
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin solubilisation and gentle fractionation in liquid ammonia†
Zea
Strassberger
a,
Pepijn
Prinsen
a,
Frits van der
Klis
b,
Daan S. van
Es
b,
Stefania
Tanase
a and
Gadi
Rothenberg
*a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: g.rothenberg@uva.nl; Web: http://hims.uva.nl/hcsc Fax: +31 20 525 5604
bWageningen UR Food & Biobased Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. Fax: (+)31 31 748 301
First published on 18th September 2014
Abstract
We present a simple method for solubilising lignin using liquid ammonia. Unlike water, which requires harsh conditions, ammonia can solubilise technical lignins, in particular kraft lignin. A commercial pine wood Kraft lignin (Indulin AT) was solubilized instantaneously at room temperature and 7–11 bars autogeneous pressure, while a commercial mixed wheat straw/Sarkanda grass soda lignin (Protobind™ 1000) was solubilized within 3 h at ambient temperature, and 30 min at. 85 °C. Hydroxide salts were not required. Wheat straw, poplar and spruce organosolv lignins, as well as elephant grass native lignin (MWL) were also solubilized, albeit at lower values. Different sequences of solubilisation and extraction were tested on the Protobind™ 1000 lignin. The remaining lignin residues were characterized by FTIR, size exclusion chromatography (SEC), elemental analysis (ICP), 2D-NMR and 31P NMR. Liquid ammonia is not an innocent solvent, as some nitrogen was incorporated in the residual lignin which then rearranged to higher molecular weight fractions. Nevertheless, the mild solubilisation conditions make liquid ammonia an attractive candidate as a solvent for lignin in future biorefinery processes.
Introduction
Biomass is our only practical renewable resource of carbon.1 Thanks to government legislation and societal pressure, its use as a source of chemicals and fuels is spreading.2–7 Terrestrial lignocellulose is the most abundant, and most easily harvested type of non-edible biomass. The three main components of lignocellulosic biomass are cellulose, hemicellulose, and lignin. Lignin is an amorphous and branched bio-polymer consisting of phenylpropane units, which represents up to 40% of the dry biomass weight.8 Since it is practically insoluble and highly complex, selective conversion of lignin into useful chemicals is one of the main challenges in biorefining.9 Today's biorefinery schemes often exclude lignin from their processes.10 For instance, the lignocellulose-to-ethanol process11 uses the cellulose and hemicelluloses, but leaves lignin as a low-added-value fuel. Lignin can be converted into fuels, but it is also the most abundant natural source of aromatics, which may be used directly or converted by the chemical intermediates sectors. With this in mind, several research groups are directing considerable efforts into converting lignin into valuable chemicals.12–16Much of today's lignin comes as a by-product of the pulp and paper industry. It can be recovered from the pulping liquor. The so-called ‘black liquor’ (produced by the Kraft pulping process) contains up to 35% of lignin, and is typically burned as a low-grade fuel in the recovery boiler.17,18 Isolating lignin from this liquor is possible, and several elegant methods were developed, such as the LignoBoost process.19 Besides lignosulfonates (sulfite pulping process), commercial kraft lignins are already being produced in large amounts, while new methods for lignin isolation are being developed within the biorefinery concept. Like Kraft, the soda process is an alkaline pulping process but without the active sulphur species, leading to sulphur-free lignins. Anthraquinone is used frequently to assist the soda pulping process. Organosolv pulping processes employ a mixture of organic acids and/or organic solvents and, like soda lignin, will probably provide the key lignin types for future biorefineries, as they are sulphur-free.20 The bottleneck in upgrading lignin to chemicals is often the solubilisation step. State-of-the art processes for dissolution and depolymerisation use either harsh acid/base treatments or high-temperature pyrolysis/gasification.15,21–23 Both methods give a plethora of products, and often an excess of salts that requires costly workup and separation. Thus, one of the main challenges in lignin valorisation is finding green, robust, and cost-effective means to dissolve lignin, which may enable its selective and efficient depolymerisation.
Here, we present a novel and gentle method for solubilizing kraft (pine wood), soda (mixed wheat straw/Sarkanda grass) and organosolv (poplar, spruce and wheat straw) lignins using pure liquid ammonia. Additional experiments confirmed that a native grass lignin (MWL from elephant grass) was also soluble. This way, we can dissolve lignin in a continuous basic environment without adding any potassium/sodium hydroxide. Barring the work of Yan et al.24 in the 1950s, this is the first report on using anhydrous liquid ammonia directly as a solvent to solubilize lignin. We discuss the fundamental similarities and differences between ammonia and water, and show that using liquid ammonia might help to overcome the drawbacks of aqueous acid or base solutions. After dissolution, the ammonia can be removed quickly by evaporation at room temperature. Note that the practical tools are in principle available as biorefineries have used liquid ammonia indirectly for isolating cellulose and hemicellulose as part of the AFEX process.25–27 Moreover, the technical-economical viability of the AFEX process differs completely from aqueous based processes, as the ammonia can be recycled almost quantitatively and the workup requires less solvent.
Water is the most abundant and eco-friendly solvent, and moving away from it to another protic polar liquid requires just cause. Ammonia and water have similar physical properties and therefore exhibit similar behaviour, but they also have some interesting differences (see ESI, Tables S1 and S2†). It is these differences that make ammonia such an effective solvent for lignin.15,28–30 To understand this, we first look at the fundamentals behind the solubilisation. Three main parameters affect the solubility of a molecular substance in liquid ammonia: the magnitude of dispersion forces, the polarity of the ammonia molecule, and its ability to form hydrogen bonds.28 The auto-ionization equilibrium of ammonia defines both the acid and base species, just like in water (eqn (1) and (2)), but the equilibrium constants are vastly different. Unlike water, ammonia practically never dissociates. Its Kd is a million times lower than the reciprocal Avogadro number,31 so from a million moles of ammonia, which is roughly one ton of ammonia, only 10 mg would dissociate.32
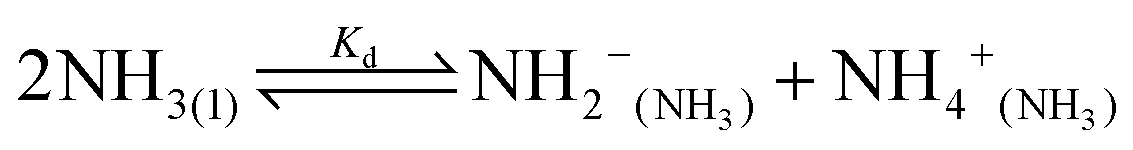 | (1) |
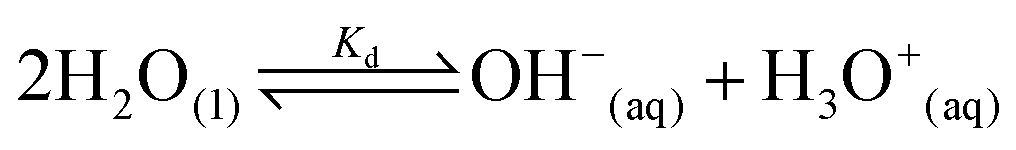 | (2) |
The dispersion forces define how two molecules will attract each other by their fluctuating dipole moment.33,34 Considering the dipole moments of ammonia and water,34 the calculation of the orientation effect and the dispersion force shows that alkyl ethers, lipophilic alcohols and aromatics will interact more strongly with ammonia (ESI, Table S2†). This, combined with the fact that ammonia is one of the most basic molecular liquids,35 makes it a uniquely effective solvent for complex aromatic biopolymers such as lignin. Water contains two protons and two electron pairs which form a symmetrical continuous hydrogen bond forming pattern, while liquid ammonia contains three protons and one electron pair. This is one of the reasons for the difference in their boiling points. Although, like water, liquid ammonia is considered to be a good hydrogen donor and acceptor, it has actually very limited hydrogen donor capacity.36 It is known that liquid ammonia dissolves both ionic compounds and organic molecules.37 Ammonia is basic, and has a stronger ionisation capacity than water.36,38 Thus, carboxylic acids, phenols and imides form ammonium ion pairs in liquid ammonia. Liquid ammonia has received attention as an alternative to aprotic polar industrial solvents, which have toxicity concerns and are difficult to recycle.37
Experimental
Lignin solubilization and extraction experiments
The solubilization experiments were carried out with a pine wood kraft lignin (Indulin AT, Sigma Aldrich), a commercial mixed wheat straw/Sarkanda grass soda lignin (Protobind™ 1000, GreenValue S.A., Granit, Switzerland), three organosolv lignins from poplar, spruce, and wheat straw (Energy Research Centre of the Netherlands, ECN), and a native grass lignin (MWL from elephant grass39). No further data on the pulping process and lignin isolation methods can be provided. The humidity of the lignins were (5.8 ± 0.3), (4.2 ± 0.5), (4.6 ± 0.6), (3.1 ± 0.4) and (3.6 ± 0.6), respectively. In a typical reaction, 250 mg of lignin was mixed with 30 mL of liquid ammonia at ambient temperature, creating an autogeneous pressure of 7–11 bars (depending on the actual room temperature).CAUTION! Working with liquid ammonia requires special attention to equipment safety. We ran all our reactions in a built-to-purpose stainless steel autoclave installed in suitable premises. A detailed description of the equipment and the safety procedures is included in the ESI.†
Besides studying the solubility of different lignins in liquid ammonia, we also ran extraction experiments of the Protobind™ 1000 lignin (P1000 soda lignin) with dichloromethane (DCM), and characterized the remaining lignin by IR, SEC, ICP and NMR. After the ammonia was released (and after cooling with ice when heated), and the autoclave was flushed with nitrogen, the solids were extracted with DCM (for detailed experimental procedures see the ESI†). The relative percentages of monomers and dimers of the extractive fractions were calculated from GC and GC/MS analyses. ICP analysis was performed by Kolbe GmbH (Germany).
Size exclusion chromatography (SEC) measurements
Lignin samples of 1 mg ml−1 dissolved in 0.5 M NaOH were injected into two serial connected columns (4.6 × 30 cm), each manually packed with a manually packed column with ethylene glycolmethacrylate copolymer TSK gel Toyopearl, HW-75F and HW-55F respectively, and eluted with the same solvent, with the following conditions: flow 1 ml min−1, column temperature 25 °C, and detection at 280 nm. The standards used for calibration of the molar mass distribution consisted of sodium polystyrene sulfonates (Mw range: 891 Da to 976 kDa) and phenol.NMR spectroscopy
Ca. 40 mg of sample was dissolved in 0.750 mL of DMSO-d6. Three samples of the P1000 soda lignin (a) were prepared by three different persons and measured at different time moments. The mean value and the corresponding standard deviation were calculated from the quantification. To explore the intrinsic variation, experiment d2 was repeated and the corresponding standard deviation on the NMR quantification was calculated. NMR spectra were recorded at 25 °C on a Bruker AVANCE III 400 MHz instrument equipped with a 5 mm BBI probe with z-gradient (5 G cm−1 A−1). Heteronuclear single quantum coherence (HSQC) experiments used Bruker's “hsqcetgpsi2” pulse program with spectral widths of 5000 Hz (from 10 to 0 ppm) and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 843 Hz (from 165 to 0 ppm) for the 1H- and 13C dimensions. The number of collected complex points was 1024 for the 1H-dimension with a recycle delay of 1.5 s. The number of transients was 32, and 256 time increments were always recorded in the 13C dimension. The 1JCH used was 145 Hz. Processing used squared cosine-bell apodization in both dimensions. Prior to Fourier transformation, the data matrixes were zero-filled up to 1024 points in the 13C-dimension. The central solvent peak was used as an internal reference (δC 39.5; δH 2.49). HSQC correlation peaks were assigned by comparing with the literature.40–43 A semi-quantitative analysis40 of the volume integrals (uncorrected) of the HSQC correlation peaks was performed using Mnova 7 processing software. In the aliphatic oxygenated region, the relative abundances of side chains involved in the various interunit linkages were estimated from the Cα–Hα correlations to avoid possible interference from homonuclear 1H–1H couplings, except for substructure I, for which Cγ–Hγ correlations had to be used. In the aromatic/unsaturated region, C6–H6 and/or C2–H2 correlations from H, G, and S lignin units and from p-coumarate (PCA) and ferulate (FA) were used to estimate their relative abundances. The PCA and FA integration volumes were not included for the calculation of the total aromatic units. For the model compounds, ca. 34 mg was dissolved in 0.500 mL of DMSO-d6. In this case, the total aromatic units were calculated from the sum of the volume integrals of the correlations signals in ring A (A2 + A5 + A6 divided by 3) and in ring B (B1 + B2 + B5 + B6 divided by 4). For the 13C experiments, the same samples from the HSQC analysis were used. In determining the threo and erythro β-O-4′ linkages abundances, the integrals of the respective C2 signals were used for the total aromatic units calculation. For the determination of the relative carbonyl abundances, the sum of all aromatic C signals (A1–6 + B1–6) were used. The 31P NMR analyses after phosphorylation of the lignin hydroxyl groups was performed according to the literature.44 The standard deviations on the corresponding hydroxyl group contents that can be expected are relatively low.45
843 Hz (from 165 to 0 ppm) for the 1H- and 13C dimensions. The number of collected complex points was 1024 for the 1H-dimension with a recycle delay of 1.5 s. The number of transients was 32, and 256 time increments were always recorded in the 13C dimension. The 1JCH used was 145 Hz. Processing used squared cosine-bell apodization in both dimensions. Prior to Fourier transformation, the data matrixes were zero-filled up to 1024 points in the 13C-dimension. The central solvent peak was used as an internal reference (δC 39.5; δH 2.49). HSQC correlation peaks were assigned by comparing with the literature.40–43 A semi-quantitative analysis40 of the volume integrals (uncorrected) of the HSQC correlation peaks was performed using Mnova 7 processing software. In the aliphatic oxygenated region, the relative abundances of side chains involved in the various interunit linkages were estimated from the Cα–Hα correlations to avoid possible interference from homonuclear 1H–1H couplings, except for substructure I, for which Cγ–Hγ correlations had to be used. In the aromatic/unsaturated region, C6–H6 and/or C2–H2 correlations from H, G, and S lignin units and from p-coumarate (PCA) and ferulate (FA) were used to estimate their relative abundances. The PCA and FA integration volumes were not included for the calculation of the total aromatic units. For the model compounds, ca. 34 mg was dissolved in 0.500 mL of DMSO-d6. In this case, the total aromatic units were calculated from the sum of the volume integrals of the correlations signals in ring A (A2 + A5 + A6 divided by 3) and in ring B (B1 + B2 + B5 + B6 divided by 4). For the 13C experiments, the same samples from the HSQC analysis were used. In determining the threo and erythro β-O-4′ linkages abundances, the integrals of the respective C2 signals were used for the total aromatic units calculation. For the determination of the relative carbonyl abundances, the sum of all aromatic C signals (A1–6 + B1–6) were used. The 31P NMR analyses after phosphorylation of the lignin hydroxyl groups was performed according to the literature.44 The standard deviations on the corresponding hydroxyl group contents that can be expected are relatively low.45
Model compound experiments
The model compounds 1-(4-hydroxy-3-methoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol (guaiacylglycerol-β-guaiacyl ether, GGGE) and 3-hydroxy-1-(4-hydroxy-3-methoxyphenyl)-2-(2-methoxyphenoxy) propan-1-one (2-(2-methoxyphenoxy)acetoguaiacone, MPAG) were synthesised as described in the ESI.† Note that in the GGGE compound both threo and erythro diastereomers were present. After drying under vacuum at room temperature, 125 mg were used under the same conditions as applied in experiment d2 with the same dry matter:NH3 ratio and dry matter:DCM ratio. The DCM extraction time was 30 min. The mass recovery was 100% for both compounds.
Results and discussion
Lignin solubilization and extraction experiments
In a typical reaction, 50–500 mg of a lignin sample was solubilised in 30 mL liquid ammonia under moderate conditions as described in the Experimental section. At room temperature, 250 mg Kraft lignin (pine wood) was solubilized instantaneously, giving a clear red solution by means of detection of the light transmittance at one reactor window of the autoclave and with the light source at the opposite window (Fig. 1a). A darker red solution (wine colour) was observed when dissolving 500 mg kraft lignin (Fig. 1b). However, with this system higher solubility ranges could not be evaluated, as it was not clear if the absence of the light transmittance at higher concentrations was due to the presence of lignin particles in suspension scattering the light (when insoluble) or to a shorter light path length or stronger light source was needed to detect light transmittance (when soluble). This can be observed very easily by checking the light transmittance at different heights of a conical extraction funnel with lignin solutions in aqueous sodium hydroxide at different lignin concentrations (Fig. 1g). Note that liquid ammonia can also be charged in a glass conical extraction funnel, but for safety reasons only at temperature ranges below its boiling point (−33 °C, 1 bar). However, at these temperatures the lignins were not soluble, even after prolonged stirring. | ||
| Fig. 1 Image of the reaction autoclave window (the light is shining through another window at the back of the autoclave), showing (a) 250 mg Indulin AT kraft lignin; (b) 500 mg Indulin AT kraft lignin; (c) 250 mg P1000 soda lignin (d) 50 mg poplar organosolv; (e) 125 mg spruce organosolv lignin and (f) 250 mg wheat straw organosolv lignin, (partially) dissolved in 30 mL liquid ammonia. (g) Light transmittance through a P1000 soda lignin aqueous solution (0.2 M NaOH) with the light source placed at different heights in a conical extraction funnel. | ||
The P1000 soda lignin (mixed wheat straw/Sarkanda grass) was also solubilized up to 500 mg in 30 mL liquid ammonia, but at a significant slower rate. At room temperature, it took almost 3 h to solubilize 250 mg P1000 soda lignin in 30 mL liquid ammonia. However, when 45 bars of argon was charged to the autoclave and the temperature was set at 85 °C (90 bars total pressure), the lignin was solubilized within 30 min, giving an orange solution (Fig. 1c). The organosolv lignins were solubilized in amounts up to 125 mg in 30 mL liquid ammonia. Fig. 1d shows 50 mg poplar organosolv lignin (orange solution) and Fig. 1e shows 125 mg spruce organosolv lignin (red solution). At higher concentrations, again, it was not clear if the lignin was insoluble or too concentrated to observe light transmittance at the fixed light distance path between the reactor windows. Fig. 1f shows 250 mg wheat straw organosolv lignin in 30 mL liquid ammonia. However, the time needed to dissolve 125 mg organosolv lignin increased as spruce < poplar < wheat straw. Spruce organosolv lignin was dissolved almost instantaneously. Finally, we explored the solubility of a native grass lignin (MWL from elephant grass). More information on these experiments is available in the ESI (Fig. S2†). 125 mg MWL was dissolved instantaneously at room temperature, although some residual MWL particles were still present. Upon heating, the MWL was dissolved completely. These results indicate that grass lignins dissolve more slowly in liquid ammonia than wood lignins. In general, these results confirmed the solubility of technical lignins (kraft, soda and organosolv) and native lignins in liquid ammonia at a moderate temperature compared to high-pressure water or steam (130–225 °C).46
By releasing the pressure, ammonia could be flushed out as a gas, avoiding any salt removal or extraction of an aqueous phase involving large volumes of organic solvents. We could then separate the low molecular weight aromatics from the solid residue by extraction with an organic solvent. The choice of solvent is important.47,48 At low pressures on a lab scale, DCM is a suitable solvent since it dissolves well aromatic monomers37 while dissolving lignin oligomers only to a limited extent, at least in the case of the P1000 soda lignin. However, it is very difficult to find a solvent which separates exclusively phenolic monomers in one single extraction step with high yield. On a larger scale and at higher pressures, methanol could be used.49
Extracting the P1000 soda lignin with DCM gave up to 20 wt% of solubles (Table 1, entry 1). Using DCM after treating with ammonia gave 16 wt% solubles (entry 3). Extracting again the DCM extracted lignin gave only 3 wt% additional solubles (entry 2), while 14 wt% were obtained when the lignin was first dissolved in liquid ammonia at room temperature (entry 6). No difference was found when dissolving the lignin in liquid ammonia before the first DCM extraction (entry 8). Fig. 2 shows the dominant coniferyl derived compounds identified by GC-MS. A detailed description of the main extractives is given in the ESI (Fig. S3†). We did not identify any free p-coumaric or ferulic acid or any of their esters in the DCM extracts. Most probably they remained in the residual lignin. However, they might also decarboxylate in the GC injector to vinylphenol and vinylguaiacol, respectively. Both of these were present in significant amounts in the DCM extracts. However, they can also be produced from normal lignin units.
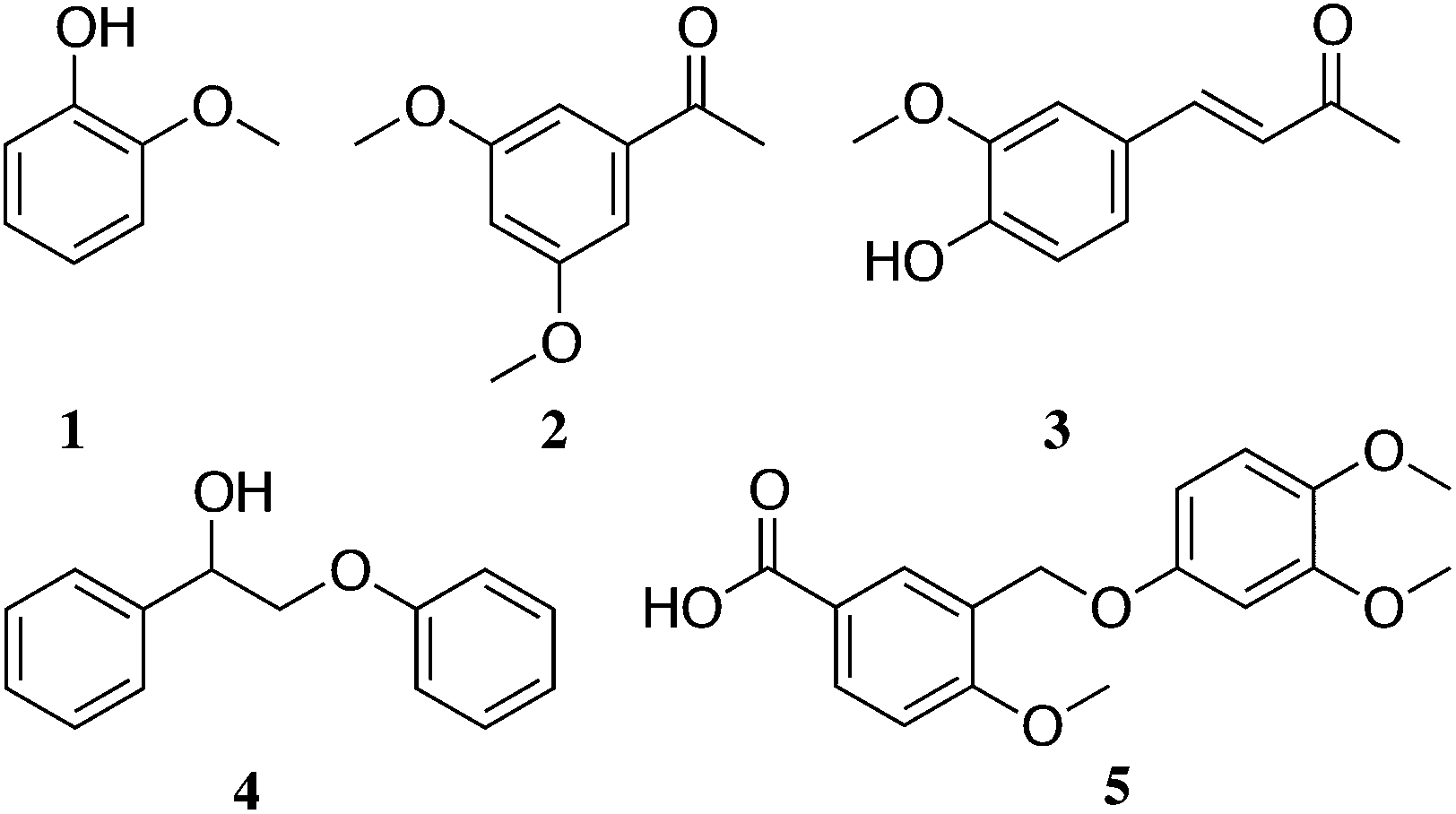 | ||
| Fig. 2 Main monomers and dimers extracted after solubilisation in ammonia (a full list is included in the ESI†): (1) guaiacol; (2) 2,5-dimethoxyacetophenone; (3) vanillideneacetone; (4) 2-phenoxy-1-phenylethanol and (5) 3-((3,4-dimethoxyphenoxy)methyl)-4-methoxybenzoic acid. | ||
| Entry | Sample | Extraction sequence | t (h) | p (bar) | T (°C) | Gas | DCM solubles | Residual lignin (wt%) | Mass balance (wt%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield (wt%) | Monomers | Dimers/trimersa (area %) | Unknownb | |||||||||
| a Peak areas of individual extractives were normalized to 100% and calculated as relative percentages. b By unknown we include compounds that could not be identify by the MS library and heavy alkane (such as hexadecane/dodecane) contamination, which came from the ammonia bottle itself, accounting for the overextended mass balance. | ||||||||||||
| 1 | b | DCM | — | — | — | — | 20 | 43 | 49 | 8 | 77 | 97 |
| 2 | c | DCM/DCM | — | — | — | — | 3 | 48 | 40 | 12 | 94 | 98 |
| 3 | d1 | NH3/DCM | 24 | 7 | 18 | NH3 | 16 | 25 | 68 | 5 | 87 | 103 |
| 4 | d2 | NH3/DCM | 3 | 90 | 85 | NH3/Ar | 24 | 26 | 41 | 22 | 80 | 104 |
| 5 | d3 | NH3/DCM | 3 | 90 | 85 | NH3/H2 | 16 | 30 | 59 | 4 | 70 | 86 |
| 6 | e1 | DCM/NH3/DCM | 24 | 7 | 18 | NH3 | 14 | 15 | 48 | 22 | 88 | 102 |
| 7 | e2 | DCM/NH3/DCM | 3 | 90 | 85 | NH3/H2 | 4 | 5 | 67 | 13 | 74 | 78 |
| 8 | f | NH3/DCM/NH3/DCM | 24 | 7 | 18 | NH3 | 14 | 33 | 36 | 25 | 55 | 68 |
Quantitative analysis of lignin extracts is very challenging, and proving actual C–C and C–O bond scission is not trivial. To study whether the residual lignin structure changes during the dissolution process, we used a combination of analytical methods: infrared spectroscopy (IR), size exclusion chromatography (SEC), elemental analysis (ICP) and NMR.
IR spectroscopy
Fig. 3 shows the IR absorption bands of the P1000 soda lignin, with characteristic bands that were assigned.50 The CO2 bands reflect the fact that samples were directly pressed using a diamond press. The dissolution of the lignin in ammonia left most of the functional groups intact. The intensity of the broad band at 3415 cm−1, corresponding to the phenol and aliphatic hydroxyl groups, increased in the NH3/DCM lignins (experiments d1–d3) compared to the P1000 soda lignin (a). At 2950 cm−1 we see aromatic methoxy groups, as well as side-chain methyl and methylene groups. Pawlak et al. studied wood components’ degradation in liquid ammonia.51 They used IR spectroscopy to characterize the modification on the structural groups of hemicellulose, cellulose and lignin, finding that the carbonyl/carboxyl region was affected. Indeed, we see differences in intensity of the bands located around 1650 cm−1 which is due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N stretching and C–N–H bending in amides, however, in much less extent than in normal ammonolysis reactions.51,52 Another broad N–H band appeared around 3170 cm−1 for all samples in contact with liquid ammonia. Bands at 1595, 1508 and 1450 cm−1 are common for all lignin sources and pertain to the aromatic ring vibration and C–H bond deformations. Other common weak vibrations in lignin pertain to the phenol OH groups and methyl C–H in methyl group, observed around 1370–1375 cm−1. Characteristic guaiacyl bands were distributed on several positions; 1269 cm−1 aromatic ring and CO stretch; and 1122 cm−1 and 809–850 cm−1 C–H deformation,47 for which no major changes were observed. Significant band intensity changes were observed in the 850–1105 cm−1 region. They most probably reflect C–O–C stretches in (substituted) xylans.53
O and C–N stretching and C–N–H bending in amides, however, in much less extent than in normal ammonolysis reactions.51,52 Another broad N–H band appeared around 3170 cm−1 for all samples in contact with liquid ammonia. Bands at 1595, 1508 and 1450 cm−1 are common for all lignin sources and pertain to the aromatic ring vibration and C–H bond deformations. Other common weak vibrations in lignin pertain to the phenol OH groups and methyl C–H in methyl group, observed around 1370–1375 cm−1. Characteristic guaiacyl bands were distributed on several positions; 1269 cm−1 aromatic ring and CO stretch; and 1122 cm−1 and 809–850 cm−1 C–H deformation,47 for which no major changes were observed. Significant band intensity changes were observed in the 850–1105 cm−1 region. They most probably reflect C–O–C stretches in (substituted) xylans.53
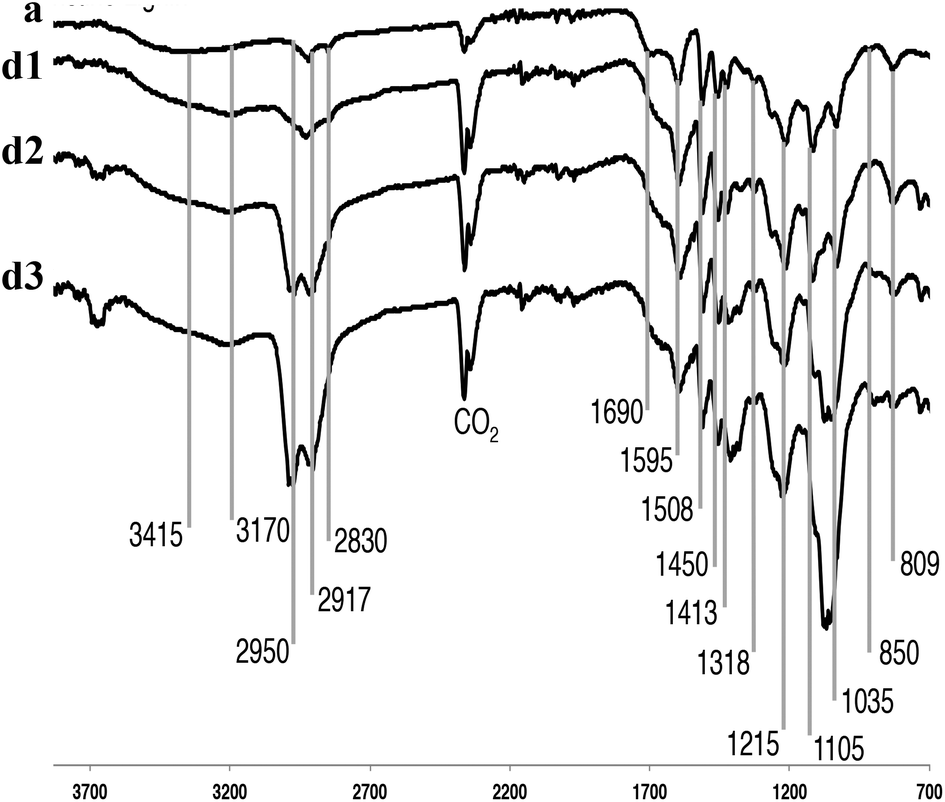 | ||
| Fig. 3 FT-IR spectra of (a) soda P1000 lignin, (d1) after extraction sequence NH3/DCM (the NH3 is at 7 bars autogeneous pressure and at 18 °C), (d2) after extraction sequence NH3/DCM (at 90 bars total pressure of NH3 and argon and at 85 °C) and (d3) after extraction sequence NH3/DCM (at 90 bars total pressure of NH3 and hydrogen and at 85 °C). | ||
Size exclusion chromatography (SEC)
To get a general picture of the molecular weight distribution of the remaining solid, we used SEC. The molecular weight distribution (Mw) and the polydispersity index (PDI) of all the lignin residues actually increased (Table 2), especially at high temperature and in the presence of H2 (entries 5 and 7). In general at low temperature, the Mw and the PDI remained more stable (entries 3 and 6) than at high temperature (entries 4, 5 and 7). At first sight, these results show that no depolymerisation took place by cleavage of lignin bonds. The extraction of low molecular weight compounds gave a residue with higher Mw. However, based on the rather strong PDI increments, we doubt if the molecular weight increments were exclusively the result from the extraction of low molecular weight compounds. However, it is difficult to observe single process effects from the SEC results, as these only show cumulative effects.| Entry | Sample | Extraction sequence | M w | M n | Polydispersity (PDI) |
|---|---|---|---|---|---|
| a 7 bars autogeneous NH3 pressure, 18 °C. b 90 bars total pressure of Ar and NH3, 85 °C. c 90 bars total pressure of H2 and NH3, 85 °C. | |||||
| 1 | a | P1000 soda lignin | 5000 | 298 | 16.8 |
| 2 | b | DCM | 5800 | 414 | 14.0 |
| 3 | d1 | NH3/DCMa | 6900 | 397 | 17.4 |
| 4 | d2 | NH3/DCMb | 9223 | 441 | 20.9 |
| 5 | d3 | NH3/DCMc | 13700 |
461 | 29.7 |
| 6 | e1 | DCM/NH3/DCMa | 8900 | 567 | 15.7 |
| 7 | e2 | DCM/NH3/DCMc | 9900 | 497 | 19.9 |
| 8 | f | NH3/DCM/NH3/DCMa | 12300 |
583 | 21.1 |
Elemental analysis (ICP)
Interestingly, the elemental composition of the solid residue of lignin changed after reaction in ammonia. Elemental analysis showed that the nitrogen content in the NH3/DCM samples (d1–d3) tripled (Table 3). These findings show, together with the SEC results, that ammonia is not an innocent solvent. It does dissolve lignin, but nitrogen atoms from the ammonia (there is no other source of nitrogen in the system) bond with the lignin and the structure rearranges to larger polymer fragments. ICP analysis of the DCM solubles showed that the oxygen content is lower and the carbon content is higher when working at 85 °C in the presence of hydrogen (see ESI, Table S3,† entry 4). Generally, using hydrogen reduces the oxygen content of the extracted monomers and dimers. We expect that at high temperature also more volatiles are produced, which would explain the lowering of the mass balance to 70–85%, depending on the vapour pressure.37 Note that the nitrogen content also increased. Only one nitrogen-containing compound was identified in the DCM soluble fraction of experiments d1–d3, in particular at high temperature (d2–d3). This compound was not present in the DCM extracted P1000 soda lignin (b). We could not identify its exact structure of this compound, but the mass fragments m/z 77, 105 and 121, indicate that it contains a benzamide moiety.| Entry | Sample | Extraction sequence | %C | %H | %N | %O |
|---|---|---|---|---|---|---|
| a 7 bars autogeneous NH3 pressure, 18 °C. b 90 bars total pressure of Ar and NH3, 85 °C. c 90 bars total pressure of H2 and NH3, 85 °C. | ||||||
| 1 | a | P1000 soda lignin | 61.7 | 5.7 | 1.0 | 5.7 |
| 2 | b | DCM | 60.2 | 6.0 | 1.1 | 6.0 |
| 3 | d1 | NH3/DCMa | 58.1 | 6.2 | 2.6 | 6.2 |
| 4 | d2 | NH3/DCMb | 57.3 | 5.8 | 2.8 | 5.8 |
| 5 | d3 | NH3/DCMc | 57.6 | 6.0 | 3.4 | 6.0 |
NMR
Further insight into the structural changes was obtained using HSQC 2D-NMR. Fig. 4 shows the HSQC spectra of the P1000 soda lignin (a) and the ammonia treated lignins (d1 and d3), Fig. 5 shows the substructures corresponding to the identified 13C–1H correlations, and Table 4 shows the corresponding abundances of the interunit linkages per 100 aromatic units. The spectra from samples c and d2 are included in the ESI (Fig. S4†). The correlations of the β–β′ linkages (C) in the spectra of samples d1–d3 are much less intense with respect to the correlation intensity of the β-O-4′ linkages. In fact, the correlation in position γ at 71.0/4.17 in the spectrum of samples d1 and d2 completely disappeared. Interestingly, in the spectrum of sample d1 six new signals appeared at 3.24/70.7, 3.70/74.9, 4.03/75.4, 4.08/72.0, 4.22/87.4 and 4.34/81.0, which were completely absent in the spectra of the P1000 soda lignin and the DCM extracted lignins (a–c) and almost completely in the NH3/DCM lignins (d2–d3). However, these signals could not be assigned yet. The solubilisation in ammonia clearly removed syringyl units, leaving a residual lignin with a lower S/G ratio (Table 4). The β-O-4′ linkages in sample d2 were less abundant than in the samples d1 and d3, in agreement with the higher extraction yield (Table 2). However, compared to the P1000 soda lignin (a) and the DCM/DCM extracted lignin (c), the β-O-4′ linkage abundances were higher in the NH3/DCM lignins. This is surprising as the extraction yield of experiments a and c were similar to the yield of experiments d1–d3.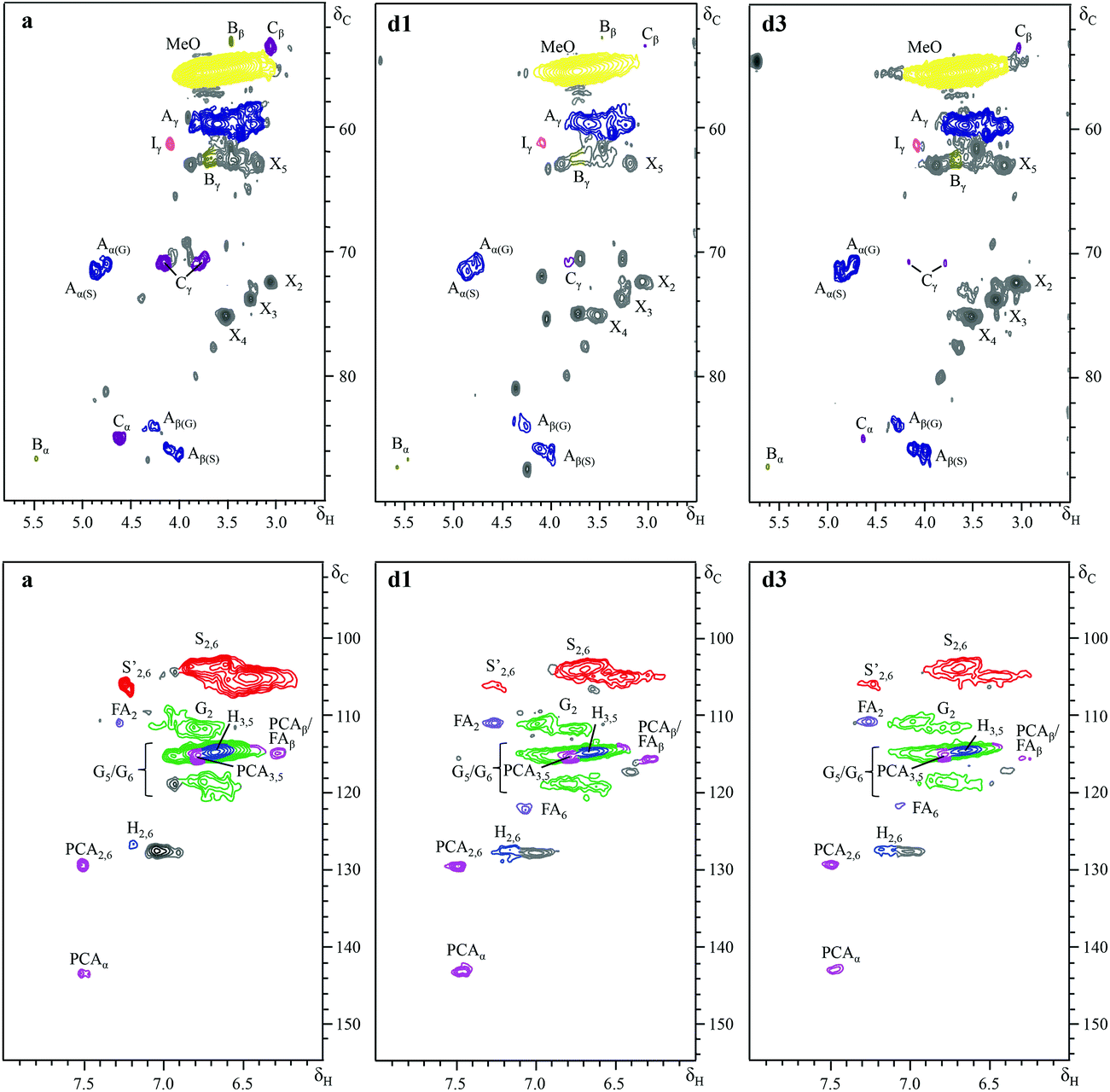 | ||
| Fig. 4 Side chain (δC/δH 50–90/2.5–5.8) and aromatic/unsaturated (δC/δH 90–155/5.5–8.0) regions in the 2D HSQC NMR spectra of (a) P1000 soda lignin, (d1) after extraction sequence NH3/DCM the NH3 is at 7 bars autogeneous pressure and at 18 °C), and (d3) after extraction sequence NH3/DCM (at 90 bars total pressure of NH3 and hydrogen and at 85 °C). | ||
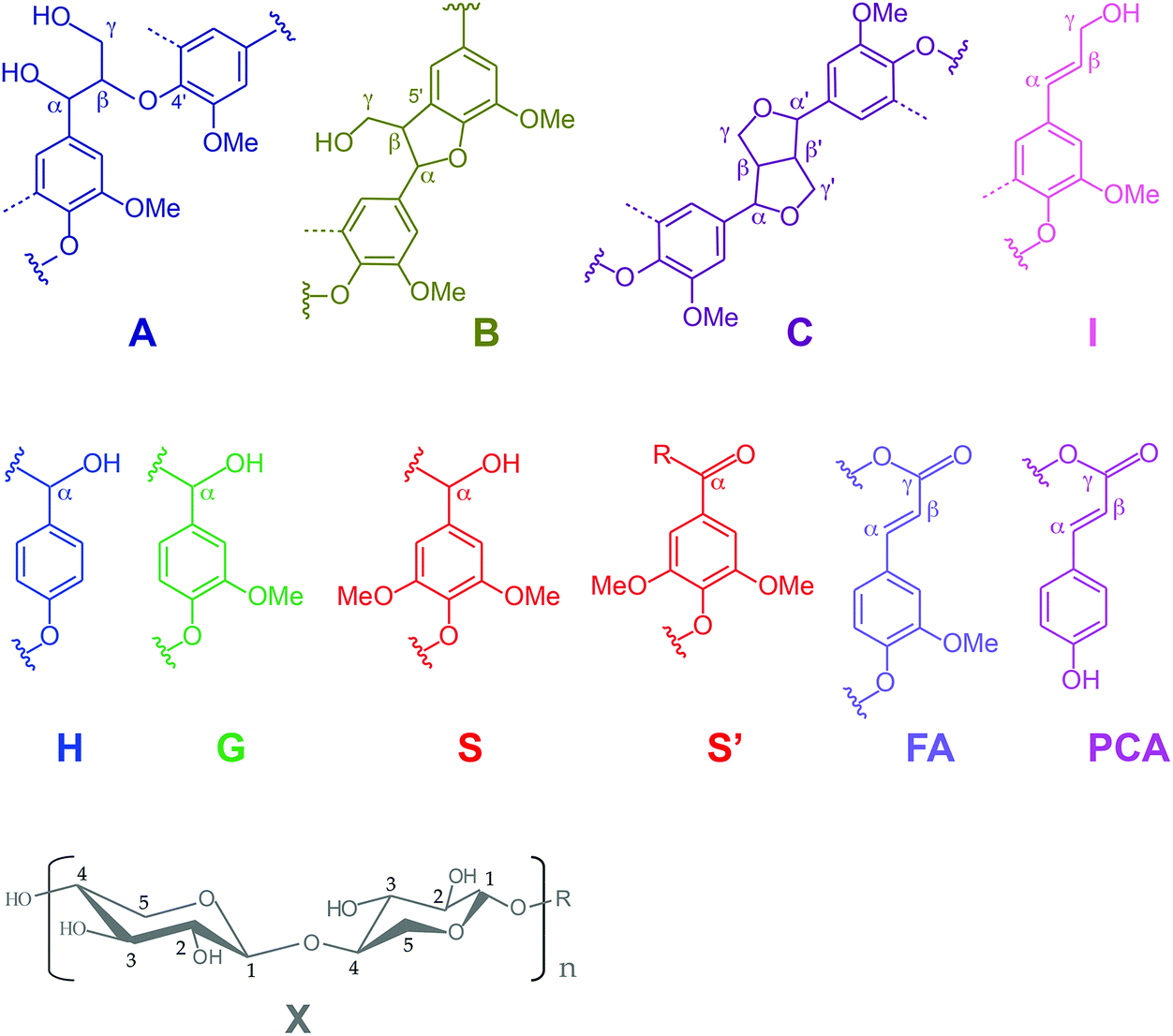 | ||
| Fig. 5 Main structures present in the (treated) P1000 soda lignins: (A) β-O-4′ alkyl–aryl ethers; (B) β-5′ phenylcoumarans; (C) β–β′ resinols; (I) cinnamyl alcohol end groups; (H) p-hydroxyphenyl units; (G) guaiacyl units; (S) syringyl units: (S′) syringyl units bearing a carbonyl group in position α; (PCA) p-coumarates: (FA) ferulates and (X) xylans. | ||
| Lignin | |||||
|---|---|---|---|---|---|
| Interunit linkages (per 100 aromatic units) | P1000 soda (a) | DCM/DCM (c) | NH3/DCM, 18 °C, 7 bars NH3 (d1) | NH3/DCM, 85 °C, 90 bars (NH3 + Ar) (d2) | NH3/DCM, 85 °C, 90 bars (NH3 + H2) (d3) |
| β-O-4′ aryl ethers (A) | 6 (±1) | 14 | 26 | 23 (±2) | 24 |
| β-5′ phenylcoumarans (B) | 0.5 (±0.2) | 2.0 | 0.8 | 1.1 (±0.3) | 0.5 |
| β–β′ resinols (C) | 1.4 (±0.1) | 1.2 | 0.4 | 0.4 (±0.2) | 0.6 |
| Cinnamyl alcohol end groups (I) | 21 (±2) | 17 | 11 | 12 (±2) | 11 |
| Lignin aromatic units (%) | |||||
| H | 2 (±0) | 1 | 5 | 4 (±0) | 3 |
| G | 31(±2) | 28 | 40 | 40 (±3) | 46 |
| S | 61(±2) | 69 | 52 | 54 (±3) | 48 |
| S′ | 5 (±1) | 2 | 3 | 2 (±1) | 3 |
| S/G | 2.2 (±0.2) | 2.5 | 1.4 | 1.4 (±0.2) | 1.1 |
| p-Hydroxycinnamates (%) | |||||
| p-Coumarates (PCA) | 3 (±1) | 3 | 7 | 6 (±2) | 4 |
| Ferulates (FA) | 3 (±0) | 1 | 8 | 7 (±1) | 8 |
| Xylans (per 100 aromatic units) (X) | 4 (±1) | 11 | 14 | 19 (±4) | 29 |
Assuming that no new β-O-4′ linkages were formed, we addressed this effect to the extraction of low molecular weight fractions, giving rise to a lower total aromatic content in the residual lignin. We consider the possibility that some low molecular weight compounds were lost during the NH3 evacuation and the N2 purging, although this was difficult to see from the mass balance due to interference with the lubricant (Table 1). Correlation signals from xylans were identified in the C1, C2, C3, C4 and C5 nuclei. The signal from the anomeric C1 at 4.27/101.5 ppm is not shown in Fig. 5. In accordance with the corresponding xylan IR bands, the ratio of xylan units per 100 aromatic lignin units increased, especially in the presence of H2 (sample d3). We also analysed the P1000 soda lignin (a), the DCM/DCM extracted lignin (c) and the lignin after the NH3//DCM treatment at room temperature (d1) with 31P-NMR. The corresponding spectra are shown in the ESI (Fig. S8†) and the quantification is shown in Table 6. The analysis showed that the residual lignin contained the same amount of aliphatic hydroxyl groups, but 8% less phenol and 6% less carboxyl groups (Table 6). However, this was also observed in the DCM/DCM extracted lignin (c). Thus, this effect was maybe only a consequence of the DCM extraction.
Model compound experiments
Although the guaiacylglycerol-β-guaiacyl ether substructure, with (MPAG) or without (GGGE) a carbonyl group in position α, is not the most representative one, especially in soda lignins, we wanted to confirm that no β-O-4′ ether linkages were cleaved during the solubilisation in ammonia. Therefore, we applied experiment d2 to the model compounds GGGE and MPAG. The corresponding HSQC and 13C spectra are shown in the ESI (Fig. S6 and S7†), and the corresponding quantification is shown in Table 5. We observed that the β-O-4′ linkage was cleaved only to a minor extent (7% based on the HSQC analysis). This is not surprising, as ammonia is a weak base. When using 13C NMR, where the signals of the carbons in the side chain as well in the C2 aromatic carbons from threo and erythro diastereomers are separated, we observed more bond cleavage (11% and 14%, respectively) (Table 5).O) abundances determined by HSQC and 13C analysis in the model compounds GGGE (guaiacylglycerol-β-guaiacyl ether, as a mixture of threo and erythro diastereomers) and MPAG (2-(2-methoxyphenoxy)acetoguaiacone), and abundances after the NH3/DCM treatment at 85 °C and 90 bars (NH3 + Ar) (identical to the conditions of experiment d2 with the P1000 soda lignin)
| Interunit linkage (per 100 aromatic units) | GGGE | MPAG | GGGE after d2 | MPAG after d2 |
|---|---|---|---|---|
|
a Calculated from the 1H–13C correlation in α relative to the sum of the correlations in the aromatic rings A and B.
b Calculated from the 13C signal area in α relative to the sum of the areas of the C2 signals in the aromatic rings A and B.
c Calculated from the 13C signal area in β relative to the sum of all signal areas in the aromatic rings A and B.
d Calculated from the sum of all 13C signal areas in the aromatic rings A and B relative to the area of the CO signal.
|
||||
| HSQC | ||||
| β-O-4′ aryl ethera | 60 | 63 | 56 | 58 |
| 13C | ||||
| threo β-O-4′ aryl etherb | 48 | — | 43 | — |
| erythro β-O-4′ aryl etherb | 51 | — | 44 | — |
| β-O-4′ aryl etherc | 47 | 47 | ||
| Aromatic C/COd |
— | 12.9 | — | 13.5 |
Although the carbonyl content cannot be determined quantitatively by 13C NMR (due to the strong differences in relaxation delays between quaternary and non-quaternary carbons), one can compare them relative to the aromatic content to see if ammonia interacted with native lignin carbonyl groups (Table 5). The carbonyl content (relative to the aromatic content) of the model compound MPAG was only 4% smaller after the NH3/DCM treatment, much smaller than under normal ammonolysis conditions where the lignin is oxidized with molecular oxygen.52 Meier et al. stated that the ammonia interacts with the lignin carboxyl groups. However, we could not confirm this as we did not achieve a satisfactory 13C spectrum of the NH3/DCM treated lignins, possibly due to their higher molecular weights. Although Meier et al. demonstrated that during ammonolysis the carboxyl groups represent the nucleation points where NH4+ salts could be formed, we believe that the 6% lower carboxyl group content in the residual lignin (Table 6) is not due to ammonolysis, as the authors clearly stated that a considerable oxygen pressure is needed for this and peroxidation (creating oxidized structures) prior to ammonolysis is not an alternative. Moreover, Table 6 shows that the lower carboxyl content might be due only to the DCM extraction. All these results show that ammonolysis is not the main factor for the higher molecular weight and polydispersity of the ammonia solubilized lignins. We feel that this can be the result of the different chemical behaviour of organic compounds in liquid ammonia, and possibly due to flash precipitation when the liquid ammonia is released.36–38
| Sample | Extraction sequence | Aliphatic OH | Phenol OH | Carboxyl OH | |||||
|---|---|---|---|---|---|---|---|---|---|
| Syringyl + condensed | Guaiacyl | Catechol | p-Hydroxyphenyl | PCA | Total | ||||
| a 7 bars autogeneous NH3 pressure, 18 °C. | |||||||||
| a | Pristine lignin | 1.59 | 2.30 | 0.86 | 0.15 | 0.44 | 0.09 | 3.85 | 1.16 |
| c | DCM/DCM | 1.66 | 1.93 | 0.70 | 0.09 | 0.39 | 0.08 | 3.19 | 1.07 |
| d1 | NH3/DCMa | 1.62 | 2.14 | 0.81 | 0.11 | 0.44 | 0.06 | 3.55 | 1.09 |
Conclusions
Our results demonstrate the effectiveness of liquid ammonia as a solvent for lignins under mild conditions, in particular for kraft and soda lignins, and somewhat less for organosolv lignins and native lignins. Kraft lignin concentrations up to at least 16.7 g L−1 were soluble in liquid ammonia (higher solubilities could not be evaluated with the present system). Apparently, liquid ammonia dissolves wood lignins quicker than grass lignins. After solubilisation in liquid ammonia and extraction with dichloromethane, the residual lignin was depleted in syringyl units, phenylcoumaran and resinol substructures, but enriched in β-O-4′ aryl ether substructures. No correlation was found with the phenol and aliphatic hydroxyl group content. Elemental analysis of the lignin residue showed that some nitrogen from the ammonia was incorporated in the residual lignin, and the lignin fragments rearranged to higher molecular weight fractions, especially at high temperature. Nevertheless, considering that ammonia is relatively cheap and can be recycled very efficiently (e.g. in the AFEX process), we feel that this lignin solubilisation method deserves more attention.Acknowledgements
This research was performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs, and the Netherlands Ministry of Education, Culture and Science. We thank Mrs J. van der Putten for the SEC measurements, Dr R. Gosselink, Dr S. Thiyagarajan and Dr G. Frissen for the NMR measurements, and D. Franciolus for the synthesis of the model compounds.References
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- B. Kamm and M. Kamm, Appl. Microbiol. Biotechnol., 2004, 64, 137–145 CrossRef CAS PubMed.
- N. R. Shiju, D. R. Brown, K. Wilson and G. Rothenberg, Top. Catal., 2010, 53, 1217–1223 CrossRef CAS.
- G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, ChemSusChem, 2009, 2, 215–217 CrossRef CAS PubMed.
- L. Durán Pachón, M. B. Thathagar, F. Hartl and G. Rothenberg, Phys. Chem. Chem. Phys., 2006, 8, 151–157 RSC.
- F. de Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef CAS PubMed.
- R. V. van Ree and E. Annevelink, Status Report Biorefinery 2007, Agrotechnology and Food Sciences Group, 2007.
- A. Gani and I. Naruse, Renewable Energy, 2007, 32, 649–661 CrossRef CAS PubMed.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- G. Gellerstedt, J. Li, I. Eide, M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 4240–4244 CrossRef CAS.
- M. Kaylen, D. L. Van Dyne, Y.-S. Choi and M. Blase, Bioresour. Technol., 2000, 72, 19–32 CrossRef CAS.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- D. Stewart, Ind. Crops Prod., 2008, 27, 202–207 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A.-S. Jönsson, A.-K. Nordin and O. Wallberg, Chem. Eng. Res. Des., 2008, 86, 1271–1280 CrossRef PubMed.
- F. Öhman, H. Theliander, P. Tomani and P. Axegard, Method for separating lignin from black liquor, 2008 Search PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- J. R. Regalbuto, Science, 2009, 325, 822–824 CrossRef PubMed.
- J. S. Shabtai, W. W. Zmierczak and E. Chornet, Process for Conversion of Lignin to Reformulated, Partially Oxygenated Gasoline, 2001 Search PubMed.
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423–424, 130–136 CrossRef CAS PubMed.
- M. M. Yan and C. B. Purves, Can. J. Chem., 1956, 34, 1747–1755 CrossRef CAS.
- B. Bals, C. Rogers, M. Jin, V. Balan and B. Dale, Biotechnol. Biofuels, 2010, 3, 1 CrossRef PubMed.
- T.-A. D. Nguyen, K.-R. Kim, S. J. Han, H. Y. Cho, J. W. Kim, S. M. Park, J. C. Park and S. J. Sim, Bioresour. Technol., 2010, 101, 7432–7438 CrossRef CAS PubMed.
- N. Sarkar, S. K. Ghosh, S. Bannerjee and K. Aikat, Renewable Energy, 2012, 37, 19–27 CrossRef CAS PubMed.
- J. J. Lagowski, Synth. React. Inorg. Met.-Org. Chem., 2007, 37, 115–153 CrossRef CAS.
- W. C. Johnson, J. Chem. Educ., 1935, 12, 249 CrossRef.
- J. B. Gill, J. Chem. Educ., 1970, 47, 619 CrossRef CAS.
- V. F. Hnizda and C. A. Kraus, J. Am. Chem. Soc., 1949, 71, 1565–1575 CrossRef CAS.
- I. Warshawsky, J. Inorg. Nucl. Chem., 1963, 25, 919–921 CrossRef CAS.
- F. London, Trans. Faraday Soc., 1937, 33, 8b–26 RSC.
- E. Burello and G. Rothenberg, Adv. Synth. Catal., 2003, 345, 1334–1340 CrossRef CAS.
- P. Ji, PhD. Thesis, University of Huddersfield, 2011.
- P. Ji, J. H. Atherton and M. I. Page, Org. Biomol. Chem., 2012, 10, 5732–5739 CAS.
- J. H. Atherton, M. I. Page and H. Sun, J. Phys. Org. Chem., 2013, 26, 1038–1043 CrossRef CAS.
- J. J. Lagowski, Acid–Base Equilibria in Liquid Ammonia, Department of Chemistry, The University of Texas, Austin, Texas, 78712, U.S.A., 1957 Search PubMed.
- J. C. del Río, P. Prinsen, J. Rencoret, L. Nieto, J. Jiménez-Barbero, J. Ralph, A. T. Martínez and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 3619–3634 CrossRef PubMed.
- J. Ralph, J. M. Marita, S. A. Ralph, R. D. Hatfield, F. Lu, R. M. Ede, J. Peng, S. Quideau, R. F. Helm, J. H. Grabber, H. Kim, G. Jimenez-Monteon, Y. Zhang, H.-J. G. Jung, L. L. Landucci, J. J. MacKay, R. R. Sederoff, C. Chapple and A. M. Boudet, Solution-state NMR of lignin, in Advances in lignocellulosics characterization, ed. D. S. Argyropoulos, Tappi Press, Atlanta, 1999, pp. 55–108 Search PubMed.
- S. A. Ralph, J. Ralph and L. Landucci, NMR database of lignin and cell wall model compounds, US Forest Prod. Lab., One Gifford Pinchot Dr., Madison, WI 53705, 2004, http://ars.usda.gov/Services/docs.htm?docid=10491, accessed January 2009 Search PubMed.
- A. T. Martínez, J. Rencoret, G. Marques, A. Gutiérrez, D. Ibarra, J. Jiménez-Barbero and J. C. del Río, Phytochemistry, 2008, 69, 2831–2843 CrossRef PubMed.
- J. Ralph and L. L. Landucci, NMR of Lignins, in Lignin and Lignans; Advances in Chemistry, ed. C. Hetiner, D. R. Dimmel and J. A. Schmidt, CRC Press (Taylor & Francis Group), Boca Raton, FL, 2010, pp. 137–234 Search PubMed.
- R. J. A. Gosselink, J. E. G. van Dam, E. de Jong, E. L. Scott, J. P. M. Sanders, J. Li and G. Gellerstedt, Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders, Holzforschung, 2010, 64(1), 193–200 CAS.
- P. Prinsen, J. Rencoret, A. Gutiérrez, T. Liitiä, T. Tamminen, J. L. Colodette, M. A. Berbis, A. T. Martínez and J. C. del Río, Ind. Eng. Chem. Res., 2013, 52, 15702–15712 CrossRef CAS.
- J. Zakzeski and B. M. Weckhuysen, ChemSusChem, 2011, 4, 369–378 CrossRef CAS PubMed.
- C. Schuerch, J. Am. Chem. Soc., 1952, 74, 5061–5067 CrossRef CAS.
- A. Arshanitsa, J. Ponomarenko, T. Dizhbite, A. Andersone, R. J. A. Gosselink, J. van der Putten, M. Lauberts and G. Telysheva, J. Anal. Appl. Pyrolysis, 2013, 103(SI), 78–85 CrossRef CAS PubMed.
- K. Wang, F. Xu and R. Sun, Int. J. Mol. Sci., 2010, 11, 2988–3001 CrossRef CAS PubMed.
- C. G. Boeriu, D. Bravo, R. J. A. Gosselink and J. E. G. van Dam, Ind. Crops Prod., 2004, 20, 205–218 CrossRef CAS PubMed.
- Z. Pawlak and A. S. Pawlak, Appl. Spectrosc. Rev., 1997, 32, 349–383 CrossRef CAS.
- D. Meier, V. Zuñiga-Partida, F. Ramírez-Cano, N.-C. Hahn and O. Faix, Bioresour. Technol., 1994, 49, 121–128 CrossRef CAS.
- A.-P. Zhang, C.-F. Liu, R.-C. Sun and J. Xiu, BioResearch, 2013, 8(2), 1604–1614 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01143k |
| This journal is © The Royal Society of Chemistry 2015 |