
The relationship between oxidation and hydrolysis in the conversion of cellulose in NaVO3–H2SO4 aqueous solution with O2†
Muge
Niu
a,
Yucui
Hou
b,
Shuhang
Ren
a,
Wenhua
Wang
a,
Qitian
Zheng
a and
Weize
Wu
*a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: wzwu@mail.buct.edu.cn; Fax: +86 10 64427603
bDepartment of Chemistry, Taiyuan Normal University, Taiyuan 030031, China
First published on 1st September 2014
Abstract
Conversion of cellulose, the most abundant biomass, shows a high selectivity to yield formic acid (FA), when oxidized by O2 in NaVO3–H2SO4 aqueous solution. This conversion involves various reactions including hydrolysis and oxidation. In this work, the relationships between these reactions were studied. There are mainly two hydrolyses and two oxidations occurring in the conversion: initial hydrolysis (from cellulose to monosaccharides) and deep hydrolysis (from monosaccharides to levulinic acid); catalytic oxidation (from monosaccharides to FA) and ordinary oxidation (from levulinic acid to acetic acid). Among these four reactions, catalytic oxidation to FA and deep hydrolysis to byproducts are competitive. The increasing rate of deep hydrolysis is faster than that of catalytic oxidation, when temperature is increased. Catalytic oxidation is promoted and deep hydrolysis is suppressed, when the O2 pressure is increased. Moreover, deep hydrolysis is promoted and catalytic oxidation is suppressed, when H2SO4 concentration is increased. The hydrolysis–oxidation pathway of this conversion was proposed. Thus, the byproducts could be inhibited, and FA-oriented transformation could be realized.
1. Introduction
Due to the consumption of fossil fuels and consequent environmental pollution, more and more attention has been drawn to renewable resources like biomass for fuel and chemical production.1,2 Formic acid (FA) is an important chemical and is widely used in chemical industries. In recent years, FA has played a new role for the efficient use of energy as fuel cells3–5 and hydrogen storage6–12 because of its unique properties. Therefore, the conversion of biomass to FA may provide a route from renewable resource not only for a fine chemical but also for energy.Recently, transforming biomass in aqueous solution using vanadium (V)-containing homogeneous catalyst with O2 as oxidant shows a high efficiency to yield FA. First, Wasserscheid et al.13 studied the biomass conversion in water using heteropoly acid H5PV2Mo10O40 as catalyst at mild temperatures (60–90 °C). The conversions of monosaccharides and disaccharides were productive (∼50% yield of FA after 26 h at 80 °C). However, cellulose, which is the most abundant biomass with highly polymerized structure, is hardly transformed even in a long reaction time (26 h). The formation of FA is negligible. Then, p-toluenesulfonic acid (TSA) was selected as an additive to the catalytic system, and the cellulose conversion and FA yield were found to increase (39% and 18.7%, respectively).14 Later, Albert et al.15 synthesized a new V-containing heteropoly acid, H8PV5Mo7O40, which showed relatively higher catalytic activity for FA formation (76% in conversion and 28% in FA yield with TSA). Fu et al.16 studied the cellulose conversion with H5PV2Mo10O40 at elevated temperatures (100–170 °C). Cellulose was totally transformed at 170 °C after 9 h, but FA yield was still very low (3%). Then, mineral acids were chosen to accelerate the cellulose hydrolysis. The FA yield increased to 34%, but 14% glucose was formed (using HCl as additive). In our previous work,17 NaVO3–H2SO4 aqueous solution with O2 was employed to yield FA from biomass. Catalyzed by VO2+ forming in acidic aqueous solution (pH < 1), the reaction produced ∼65% yield of FA using monosaccharides as substrates in 1 min at 160 °C. Cellulose could be fully converted and FA yield could reach ∼60% in a prolonged time (2 h).
All the above mentioned conversions of cellulose were performed in acidic solutions with O2. It is easy to infer that the conversion of cellulose to FA undergoes at least two reactions: hydrolysis from highly polymerized cellulose to monosaccharides and oxidation from monosaccharides to FA. This assumption can be evidenced by the following facts: (1) monosaccharides (glucose) are detected during the conversion;16 (2) adding TSA14 or mineral acids significantly accelerates the conversion. Moreover, the oxidation of cellulose in acidic solution could produce 5-hydroxymethylfurfural (HMF) and levulinic acid.18 It is known that HMF and levulinic acid are the products of glucose transformation in acidic solution. Therefore, the deep hydrolysis of monosaccharides may occur in the cellulose conversion to FA. In addition, the formation of glycolic acid18 indicates that the oxidation of hydrolysis products may also occur during the conversion of cellulose to FA.
Based on the above discussion, it can be inferred that the conversion of biomass into FA is a complex system with several simultaneous and/or successive reactions. These reactions affect each other, and thus affect the whole conversion process and product distribution. To date, the relationships between these reactions have not been thoroughly studied. Only with a better understanding of these relationships, we can tune these reactions to avoid byproducts and produce FA in a more effective way, where all the reactions of the biomass conversion are well coordinated to form FA.
In this work, the relationships between the reactions in cellulose conversion were studied in high-efficiency NaVO3–H2SO4 aqueous solution with O2 as oxidant, and the hydrolysis–oxidation pathway and competition relationship were suggested. Based on byproduct analysis and residue studies at different temperatures, two hydrolysis and two oxidations were found to occur. Among the four reactions, the deep hydrolysis, which is followed by oxidation to form byproduct, has a competitive relationship with catalytic oxidation. The experiments with different O2 pressures and H2SO4 concentrations further verified the conversion pathways. This understanding of relationships between the reactions in cellulose conversion could provide guidance to effectively drive the cellulose conversion towards the direction of forming FA.
2. Experimental section
2.1. Chemicals
Microcrystalline cellulose (96%), HMF (98%), furfural (99%), levulinic acid (99%), succinic acid (AR) and sodium metavanadate (NaVO3, AR) were purchased from Aladdin Reagent Inc. (Shanghai, China). D-(+)-Glucose anhydrous (99%) was purchased from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). D-Fructose (98%) was purchased from Bio-Basic Inc. (Canada). Sulfuric acid (H2SO4, 98%) was purchased from Beijing Modern Oriental Fine Chemicals Co., Ltd. (Beijing, China). Formic acid (98%) was purchased from Tianjin Fuchen Chemical Reagents Factory. Acetic acid (99.5%) was purchased from Beijing Tongguang Fine Chemical Company. Oxygen (O2, 99.995%) and nitrogen (N2, 99.999%) were supplied by Beijing Haipu Gas Industry Co., Ltd. (Beijing, China). All the reagents were analytical grade and used without further purification.2.2. Conversion of substrates
A series of NaVO3–H2SO4 aqueous solutions with various mass fractions (NaVO3: 0.35%, H2SO4: 0%, 0.7%, 1%, 2% and 3%) were prepared before the conversion as follows: a known amount of NaVO3 (0.875 g) was dissolved into distilled water (50 cm3) under ultrasonic heating. Then, a known amount of H2SO4 (0 g, 1.75 g, 2.5 g, 5 g and 7.5 g) was diluted by distilled water (50 cm3) and mixed with the NaVO3 solution. Finally, the mixed solution was diluted to a known volume (250 cm3) with distilled water to meet the above-listed mass fraction.The conversion was carried out in a 25 cm3 batch reactor of Hastelloy alloy (HC 276) with a magnetic stirrer. In a typical procedure, a certain amount of substrate and 6.0 cm3 of NaVO3–H2SO4 aqueous solution were loaded into the reactor. Then, the reactor was sealed and purged with O2. Subsequently, O2 was charged into the reactor to a desired pressure. Two heating furnaces were used together to reduce the heat-up time. They were heated up to a temperature that was 130 degrees higher and 30 degrees higher than the desired temperature, respectively. Next, the reactor was put into the heating furnace at a higher temperature. The desired temperature was reached in 5 min, and the reaction time was recorded. Then, the reactor was quickly transferred into the heating furnace at a lower temperature to prevent further increase in the temperature. The furnace temperature was controlled in order to maintain the reactor temperature around the desired value. The reactor was stirred at a speed of 1000 r min−1. The pressure and temperature of the reactor were measured by a pressure transducer with an uncertainty of ±0.025 MPa and a thermocouple with an uncertainty of ±0.5 °C, respectively. After the reaction, the reactor was quenched by cold water. When the temperature of reactor reached room temperature, the gas was released and the liquid mixture was filtered. The residue was washed with distilled water and dried in an oven at 60 °C for 24 h before further use. The liquid sample was analyzed by HPLC.
2.3. Analysis of products
The liquid sample was analyzed by high-performance liquid chromatography (HPLC, Waters 2695, USA) with a Shodex SH 1011 column (Shodex, Japan). A diode array detector (Waters 2998, USA) was employed to analyze the furan compounds (HMF and furfural). A differential refractive index detector (Waters 4110, USA) was employed to analyze other products. The column oven temperature was set at 55 °C and the mobile phase was 0.01 mol dm−3 diluted H2SO4 aqueous solution at a flow rate of 0.5 cm3 min−1. All the yields of products were calculated on a carbon base. The amounts of total organic carbon of the liquid samples were measured with a TOC analyzer (Shimadzu TOC-L CPN, Japan). Cellulose conversion was calculated by the difference in solid weight, before and after the reaction. The surface morphology of residues after the conversion of cellulose was studied by scanning electron microscopy (SEM, Zeiss Supra 55, Germany) with an accelerating voltage of 5 kV.3. Results and discussion
3.1. Conversion of cellulose into FA at various temperatures
First, the conversion of cellulose in NaVO3–H2SO4 aqueous solution at temperatures from 140 °C to 180 °C was studied. Fig. 1 shows the conversion of cellulose and yield of FA. As expected, when temperature was increased, both the conversion and the FA yield increased in a short time. For example, when the temperature was raised from 140 °C to 180 °C, the conversion of cellulose was increased from 30% to 100% and the yield of FA was increased from 22.1% to 52.3% in 2 min. At a low temperature of 140 °C, even a long reaction time of 60 min could not obtain a high conversion and yield (48.9% in conversion and 32.4% in yield). The decrease in FA yield at elevated temperatures after a long period of time is due to the low stability of FA under the same conditions. As a result, an increase in the reaction temperature could produce FA in a higher yield in a shorter time.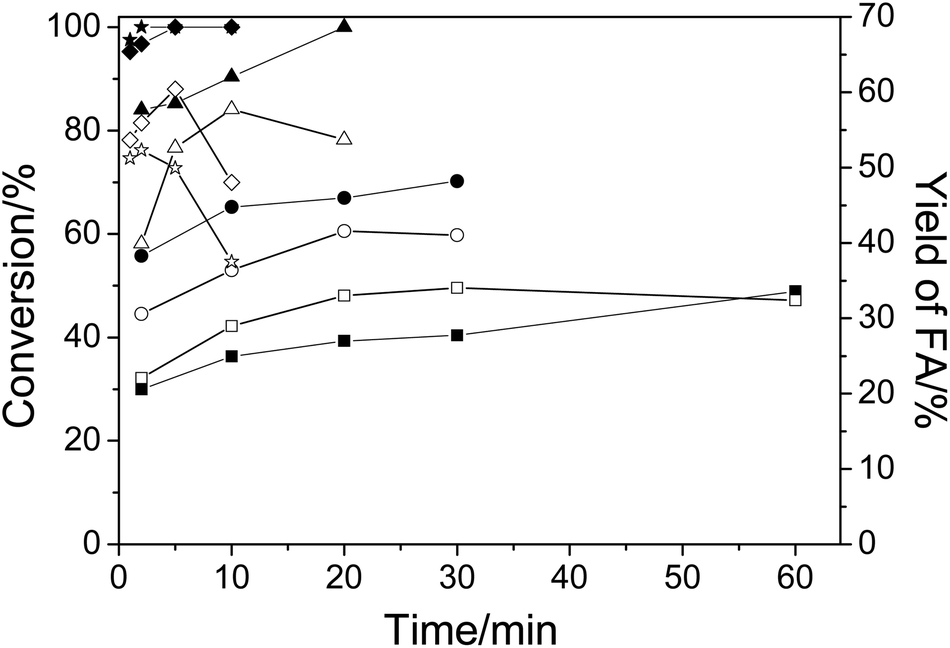 | ||
| Fig. 1 The conversion of cellulose and the yield of FA in cellulose conversion at various temperatures. Conditions: cellulose, 0.1 g; H2SO4, 2 wt%; NaVO3, 0.35 wt%; H2O, 6.0 cm3; O2, 3 MPa. ■, ●, ▲, ◆ and ★ represent the conversion of cellulose at 140 °C, 150 °C, 160 °C, 170 °C and 180 °C, respectively. □, ○, △, ◇ and ☆ represent the yield of FA at 140 °C, 150 °C, 160 °C, 170° C and 180 °C, respectively. | ||
3.2. Byproducts analysis
As mentioned in section 3.1, the elevated temperature accelerated the reaction and helped to produce high-yield FA in a short time. However, the byproduct—acetic acid—was obtained in high-temperature reactions. The amounts of acetic acid at different temperatures are listed in Table S1.† Although the amount of acetic acid was considerably lower than that of the main product FA, the formation of acetic acid suggested a significant side reaction occurring at elevated temperature. In order to investigate this side reaction, the source of acetic acid should be first identified.The product distributions of cellulose conversion from 140 °C to 180 °C under an O2 pressure of 3 MPa after 10 min are shown in Table 1, entries 1–5. Only FA, acetic acid and levulinic acid were detected in the product mixtures. The yield of acetic acid significantly increased with temperature. The results of oxidation of cellulose without the addition of NaVO3 at various temperatures are shown in Table 1, entries 6–10. With the increase of temperature, acetic acid yield increased. This trend is similar to that of the reaction with NaVO3 (Table 1, entries 1–5). It was reported that cellulose and carbohydrates could produce acetic acid in hydrothermal oxidations without catalyst.19–21 This result suggests that acetic acid is not because of the catalytic oxidation.
| Entry | Temperature/°C | Formic acid/% | Acetic acid/% | Succinic acid/% | Glucose/% | Fructose/% | HMF/% | Furfural/% | Levulinic acid/% |
|---|---|---|---|---|---|---|---|---|---|
| Conditions: cellulose, 0.1 g; H2SO4, 2 wt%; NaVO3, 0.35 wt%; H2O, 6.0 cm3; O2, 3 MPa; time, 10 min.a NaVO3 was not added in the reaction mixture.b NaVO3 was not added in the reaction mixture and O2 was replaced by N2 with the same pressure. | |||||||||
| 1 | 140 | 29.0 | 0.3 | 1.5 | |||||
| 2 | 150 | 36.4 | 0.3 | 1.9 | |||||
| 3 | 160 | 57.7 | 1.4 | 0.9 | |||||
| 4 | 170 | 48.0 | 5.2 | 1.0 | |||||
| 5 | 180 | 37.5 | 9.2 | 0.8 | |||||
| 6a | 140 | 7.6 | 0.7 | 2.1 | 0.7 | ||||
| 7a | 150 | 17.3 | 2.3 | 1.1 | 1.0 | 1.5 | |||
| 8a | 160 | 24.0 | 3.0 | 0.6 | |||||
| 9a | 170 | 44.7 | 6.6 | 1.2 | |||||
| 10a | 180 | 13.5 | 5.1 | 0.4 | |||||
| 11b | 140 | 8.4 | 1.2 | 0.9 | 0.6 | 1.3 | |||
| 12b | 150 | 1.7 | 20.5 | 1.0 | 2.9 | 3.2 | 8.5 | ||
| 13b | 160 | 5.9 | 16.5 | 1.4 | 3.7 | 3.1 | 20.8 | ||
| 14b | 170 | 7.9 | 2.7 | 1.7 | 3.3 | 30.7 | |||
| 15b | 180 | 6.2 | 0.3 | 3.0 | 34.6 | ||||
Cellulose conversion is performed in acidic solution, thus hydrolysis occurs simultaneously with catalytic oxidation. In the catalytic reaction, very small amounts of levulinic acid were detected (see Table S1†). Thus, experiments without NaVO3 and O2 (replaced by N2 with the same pressure) were carried out (Table 1, entries 11–15). With only H2SO4 in the reaction mixture, the conversion turned out to be hydrolysis. With the increase of temperature, the amount of glucose first increased then decreased. Whereas the dehydration products, HMF and furfural, were gradually forming. When the temperature reached 170 °C or 180 °C, levulinic acid became the main product (the production of FA is due to the hydrolysis of HMF or furfural22). The results show that high temperature accelerates the hydrolysis of cellulose.
It can be observed that at elevated temperatures, acetic acid and glucose degradation products (HMF, furfural and levulinic acid) were largely formed in experiments with and without O2, respectively. This phenomenon indicates that acetic acid was probably formed from the oxidation of the degradation products from glucose. Thus, a series of model compounds were selected as substrates for catalytic conversion to find out the precursor of acetic acid. The results are shown in Table 2. Oxidation of glucose and fructose both gave a high selectivity to FA (entries 1 and 2). However, because of the oxidation of HMF and furfural, low yields of FA and relatively high yields of acetic acid were obtained (entries 3 and 4). Levulinic acid is obtained from the hydrolysis of HMF and furfural. Succinic acid is probably obtained from the oxidation of HMF.23 When levulinic acid acted as the substrate, there was no FA produced but a high yield of acetic acid was formed (entry 5). The selectivity to acetic acid was the highest (25.6%) among the five compounds listed in the table. Succinic acid and acetic acid were chosen to be the substrates, and no other products in the liquid phase were found (entries 6 and 7). Thus, it can be concluded that the acetic acid in the catalytic conversion was formed by the oxidation of deep hydrolysis products: HMF, furfural and levulinic acid. Considering that levulinic acid has the dominant position in the hydrolysis products at high temperatures and has a high selectivity to form acetic acid in the catalytic system, levulinic acid is regarded as the main source of acetic acid at elevated temperatures.
| Entry | Substrate | Conversion/% | Formic acid/% | Acetic acid/% | Levulinic acid/% | Succinic acid/% | Selectivity to FA/% | Selectivity to AA/% |
|---|---|---|---|---|---|---|---|---|
| a Conditions: substrate, 0.05 g; H2SO4, 2 wt%; NaVO3, 0.35 wt%; H2O, 6.0 cm3; O2, 3 MPa; temperature, 160 °C; time, 1 min; AA stands for acetic acid. | ||||||||
| 1 | Glucose | 93.8 | 44.3 | 1.2 | <0.1 | <0.1 | 47.2 | 1.3 |
| 2 | Fructose | 100 | 44.1 | 2.8 | <0.1 | 0.7 | 44.1 | 2.8 |
| 3 | HMF | 89.3 | 25.5 | 2.4 | 7.8 | 7.0 | 28.6 | 2.7 |
| 4 | Furfural | 62.7 | 18.0 | 2.6 | 6.1 | 28.7 | 4.1 | |
| 5 | Levulinic acid | 23.8 | 6.1 | 25.6 | ||||
| 6 | Succinic acid | 12.2 | ||||||
| 7 | Acetic acid | 12.0 | ||||||
3.3. SEM studies of residues after conversion
The ratio of acetic acid to FA increases with increasing temperature (shown in Table S2†). This indicates that when the temperature is increased, both the FA and acetic acid increase but acetic acid increases faster than FA. As proven in section 3.2, acetic acid is mainly obtained from the oxidation of levulinic acid, which is the deep hydrolysis product of cellulose. All these results provide a hint that (1) there are two reactions, which could be called “catalytic oxidation” and “deep hydrolysis”, leading to the main product (FA) and the main byproduct (acetic acid), respectively; (2) the increasing rate of deep hydrolysis is faster than that of catalytic oxidation, when the temperature is increased. To further verify this assumption, SEM images for residues after catalytic conversion and hydrolysis at various temperatures were studied.SEM images of different residues after catalytic conversion are shown in Fig. 2 (SEM images with different magnifications are shown in Fig. S1†). For the purpose of reflecting the residues morphology, a low magnification (×200 and ×500) was employed. In Fig. 2, part (a) is for untreated cellulose; parts (b), (c) and (d) are for residues after conversion at 140 °C, 150 °C and 160 °C, respectively. After a low-temperature conversion at 140 °C, the cellulose particle size became smaller (Fig. 2b). This phenomenon is easily understandable for the degradation of cellulose. However, when the temperature was increased to 150 °C, the residue began to agglomerate (Fig. 2c). When the temperature was further increased to 160 °C, the residue significantly agglomerated and larger blocks were formed (Fig. 2d). To explain this unexpected agglomeration of residues in catalytic conversion with increasing temperature, the SEM images of residues after hydrolysis at corresponding temperatures (140 °C, 150 °C and 160 °C) were studied. Fig. 2e, f and g represent residues after hydrolysis at 140 °C, 150 °C and 160 °C. More severe agglomeration occurred, when the temperature was increased. A similar agglomeration was reported by Zhao et al.24 when cellulose hydrolysis was performed in hot water. This phenomenon is probably caused by the interaction between the hydrolysis products (mainly HMF and furfural) and unreacted cellulose.25–27 This indicates that the agglomeration occurring in catalytic conversion is due to the hydrolysis. After hydrolysis at 140 °C (Fig. 2e), the particles were smaller compared to the untreated cellulose (Fig. 2a), but larger than those of catalytic conversion (Fig. 2b), and some particles appeared as blocks. From the comparison of these SEM images, we could see that, in addition to transforming cellulose and glucose to FA, the O2 and catalyst (NaVO3) could prevent the cellulose from agglomeration. Furthermore, these results indicate two reactions in catalytic conversion: (1) initial hydrolysis makes cellulose particles smaller; (2) catalytic oxidation transforms hydrolysis product—glucose—to FA and prevents the further degradation of glucose to HMF and furfural, which led to agglomeration. When the temperature was raised from 140 °C to 150 °C, the cellulose agglomerated to larger blocks after hydrolysis (Fig. 2f). For the catalytic oxidation at 150 °C, small particles still remained but large blocks were also formed (Fig. 2c). This indicates that in addition to the two reactions occurring at 140 °C, the third reaction—deep hydrolysis (further degradation of glucose mentioned before)—had also occurred. The catalytic oxidation cannot stop the agglomeration caused by the deep hydrolysis of cellulose. When the temperature was raised to 160 °C, the residues of catalytic conversion and hydrolysis both highly agglomerated (Fig. 2d and g). The images are almost identical. This demonstrates that the catalytic oxidation hardly inhibits deep hydrolysis.
 | ||
| Fig. 2 SEM images of residues after cellulose conversion. (a) represents cellulose without treatment; (b), (c) and (d) represent the catalytic conversion of cellulose at 140 °C, 150 °C and 160 °C, respectively; (e), (f) and (g) represent the hydrolysis of cellulose at 140 °C, 150 °C and 160 °C, respectively. Treatment conditions: cellulose, 0.1 g; H2SO4, 2 wt%; NaVO3, 0.35 wt% (only for catalytic conversion); H2O, 6.0 cm3; O2, 3 MPa (only for catalytic conversion); N2, 3 MPa (only for hydrolysis); time, 1 min. | ||
SEM studies indicate that deep hydrolysis and catalytic oxidation are competitive. With the increased temperature, the increasing rate of deep hydrolysis is faster than that of catalytic oxidation. This rule is in agreement with the rule that the yield of acetic acid increases with increasing temperature. The conclusion that acetic acid comes from the products of deep hydrolysis has been further verified.
3.4. Hydrolysis–oxidation pathway
Based on the above discussions, a reaction pathway of cellulose conversion in NaVO3–H2SO4 aqueous solution with O2 is proposed (see Fig. 3). Cellulose is first converted into monosaccharides (glucose and fructose) by hydrolysis. Then, the monosaccharides go through two parallel reactions: (1) catalytic oxidation to FA with high selectivity and (2) deep hydrolysis to HMF, furfural, and levulinic acid, which further produce acetic acid by oxidation. Among the deep hydrolysis products, levulinic acid is most likely to be oxidized to acetic acid, the main byproduct in the catalytic conversion of cellulose. Very small amount of succinic acid is probably obtained from oxidation of HMF.23 When HMF and furfural are significantly formed, condensation occurs and unknown black substances (so called “humin” or “humic acid”) are finally formed.26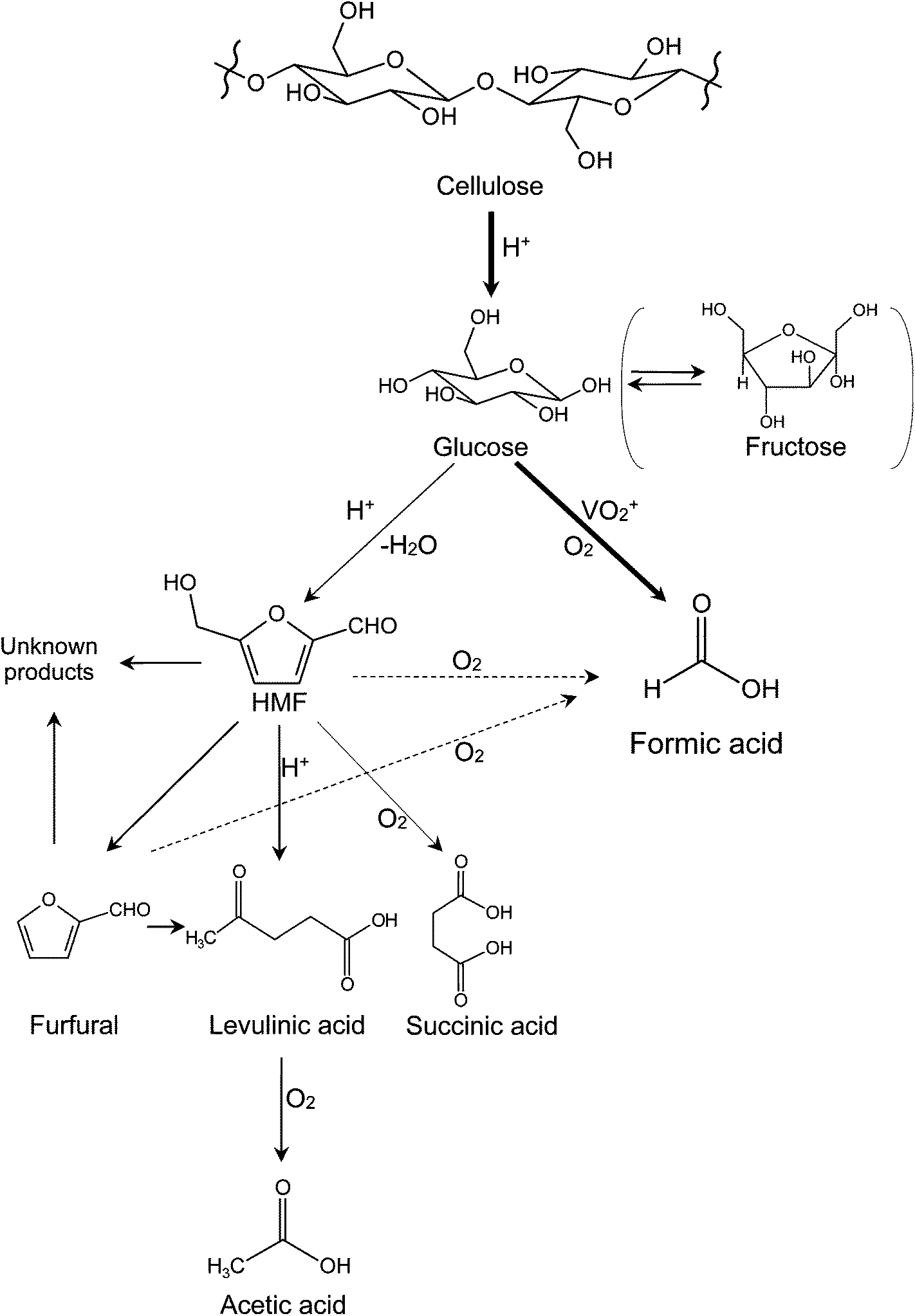 | ||
| Fig. 3 Proposed pathway of cellulose conversion in NaVO3–H2SO4 aqueous solution with O2 as oxidant. | ||
This hydrolysis–oxidation pathway shows both synergistic and competitive relationships between oxidation and hydrolysis. Initial hydrolysis (from cellulose to monosaccharides) and catalytic oxidation are synergistic. The increasing rate of initial hydrolysis produces more glucose that can be the substrate for FA. Deep hydrolysis and catalytic oxidation are competitive. When deep hydrolysis accelerates, less glucose remains to yield FA; when catalytic oxidation accelerates, less glucose remains to deep hydrolyze to yield levulinic acid. Deep hydrolysis and ordinary oxidation (from levulinic acid to acetic acid) are synergistic. The increasing rate of deep hydrolysis leads to more levulinic acid that can be further oxidized to acetic acid.
Based on this reaction pathway, it is promising to move the balance between FA and acetic acid by varying related reaction parameters. Then, two sets of experiments were carried out to move this balance by varying O2 pressure and H2SO4 concentration, respectively.
The effect of O2 pressure on the product distributions is shown in Fig. 4a (the conversion data are shown in Fig. S2†). An increase in O2 pressure accelerated both catalytic oxidation and ordinary oxidation, thus the yields of FA and acetic acid both increased. When the O2 pressure was further increased, the catalytic oxidation was still promoted and the deep hydrolysis was suppressed. Therefore, the FA yield was still increasing but the levulinic acid yield was decreasing. Because acetic acid is the product of levulinic acid, yield of acetic acid was consequently decreasing.
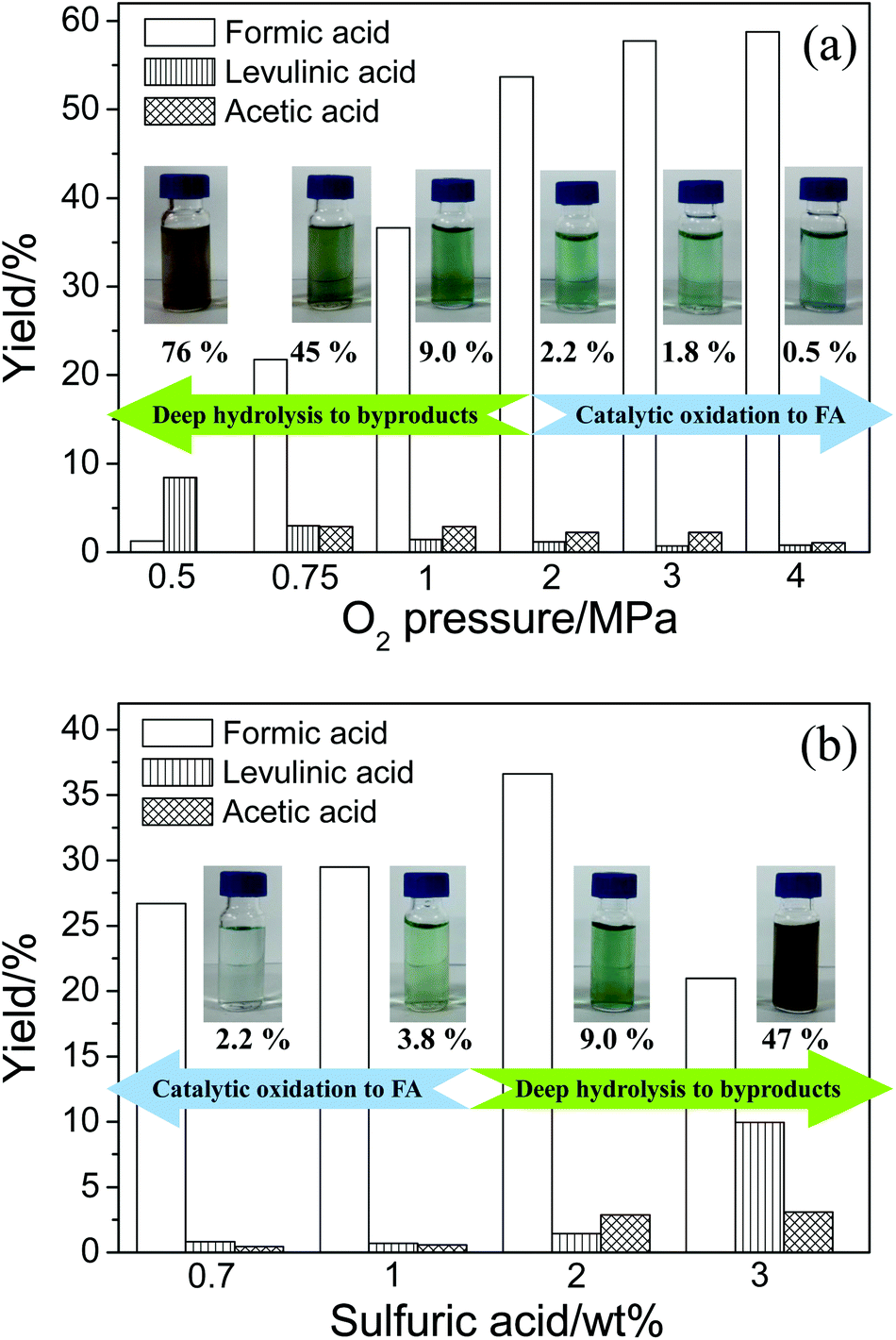 | ||
| Fig. 4 The yield of products in cellulose conversion with (a) various O2 pressures (conditions: cellulose, 0.1 g; NaVO3, 0.35 wt%; H2SO4, 2%; H2O, 6.0 cm3; time, 10 min) and (b) various H2SO4 concentrations (conditions: cellulose, 0.1 g; NaVO3, 0.35 wt%; H2O, 6.0 cm3; O2, 1 MPa; time, 10 min), respectively. The images of liquid samples are shown beside the corresponding data columns. The ratios of carbon in the unknown products to TOC in liquid samples are listed below the images. | ||
The effect of H2SO4 concentration on the product distributions is shown in Fig. 4b (the conversion data are shown in Fig. S3†). The addition of H2SO4 first accelerated the initial hydrolysis of cellulose. The glucose was soon formed and the FA from the catalytic oxidation of glucose was increasing. When H2SO4 concentration was further increased, the deep hydrolysis of glucose was promoted. As a consequence, the catalytic oxidation was suppressed. Thus, the levulinic acid yield was increasing and the FA yield was decreasing. Because acetic acid is the product of levulinic acid, yield of acetic acid was consequently increasing.
The images of liquid samples after conversion with different O2 pressures and H2SO4 concentrations are shown beside the corresponding data columns in Fig. 4. When O2 pressure was decreasing or H2SO4 concentration was increasing, the color of liquid samples gradually changed from light green blue (the color of reduced vanadium) to dark brown. This phenomenon is probably due to the formation of unknown products, which is often found after cellulose hydrolysis. In other words, the amount of unknown products could reflect the extent of hydrolysis. Researchers often analyze the unknown products in aqueous phase by determining the total organic carbon (TOC).28–30 Thus, the TOC in the liquid sample was measured. The difference of TOC and carbon of the known products could refer to the amount of the unknown products. The ratios of carbon in unknown products to TOC in liquid samples are listed below the images. The amount of unknown products significantly increased with decreasing O2 pressure or increasing H2SO4 concentration. This indicates the deep hydrolysis actually occurred. The changes in color and amount of unknown products in liquid samples also give evidence to the competitive relationship between catalytic oxidation and deep hydrolysis.
4. Conclusions
In this work, the relationships between various reactions in cellulose conversion in NaVO3–H2SO4 aqueous solution with O2 as oxidant were studied. A pathway to byproducts has been found: the hydrolysis of cellulose to levulinic acid, which then is oxidized to acetic acid. The catalytic oxidation and deep hydrolysis, which lead to the formation of FA and the main byproduct acetic acid respectively, are competitive. Based on this competitive relationship, the enhancement of oxidation conditions can promote catalytic oxidation to the main product FA and suppress the formation of byproducts. In contrast, if the hydrolysis conditions are intensified, deep hydrolysis will be promoted and byproducts will be significantly formed.Acknowledgements
We thank professors Zhenyu Liu and Qingya Liu for their help. This work was supported by the Natural Science Foundation of China (21076138), the National Basic Research Program of China (2011CB201303), and the Program for New Century Excellent Talents in University (NCET-08-0710).References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- N. V. Rees and R. G. Compton, J. Solid State Electrochem., 2011, 15, 2095–2100 CrossRef CAS PubMed.
- C. Rice, S. Ha, R. I. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83–89 CrossRef CAS.
- M. Weber, J. T. Wang, S. Wasmus and R. F. Savinell, J. Electrochem. Soc., 1996, 143, 158–160 CrossRef PubMed.
- A. Boddien, D. Mellmann, F. Gartner, R. Jackstell, H. Jungle, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- S. D. Senanayake and D. R. Mullins, J. Phys. Chem. C, 2008, 112, 9744–9752 CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- Z. Wang, J. Yan, Y. Ping, H. Wang, W. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed.
- R. Wölfel, N. Taccardi, A. Bösmann and P. Wasserscheid, Green Chem., 2011, 13, 2759–2763 RSC.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962 CAS.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Green Chem., 2014, 16, 226–237 RSC.
- J. Li, D. Ding, L. Deng, Q. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS PubMed.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 2883–2890 CrossRef CAS.
- F. Jin, Z. Zhou, T. Moriya, H. Kishida, H. Higashijima and H. Enomoto, Environ. Sci. Technol., 2005, 39, 1893–1902 CrossRef CAS.
- H. R. Holgate, J. C. Meyer and J. W. Tester, AIChE J., 1995, 41, 637–648 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS PubMed.
- H. Zhao, J. H. Kwak, Z. C. Zhang, H. M. Brown, B. W. Arey and J. E. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS PubMed.
- B. F. M. Kuster and H. S. van der Baan, Carbohydr. Res., 1977, 54, 165–176 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- I. Sereewatthanawut, S. Prapintip and K. Watchiraruji, Bioresour. Technol., 2008, 99, 555–561 CrossRef CAS PubMed.
- K. Yang, D. Lin and B. Xing, Langmuir, 2009, 25, 3571–3576 CrossRef CAS PubMed.
- R. J. Chimentão, E. Lorente, F. Gispert-Guirado, F. Medina and F. López, Carbohydr. Polym., 2014, 111, 116–124 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00970c |
| This journal is © The Royal Society of Chemistry 2015 |