
Palladium-heterogenized porous polyimide materials as effective and recyclable catalysts for reactions in water†
E.
Rangel Rangel
a,
E. M.
Maya
a,
F.
Sánchez
*b,
J. G.
de la Campa
a and
M.
Iglesias
*c
aDept. Química Macromolecular Aplicada. Instituto de Ciencia y Tecnología de Polímeros, CSIC., C/Juan de la Cierva, 3, 28006 Madrid, Spain
bDept. Síntesis, Estructura y Propiedades de Compuestos Orgánicos. Instituto de Química Orgánica General, CSIC, C/Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: felix-iqo@iqog.csic.es
cDept. Nuevas Arquitecturas en Química de Materiales. Instituto de Ciencia de Materiales de Madrid, CSIC, C/Sor Juana Inés de la Cruz, 3. Cantoblanco, 28049 Madrid, Spain. E-mail: marta.iglesias@icmm.csic.es
First published on 12th September 2014
Abstract
New functionalized porous polyimides (PPIs-NO2, PPIs-NH2, and PPIs-NPy) were synthesized and characterized and the PPI-NPy materials were applied as supports to obtain heterogenized palladium-complexes (PPI-NPy-Pd). The PPI-NPy-Pd hybrid materials have behaved as very efficient heterogeneous catalysts in the Suzuki coupling reaction in water, affording the corresponding cross-coupling products in excellent yields. Furthermore, the catalysts have shown excellent chemical and thermal stability and good recyclability. No evidence of the leaching of Pd from the catalyst during the course of reaction was observed, suggesting true heterogeneity in our catalytic systems.
Introduction
Heterogeneous catalysts can be prepared by heterogenization of the homogeneous counterpart (transition metal complexes, metal salts) over a solid matrix which leads to the formation of solid recyclable molecular catalysts with well-defined active centers. Therefore, the design and development of a suitable support capable of improving the catalyst characteristics by a cooperative effect between the metal complex and the support is still an important challenge for successful catalysts.1 In recent decades, many inert solid materials such as mesoporous solids (MCM-41, SBA-15 or related mesoporous silicas),2 hybrid organic–inorganic structured porous materials,3 polymers,4 and coordination polymers5 as metal–organic frameworks (MOFs),6 which could originate through the coordination between transition metal centers and chelating organic ligands have been used as supports. Porous organic polymers (POFs) have been developing as very promising functional materials for gas storage, separation and heterogeneous catalysis and sensing, due to their surface area, porosity and chemical stability. For this reason, the interest in using porous organic polymers as transition metal supports remains a challenge.7A new class of attractive porous materials that have emerged recently is the family of aromatic porous polyimides (PPIs) because they exhibit large surface areas and interesting properties in gas storage or gas separation.8 But so far they have not been used for catalytic applications.
The palladium-catalyzed reaction of haloarenes with aryl-boronic acids (Suzuki coupling) is one of the most extensively studied organic transformations for the synthesis of biaryl compounds and this reaction is important from both the academic and industrial point of view. However, palladium residues are highly polluting because of its difficulty to be removed and recycled and this causes serious problems in large-scale synthesis.9 To solve these problems, palladium has been attached to different types of supports, such as mesoporous silica supports,10 activated carbon,11 zeolites, molecular sieves, metal oxides, metal–organic frameworks (MOFs)12 and organic polymers.13,9c However, few examples of porous polymer palladium catalysts can be found. In 2011, Ding et al. reported the first porous polyimine Pd catalyst to be used in the Suzuki–Miyaura reaction, using p-xylene as the solvent.14 Recently palladium Covalent Organic Framework (TpPa-1) has also been anchored to porous polydivinylbenzene and a TpPa-1 matrix and used as a catalyst in cross-coupling reactions showing good stability in both acidic and alkaline media, high activity and recyclability, but the reactions have to be performed in ethanol, ethanol–water or N-methyl pyrrolidinone.15 The use of water as the solvent has gained special importance in past few years due mainly to the economic and environmental advantages, because it is abundant, cheap, nontoxic, noncorrosive, non-flammable and has unique properties in solvating molecules which can improve the rate and selectivity of the reactions.16,13b Thus, the interest in using water as a solvent in organic reactions is evident because most of the conventional organic solvents are toxic and volatile, particularly chlorinated hydrocarbons which contribute significantly to pollution.17
These porous hydrophobic polyimide materials are suitable for functionalization with different transition metal complexes useful as heterogeneous catalysts. Herein, as a preliminary application, heterogenized palladium complexes (Fig. 1) were easily prepared by amino functionalization of porous polyimide networks and subsequent incorporation of bidentate nitrogen-binding ligands able to coordinate the metal centre.
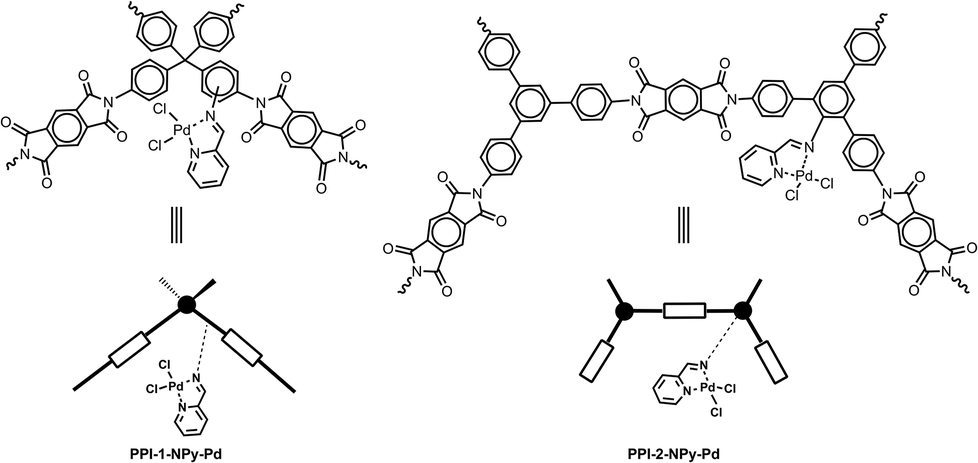 | ||
| Fig. 1 Structure of the repeat unit and schematic representation of heterogeneous Pd catalysts based on porous polyimide networks. | ||
These as-synthesized hydrophobic PPI-NPy-Pd(II) materials exhibited outstanding catalytic activity in Suzuki cross-coupling reactions under mild conditions using water as the solvent. The effectiveness and recyclability of these hydrophobic catalysts was proved after at least four cycles.
Results
Amino functionalized porous polyimides
Amino functionalized porous polyimides, PPI-1-NH2 and PPI-2-NH2 (Fig. 2), were prepared in two steps from the corresponding porous polyimides networks, PPI-1 and PPI-2. These starting materials were synthesized according to the methods previously reported,8f,j by the reaction of pyromellitic dianhydride (PMDA) with tetra-(4-aminophenyl)methane (Th)18 or 1,3,5-tris-(4-aminophenyl)benzene (C3v)19 at 200 °C using m-cresol as the solvent and quinoline as the catalyst. Then, PPI-1 and PPI-2 were treated with a mixture of nitric acid and sulphuric acid20 to yield the nitro-functionalized polyimides PPI-1-NO2 and PPI-2-NO2.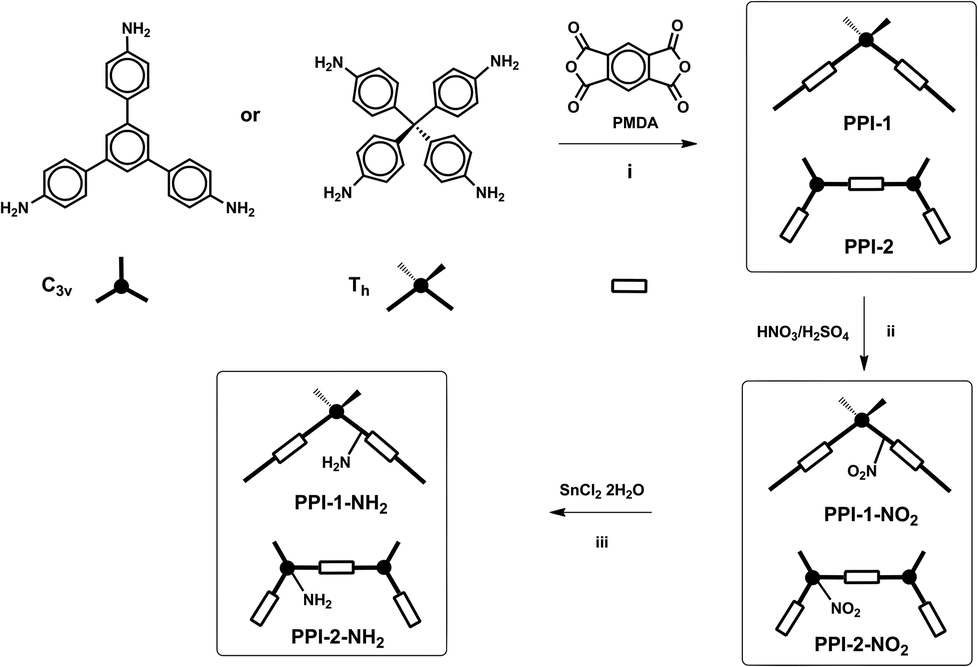 | ||
| Fig. 2 Synthesis of amino functionalized porous polyimides. (i) m-cresol/quinoline, 200 °C; (ii) HNO3/H2SO4, −10 °C (iii) SnCl2·2H2O, THF, reflux, 24 h. | ||
Mononitration of 1,3,5-triphenylbenzene yields three isomers with nitro groups located on the central ring (62%) and on the peripheral-rings, in ortho and para positions (23 and 15%, respectively).21 However, the nitration of PPI-2 yields a main isomer (PPI-2-NO2) with NO2 groups located on the central ring of the C3v amine because the para positions were occupied and the ortho ones were deactivated.
In the next step, the nitro groups were reduced with SnCl2·2H2O in THF,22 to yield the corresponding amino functionalized polyimides, PPI-1-NH2 and PPI-2-NH2.
The reduction was followed by IR spectroscopy and it was considered finished when the band attributed to the nitro groups at 1530 cm−1 disappeared. The other characteristic band of the nitro groups at 1350 cm−1 was overlapped with the bands of the polyimide skeleton. In the spectra of the amino functionalized polyimides, two new bands emerged, one broad of low intensity at 3500–3300 cm−1 and other of higher intensity at 1615 cm−1 that were assigned to the presence of –NH2 groups. As an example, Fig. 3 shows the IR spectra of the amino and nitro functionalized porous polyimides compared with the corresponding starting material PPI-2. The thermal stability was evaluated by thermogravimetric analysis (TGA) in air and nitrogen atmosphere (Fig. S1†). It can be observed as the weight loss attributed to the nitro groups disappears in the thermograms of amino derivatives (Fig. S1b†); PPI-1-NH2 and PPI-2-NH2 exhibited lower thermal stability than the starting polyimides (Table 1).
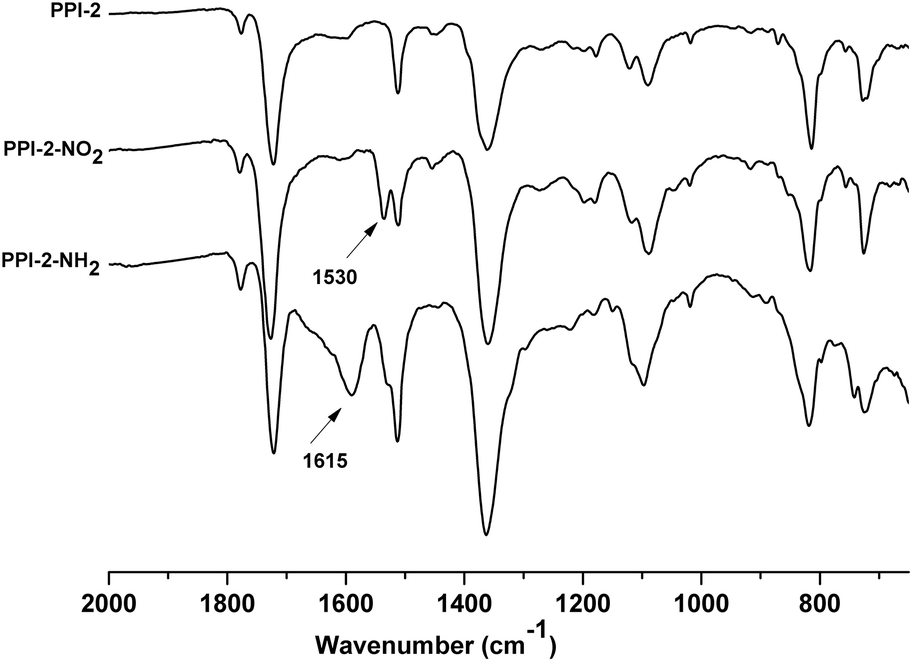 | ||
| Fig. 3 FT-IR spectra of PPI-2, PPI-2-NO2 and PPI-2-NH2. | ||
| Polyimide | Tdonset (°C) | S BET (m2 g−1) | Aver. pore size (nm) |
|---|---|---|---|
| PPI-1 | 570 | 474 | 3.8 |
| PPI-1-NH2 | 405 | 206 | 10.3 |
| PPI-2 | 540 | 604 | 5.9 |
| PPI-2-NH2 | 410 | 274 | 10.8 |
The porosity of the new materials was characterized by nitrogen sorption after heating the samples at 100 °C. The N2 isotherms (Fig. S2†) were typical for microporous materials. The porosity parameters obtained are recorded in Table 1 and the pore-size distribution is depicted in Fig. S3.† The specific surface area of the polyimide PPI-2 was slightly higher than that of PPI-1, which indicated that in this case, the growth of the network has been easier from the monomer with trigonal geometry. The amino-functionalized polyimides showed a considerable decrease of the specific surface areas compared with the starting polyimides. The reduction of the specific surface area after post-functionalization of porous aromatic frameworks (PAFs) has been also reported.7
The SEM images of both amino functionalized polyimides (Fig. 4) were similar to those of the precursors and of the nitro derivatives (Fig. S4†) displaying the typical morphology of porous cross-linked polymers with spherical structures and agglomerates.
 | ||
| Fig. 4 SEM images of PPI-1-NH2 (left) and PPI-2-NH2(right). | ||
Heterogeneous palladium catalysts
The incorporation of palladium on the porous polyimides was carried out in two steps (Fig. 5), following the same strategy used to incorporate rhodium and copper into amino functionalized porous aromatic frameworks (PAFs).22 Thus, in the first step, the corresponding amino support, PPI-1-NH2 or PPI-2-NH2, was reacted with picolinaldehyde to yield the imino pyridine ligands PPI-1-NPy and PPI-2-NPy. In the second step, these derivatives were reacted with bis(benzonitrile)palladium(II) chloride in acetonitrile to obtain the palladium-incorporated heterogenized catalysts PPI-1-NPy-Pd and PPI-2-NPy-Pd.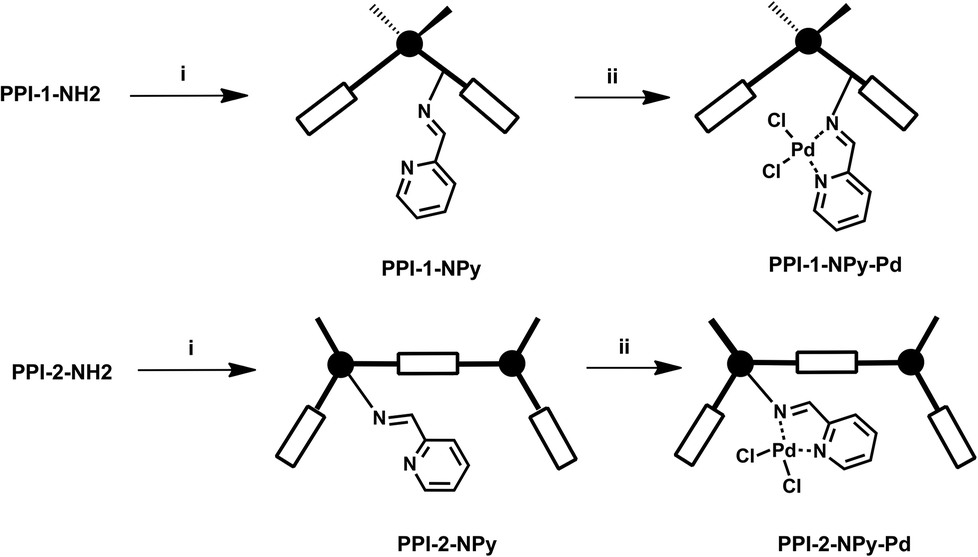 | ||
| Fig. 5 Synthesis of heterogenized palladium catalysts. (i) picolinaldehyde, ethanol, reflux; (ii) PdCl2(C6H5CN)2, acetonitrile. | ||
The analysis by ICP of the palladium content incorporated into the supports revealed that the PPI-2-NPy ligand showed higher metal binding capacity than PPI-1-NPy. Thus, the amount of palladium in PPI-2-NPy-Pd was 3.42% while PPI-1-NPy-Pd showed 1.76%.
The specific surface areas of PPIs-NPy-Pd significantly decreased (10–30 m2 g−1), which could be due to a partial blockage of the pores or interpenetration of the network. The latter behavior has been recently observed in urea-based porous organic frameworks and despite the low surface areas (91–113 m2 g−1), the palladium catalyst prepared from these materials exhibited high catalytic activity and selectivity in reactions in water.23
The SEM images of palladium heterogenized catalysts (Fig. 6) were similar to those of the amino functionalized polyimides, showing the characteristic morphology of porous polymers, despite the apparent loss of porosity observed by BET.
 | ||
| Fig. 6 SEM images of PPI-1-NPy-Pd (left) and PPI-2-NPy-Pd (right). | ||
The thermal stability of imino pyridine ligands and the Pd catalysts was also evaluated by TGA and compared with that of the starting amino functionalized polyimides (Fig. S5†). Compared with the precursors PPI-1-NH2 and PPI-2-NH2 it can be observed that the bidentate ligand increased the thermal stability around 35–55 °C. The derivative thermogravimetric curve of PPI-1-NPy-Pd showed a main peak accompanied by a shoulder, and a second peak of very low intensity. The thermal degradation of PPI-2-NPy-Pd occurred also in two stages at initial decomposition temperatures of 355 and 430 °C. Thus, as happened with their precursors, the degradation of both palladium catalysts followed different mechanisms. Compared with PPI-1-NPy and PPI-2-NPy it should be noted that palladium decreases the thermal stability around 55 °C in the case of PPI-1-NPy-Pd and 80 °C for PPI-1-NPy-Pd (Fig. S5†).
The imino pyridine ligands and palladium heterogenized catalysts were also characterized by 13C-NMR and elemental analysis and compared with the corresponding parent supports PPI-1-NH2 and PPI-2-NH2. The spectra of the imino pyridine ligands and palladium catalysts were very similar to those of the starting materials (Fig. S6†). Thus, the three spectra showed two signals of low intensity at 165.9 and 141.7 ppm attributed to the carbonyl groups and to the quaternary aromatic carbons, respectively, and a group of signals of high intensity, between 138 and 125 ppm, characteristic of the rest of aromatic carbons. The signals of the pyridine groups were indistinguishable from these signals. Elemental analysis confirms the functionalization (Table S1†).
Reference palladium complex for homogeneous catalysis
For comparative purposes, a palladium complex NPy-Pd was prepared following the route depicted in Fig. 7. Thus, 4-tritylaniline, prepared from triphenylmethylchloride, was reacted with 2-Pyridinecarboxaldehyde to obtain quantitatively (E)-N-(pyridin-2-ylmethylene)-4-tritylaniline (NPy).7b Then, NPy was reacted with bis(benzonitrile)palladium(II) chloride in acetonitrile to obtain the palladium complex NPy-Pd.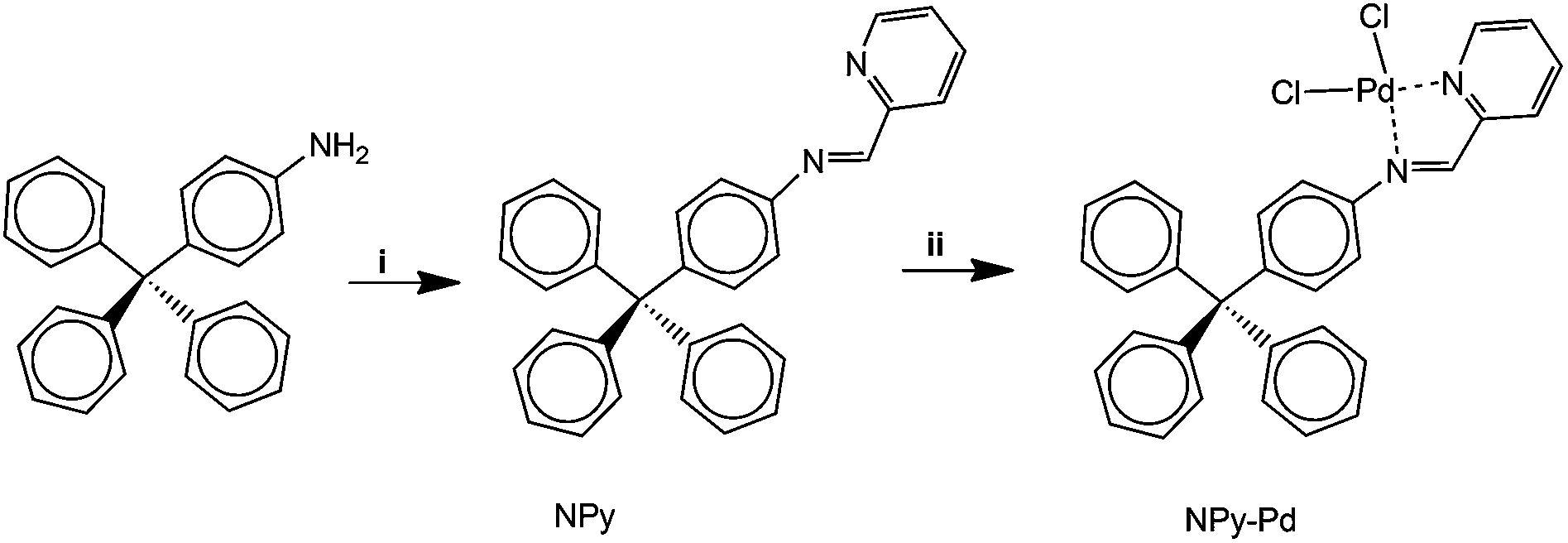 | ||
| Fig. 7 Synthesis of homogeneous palladium catalyst: (i) picolinaldehyde, ethanol, reflux; (ii) PdCl2(C6H5CN)2, acetonitrile. | ||
NPy and NPy-Pd were characterized by NMR and FTIR spectroscopies. The 1H-NMR (Fig. S7†) of both compounds showed a group of signals between 7 and 7.4 ppm, attributed to the aromatic protons of the tetra-phenyl units. The signal corresponding to the proton of the imine group (j) appeared at 8.6 ppm in the NPy spectrum whereas in the NPy-Pd spectrum this signal was shifted to 8.8 ppm due to the metal coordination. Similar displacement was observed in the signals corresponding to the protons of the pyridine ligands. 13C-NMR (Fig. S8†) spectra showed a remarkable displacement of the carbon of the imine bond from 160 to 175 ppm and the carbons meta to the nitrogen atoms when the metal was coordinated. On comparing the FT-IR spectra of both compounds (Fig. S9†) it can be also observed that imine group shifts to lower frequency (Δν = 37 cm−1) when coordination with palladium occurs.
Catalytic activity
Recently, organic reactions in aqueous media have attracted a great deal of attention because water is benign, cheap, and easily available. The separation of products, however, should be the key consideration when employing water as the solvent for organic reactions. Among the various reactions developed, the Suzuki reaction has strongly benefited from aqueous chemistry.24 Our objective was the application of hydrophobic PPI-n-NPy-Pd polymers as catalysts for Suzuki cross-coupling reaction in pure water (Scheme 1). | ||
| Scheme 1 Suzuki coupling reaction of halobenzenes with boronic acids. | ||
Preliminary experiments showed that the model reaction between iodobenzene (1 mmol) and phenyl boronic acid (1.5 mmol) using 1 mmol% of PPI-1-NPy-Pd (based on Pd) as the catalyst and excess of K2CO3 (2.0 mmol) as the base proceeded smoothly in H2O at 100 °C affording the desired biphenyl within 24 h in excellent isolated yield (90%). At this point, we performed a series of experiments in order to optimize the reaction conditions under aerobic conditions. Thus, the reaction in the absence of any base resulted in a negligible conversion (Table S2,† entry 2). Among a variety of bases that showed satisfactory conversions (e.g., K2CO3, K3PO4, Et3N and (i-Pr)2NH) (Table S2,† entries 3–5), we chose to proceed with (i-Pr)2NH. In terms of solvent, H2O afforded the desired product with the highest yield. Other solvents such as xylene resulted in poorer yields (Table S2†). At 70 °C temperature a low amount of coupled product was obtained. Increasing the temperature from 70 to 90 °C improved significantly the conversion (Table S2†). The base–substrate ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was the best for this catalytic system. Then, reactions were carried out at 100 °C in air using (i-Pr)2NH as base and water as the reaction medium. Further, as expected, no product formation was observed in the control experiments in the absence of PPI-1-NPy-Pd (Table S2,† entry 8). Based on the optimized experimental conditions, we explored the substrate scope using iodo and bromobenzene and different arylboronic acids. The results for Suzuki PPI-n-NPy-Pd-catalyzed reactions are summarized in Table 2 and Fig. S10 and S11.† In general, all the reactions were very clean and the corresponding biaryl products were isolated in good to excellent yields. As expected, iodobenzene was rapidly converted to the corresponding biaryl products with excellent yields (Table 2, entries 1 and 11). The less reactive chlorobenzene displayed a poor conversion. PPI-2-NPy-Pd presents a better catalytic activity for Suzuki reaction than PPI-1-NPy-Pd. This fact confirms that both amines C3v and Th yield completely different networks, the Pd of the C3v derivative being more available. The greater accessibility to PPI-2 type network is consistent with the higher metal binding capacity of PPI-2-NPy and the higher specific surface areas of the precursors, PPI-2 and PPI-NH2.
:1 was the best for this catalytic system. Then, reactions were carried out at 100 °C in air using (i-Pr)2NH as base and water as the reaction medium. Further, as expected, no product formation was observed in the control experiments in the absence of PPI-1-NPy-Pd (Table S2,† entry 8). Based on the optimized experimental conditions, we explored the substrate scope using iodo and bromobenzene and different arylboronic acids. The results for Suzuki PPI-n-NPy-Pd-catalyzed reactions are summarized in Table 2 and Fig. S10 and S11.† In general, all the reactions were very clean and the corresponding biaryl products were isolated in good to excellent yields. As expected, iodobenzene was rapidly converted to the corresponding biaryl products with excellent yields (Table 2, entries 1 and 11). The less reactive chlorobenzene displayed a poor conversion. PPI-2-NPy-Pd presents a better catalytic activity for Suzuki reaction than PPI-1-NPy-Pd. This fact confirms that both amines C3v and Th yield completely different networks, the Pd of the C3v derivative being more available. The greater accessibility to PPI-2 type network is consistent with the higher metal binding capacity of PPI-2-NPy and the higher specific surface areas of the precursors, PPI-2 and PPI-NH2.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Catalyst | Mol.b (%) | X | R | Yieldc (%) (h) | TONd | TOFe (h−1) | |
| a Reaction conditions: arylhalide (1.0 mmol), boronic acid (1.5 mmol), (i-Pr)2NH (2.0 mmol), T: 100 °C, water (1 ml). b Based on Pd. c Yield determined by GC and GCMS analysis. d TON = (mol of product/mol of catalyst). e mmol subs. per mmol cat. h. f Decomposition of catalyst. | |||||||
| 1 | PPI-1-NPy-Pd | 0.3 | I | H | 88 (3) | 293 | 462 |
| 2 | 0.5 | Br | H | 88 (2) | 176 | 450 | |
| 3 | 0.5 | Br | OMe | 93 (3) | 186 | 96 | |
| 4 | 0.4 | Br | NO2 | 82 (4) | 205 | 125 | |
| 5 | 0.4 | Br | Me | 98 (2) | 245 | 126 | |
| 6 | 0.4 | Br | COH | 85 (1.5) | 212 | 145 | |
| 7 | 0.8 | Br | CN | 93 (5) | 116 | 57 | |
| 8 | NPy-Pd | 0.4 | Br | COH | 49 (2)f | 122 | 95 |
| 9 | 0.5 | Br | OMe | 43 (3)f | 86 | 40 | |
| 10 | 0.8 | Br | CN | 4 (5)f | 6 | — | |
| 11 | PPI-2-NPy-Pd | 0.3 | I | H | 96 (0.5) | 319 | 685 |
| 12 | 0.5 | Br | H | 100 (0.5) | 200 | 580 | |
| 13 | 0.5 | Br | OMe | 100 (0.5) | 200 | 400 | |
| 14 | 0.5 | Br | NO2 | 85 (3.0) | 170 | 168 | |
| 15 | 0.5 | Br | Me | 100 (1.5) | 200 | 159 | |
| 16 | 0.5 | Br | CHO | 100 (1.5) | 200 | 168 | |
| 17 | 0.5 | Br | CN | 94 (7.0) | 188 | 55 | |
Next, to demonstrate the versatility of the catalytic system for the Suzuki coupling reaction, we tested 4-substituted boronic acids, bearing either electron-donating or electron-withdrawing groups, such as –OMe, –CHO, –CN, –Me and –NO2, which provided selectively the corresponding products in good to excellent yields within 0.5–7 h. The (4-cyanophenyl)boronic acid required the longest reaction times (Table 2, entries 7 and 17). Homocoupled product of arylboronic acid was not detected under the reaction conditions.
In order to find the effect of heterogenization, the activity of the polyimide anchored Pd(II) catalyst (PPI-1-NPy-Pd) was compared to that of the homogeneous analogue in the Suzuki coupling reaction. From the results (Table 2) it is seen that the present heterogeneous catalyst is more reactive than the corresponding homogeneous one. The reaction using a homogeneous catalyst with bromobenzene and 4-methoxyphenylboronic acid in H2O medium for 3 h resulted in 43% conversion (Table 2, entry 9), while the analogous heterogeneous reaction was performed successfully with 93% conversion by GC analysis. The reaction with 4-formylphenylboronic affords only 49% conversion (Table 2, entry 8) and the reaction with (4-cyanophenyl)boronic acid does not occurs. Besides, decomposition of the Pd-catalyst was clearly observed. The activity and selectivity of the catalyst was also comparable with other reported heterogeneous (polystyrene-, sepiolite-supported Pd(II)) catalysts25 for the reaction of aryl halides under aerobic conditions.
For preparative task we scaled up the reaction with ten times the amount of reagents and catalyst, GC conversion after 30 min was 100% and the final products were isolated with a yield of 85%.
An important point concerning the use of the heterogeneous catalyst is its lifetime, particularly for industrial and pharmaceutical applications. To determine whether the catalyst is actually working in a heterogeneous manner, a hot-filtration test was performed in the Suzuki coupling reaction of bromobenzene and 4-methoxyphenylboronic acid (Fig. 8). During catalytic reaction, the solid catalyst was separated from the reaction mixture by filtration after 5 min (conversion 10%) and 15 min (conversion 46%); the reaction was carried out for a further 3 h. The gas chromatographic analysis showed no increment in the conversion. Atomic absorption spectrometric analysis of the liquid phase of the reaction mixtures collected by filtration confirms that Pd is absent in the reaction mixture. These results suggested that no leaching of palladium takes place during the reaction and that the catalyst is purely heterogeneous in nature.
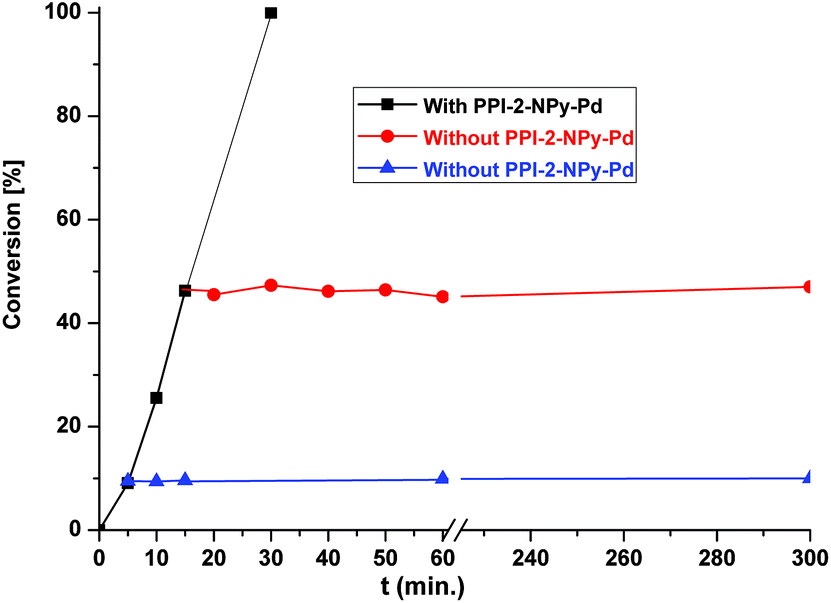 | ||
| Fig. 8 Hot filtration experiments. | ||
As an important aspect of heterogeneous catalysis, we further studied the recyclability of the catalyst for a model Suzuki reaction. After each run, the catalyst was separated by simple filtration, washed thoroughly with water, and ethyl acetate, and dried at 100 °C under vacuum for 8 h. The dried catalyst was then reused with a fresh reaction mixture. In spite of a small catalyst deactivation (Fig. 9, Table S4†), the results showed a good recyclability of both catalysts, being better for the reaction of iodobenzene with phenylboronic acid (PPI-1-NPy-Pd, up to seven runs) than when bromobenzene is the reactant (PPI-2-NPy-Pd, Fig. S12, Table S5†). In general, the average catalyst productivity and activity were identified in Suzuki reactions (TON 110–465; TOF 55–685 h−1). ICP analysis for the reused PPI-1-NPy-Pd (after seven cycles) catalyst reveals that 20% of Pd was lost, probably due to the washings of the catalyst.
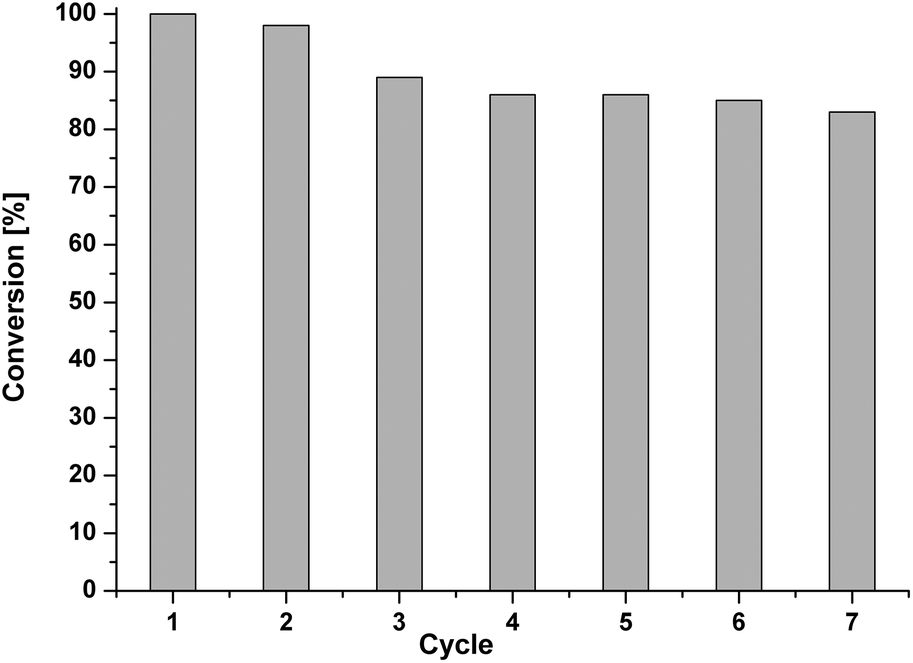 | ||
| Fig. 9 Recycling experiments for Suzuki reaction between iodobenzene and phenylboronic acid. | ||
The chemical structure of the catalysts was checked after the recycling by IR spectroscopy and WAXS (Fig. S13 and S14†). No differences were found between the materials before and after recycling which indicated that the chemical structure was maintained after the catalysis. By TGA (Fig. 10) a slight reduction in the thermal stability (around 10–15 °C) was observed after recycling.
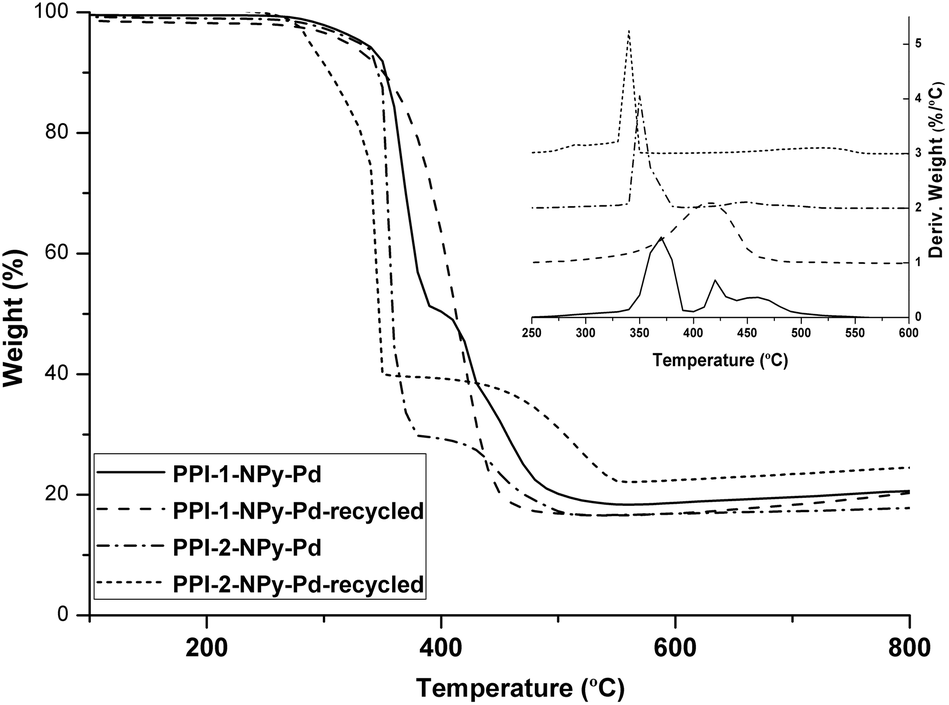 | ||
| Fig. 10 TGA and DTG (vertically shifted) of PPI-1-NPy-Pd, PPI-2-NPy and the corresponding recycled catalysts. | ||
Experimental
Materials
The syntheses of tetra(4-aminophenyl)methane (Th) and 1,3,5-tris(4-aminophenyl)benzene (C3v) were carried out according to the methods reported.18,19 Pyromellitic dianhydride (PMDA) (Aldrich) was purified by sublimation. 2-Pyridinecarboxaldehyde (Aldrich), bis(benzonitrile)palladium(II) chloride (Aldrich), ethanol (Prolabo), m-cresol (Aldrich) and acetonitrile (Aldrich) were used without purification.Porous polyimides (PPI-n)
The products were characterized by elemental analysis, ATR-FTIR, solid 13C-NMR, TGA and SEM. The specific surface area and pore size were determined by BET.
Amino functionalized porous polyimides (PPI-n-NH2).
PPI-n-NO2 and PPI-n-NH2 were characterized by elemental analysis, ATR-FTIR, solid 13C-NMR, TGA and SEM. The specific surface area and pore size were determined by BET.
Imino pyridine ligands (PPI-n-NPy)
Heterogeneous palladium catalysts (PPI-n-NPy-Pd)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 1H-NMR (400 MHz, DMSO-d6): 8.72 (d, 1 H), 8.63 (s, 1 H), 8.14 (d, 1 H), 7.95 (td, 1 H), 7.53 (m, 1 H), 7.4–7.0 (m, 19 H); 13C-NMR (100 MHz, DMSO-d6): δ (ppm) 160.97, 154.40, 150.06, 148.28, 146.68, 145.44, 137.43, 131.78, 130.82, 128.15, 126.42, 126.01, 121.54, 120.97, 64.60. HR-MS calculated for C31H24N2 424.53, found [M+] 424.40.
N). 1H-NMR (400 MHz, DMSO-d6): 9.06 (d, 1 H), 8.78 (s, 1 H), 8.38 (td, 1 H), 8.17 (d, 1 H), 7.93 (m, 1 H), 7.4–7.0 (m, 19 H); 13C-NMR (100 MHz, DMSO-d6): δ (ppm) 173.06, 156.24, 150.50, 147.28, 146.50, 145.25, 141.73, 130.89, 135.59, 129.90, 129.51, 128.32, 126.66, 123.84, 64.84. HR-MS calculated for C31H24N2PdCl2 601.85, found [M+] 602.1.
N). 1H-NMR (400 MHz, DMSO-d6): 8.72 (d, 1 H), 8.63 (s, 1 H), 8.14 (d, 1 H), 7.95 (td, 1 H), 7.53 (m, 1 H), 7.4–7.0 (m, 19 H); 13C-NMR (100 MHz, DMSO-d6): δ (ppm) 160.97, 154.40, 150.06, 148.28, 146.68, 145.44, 137.43, 131.78, 130.82, 128.15, 126.42, 126.01, 121.54, 120.97, 64.60. HR-MS calculated for C31H24N2 424.53, found [M+] 424.40.
N). 1H-NMR (400 MHz, DMSO-d6): 9.06 (d, 1 H), 8.78 (s, 1 H), 8.38 (td, 1 H), 8.17 (d, 1 H), 7.93 (m, 1 H), 7.4–7.0 (m, 19 H); 13C-NMR (100 MHz, DMSO-d6): δ (ppm) 173.06, 156.24, 150.50, 147.28, 146.50, 145.25, 141.73, 130.89, 135.59, 129.90, 129.51, 128.32, 126.66, 123.84, 64.84. HR-MS calculated for C31H24N2PdCl2 601.85, found [M+] 602.1.
General experimental procedure for Suzuki cross-coupling reaction in water medium
The reactions were carried out in a micro reaction vessel (5 mL). A mixture of aryl halide (1.0 mmol), arylboronic acid (1.5 mmol), diisopropyl amine (2 mmol), H2O (1 ml) and PPI-n-NPy-Pd catalyst (1.4–0.3 mol%) was stirred at 100 °C under air. To study the progress of the reaction, aliquots were taken from the reaction mixtures at different times, extracted with ethyl acetate and quantified by GC-MS analysis.Recycling experiments
Once the reaction was complete, the heterogenized PPI-n-NPy-Pd complex was recovered by washing with ethyl acetate and water, with stirring, for 1 day. Then, the catalyst was centrifuged and dried in an oven at 100 °C.Conclusions
In summary, we have reported the preparation of new functionalized porous polyimides (PPIs) with two different 2D and tetrahedral topologies with high specific surfaces and thermal and chemical stabilities. These new materials have been used as effective supports for heterogeneous palladium complexes which were efficiently used as catalysts for Suzuki cross-coupling reactions in water medium under aerobic conditions. The catalysts show not only high catalytic activity, but also offer many practical advantages such as oxygen insensitivity, thermal stability, and fair recyclability. The excellent catalytic performance in water medium and the easy preparation and separation of the catalyst make them good recyclable heterogeneous catalytic systems in water and a useful alternative to other heterogeneous palladium catalysts.Acknowledgements
The financial support provided by the Spanish Ministerio de Economía y Competitividad (Project MAT2010-20668, MAT2011-29020-C02-02 and Consolider-Ingenio 2010-(CSD-0050-MULTICAT) is gratefully acknowledged. A predoctoral JAE fellowship from the Consejo Superior de Investigaciones Científicas (CSIC) to E. Rangel is also acknowledged.Notes and references
- (a) F. Averseng, M. Vennat and M. Che, Grafting and anchoring of transition metal complexes to inorganic oxides. Handbook of Heterogeneous Catalysis, Wiley, 2008, vol. 2, pp. 522–539 Search PubMed; (b) A. Corma, Catal. Rev. Sci. Eng., 2004, 46, 369 CrossRef CAS PubMed.
- (a) L. F. Bobadilla, T. Blasco and J. A. Odriozola, Phys. Chem. Chem. Phys., 2013, 15, 16927 RSC; (b) V. Ayala, A. Corma, M. Iglesias, J. A. Rincón and F. Sánchez, J. Catal., 2004, 224, 170 CrossRef CAS PubMed; (c) A. Corma, E. Gutiérrez-Puebla, M. Iglesias, A. Monge, S. Pérez-Ferreras and F. Sánchez, Adv. Synth. Catal., 2006, 348, 1899 CrossRef CAS; (d) A. Corma, C. González-Arellano, M. Iglesias, M. T. Navarro and F. Sánchez, Chem. Commun., 2008, 6218 RSC; (e) C. del Pozo, N. Debono, A. Corma, M. Iglesias and F. Sánchez, ChemSusChem, 2009, 2, 650 CrossRef CAS PubMed; (f) A. Arnanz, C. González-Arellano, A. Juan, G. Villaverde, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2010, 46, 3001 RSC; (g) C. del Pozo, A. Corma, M. Iglesias and F. Sánchez, Green Chem., 2011, 13, 2471 RSC; (h) C. Del Pozo, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2012, 291, 110 CrossRef CAS PubMed; (i) J. Mondal, A. Modak, A. Dutta, S. Basu, S. N. Jha, D. Bhattacharyya and A. Bhaumik, Chem. Commun., 2012, 48, 8000 RSC; (j) K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS.
- (a) C. Baleizao and H. García, Chem. Rev., 2006, 106, 3987 CrossRef CAS PubMed; (b) A. Corma, U. Díaz, T. García, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011 CrossRef CAS PubMed.
- (a) S. M. Islam, A. S. Roy, P. Mondalm and N. Salam, J. Mol. Catal. A: Chem., 2012, 358, 38 CrossRef CAS PubMed; (b) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS PubMed; (c) A. D. Pomogailo, in Catalysis by Polymer-Immobilized Metal Complexes, OPA, Amsterdam, Netherland, 1998 Search PubMed.
- (a) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; (b) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS PubMed; (c) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (d) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- (a) X. Zhang, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2009, 265, 155 CrossRef CAS PubMed; (b) A. Arnanz, M. Pintado-Sierra, A. Corma, M. Iglesias and F. Sánchez, Adv. Synth. Catal., 2012, 354, 1347 CrossRef CAS; (c) M. Pintado-Sierra, A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2013, 299, 137 CrossRef CAS PubMed; (d) A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2013, 5, 3092 CrossRef CAS.
- (a) E. Verde-Sesto, E. M. Maya, A. E. Lozano, J. G. de la Campa, F. Sánchez and M. Iglesias, J. Mater. Chem., 2012, 22, 24637 RSC; (b) E. Verde-Sesto, M. Pintado-Sierra, A. Corma, E. M. Maya, J. G. de la Campa, M. Iglesias and F. Sánchez, Chem. – Eur. J., 2014, 20, 5111 CrossRef CAS PubMed.
- (a) J. Weber, Q. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871 CrossRef CAS; (b) J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880 CrossRef CAS; (c) Y. Luo, B. Li, L. Liang and B. Tan, Chem. Commun., 2011, 47, 7704 RSC; (d) O. K. Farha, Y. S. Bae, B. G. Hausser, A. M. Spokoyny, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Chem. Commun., 2010, 46, 1056 RSC; (e) Z. Wang, B. Zhang, H. You, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730 RSC; (f) Z. Wang, B. Zhang, H. Yu, G. Li and Y. Bao, Soft Matter, 2011, 7, 5723 RSC; (g) K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Chem. Mater., 2012, 24, 969 CrossRef CAS; (h) G. Li and Z. Wang, Macromolecules, 2013, 46, 3058 CrossRef CAS; (i) C. Shen, Y. Bao and Z. Wang, Chem. Commun., 2013, 49, 3321 RSC; (j) E. Rangel-Rangel, E. M. Maya, F. Sánchez, J. G. de la Campa and J. de Abajo, J. Membr. Sci., 2013, 447, 403 CrossRef CAS PubMed.
- (a) C. E. Tucker and J. G. De Vries, Top. Catal., 2002, 19, 11 CrossRef; (b) B. M. Bhanage and M. Ari, Catal. Rev., 2001, 43, 315 CrossRef CAS PubMed; (c) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed.
- (a) W.-Y. Wu, T.-Ch. Lin, T. Takahashi, F.-Y. Tsai and Ch.-Y. Mou, ChemCatChem, 2013, 5, 1011 CrossRef CAS; (b) H. Lin and P. K. Wong, Curr. Org. Synth., 2010, 7, 599 CrossRef; (c) S. Banerjee, V. Balasanthiran, R. T. Koodali and G. A. Sereda, Org. Biomol. Chem., 2010, 8, 4316 RSC; (d) C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Adv. Synth. Catal., 2004, 346, 1758 CrossRef; (e) M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190 RSC.
- J. Yang, X. Tan, Y. Wang and X. Wang, J. Porous Mater., 2013, 20, 501 CrossRef CAS.
- A. S. Roya, J. Mondal, B. Banerjee, P. Mondal, A. Bhaumik and S. M. Islam, Appl. Catal., A, 2014, 469, 320 CrossRef PubMed.
- (a) B. Tamami, F. Farjadian, S. Ghasemia and H. Allahyar, New J. Chem., 2013, 37, 2011 RSC; (b) M. Lamblin, L. Nassar-Hardy, J. Hierso, E. Fouket and F. Felpin, Adv. Synth. Catal., 2010, 352, 33 CrossRef CAS; (c) J. H. Kim, J. W. Kim, M. Shokouhimehr and Y. S. Lee, J. Org. Chem., 2005, 70, 6714 CrossRef CAS PubMed; (d) F. Ciardelli, E. Tsushida and D. Wohrle, Macromolecule metal complexes, Springer, Berlin, 1996 Search PubMed; (e) M. R. Buchmeiser, Polymeric materials in organic synthesis and catalysis, Wiley-VCH, Germany, 2003 Search PubMed.
- S. Ding, J. Gao, Q. Wang, Y. Zhang, W. Song, C. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- (a) Q. Sun, L. Zhu, Z. Sun, X. Meng and F. Xiao, Sci. China. Chem., 2012, 55, 2097 Search PubMed; (b) P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. Díaz Díaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944 RSC.
- M. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- S. Liu and J. Xiao, J. Mol. Catal. A.: Chem., 2007, 270, 1 CrossRef CAS PubMed.
- H. Yabing, B. Zheng, K. Chuanqing, C. Yanquin and G. Lianxun, Tetrahedron, 2012, 66, 3553 Search PubMed.
- P. Ganesan, X. Yang, J. Loos, T. Savenije, R. Abellon, H. Zuilhof and E. Sudhölter, J. Am. Chem. Soc., 2005, 127, 14530 CrossRef CAS PubMed.
- J. G. Young and W. Onyebuagu, J. Org. Chem., 1990, 55, 2155 CrossRef CAS.
- G. Ed. Lewis, J. Org. Chem., 1965, 30, 2798 CrossRef.
- (a) E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sánchez and A. Corma, Chem. Mater., 2013, 25, 981 CrossRef CAS; (b) E. Merino, E. Verde, E. M. Maya, A. Corma, M. Iglesias and F. Sánchez, Appl. Catal., A, 2014, 469, 206 CrossRef CAS PubMed.
- L. Li, Z. Chen, H. Zhong and R. Wang, Chem. – Eur. J., 2014, 20, 3050 CrossRef CAS PubMed.
- (a) V. Polshettiwar, A. Decottignies, Ch. Len and A. Fihri, ChemSusChem, 2010, 3, 502 CrossRef CAS PubMed; (b) Ch. Röhlich, A. S. Wirth and K. Köhler, Chem. – Eur. J., 2012, 18, 15485 CrossRef PubMed; (c) A. N. Marziale, D. Jantke, S. H. Faul, T. Reiner, E. Herdtweck and J. Eppinger, Green Chem., 2011, 13, 169 RSC; (d) J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060 CrossRef CAS PubMed; (e) J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346 CrossRef CAS PubMed; (f) Ch. Liu, Y. Zhang, N. Liu and J. Qiu, Green Chem., 2012, 14, 2999 RSC.
- (a) S. M. Islam, P. Mondal, A. Singha Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS PubMed; (b) K. Shimizu, T. Kan-no, T. Kodama, H. Hagiwara and Y. Kitayama, Tetrahedron Lett., 2002, 43, 5653 CrossRef CAS; (c) L. Liu, Y. Zhang and Y. Wang, J. Org. Chem., 2005, 70, 6122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01326c |
| This journal is © The Royal Society of Chemistry 2015 |