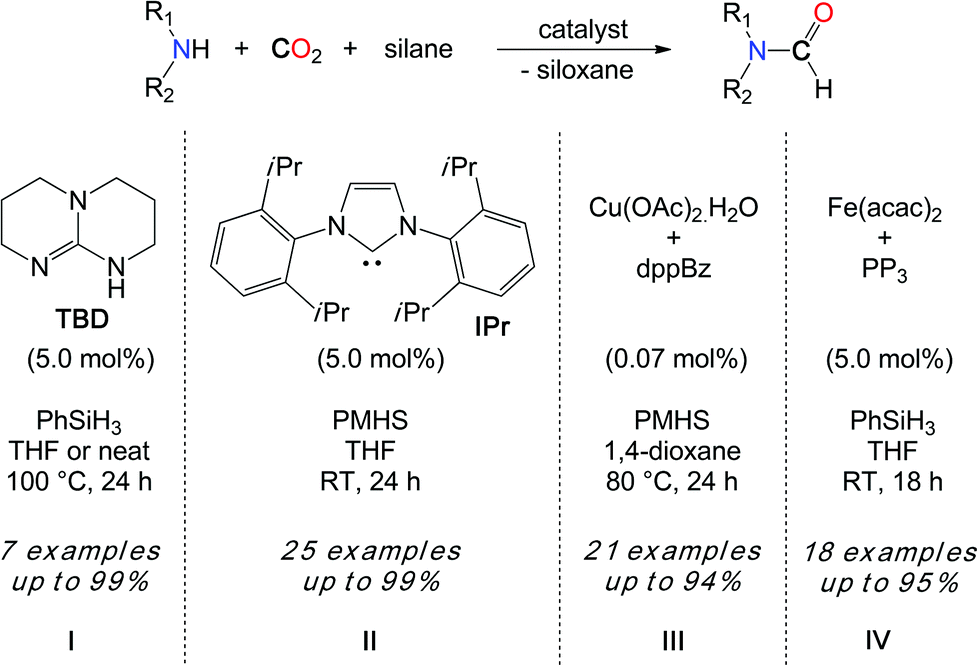 | ||
| Scheme 8 Formylation of amines using CO2 and hydrosilane reductants. | ||
From a mechanistic perspective, preliminary stoichiometric transformations suggested that TBD is able to activate the CO2 molecule to yield adduct 3 (Scheme 9).34 In the presence of morpholine, 3 is transformed into salt 4 in which the carbamate anion is efficiently stabilized by the guanidinium cation TBDH+ through H-bonds. Reacting 4 with one equivalent of phenylsilane affords the formamide product, the silanol as a by-product and the active TBD base.33 For comparison, alcohols (MeOH, iPrOH and PhCH2OH) also react with 3 and the formation of the corresponding carbonate salt was observed. Unfortunately, the reduction of these carbonates to the alkylformates was not achieved and the alcohol function was released as silyl ether.
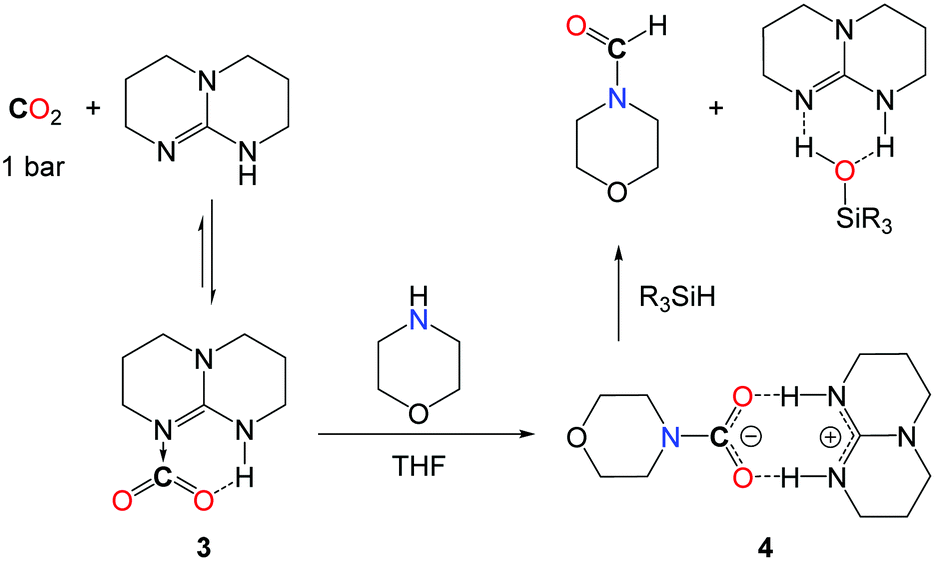 | ||
| Scheme 9 Mechanistic investigation for the TBD-catalyzed formylation of morpholine. | ||
Importantly, Zhang, Ying et al. demonstrated in 2009 that N-heterocyclic carbenes (NHCs) were efficient catalysts in the hydrosilylation of CO2 to methoxysilanes, also at room temperature.35 Inspired by this seminal work, our group has demonstrated that NHCs are able to promote the formylation of a wide scope of N–H bonds, with CO2 and hydrosilanes (Scheme 8, II).36 Using IPr as the most reactive organocatalyst, primary and secondary amines were quantitatively converted to their formamides, at room temperature. Notably, both N–H bonds of aniline derivatives could be formylated and the transformation was also extended to more fragile substrates such as imines, hydrazines, hydrazones and N-heterocycles. Overall, IPr is about 2000 times more reactive than TBD and this increased catalytic activity enabled the use of less reactive hydrosilanes in the formylation reaction. In fact, polymethylhydrosiloxane (PMHS) is a highly attractive reductant in hydrosilylation chemistry, as it is a cost-efficient, non-toxic and air-stable by-product of the silicone industry. Importantly, PMHS proved to be an efficient reductant in the NHC-catalyzed formylation of N–H bonds with CO2. More recently, a copper/diphosphine catalytic system has been developed by the group of Baba.37 This system has the advantage of using an earth abundant metal catalyst, namely a 1:3 mixture of Cu(OAc)2·H2O and dppBz (dppBz: 1,2-bis(diphenylphosphino)benzene). With low catalyst loadings (<0.1 mol%), TONs up to 9000 could be achieved. Moreover, an atmospheric pressure of CO2 (1 bar) was sufficient for the formylation of amines using PMHS as a reductant, at 80 °C (Scheme 8, III). Primary and secondary amines could be reacted in moderate to very good yields. Although our group has shown that Fe(acac)2 supported by P(C2H4PPh2)3 is an efficient formylation catalyst at room temperature with 1 bar CO2, this iron system is limited to PhSiH3 and milder PMHS and TMDS hydrosilanes are unreactive under these conditions (Scheme 8, IV).38
In 2013, the group of Mizuno followed a two step procedure to convert CO2 to formamides (Scheme 10, I).39 The hydrosilylation of CO2 was first accomplished with dimethylphenylsilane, using a catalytic amount of Rh2(OAc)4 (0.25 mol%) and K2CO3 (0.5 mol%). The resulting formoxysilane was subsequently trapped by addition of an amine. Although this reaction afforded formamides in low to moderate yields, the two step strategy allowed for the formation of a secondary alcohol by replacing the amine with a Grignard reagent.
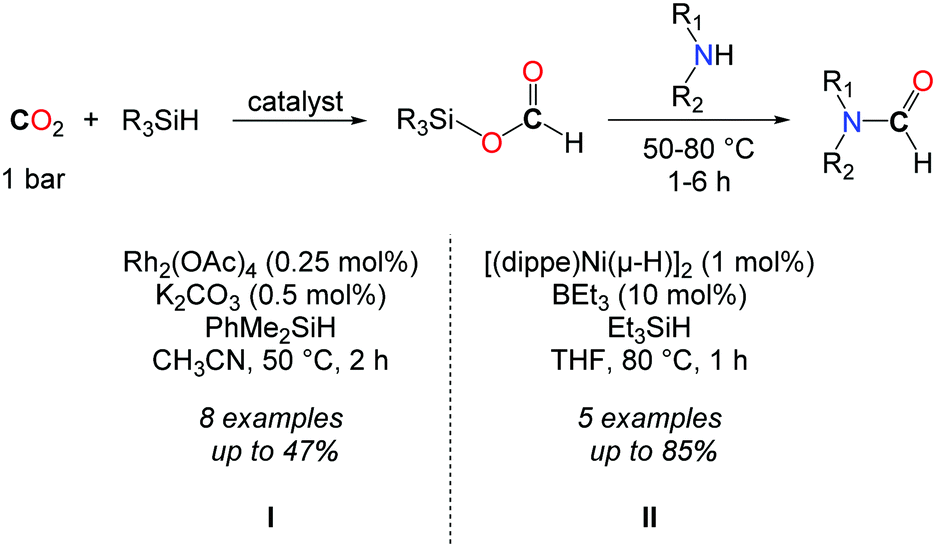 | ||
| Scheme 10 Two-step procedure for the catalytic formylation of amines using CO2 and hydrosilanes. | ||
In continuity with the work of Mizuno, the group of Garcia reported afterwards a Ni-based catalyst for the hydrosilylation of CO2.40 [(dippe)Ni-(μ-H)]2 (dippe: 1,2-bis(diisopropylphosphino)ethane) was used in THF at 80 °C as a well-defined Ni-catalyst. In order to achieve higher activity towards the hydrosilylation of CO2, catalytic amounts of a Lewis base (Et3B, 10 mol%) were needed, so as to polarize the Si–H and/or the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. Under these conditions, formamides could be obtained in very good yields starting from primary amines (Scheme 10, II). Nevertheless, secondary amines proved to be much less reactive in this approach and the formylation of dibenzylamine was limited to 47%, after 5 h.
O bonds. Under these conditions, formamides could be obtained in very good yields starting from primary amines (Scheme 10, II). Nevertheless, secondary amines proved to be much less reactive in this approach and the formylation of dibenzylamine was limited to 47%, after 5 h.
The reduction of CO2 using hydroboranes (namely 9-BBN, pinBH and catBH) has been recently discovered by the group of Guan, using nickel(II) complexes supported by phosphine–pinceur ligands (Scheme 11, I).41 In 2012, Sabo-Etienne, Bontemps and co-workers reported an active ruthenium(II) hydride complex able to promote the same reaction and reaction intermediates such as formoxyboranes, formaldehyde and a C2-compound (pinBOCH2OCOH) were identified in the reduction of CO2 to methoxyborane (Scheme 11, II).42 Remarkably, the authors were able to trap the transient formaldehyde intermediate with the bulky 2,6-bis(diisopropyl)aniline to afford the corresponding imine (2,6-iPr2–C6H3)NCH2.43
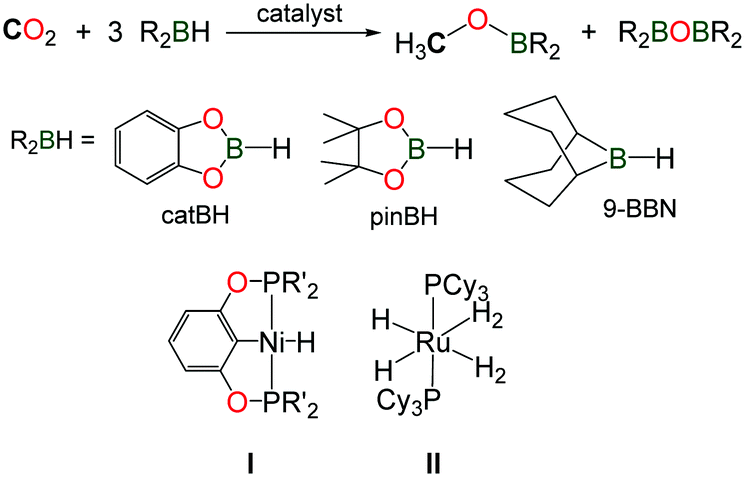 | ||
| Scheme 11 Reported catalysts for the reduction of CO2 to methanol using hydroboranes. | ||
In this context, Shintani and Nozaki showed that hydroboranes could serve as efficient reductants for the formylation of amines with CO2, albeit within a two-step strategy.44 Thus, a NHC/CuI system has been successfully employed for the mono formylation of primary as well as secondary amines in very good to excellent isolated yields (Scheme 12). The system relies on the use of pinacolborane in addition to the discrete IPrCu(OtBu) complex, present in 10 mol%. The reactions were carried out in THF at 35 °C for 24 h followed by the addition of the amine in the presence of an additional base (NEt3). The corresponding formamides were obtained after 45 h at 65 °C.
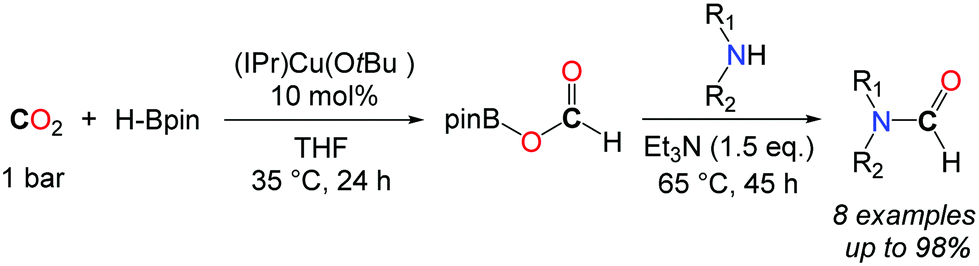 | ||
| Scheme 12 Formylation of amine via a catalytic hydroboration of CO2. | ||
3. Synthesis of formamidine derivatives using CO2
In the search for novel catalytic processes able to utilize CO2 as a C1 building block in organic synthesis, the complete deoxygenation of CO2 is of interest to access non-carbonyl compounds. Indeed, the scope of chemical functions available from CO2 is mostly limited to molecules in which at least one C–O bond from CO2 is retained and the complete reduction of CO2 to CH4 is one of the rare catalytic processes able to promote the deoxygenation of CO2.45 The recent success obtained for the conversion of CO2 to formamides, under mild conditions, enabled the utilization of this transformation in cascade strategies. Indeed, formamide derivatives can act as electrophiles and, for example, the condensation of a primary amine RNH2 to a formamide R′2NCHO is a route to the synthesis of formamidines RNC(H)NR′2, in the presence of hard drying agents, such as phosphorus oxychloride, trifluoroacetic anhydride and thionyl chloride. In 2013, our group and the group of Liu demonstrated independently that, starting from o-phenylenediamines, benzimidazoles were formed by two different catalytic systems (Schemes 13 and 14).46
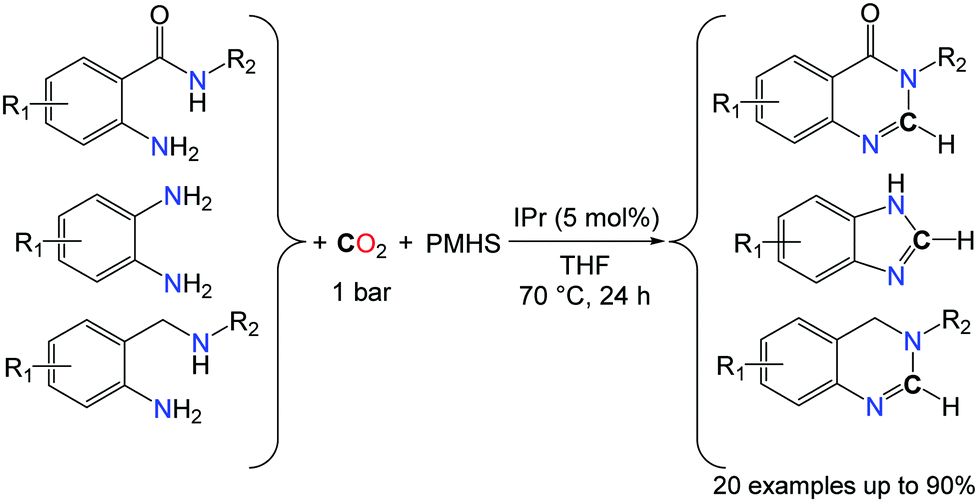 | ||
| Scheme 13 Synthesis of formamidine derivatives using CO2 and hydrosilanes as reductants. | ||
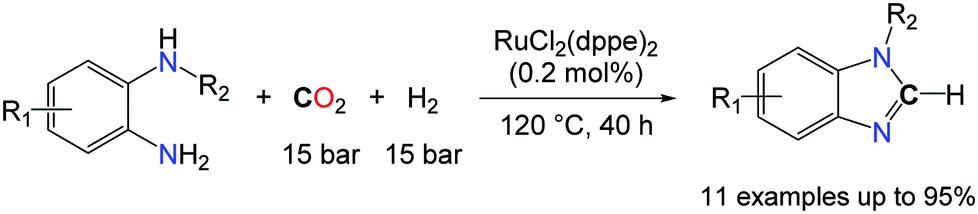 | ||
| Scheme 14 Synthesis of formamidine derivatives using CO2 and H2 as reductants. | ||
Using the NHC-catalyzed formylation of amines with CO2 and hydrosilanes, very good yields and selectivities could be achieved for the synthesis of benzimidazole rings, after the optimisation of the reaction temperature (70 °C instead of room temperature) in the presence of the PMHS as the reductant. The reactions were carried out in THF under 1 bar of CO2 (Scheme 13).46a It is noteworthy that the methodology is not limited to the formation of benzimidazole derivatives and 6-membered N-heterocyclic compounds could be obtained in good yields with anthranilamide and 2-aminobenzylamine derivatives. In fact, both amide and amine N–H bonds were found reactive in anthranilamides, leading to the formation of quinazolinones in the presence of CO2 and PMHS. Similarly, 3,4-dihydroquinazoline heterocycles were synthesized from their corresponding 2-aminobenzylamines.
In contrast, Liu and co-workers capitalized on the formylation of N–H bonds with CO2/H2 as an entry to formamidine derivatives.46b Utilizing RuCl2(dppe)2 (0.2 mol%) as a catalyst, benzimidazoles were formed in very good to excellent yields from o-phenylenediamines substituted with either electron withdrawing or donating groups. Nonetheless, the utilization of H2 as a reductant comes with a kinetic prize and enables only the preparation of benzimidazoles. It also imposes harsh reaction conditions, as the catalytic transformation proceeds at 120 °C under elevated gas pressures (mixture of CO2 and H2: 150 bar). The intermolecular version of these transformations would be desirable to access formamidines from two equivalents of primary amines and CO2, in the presence of a reductant. So far, hydrosilanes have appeared as the most promising reductant to tackle this challenge and N,N′-diphenyl-formamidine could be successfully obtained in 44% yield, from two equivalents of PhNH2 in the presence of an atmosphere of CO2 and 0.6 equiv. PhSiH3, using 5.0 mol% TBD.46a It is likely, however, that this transformation follows a different route as TBD is unable to convert aniline to its formamide. Given the recent developments in the successful reduction of urea derivatives to formamidines,47 it is probable that TBD favours the formation of N,N′-diphenyl-urea which is consecutively reduced by the hydrosilane.
4. Methylation of N–H bonds with carbon dioxide
Among nitrogen organic chemicals featuring C1 chemical groups, methylamines are the most reduced species with a N–CH3 carbon atom in the –II oxidation state. Current methylation methodologies rely on the use of formaldehyde in the presence of a reductant for the formation of the N–CH3 backbones and, in the laboratory, hazardous alkylating agents such as methyliodide, dimethylsulfate and dimethylcarbonate are preferred.48 Utilizing CO2 as a carbon source for the methylation of N–H bonds would therefore improve the sustainability of methylamines by providing an alternative route to their petrochemical synthesis.49 Such a transformation requires the use of a mild reductant and an efficient catalyst able to promote the 6-electron reduction of CO2 and facilitate the formation of a C–N bond. Early investigations on the synthesis of DMF from CO2, H2 and dimethylamine led to the observation of trimethylamine (TMA) as a side-product.17 The authors assumed that TMA resulted from the catalytic hydrogenation of DMF. Yet, as proposed by Vaska and co-workers, the scrambling of an alkyl group between two amine molecules or between an amine and an amide is possible and could account for the formation of TMA, upon reductive functionalization of CO2 with Me2NH and H2.In 1995, Baiker et al. reported the first synthesis of methylamines, in a fixed-bed microreactor, using a mixture of ammonia, H2 and CO2 and heterogeneous Cu/Al2O3 catalysts.50 Nevertheless, very harsh reaction conditions were required, as the experiments were conducted between 240 and 280 °C under 6 bar of NH3–CO2–H2 (ratio 1/1/3) (Scheme 15). The formation of methylamine (MA) is favoured at high temperatures together with the production of CO via the reverse water gas shift reaction. Importantly, the concentration of ammonia in the gas mixture has a significant impact on the selectivity and, at 240 °C, MA is favoured at high concentrations while low concentrations of NH3 facilitate the accumulation of TMA.
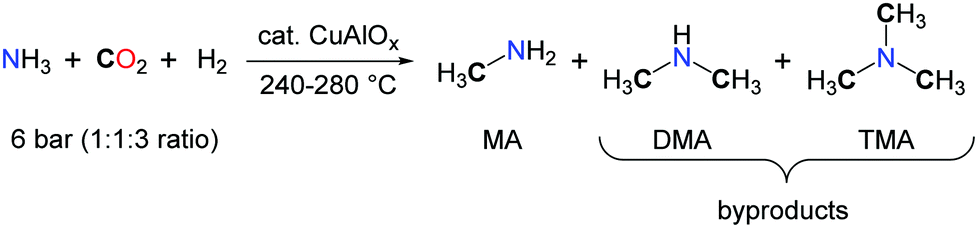 | ||
| Scheme 15 Methylation of ammonia catalyzed by a CuAlOX catalyst. | ||
Two plausible pathways were proposed for the catalytic mechanism depending on the carbon species involved in the formation of the C–N bond.51 Indeed, methanol, obtained by hydrogenation of CO2, can undergo an amination step to afford the methylamine product and a molecule of water. Alternatively, a C0 intermediate corresponding to the adsorption of formaldehyde at the catalyst surface can account for the formation of an imine intermediate, which is readily hydrogenated to its methylamine. A subsequent work was devoted to testing various types of metal–alumina catalysts (Cu, Ag, Pt, Ni, Co, Fe), under the same reactions conditions, so as to improve the catalytic activities and selectivities.52 The best amine production rates were obtained with Cu-based catalysts. Cu–Mg–Al mixed oxides showed interesting activities in this reaction at 280 °C for the same gas composition.53 While good selectivities were obtained in all cases for the formation of MA (>79%), the catalytic activity is directly controlled by the content of aluminium in the catalytic system. The rate of methylamine production is tripled when the molecular ratio of aluminium is increased from 25 to 33% in the catalyst (0.24 vs 0.67 mol kgcata–1 h–1).
In 2013, our group demonstrated that replacing H2 with a more polar hydrosilane reductant enables the methylation of a large scope of amines, under mild reaction conditions, namely with 1 to 5 bar CO2 at 100 °C (Scheme 16, I).54 Using a homogeneous system, based on a discrete NHC/Zn catalyst a variety of primary and secondary aromatic and aliphatic amines were successfully methylated, from CO2 as the carbon source and PhSiH3 as the reductant. The source of the extra methyl group was established unambiguously using 13C-labelled CO2. While bare ZnCl2 and ZnEt2 salts showed a modest catalytic activity in the methylation of N-methylaniline, a set of NHCs, phosphines and N-heterocycles were screened as supporting ligands to enhance the catalytic activity and NHCs afforded the highest conversions. It is likely however that the carbene ligands increase the stability of the Zn catalyst rather than its activity, as it was recently demonstrated that ZnH2 is efficiently stabilized using NHCs.55
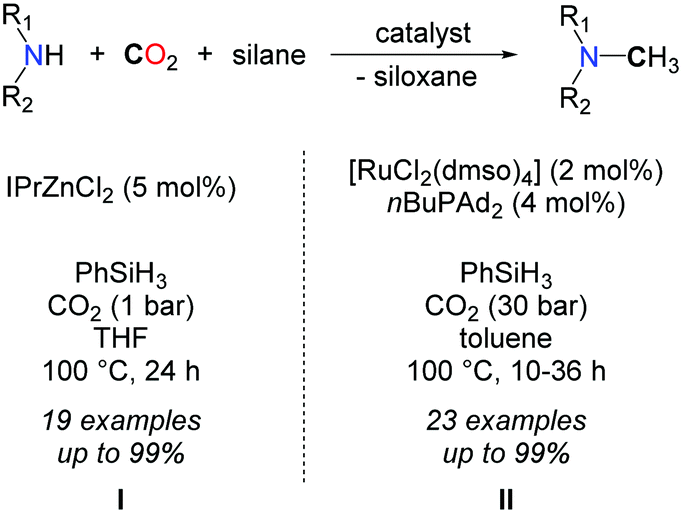 | ||
| Scheme 16 Catalytic systems for the methylation of amines using CO2 and hydrosilanes. | ||
Preliminary mechanistic investigations were conducted so as to identify the main intermediates involved in the methylation reaction. Kinetic data and monitoring of the product distribution over time showed that formamides are the main intermediates. In fact, the Hammett correlation, studied for substituted N-methylaniline reagents, reveals that the mechanism involves two steps with opposite electronic demands at the nitrogen center. While the formylation of the N–H bond is favoured by electron-donating groups, the reduction of the resulting formamide is facilitated in the presence of electron-withdrawing groups on the aryl ring (Scheme 17). Notably, this mechanism is consistent with the known catalytic activity of zinc complexes in the hydrosilylation of amides.56 As a result of this mechanism, secondary aliphatic amines are less reactive than aromatic amines and require longer reaction times (up to 72 h) to undergo the methylation of N–H bonds, because the hydrosilylation of the corresponding formamide is significantly slower.
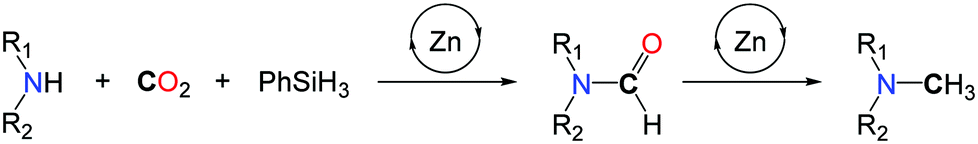 | ||
| Scheme 17 Proposed mechanism for the methylation of amines with IPrZnCl2. | ||
Shortly afterwards, an elegant procedure has been published by the group of Beller to promote the same reaction.57 Once again hydrosilanes (namely PhSiH3) were used as reductants in this case in association with the ruthenium complex RuCl2(dmso)4 (2 mol%) and CataCXium®A as a ligand (nBuPAd2, 4 mol%). Overall, the scope and reaction conditions are very similar to the zinc system and the reactions were carried out in toluene at 100 °C. 30 bar of CO2 were required to obtain good to excellent yields with primary and secondary amines (Scheme 16, II). Moreover, the authors successfully demonstrated that their procedure enables the selective methylation of the amine function when amino alcohols are used. Indeed, N-methylephedrine was obtained in good yields (73%) and high selectivity (>7:1) starting from ephedrine.
Based on mechanistic investigations, the authors postulate two possible pathways (Scheme 18). The ruthenium(II) catalyst may follow a route analogous to the zinc system and promote, first, the formylation of the N–H bond, followed by reduction of the formamide with PhSiH3. Alternatively, functionalization of CO2 with two equivalents of the amine reagent can afford a urea derivative, under dehydrating conditions. Consecutive hydrosilylation promoted by the Ru catalyst results in the formation of the methylamine product.
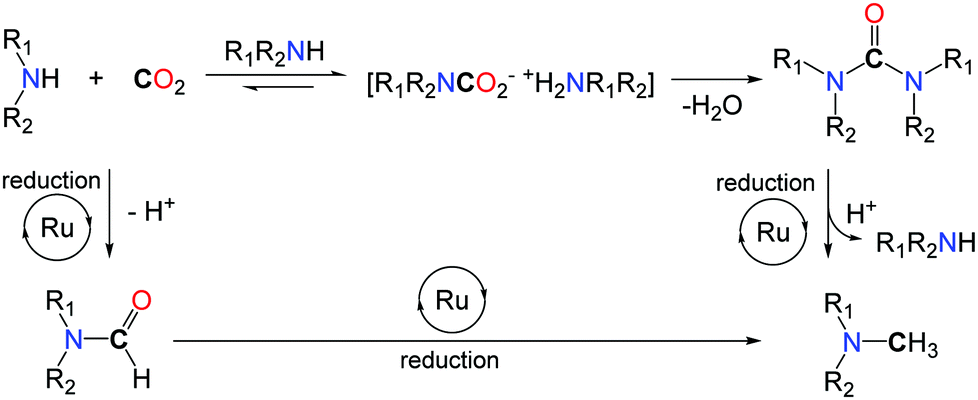 | ||
| Scheme 18 Proposed mechanism for the Ru-catalyzed methylation of amines. | ||
In this context control experiments were performed, starting from 1,2-ethylenediamines, and the two pathways were found viable. Additionally, the catalyst developed for the methylation of amines showed a catalytic activity in the hydrosilylation of ureas, under the applied reaction conditions.
Searching for earth abundant catalysts for the reduction of CO2, our group reported in 2014 the first iron catalysts able to promote the hydrosilylation of carbon dioxide. In the presence of P(C2H4PPh2)3 as a supporting ligand, Fe(acac)2 catalyzes the formylation of amines with PhSiH3 and 1 bar CO2 at room temperature while it facilitates the methylation of aniline derivatives at 100 °C.38
The use of H2 as a reductant for the methylation of amines with CO2 is highly desirable for large scale applications and the production of bulk chemicals from CO2. Yet, beyond the promising results obtained by the Baiker group (vide supra), efficient catalysts able to promote the methylation of a large scope of N–H bonds have remained elusive, until very recently. In 2013, Klankermayer et al. and Beller et al. reported independently the first homogeneous catalysts able to convert amines to methylamines using CO2/H2 as a source of the N–CH3 group (Scheme 19).58 The two catalytic systems are very similar and rely on ruthenium triphospine complexes activated by a catalytic amount of an acid, using the same ratio of CO2 and hydrogen (20/60 bar). The reactions for the mono or double methylations starting from secondary and primary amines are performed at nearly the same temperature (140–150 °C). Based on detailed mechanistic investigations, Klankermayer and Leitner developed over the last years a highly efficient molecular system, in catalytic reduction chemistry, consisting of the activation of a well-defined (triphos)Ru(tmm) (tmm=trimethylenemethane) complex by an acid co-catalyst such as trifluoromethanesulfonimide (HNTf2). This system showed a very good catalytic activity in the methylation of amines with CO2/H2.59
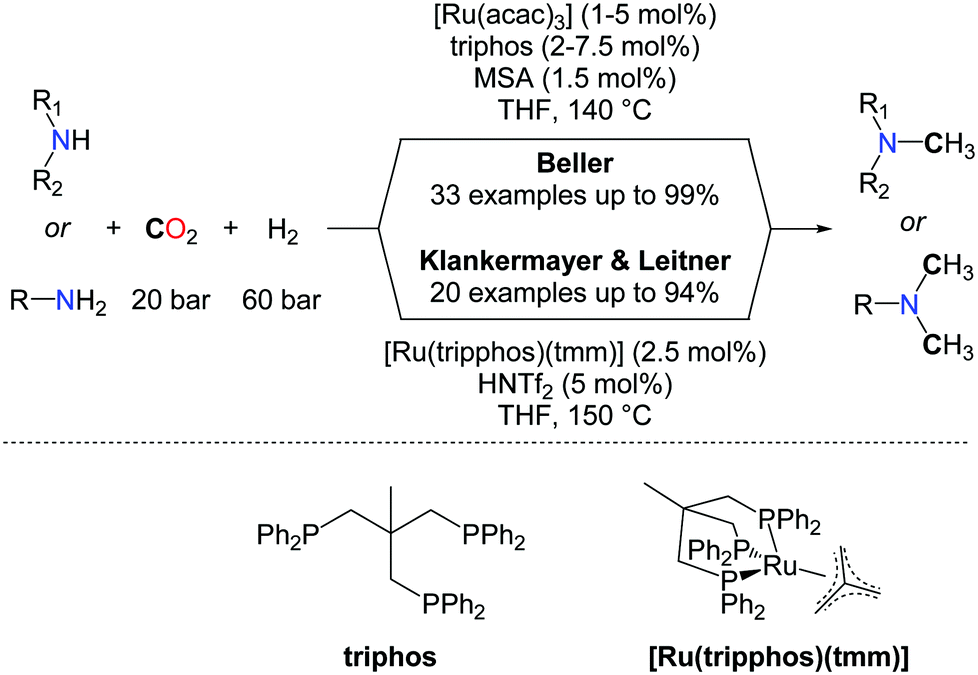 | ||
| Scheme 19 Ru-catalyzed methylation of amines using H2 and CO2. | ||
In the system developed by Beller and co-workers, Ru(acac)2 mixed with triphos(1,1,1-tris(diphenylphosphinomethyl)ethane) as the ligand is used in the presence of catalytic amounts of methanesulfonic acid (MSA, 1.5 mol%) to obtain the methylated amines in good to excellent yields (Scheme 19). Notably, several other ligands were tested under the same reaction conditions but only traces of the product were observed. Moreover the mono-methylation of amines could be achieved in the system developed by Beller by taking advantage of the electronic and steric hindrance of substituted anilines.
From a mechanistic perspective, both groups conclude that a sequential formylation/amide reduction is the most favourable route in the methylation reaction, and the formation of formamide intermediates was clearly demonstrated. As a consequence, aliphatic amines exhibit a lower reactivity than aromatic amines, presumably because the reduction of the corresponding formamide intermediate is more difficult, in the absence of an aromatic substituent. However, the Beller's group found that replacing the acid promoter with LiCl salt, together with increased catalyst loadings, enabled the quantitative bismethylation of aliphatic primary amines, such as benzylamine. In parallel, Klankermayer et al. successfully demonstrated the tandem hydrogenation and N-methylation of acetanilide yielding the unsymmetrical methyl/alkyl anilines. The chemoselectivity of the reaction was investigated and the mono-methylation of diamines shows promising selectivity. Despite the reductive conditions, the methylation of primary anilines is tolerant to ether, ester, hydroxyl, and carbon–halogen functionalities. Nonetheless, CC double bonds might be reduced under the applied conditions, depending on the substitution scheme of the unsaturation. Indeed, Klankermayer et al. observed that the CC bond in indole is hydrogenated upon methylation. In contrast, Beller's system enables methylation of nortriptyline, leaving the trisubstituted olefin untouched, and this latter reaction was utilized for the synthesis of 13C-labelled amitriptyline from 13CO2.
More recently, the group of Shi reported a CuAlOx catalyst able to promote the methylation of amines with CO2 and H2, albeit with high catalyst loadings.60 Primary and secondary aromatic and aliphatic amines as well as nitriles and nitroarenes were converted to methylamines, at elevated temperatures (up to 170 °C) and pressures (H2/CO2: 60–70/30 bar) (Scheme 20).
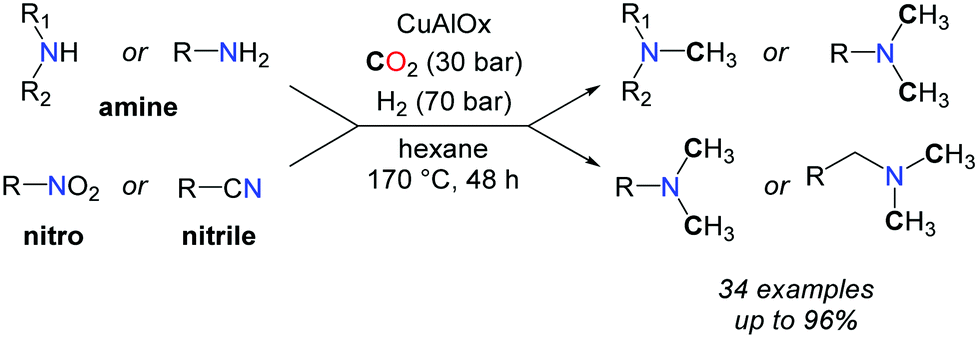 | ||
| Scheme 20 Methylation of amines with a heterogeneous CuAlOx system. | ||
5. Conclusions and outlook
The utilization of amines as bases to facilitate the hydrogenation of CO2 to formates led to the recognition that CO2 could serve as a C1 building block for the formylation of N–H bonds. Developed in the 1980s, this reaction has been mostly utilized for the conversion of dimethylamine to DMF and its scope remained quite limited, despite the design of novel metal catalysts. In 2012, the use of hydrosilanes in the formylation of amines with CO2 was unveiled. The hydrosilane reductant exhibits a kinetic advantage over H2 and enables the formylation of a wide scope of amines with organic catalysts working at room temperature and 1 bar CO2. Hydrosilanes and hydroboranes have a limited availability and must be utilized as non-renewable stoichiometric reagents. Nevertheless, they possess a mild reduction potential well-suited for CO2 reduction and they exhibit an increased chemoselectivity with respect to H2. This chemical behaviour allowed for the design of novel processes to promote the complete deoxygenation of CO2 to formamidine and methylamine derivatives, and the H2-versions of these transformations were unveiled shortly afterwards, using ruthenium catalysts. Overall, while organic ureas were privileged targets for CO2 conversion with amines, the reductive functionalization of CO2 has significantly opened the spectrum of nitrogen compounds available from this oxidized carbon feedstock.Future efforts will surely concern the design of novel catalysts based on earth abundant metal ions or organic molecules, with the aim of increasing the catalytic activity of these new transformations, at mild temperature and pressure. A better understanding of the mechanisms at play in the reductive functionalization of CO2 is a venue to explore, and to facilitate the design of next generation catalysts. Such knowledge would also help improve the chemo-selectivity of the reduction of CO2, as the presence of reducible chemical groups, such as aldehydes and ketones, has been problematic so far. Nonetheless, these efforts are motivated by the number of potential applications involving these novel reactions. While the conversion of NH3–CO2–H2 mixtures could provide meaningful alternatives to the production of bulk chemicals such as methylamine, dimethylamine, trimethylamine, and their formamide derivatives, these technologies will also be relevant in the synthesis of fine chemicals. Although challenging, 11C and 14C labelled molecules are important targets, as 11CO2 and 14CO2 are the primary sources of these C-isotopes and N–CHO, N–CHN and N–CH3 backbones are commonly found in nitrogen heterocycles and bioactive compounds.
Acknowledgements
For financial support of this work, we acknowledge the CEA, CNRS, ADEME, the CHARMMMAT Laboratory of Excellence and the European Research Council (ERC Starting Grant Agreement n.336467). T.C. thanks the Foundation Louis D. – Institut de France for its formidable support.Notes and references
- Carbon Dioxide: Projected emissions and concentrations, International Panel on Climate Change, 2013 Search PubMed.
- (a) P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281 RSC; (b) D. Stolten and V. Scherer, Efficient Carbon Capture for Coal Power Plants, Wiley-VCH, 2011 Search PubMed.
- Tracking industrial energy efficiency and CO2 emissions International Energy Agency, Paris, France, 2007 Search PubMed.
- (a) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (c) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (d) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed; (e) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (f) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS PubMed; (g) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC.
- Z. Z. Yang, L. N. He, J. Gao, A. H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602 CAS.
- (a) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC; (b) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC; (c) R. E. Rodriguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grutzmacher, Nat. Chem., 2013, 5, 342 CrossRef CAS PubMed; (d) M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85 CrossRef CAS PubMed; (e) N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645 RSC; (f) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS; (g) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 CrossRef CAS; (h) A. Boddien, D. Mellmann, F. Gartner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733 CrossRef CAS PubMed; (i) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777 CrossRef CAS PubMed; (j) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168 CrossRef CAS PubMed; (k) K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165 RSC; (l) E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702 CrossRef CAS PubMed; (m) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609 CrossRef CAS PubMed; (n) R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884 RSC.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, Chemsuschem, 2011, 4, 1194 CrossRef CAS PubMed.
- Carbon Recycling International, 2014. See http://www.carbonrecycling.is.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Catal. Commun., 2011, 15, 88 CrossRef CAS PubMed.
- (a) P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747 CrossRef CAS; (b) K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226 CrossRef CAS PubMed.
- H. Bipp and H. Kieczka, Formamides, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000 Search PubMed.
- M. W. Farlow and H. Adkins, J. Am. Chem. Soc., 1935, 57, 2222 CrossRef CAS.
- P. Haynes, L. H. Slaugh and J. F. Kohnle, Tetrahedron Lett., 1970, 11, 365 CrossRef.
- K. Kudo, H. Phala, N. Sugita and Y. Takezaki, Chem. Lett., 1977, 1495 CrossRef CAS.
- Y. Morimoto, Y. Fujiwara, H. Taniguchi, Y. Hori and Y. Nagano, Tetrahedron Lett., 1986, 27, 1809 CrossRef CAS.
- (a) S. Schreiner, J. Y. Yu and L. Vaska, Inorg. Chim. Acta, 1988, 147, 139 CrossRef CAS; (b) S. Schreiner, J. Y. Yu and L. Vaska, J. Chem. Soc., Chem. Commun., 1988, 602 RSC.
- L. Vaska, S. Schreiner, R. A. Felty and J. Y. Yu, J. Mol. Catal., 1989, 52, L11 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1994, 116, 8851 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344 CrossRef CAS.
- O. Krocher, R. A. Koppel and A. Baiker, Chem. Commun., 1997, 453 RSC.
- (a) O. Kröcher, R. A. Köppel and A. Baiker, J. Mol. Catal. A: Chem., 1999, 140, 185 CrossRef; (b) O. Krocher, R. A. Koppel, M. Froba and A. Baiker, J. Catal., 1998, 178, 284 CrossRef CAS; (c) L. Schmid, M. Rohr and A. Baiker, Chem. Commun., 1999, 2303 RSC.
- F. Liu, M. B. Abrams, R. T. Baker and W. Tumas, Chem. Commun., 2001, 433 RSC.
- M. Minato, D. Y. Zhou, K. Sumiura, R. Hirabayashi, Y. Yamaguchi and T. Ito, Chem. Commun., 2001, 2654 RSC.
- J. Liu, C. Guo, Z. Zhang, T. Jiang, H. Liu, J. Song, H. Fan and B. Han, Chem. Commun., 2010, 46, 5770 RSC.
- C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem. – Eur. J., 2012, 18, 72 CrossRef CAS PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701 CrossRef CAS PubMed.
- G. Süss-Fink, M. Langenbahn and T. Jenke, J. Organomet. Chem., 1989, 368, 103 CrossRef.
- (a) L. Schmid, A. Canonica and A. Baiker, Appl. Catal., A, 2003, 255, 23 CrossRef CAS; (b) L. Schmid, M. Schneider, D. Engel and A. Baiker, Catal. Lett., 2003, 88, 105 CrossRef CAS.
- P. Munshi, D. J. Heldebrant, E. P. McKoon, P. A. Kelly, C. C. Tai and P. G. Jessop, Tetrahedron Lett., 2003, 44, 2725 CrossRef CAS.
- X. Cui, Y. Zhang, Y. Deng and F. Shi, Chem. Commun., 2014, 50, 189 RSC.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, Chem. Lett., 1987, 597 CrossRef CAS.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187 CrossRef CAS PubMed.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465 CrossRef CAS PubMed.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322 CrossRef CAS PubMed.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934 CrossRef CAS PubMed.
- K. Motokura, N. Takahashi, D. Kashiwame, S. Yamaguchi, A. Miyaji and T. Baba, Catal. Sci. Technol., 2013, 3, 2392 CAS.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529 CAS.
- S. Itagaki, K. Yamaguchi and N. Mizuno, J. Mol. Catal. A: Chem., 2013, 366, 347 CrossRef CAS PubMed.
- L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 7186 CrossRef.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872 CrossRef CAS PubMed.
- (a) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671 CrossRef CAS PubMed; (b) S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253 CrossRef CAS PubMed.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419 CrossRef CAS PubMed.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459 CrossRef CAS.
- (a) A. A. Peterson and J. K. Norskov, J. Phys. Chem. Lett., 2012, 3, 251 CrossRef CAS; (b) C. Oloman and H. Li, Chemsuschem, 2008, 1, 385 CrossRef CAS PubMed; (c) T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2006, 128, 12362 CrossRef CAS PubMed; (d) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS PubMed; (e) S. Park, D. Bézier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404 CrossRef CAS PubMed; (f) M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323 CrossRef CAS PubMed; (g) P. G. Jessop, F. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS PubMed.
- (a) O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117 CrossRef CAS; (b) B. Yu, H. Zhang, Y. Zhao, S. Chen, J. Xu, C. Huang and Z. Liu, Green Chem., 2013, 15, 95 RSC.
- J. Pouessel, O. Jacquet and T. Cantat, ChemCatChem, 2013, 5, 3552 CrossRef CAS.
- (a) W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 880 CrossRef; (b) H. T. Clarke, H. B. Gillespie and S. Z. Weisshaus, J. Am. Chem. Soc., 1933, 55, 4571 CrossRef CAS; (c) E. Farkas and C. J. Sunman, J. Org. Chem., 1985, 50, 1110 CrossRef CAS; (d) F. Fache, L. Jacquot and M. Lemaire, Tetrahedron Lett., 1994, 35, 3313 CrossRef CAS; (e) J. R. Harding, J. R. Jones, S.-Y. Lu and R. Wood, Tetrahedron Lett., 2002, 43, 9487 CrossRef CAS.
- A. Tlili, X. Frogneux, E. Blondiaux and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 2543 CrossRef CAS PubMed.
- S. V. Gredig, R. A. Koeppel and A. Baiker, J. Chem. Soc., Chem. Commun., 1995, 73 RSC.
- S. V. Gredig, R. Koeppel and A. Baiker, Catal. Today, 1996, 29, 339 CrossRef CAS.
- S. V. Gredig, R. Koeppel and A. Baiker, Appl. Catal., A, 1997, 162, 249 CrossRef CAS.
- S. M. Auer, S. V. Gredig, R. A. Köppel and A. Baiker, J. Mol. Catal. A: Chem., 1999, 141, 193 CrossRef CAS.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127 RSC.
- A. Rit, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2013, 52, 4664 CrossRef CAS PubMed.
- (a) S. Das, D. Addis, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 12186 CrossRef CAS PubMed; (b) S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568 CrossRef CAS PubMed.
- (a) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156 CrossRef CAS PubMed; (b) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554 CrossRef CAS PubMed.
- (a) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499 CrossRef CAS PubMed; (b) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem. – Eur. J., 2013, 19, 11039 CrossRef CAS PubMed; (c) A. A. Nunez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154 RSC; (d) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem. Int., Ed., 2010, 49, 5510 CrossRef CAS PubMed; (e) F. M. A. Geilen, B. Engendahl, M. Holscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349 CrossRef CAS PubMed.
- X. J. Cui, X. C. Dai, Y. Zhang, Y. Q. Deng and F. Shi, Chem. Sci., 2014, 5, 649 RSC.
| This journal is © The Royal Society of Chemistry 2015 |