
Low-temperature microwave-assisted pyrolysis of waste office paper and the application of bio-oil as an Al adhesive†
Zhanrong
Zhang
a,
Duncan J.
Macquarrie
a,
Mario
De bruyn
a,
Vitaliy L.
Budarin
a,
Andrew J.
Hunt
a,
Mark J.
Gronnow
b,
Jiajun
Fan
a,
Peter S.
Shuttleworth
c,
James H.
Clark
a and
Avtar S.
Matharu
*a
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, England. E-mail: avtar.matharu@york.ac.uk
bBiorenewables Development Centre, the Biocentre, York Science Park, York, YO10 5NY, England
cDepartamento de Física de Polímeros, Elastómeros y Aplicaciones Energéticas, Instituto de Ciencia y Tecnología de Polímeros, CSIC, calle Juan de la Cierva, 3, 28006 Madrid, Spain
First published on 15th August 2014
Abstract
The conversion of waste office paper (printed or photocopied) to bio-oil via low temperature (<200 °C) microwave-assisted pyrolysis, and its utilisation as an adhesive for aluminium–aluminium bonding are reported. The yields for the organic and aqueous phase bio-oil are 19% and 23%, respectively. The pyrolysis products were characterized by ICP-MS, ATR-IR, GC-MS and NMR to reveal broad categories of compounds indicative of sugars (carbohydrates), aromatics and carbonyl-containing moieties. Application of the organic phase bio-oil (70 mg) to Al plates (50 mm × 50 mm) followed by curing at different temperatures and time periods revealed that a maximum tensile strength of approximately 2300 N could be attained at 160 °C for 8 h cure. Also, at a fixed temperature, the tensile strength increased with increasing curing time. To gain an in-depth understanding of the adhesive properties of bio-oil, a liquid–liquid fractionation of the organic phase bio-oil was conducted. The ‘acidic’ fraction showed far better adhesion properties than the ‘neutral’ fraction with no bonding achieved for the aqueous fraction. A combination of the ‘acidic’ and ‘neutral’ fraction gave better adhesion, thus suggesting a possible synergistic or co-operative effect.
1. Introduction
Globally, paper is one of the most widely recycled materials. For example, in 2011 the recycling rate in the EU was over 70%.1 In theory, paper can be recycled up to six or seven times but in reality this is far from true. Paper cannot be recycled indefinitely as fibres become too short and worn to be useful in creating a new sheet of paper or cardboard box. The current average rate in Europe is 3.5, while world-wide the average is 2.4 times.1 In an era of confidentiality and data protection, document shredding and/or milling of paper is on the rise, which can render it ineffective for recycling as the resulting fibres become too short. Finely chopped or ground paper also causes maintenance problems and fire hazards when fed into certain types of paper mill machinery, and some mill machines simply cannot recognise small or shredded materials detrimental to paper quality.2Thus, a significant amount of waste paper and board ends up in landfill or incineration, which is a loss of valuable cellulosic raw material and might have negative impacts on the environment. The recognition of the value of this rejected material is now important, as the economic differential between the price of paper for recycling and the cost of reject management is becoming marginal. Paper destined for recycling should be seen as a source for many valuable components to produce additional high value products alongside paper.3
The production of biofuels and other sugar derivatives (such as gluconic acid,4 lactic acid,5etc.) from waste paper through biochemical conversion has been reported. Also, it has been estimated that the annual global production of bio-ethanol can be as much as 82.9 billion litres from waste paper, replacing 5.36% of petroleum consumption, with accompanying greenhouse gas (GHG) emission savings between 29.2% and 86.1%.3 However, one disadvantage of biochemical conversion, which generally involves enzymatic hydrolysis, is the low enzymatic degradation rate of lignocellulosic materials due to the resistant crystalline structure of cellulose and the physical barrier formed by lignin that is associated with the cellulose.6 Also, the high enzyme cost is another disadvantage for biochemical conversion from an economic point of view.7
Pyrolysis is considered to be a promising method for converting biomass into value-added products and aims to obtain char, gas and a liquid product (bio-oil) through thermal decomposition of biomass in the absence of oxygen. The pyrolysis process not only recovers the energy value of waste biomass, but also the chemical value. There are several comprehensive reviews existing in the literature on pyrolysis of biomass/waste material for generation of value-added chemicals, fuels, materials and upgrading methods for pyrolysis products.8–15 Bio-oil is potentially one of the most valuable products. It can be readily stored and transported, and can not only be used as a feedstock for the production of valuable chemicals, but also as a liquid fuel after upgrading.8 Even though the recovery of pure chemical compounds from the complex bio-oil is technically feasible, it is unattractive economically due to its high costs and low concentrations of specific chemicals.16 Also, although bio-oils have been tested successfully in engines, turbines and boilers, and have been upgraded to high-quality hydrocarbon fuels, its high energetic and financial costs are still presently unacceptable due to its high oxygen content and high viscosity and many other reasons.8
Recently, the combination of microwave with pyrolysis has attracted much attention due to the nature and many advantages of microwave heating, such as uniform and rapid internal heating of large biomass particles, instantaneous response for rapid start-up and shut down, energy efficiency, no need for agitation and controllability etc.17 Also, microwave processing has now been widely accepted as an effective technique for both pilot scale and large continuous processing for waste treatment.18 Luque et al. pointed out that microwave-assisted pyrolysis, especially at low temperatures, is a promising alternative for valorisation and processing of biomass and waste materials, and has potential to be integrated into future biorefineries.19 In this context, Lam and Chase reviewed existing processes for converting waste to energy using microwave pyrolysis,15 and Yin reviewed microwave-assisted pyrolysis of biomass for the production of biofuels.20 In recent years, significant efforts have been devoted to the application of microwave pyrolysis to a vast array of biomass/waste materials, such as sewage sludge,21–23 algae,24–27 coffee hulls,28 wheat straw,29 cellulose,30,31 and other organic municipal solid wastes,32–36 for valorisation. However, to our knowledge, there is no existing study on microwave-assisted pyrolysis of waste office paper to produce a bio-oil adhesive for Al–Al bonding.
Herein, we report our preliminary research on low temperature (<200 °C) microwave-assisted pyrolysis of milled waste office paper to yield bio-oil which may be used as a bio-based adhesive for metal-to-metal bonding. It is known that bio-oils can be used to generate adhesives.37,38 Indeed, several publications mentioned the use of the phenolic fraction of bio-oil, which derived mainly from lignin, as a replacement for the phenol needed in the phenol/formaldehyde resin formulations, used to produce oriented strand board (OSR) and plywood.39,40 The milled waste office paper derived bio-oil was analysed and characterized by Fourier transform-infrared spectroscopy (FT-IR), gas chromatography-mass spectroscopy (GC-MS), 13C nuclear magnetic spectroscopy (13C NMR) and inductively coupled plasma-mass spectrometry (ICP-MS). The adhesive properties of bio-oil were investigated by curing at various temperatures and time on Al plates. Also, an attempt was made to investigate the adhesive properties of bio-oil by liquid–liquid fractionation. The cured bio-oil polymers on the Al/bio-oil/Al interface was characterized with FT-IR and solid-state 13C CP/MAS NMR with and without dipolar dephasing.
2. Experimental
2.1. Samples
Waste office papers (printed or photocopied) were obtained from the Green Chemistry Centre of Excellence, University of York and milled using a Farm Feed Solutions (UK) 12 kW hammer mill with 5 mm screen. The standard commercial samples and DMSO-d6 (>99% purity) for 13C NMR analysis were purchased from Sigma Aldrich (UK) and used without further purification.2.2. Low-temperature microwave-assisted pyrolysis
Low-temperature microwave-assisted pyrolysis of milled waste office paper was conducted on a Milestone ROTO-SYNTH Rotative Microwave Reactor (Milestone Srl., Italy) fitted with a vacuum system. Thus, the condensable volatile compounds were collected as two phases (organic and aqueous) as shown in Fig. 1. In the current work, the incondensable gas products were depleted from the system. Prior to microwave treatment at fixed power (1200 W, 2.45 GHz), milled office paper (150 g per run) was wet pressed into the microwave glass vessel and dried in an oven at 105 °C for 48 h until a fixed weight was reached (moisture content less than 3% with a density of 0.3 g cm−3). The process temperature was measured via an on-line infrared temperature probe and maintained below 200 °C, whilst the process pressure was monitored in the control panel throughout the process. | ||
| Fig. 1 Experimental set-up for microwave-assisted pyrolysis of milled waste office paper. | ||
2.3. Characterization methods
Proximate analysis was conducted to determine the moisture, volatile matter, fixed carbon and ash content of milled waste office paper and bio-char using a Netzsch 409 Simultaneous Thermal Analyzer (STA). The detailed procedure was according to the literature.41 Ash content of bio-oils was determined as the residue material after heating bio-oil samples (100 mg) at 900 °C for 1 h in the Netzsch 409 STA. The temperature program was ramped from 30 °C to 900 °C at 50 °C min−1 and maintained at 900 °C for 1 h under an air atmosphere (100 ml min−1).Elemental analysis (C, H and N) was conducted on an Exeter Analytical CE440 Elemental Analyser. Sulfur contents of milled office paper and its MW-assisted pyrolysis products, together with total organic carbon (TOC) of aqueous phase bio-oil, were determined by Yara Analytical Services, York.
The calorific value (CV) of milled waste office paper and its pyrolysis products were determined using a Parr 6200 bomb calorimeter (Parr Instrument Company, Moline, IL, USA). CV measurements were carried out in triplicate and mean values were reported.
The water content of the organic phase bio-oil was determined on a GR Scientific Cou-Lo Aquamax Karl Fisher Titrator, using HYDRANAL®-Coulomat AK (anolyte solution) and HYDRANAL®-Coulomat CG-K (catholyte solution) as coulometric reagents, to avoid any interferences from ketones, amines or aldehydes.
The ATR-IR analysis of pyrolysis products (except the gas fraction) and scrapings of cured bio-oil polymers was performed on a Bruker Vertex 70 spectrometer in attenuated total reflectance (ATR) mode, with a resolution of 2 cm−1 and 64 scans.
The 13C NMR characterization of bio-oils (both aqueous and organic phases) were performed on a Jeol ECX-400 NMR spectrometer at 100 MHz for 1024 scans, using the central resonance of DMSO-d6 (δC, 39.52 ppm) as the internal reference.
The GC-MS characterization of liquid products (bio-oils) was conducted on a Perkin Elmer Claus 500 gas chromatograph equipped with a Perkin Elmer Claus 560 s mass spectrometer. The column was a non-polar ZB-5HT (30 m × 0.25 mm id × 0.25 μm film thickness) from Phenomenex (UK). The oven temperature programme was maintained at 60 °C for 1 min, then ramped at 8 °C min−1 to 360 °C and held for 1 min. The injection volume of sample is 1 μl. The identification of chromatographic peaks was by comparison with the NIST 2008 library.
Inductively coupled plasma-mass spectrometry (ICP-MS) of milled office paper and its MW-assisted pyrolysis products were conducted on an Agilent 7700× ICP-MS spectrometer to determine their metal contents. Prior to analysis, solid samples were acid digested in HCl and HNO3 at a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Liquid samples (bio-oils) were diluted in acetone.
:1. Liquid samples (bio-oils) were diluted in acetone.
Solid-state 13C CP/MAS NMR spectra were acquired using a 400 MHz Bruker Avance III HD spectrometer equipped with a Bruker 4 mm H(F)/X/Y triple-resonance probe and a 9.4 T Ascend® superconducting magnet. The CP experiments employed a 1 ms linearly-ramped contact pulse, spinning rates of 10000 ± 2 Hz, optimized recycle delay of 3 seconds, spinal-64 heteronuclear decoupling (at νrf = 85 kHz) and are a sum of 1024 co-added transients. The dipolar dephasing experiments added a rotor-synchronized 180 μs dephasing delay with a pi refocusing pulse to minimize baseline distortion. Chemical shifts are reported with respect to TMS, and were referenced using adamantane (29.5 ppm) as an external secondary reference.
2.4. Liquid–liquid fractionation of bio-oil
The organic phase bio-oil (10 g) from low-temperature MW-assisted pyrolysis of office paper was dissolved in ethyl acetate at a 1:1 weight ratio and transferred to a separating funnel containing an equivolume of aqueous sodium hydroxide solution (2 M). After shaking and settling of the two layers, the aqueous layer was set aside whilst the organic layer was further extracted with aqueous 2 M sodium hydroxide solution (5 × 100 mL). The organic layer was isolated, dried and the solvent removed in vacuo to give the ‘neutral’ fraction as a brown oil (1.9 g). The combined aqueous extract was acidified (1 M HCl), extracted with ethyl acetate (5 × 100 mL), dried (MgSO4) and the solvent removed in vacuo to afford the ‘acidic’ fraction as a red-brown oil (2.5 g).
2.5. Curing experiments
Bio-oil (70 mg) was applied to two shot-blasted aluminium plates (50 mm × 50 mm) which were then pressed together using light pressure (finger-strength), weighed down with a stainless steel block (350 g) to mimic constant pressure and cured at various temperatures: 120 °C, 140 °C, 160 °C and 180 °C, for 4 and 8 h.2.6. Mechanical strength tests
The tensile strength of the adhesive bond formed between the two aluminium plates post cure was measured using an Instron 3367 Dual Column System fitted with 3000 N capacity load cell, at a cross-head speed of 5 mm min−1. The results reported were the averages of four measurements.3. Results and discussion
3.1. Microwave assisted pyrolysis of milled office paper
The distribution of products as a result of low-temperature microwave-assisted pyrolysis of milled waste office paper is shown in Fig. 2. This distribution is typical of waste biomass such as wheat straw.29 The yields for the organic and aqueous phase bio-oils are 19% and 23%, respectively. This microwave-assisted pyrolysis process also yields a black char-like material with yield up to 43%. As the gas fraction was depleted from the microwave-assisted pyrolysis system by the vacuum pump, gas yield (15%) was calculated by difference.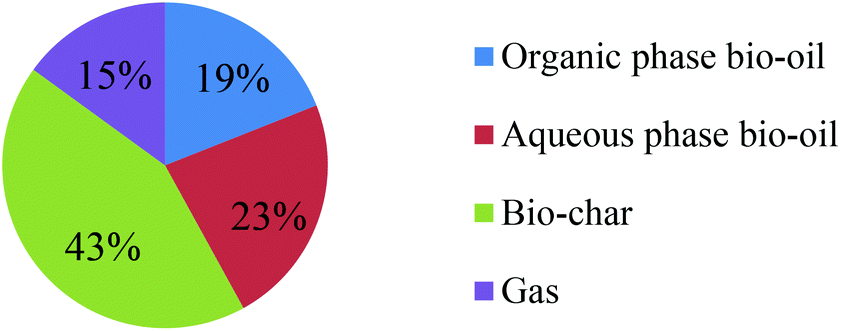 | ||
| Fig. 2 Yield of low temperature microwave-assisted pyrolysis products. | ||
The results for proximate and ultimate analysis and mineral elemental composition determined by ICP-MS, of the milled waste office paper and its low temperature MW-assisted pyrolysis products are summarized in Tables 1 and 2, respectively. As expected compared with the original milled waste paper (13.6 MJ kg−1) the resulting organic phase bio-oil has a higher calorific value (21.8 MJ kg−1), i.e., energy densified. The calorific value of the biochar (11.7 MJ kg−1) is lower than that of the original waste paper raw material (13.6 MJ kg−1), which is due to its elevated content of inorganic matter, namely calcium carbonate (CaCO3) and clays, commonly added during paper manufacturing.42 The ICP-MS data (Table 2) show very high levels of calcium in the biochar and the ATR-IR spectrum of the biochar (Fig. 3D) shows prominent absorbance bands for calcium carbonate. In addition, the bio-char has a higher ash (inorganic) content, as determined by TGA (33.8%), compared with that of milled office paper (12.8%). Also, a significant amount of silica/silicon was present in both waste office paper as well as its MW-pyrolysis products. The high silica/silicon content most likely originates from kaolinite (aluminosilicate), water-soluble silica-based sizing agents and potential organosilane sizing agents that are added during manufacturing of high-quality coated paper.
 | ||
| Fig. 3 ATR-IR spectra of (A) milled waste office paper, (B) organic phase bio-oil, (C) aqueous phase bio-oil and (D) bio-char. | ||
| Waste office paper | Organic phase bio-oil | Aqueous phase bio-oil | Bio-char | |
|---|---|---|---|---|
| a wt%. b N.D.: not detected. c Calculated by difference. d TOC: total organic carbon. e Determined by Karl Fisher titration. | ||||
| Proximate analysis (wt%) | ||||
| Moisture | 4.5 | — | — | 1.1 |
| Volatile matter | 71.9 | — | — | 44 |
| Fixed carbon | 10.8 | — | — | 21.1 |
| Ash | 12.8 | 1.2 | 8.3 | 33.8 |
| Ultimate analysis | ||||
| Ca | 34.56 | 49.88 | 16.89 | 38.89 |
| Ha | 4.30 | 5.84 | 4.22 | 4.0 |
| Na | N.D.b | N.D. | N.D. | N.D. |
| Sa | 0.09 | 0.04 | 0.03 | 0.17 |
| Oa,c | 48.25 | 44.24 | 78.86 | 23.14 |
| Heating value (MJ kg−1) | 13.6 | 21.8 | — | 11.7 |
| TOC (mg l−1)d | — | — | 180000 |
— |
| Water content (%)e | — | 2.6 | — | — |
| Element (ppma) | Milled office paper | Organic phase bio-oil | Aqueous phase bio-oil | Bio-char |
|---|---|---|---|---|
| a ppm refers to parts per million in mass. b N.D. = not detected. | ||||
| Na | 2463.83 | 194.33 | 123.36 | 3790.26 |
| Mg | 919.61 | 2.90 | 12.49 | 2414.23 |
| Al | 328.49 | 3.56 | 8.76 | 532.93 |
| Si | 27168.06 |
20668.25 |
23239.69 |
4956.24 |
| K | 1102.36 | 199.48 | 131.59 | 539.56 |
| Ca | 92877.53 |
712.27 | 411.83 | 243719.67 |
| Cr | 12.73 | 0.29 | 0.57 | 7.35 |
| Mn | 20.89 | N.D.b | N.D.b | 46.56 |
| Fe | 936.27 | 1.10 | 4.03 | 929.95 |
| Cu | 10.81 | 0.25 | 0.26 | 21.63 |
| Zn | 7.74 | 12.31 | N.D.b | 13.86 |
| As | 4.64 | 6.01 | 2.51 | 11.32 |
| Nb | 0.45 | 0.04 | 0.52 | 0.05 |
| Pd | 0.31 | 0.02 | 0.11 | 0.63 |
| Sn | 1.13 | 18.84 | 1.69 | 16.53 |
| Ir | 0.16 | 0.03 | 0.05 | 0.04 |
| Pt | 0.05 | 0.03 | 0.05 | 0.06 |
| Au | 5.52 | 0.49 | 0.25 | 3.88 |
| Pb | 0.22 | 0.02 | 0.07 | 1.17 |
3.2. ATR-IR analysis of pyrolysis products
The ATR-IR spectral analysis of the virgin milled office paper and subsequent pyrolysis products (except for gas) are shown in Fig. 3. As can be seen in the spectrum of milled waste office paper (Fig. 3A), the broad absorbance band between 3600 cm−1 and 3200 cm−1 is attributed to the O–H stretching vibration associated with cellulosic matter and sugars. Bands between 3000 cm−1 and 2800 cm−1 originated by C–H stretching vibrations associated with saturated or aliphatic structures. A very weak but slightly broad absorption band centred at 1630 cm−1 is due to the O–H deformation most likely from residual water or cellulosic matter. The very broad, strong band ranging from 1500 cm−1 to 1250 cm−1 is a complex mixture of many absorbances: the C–H deformation (1370 cm−1); the C–OH stretch (1315 cm−1); and the C–O–C stretch (1245 cm−1), but is mainly due to CaCO3 vibrations seen centred at 1415 cm−1.43 The C–O–C vibrations are further evidenced at 1160 cm−1 and 1100 cm−1. The sharp band at 870 cm−1 also evidences the presence of CaCO3, the content of which can be up to 8% in office paper.42,43 The band at 870 cm−1 may also correspond to torsional vibrations of methylene groups.44A vast number of structural changes during the pyrolysis process can be seen from the spectra of the two fractions of bio-oil. From the spectrum of the organic phase bio-oil (Fig. 3B), the most significant change with respect to milled waste office paper (Fig. 3A) is the appearance of a medium intensity band centred at 1715 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, indicating the presence of carbonyls. Also, the O–H stretching vibration drifts upward to a higher wavenumber (3400 cm−1) when compared with that of the raw material (3320 cm−1), indicating a reduction in hydrogen bonding. The absorption band at 1623 cm−1 is due to the CC stretching of alkene or aromatic compounds, but may also be associated with residual water. Fig. 3C shows the IR spectrum of the aqueous phase bio-oil. The intense, broad O–H stretching vibration band between 3700 cm−1 and 3000 cm−1, together with the intense carbonyl stretching vibration band (centred at 1715 cm−1), suggests the presence of carboxylic acids. The band at 1640 cm−1 and the broad O–H stretching band confirm the presence of water. The aqueous phase bio-oil may also contain trace amounts of sugars and other compounds from the various bands in the region from 1500 cm−1 to 800 cm−1.
O stretching vibration, indicating the presence of carbonyls. Also, the O–H stretching vibration drifts upward to a higher wavenumber (3400 cm−1) when compared with that of the raw material (3320 cm−1), indicating a reduction in hydrogen bonding. The absorption band at 1623 cm−1 is due to the CC stretching of alkene or aromatic compounds, but may also be associated with residual water. Fig. 3C shows the IR spectrum of the aqueous phase bio-oil. The intense, broad O–H stretching vibration band between 3700 cm−1 and 3000 cm−1, together with the intense carbonyl stretching vibration band (centred at 1715 cm−1), suggests the presence of carboxylic acids. The band at 1640 cm−1 and the broad O–H stretching band confirm the presence of water. The aqueous phase bio-oil may also contain trace amounts of sugars and other compounds from the various bands in the region from 1500 cm−1 to 800 cm−1.
Compared with the spectrum of milled waste office paper, significant changes can be observed in the spectrum of bio-char (Fig. 3D). The broad O–H stretching band between 3500 cm−1 and 3200 cm−1 has almost disappeared, indicating that the oxygen in the parent material was removed during the pyrolysis process and any acidic structures were cracked to leave a carbonaceous solid product. The intense band at 1410 cm−1 and the narrow band at 870 cm−1 and 710 cm−1 were assigned to CaCO3. As carbon is formed during the pyrolysis process, once the char gets dark in colour, it acts as a black body and absorbs radiation over all frequencies, making most bands appear relatively weak. The presence of remaining organics is still evidenced by bands at 1700 cm−1, 1580 cm−1 and 1035 cm−1.
3.3. 13C NMR characterization of bio-oils
In order to obtain more structural information, both the organic and aqueous phase bio-oils were investigated by 13C NMR spectroscopy. Fig. 4 shows the 13C NMR spectra of the organic (Fig. 4A) and aqueous phase bio-oil (Fig. 4B). Due to the complex nature of bio-oil, the carbon assignments of bio-oils were broadly categorized into several groups according to chemical shifts, as summarized in Table 3. | ||
| Fig. 4 13C NMR spectra of (A) organic phase bio-oil, (B) aqueous phase bio-oil. (All spectra used the central resonance of DMSO-d6 (δC, 39.52 ppm) as the internal reference.) *Possible artefacts. | ||
| Chemical shift (ppm) | Carbon assignments |
|---|---|
| 0–28 | Short aliphatic structures |
| 28–55 | Long and branched aliphatic structures |
| 55–105 | Carbohydrate sugars, alcohols, ethers |
| 110–165 | Aromatics, olefins |
| 165–180 | Carboxylic acids, esters |
| 180–220 | Aldehydes, ketones |
The spectrum of organic phase bio-oil shows more signals and was of greater intensity than that of the aqueous phase bio-oil between 55 and 110 ppm, suggesting more carbohydrates and its derivatives (e.g., levoglucosan, levoglucosenone) were trapped in the organic phase bio-oil.
A similar trend was observed in the aromatic region (110–165 ppm), which implies the organic phase bio-oil contains more aromatic compounds than the aqueous phase bio-oil. Although the true identity has not been ascertained, possible compounds in this region could include 5-(hydroxymethyl)-2-furaldehyde (HMF) and phenolics. It is well documented in the literature that simple phenols are present in bio-oils obtained from pyrolysis of cellulose and hemicellulose.45,46 These relatively small amounts of phenolics may come either from gas phase polymerization of small molecular weight unsaturated species46 or from hydrothermolysis of HMF or other furan derivatives.47 Luijkx et al. studied the reaction pathway for hydrothermal transition of HMF to 1,2,4-trihydroxybenzene, which may also be present in the organic phase bio-oil.47 A small portion of signals in this region may also be attributed to phenolics derived from lignin pyrolysis, as lignin content in office paper is up to 6%, but this is yet to be fully ascertained.42 The presence of (hetero-) aromatics is also evidenced by the 1H NMR of organic phase bio-oil (see ESI, Fig. S1†).
Both the organic and aqueous phase bio-oils contain carbonyl carbons: carboxylic acids and esters (165–180 ppm); ketones and aldehydes (180–215 ppm). The types of carbonyl carbons in the organic phase bio-oil are more varied and complex than those of the aqueous phase bio-oil. Among these, levulinic acid (4-oxopentanoic acid) is a common acid derived from HMF during pyrolysis of biomass. Signals between 0 and 55 ppm are attributed to aliphatic structures within the bio-oil.
The compositions of pyrolysis products of milled waste office paper are similar to those obtained from pyrolysis of cellulose and hemicellulose. Some typical pyrolysis products, such as levoglucosan, levoglucosenone, HMF, and possibly 1,2,4-trihydroxybenzene, were identified by comparison of the 13C NMR chemical shifts of the organic phase bio-oil and commercial samples (see ESI, Table S1†).
3.4. GC-MS characterization of bio-oils
Gas chromatography-mass spectrometry (GC-MS) characterization was performed to identify the compounds obtained in the organic and aqueous phase bio-oil. Fig. 5 shows the GC traces of organic (Fig. 5A) and aqueous phase bio-oil (Fig. 5B). Due to the complex composition of bio-oils, many peaks were detected for both phases even though generally only 25% of bio-oil compounds can be observed by GC as most lignin and carbohydrate oligomers have insufficient volatility under the operating conditions of the instrument.10,48 As a result, complete identification of peaks is very difficult and only those separated products in considerable amounts were qualified. Tables 4 and 5 show the major identified compounds in the organic and aqueous phase bio-oil, respectively. | ||
| Fig. 5 GC-MS spectra of (A) organic and (B) aqueous phase bio-oils derived from MW-assisted pyrolysis of milled waste office paper. *Possible artefacts. | ||
| Peak number | Retention time (min) | Identified compound |
|---|---|---|
| a According to the NIST 2008 database. b Tentative assignment. | ||
| 1 | 3.13 | Acetic acid |
| 2 | 3.56 | Furfural |
| 3 | 4.15 | Tetrahydro-2,5-dimethoxyfuran |
| 4 | 4.57 | 2(5H)-Furanone |
| 5 | 4.85 | 1,2-Cyclopentanedione |
| 6 | 6.04 | 1-Hydroxy-2-pentanone |
| 7 | 6.58 | 3-Methyl-1,2-cyclopentanedione |
| 8 | 7.59 | Guaiacolb |
| 9 | 8.03 | Levoglucosenone |
| 10 | 8.12 | Benzenetriolb |
| 11 | 9.31 | 2-Furanmethanol |
| 12 | 9.76 | 1,2-Benzenediol (Catechol)b |
| 13 | 9.82 | 1,4:3,6-Dianhydro-π-D-glucopyranose |
| 14 | 10.09 | 3,4-Anhydro-D-galactosan |
| 15 | 10.34 | 3-Methylbenzaldehyde |
| 16 | 10.6 | 5-(Hydroxymethyl)-2-furaldehyde (HMF) |
| 17 | 12.96 | 5-Hydroxy-9-oxabicyclo[3.3.1]nonan-2-one |
| 18 | 14.7 | Levoglucosan |
| 19 | 23.3 | 4,4′-(1-Methylethylidene)bisphenol |
| Peak number | Retention time (min) | Identified compound |
|---|---|---|
| a According to the NIST 2008 database. | ||
| 1 | 3.13 | Acetic acid |
| 2 | 3.46 | Furfural |
| 3 | 4.09 | Tetrahydro-2,5-dimethoxyfuran |
| 4 | 4.49 | 2(5H)-Furanone |
| 5 | 4.76 | 1,2-Cyclopentanedione |
| 6 | 6.04 | 1-Hydroxy-2-pentanone |
| 7 | 6.46 | 3-Methyl-1,2-cyclopentanedione, |
| 8 | 6.88 | 1,2,3-Butanetriol |
| 9 | 7.05 | 2,2-Dimethyl-acetoacetic acid ethyl ester |
| 10 | 7.46 | 2,5-Dimethyl-4-hydroxy-3(2H)-furanone |
| 11 | 9.69 | 1,4:3,6-Dianhydro-π-D-glucopyranose |
| 12 | 12.95 | 5-Hydroxy-9-oxabicyclo[3.3.1]nonan-2-one |
The main difference between the organic phase bio-oil and the aqueous phase bio-oil is the absence of levoglucosan (peak marked ‘18’ in Fig. 5A) in the latter. The compounds identified in the organic phase bio-oil can be broadly categorized into several groups: carbohydrates and their derivatives such as 1,4:3,6-dianhydro-πD-glucopyranose, 3,4-anhydro-D-galactosan and levoglucosan; possible phenolic compounds such as guaiacol, benzenetriol and 1,2-benzenediol (catechol) but their true identity is still to be ascertained; furanic structures such as furfural, tetrahydro-2,5-dimethoxyfuran and 5-(hydroxymethyl)-2-furaldehyde (HMF). The organic phase bio-oil also contains acetic acid and 4,4′-(1-methylethylidene)bisphenol (bisphenol A) which is an additive in paper products.49 The aqueous phase bio-oil (Fig. 5B and Table 5) comprises a similar compositional profile in the range 3–10 min albeit with fewer components. Water soluble acids are noted together with polar small molecules.
3.5. Yield of fractionation of organic phase bio-oil
As the bio-oil is a complex mixture of sugar and its derivatives, (hetero-) aromatics, hydrocarbons and carbonyl compounds, to gain some understanding of the bonding behaviour of bio-oil, the organic phase bio-oil (10 g) was subjected to liquid–liquid fractionation, from which three different bio-oil fractions (i. “neutral” ethyl acetate fraction, ii. “acidic” ethyl acetate fraction, and iii. aqueous fraction) were obtained. As shown in Fig. 6, the yields for the “neutral” and “acidic” ethyl acetate fractions are 19% and 25%, respectively. For the aqueous fraction, as NaCl was generated during the acidification process, the yield for this fraction (56%) was calculated by difference. | ||
| Fig. 6 Yield of liquid–liquid separation of organic phase bio-oil. | ||
3.6. 13C NMR analysis of different bio-oil fractions
Fig. 7 illustrates the 13C NMR spectra of “neutral”, “acidic” ethyl acetate fraction and aqueous fraction bio-oil.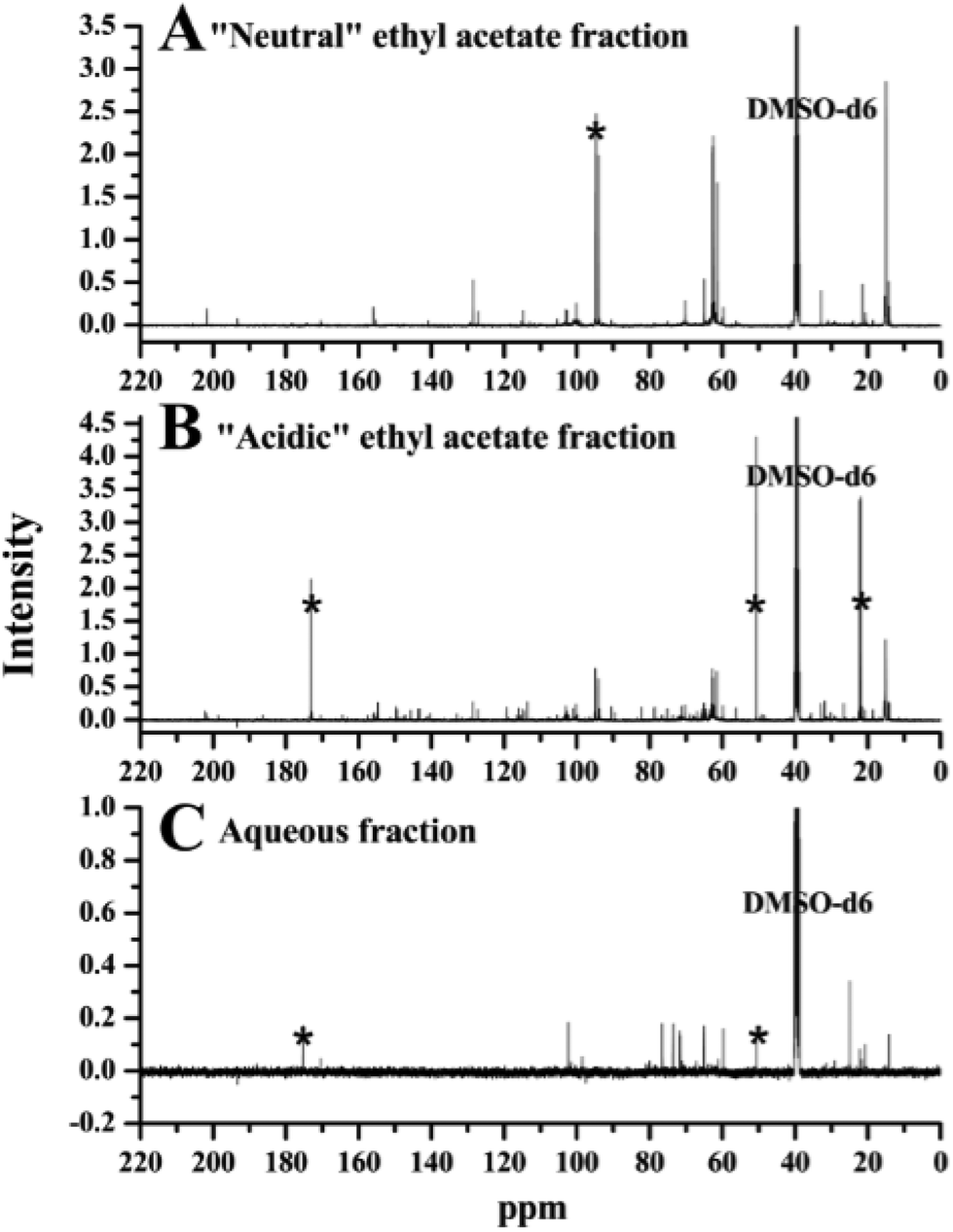 | ||
| Fig. 7 13C NMR spectra of (A) “neutral” ethyl acetate fraction, (B) “acidic” ethyl acetate fraction and (C) aqueous fraction bio-oil. (All spectra used the central resonance of DMSO-d6 (δC, 39.52 ppm) as the internal reference.) *Possible artefacts. | ||
Due to the complex chemical composition of the organic phase bio-oil, it is very hard to achieve complete separation. Basically, a relatively large percentage of (hetero-) aromatic compounds were separated into the “acidic” ethyl acetate fraction, with many peaks detected in the (hetero-) aromatic portion of the spectrum of “acidic” ethyl acetate fraction bio-oil, ranging from 110 to 165 ppm. The sharp signals (centred at 94 ppm, 62 ppm and 15 ppm) shown in the spectrum of “neutral” ethyl acetate fraction bio-oil (Fig. 7A) were also detected in the “acidic” ethyl acetate fraction bio-oil (Fig. 7B), but with lower intensities in the spectrum of the “acidic” ethyl acetate fraction. These peaks are possibly due to ethers with relatively short carbon chains. It is noteworthy that the spectrum of aqueous fraction bio-oil (Fig. 7C) is almost ‘clean’ in the aromatic portion (105–165 ppm), with most peaks detected between 55 and 105 ppm (mostly carbohydrate sugars).
3.7. GC-MS characterization of different bio-oil fractions from liquid–liquid separation
Fig. 8 shows the GC traces for the “neutral” (Fig. 8A), “acidic” ethyl acetate fraction (Fig. 8B) and aqueous fraction bio-oil (Fig. 8C) resulting from liquid–liquid separation of the organic phase bio-oil. | ||
| Fig. 8 GC-MS spectra of (A) “neutral”, (B) “acidic” ethyl acetate fraction and (C) aqueous fraction bio-oil resulted from liquid–liquid separation of organic phase bio-oil. *Possible artefacts. | ||
It is noteworthy that compared with the GC trace of aqueous fraction bio-oil (Fig. 8C), sugars were not detected in the two ethyl acetate fractions (Fig. 8A and 8B), suggesting most carbohydrate sugars were remaining in the aqueous fraction. Most furans and acidic compounds were separated into the “acidic” ethyl acetate fraction; while some furans and acidic compounds were also detected in the “neutral” ethyl acetate fraction.
3.8. Mechanical strength tests and bonding mechanism analysis
Fig. 9 shows the average tensile strengths obtained with the organic phase bio-oil cured shot-blasted aluminium plates, oven cured at 120 °C, 140 °C, 160 °C and 180 °C for 4 and 8 h. In general, the bio-oil adhesive could bond the two Al plates with acceptable bond strengths at all curing temperatures and time periods. The maximum tensile strengths were obtained when cured at 140 °C and 160 °C, reaching around 2300 N (160 °C, 8 h). In contrast, the minimum tensile strengths (around 900 N) were obtained when cured at 120 °C for 4 h. Also, the longer the curing time, the higher the Al–bio-oil–Al bond tensile strengths. This effect is more significant when cured at lower temperatures. For example, when cured at 120 °C, the tensile strength increased by ∼50% to 1360 N (for 8 h cure) from around 900 N (for 4 h cure). However, at higher temperatures, the increase of tensile strength was only around 15%.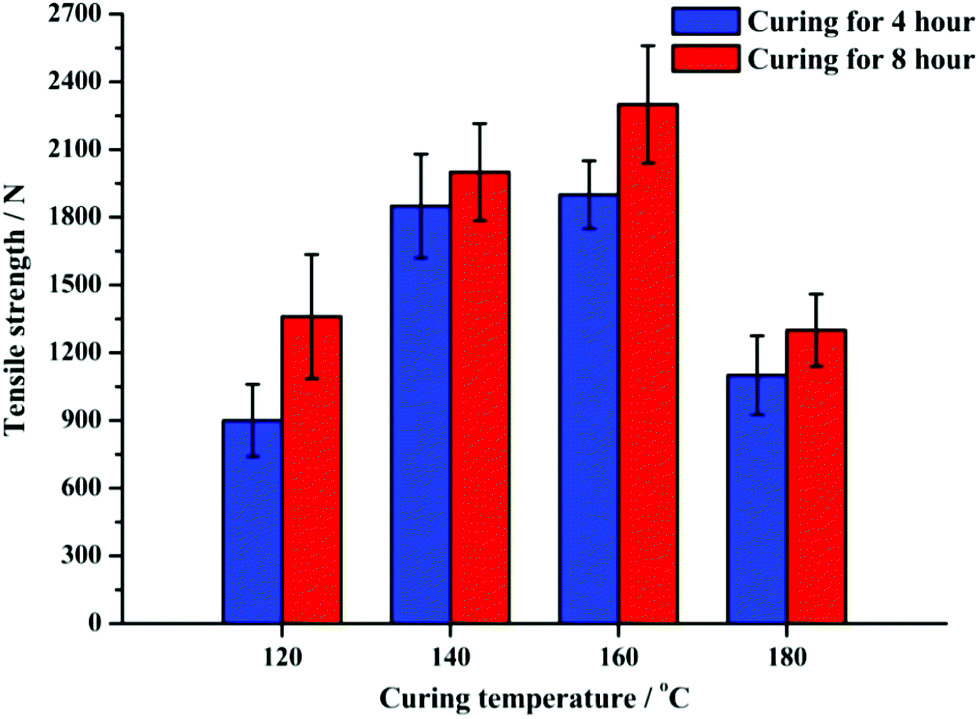 | ||
| Fig. 9 Tensile strengths of organic phase bio-oil cured shot-blasted Al plates. | ||
To get more structural information, the ATR-IR spectra of the organic phase bio-oil and scrapings from the organic/Al interface were compared (Fig. 10). Many significant changes of functional groups occurred before and after curing of bio-oil. Compared with the original organic phase bio-oil, the cured bio-oil polymer is characterized by a lower CO:CC ratio, suggesting polymerization occurred in carbonyl groups. Also, the CC stretching frequency at 1617 cm−1 is characteristic of olefinic double bonds in five membered rings. The intensity of this peak increased after curing of the organic phase bio-oil on the aluminium surface, suggesting the formation of conjugated/aromatic structures. This effect is also evidenced by a significant number of changes between 800 cm−1 and 900 cm−1, which represents the C–H out of plane bending vibrations in substituted alkenes and aromatics. This may be caused by changes in the substitution patterns of such molecules during polymerization.
 | ||
| Fig. 10 ATR-IR spectra of organic phase bio-oil, scrapings of the Al–bio-oil–Al interface polymer formed at 160 °C for 4 and 8 h cure. | ||
Another significant change is observed in the C–O stretching region (900 cm−1–1200 cm−1): the intensity of this band dramatically decreased, and many subtle changes occurred after curing. However, for bands centred at 1443 cm−1, 1184 cm−1, 1138 cm−1, 1075 cm−1, 1012 cm−1, 918 cm−1, 890 cm−1 and 856 cm−1, they are either remaining or becoming more obvious in the spectra of scrapings. These bands are characteristic absorptions for levoglucosan, which is a common sugar derivative during pyrolysis of biomass. So it is possible that carbohydrate sugars were not significantly involved in the polymerization reactions during curing. This hypothesis is further investigated in the following discussions.
The scrapings from the bio-oil–Al interface (sample cured at 160 °C for 8 h, under which condition the maximum tensile strength was achieved) were further characterized with CP/MAS solid-state 13C NMR. Also, a solid-state 13C CP/MAS NMR with a dipolar dephasing spectrum was recorded on this scraping to allow better assignment of resonance bands. In the latter case, immobile carbons with directly attached hydrogens were significantly attenuated, so the spectrum will largely consists of the quaternary carbons and mobile carbons such as methyl groups. The spectra are shown in Fig. 11 and overlapped with the liquid-state 13C NMR of the organic phase bio-oil. From the 13C CP/MAS NMR spectrum, the broad intense band between 50 ppm and 100 ppm is assigned to sugar carbons, which was almost not observed in the 13C NMR spectrum with dipolar dephasing. The various resonance bands between 105 ppm and 165 ppm verify the presence of complicated aromatic structures. The resonance bands between 140 ppm and 160 ppm are attributed to quaternary aromatic carbons, which still exists in the dipolar dephasing 13C spectrum. Bands between 105 ppm and around 140 ppm are aromatic carbons with one hydrogen atom attached, so the intensities of these bands decreased in the 13C dipolar dephasing spectrum. The band between 100 ppm and 105 ppm in the 13C NMR spectrum is a characteristic resonance band for anomeric carbons in carbohydrate compounds. The scrapings also contain various carbonyl structures as proved by bands centered at 210 ppm and 175 ppm. In both cases the bands still remain in the spectrum of 13C NMR with dipolar dephasing, suggesting that they are mainly ketone (210 ppm) and ester (175) structures. Alkyl groups are also present in the scrapings: methyl groups (0–20 ppm) and CH2 structures (20 ppm–50 ppm). The fact that there is no significant change seen in the spectra of the post-cure scrapings is indicative of little or no decomposition of the main structural units, i.e., the aromatic and sugar moieties largely remain intact, thus undergoing possible homo- and cross-coupling polymerisation reactions that help adhesion.
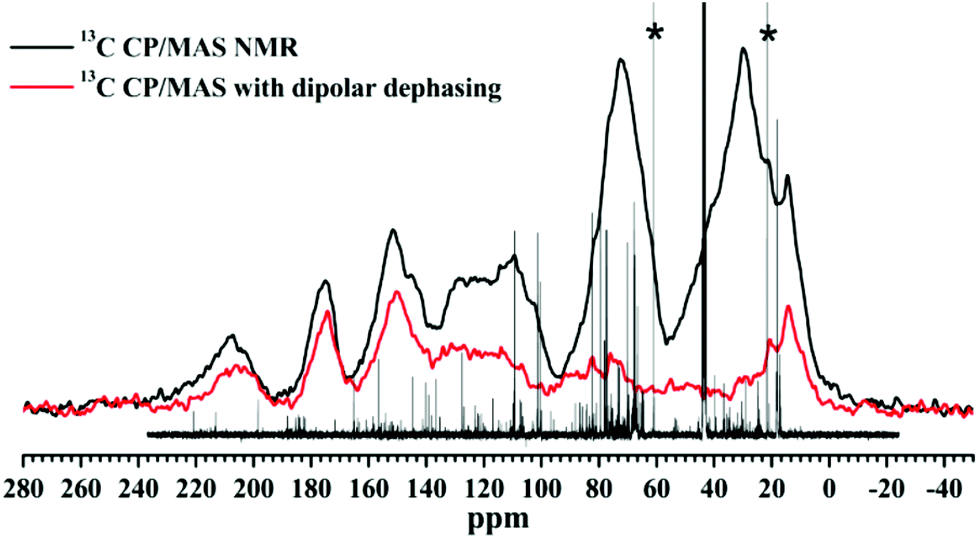 | ||
| Fig. 11 Solid-state 13C CP/MAS NMR spectrum of bio-oil/Al scraping (black) and 13C CP/MAS NMR spectrum of bio-oil/Al scraping with dipolar dephasing (red), overlapped with the liquid-state 13C NMR of organic phase bio-oil. *Possible artefacts. | ||
The tensile strengths of the organic phase bio-oil, “neutral” and “acidic” ethyl acetate fraction bio-oil, the aqueous fraction solids, and the mixture of the two ethyl acetate fraction bio-oil cured Al plates are shown in Fig. 12 (specimens cured with 70 mg bio-oil at 160 °C for 4 h). The tensile strengths obtained with different fractions of bio-oil are all lower than that obtained with organic phase bio-oil. The tensile strength of the Al-“acidic” ethyl acetate fraction bio-oil–Al bond was about 1260 N, while the “neutral” ethyl acetate fraction could also bond the two Al plates but with very weak tensile strength (300 N). For the solids obtained from the aqueous fraction (water removed), they cannot bond the Al plates at all. As these solids are probably composed of carbohydrates mainly, it is obvious that the sugars by themselves could not bond substrates. An interesting observation is that when the “neutral” and “acidic” ethyl acetate fraction bio-oils were mixed by a weight ratio of 1:1.25 (the same ratio as in the organic phase bio-oil), the tensile strengths of this mixture bonded Al plates (1460 N) were higher than those when bonded separately, but lower than that of organic phase bio-oil (1900 N). So it is possible that there is a co-operative or synergistic effect between the compounds from “neutral” and “acidic” ethyl acetate fraction and those from aqueous fraction that is responsible for the bonding properties of bio-oil, with the aromatic (mainly furanic) and acidic compounds mainly from “acidic” ethyl acetate fraction contributing more towards bonding. Bonding may be similar to phenol–formaldehyde cross-linking and this is being further investigated with a model compound study using catechol, HMF and levoglucosan, three components which have been tentatively identified in our pyrolysis products.
 | ||
| Fig. 12 Tensile strengths of organic phase bio-oil, “neutral” and “acidic” ethyl acetate and aqueous fraction bio-oil, and a mixture of the “neutral” and “acidic” ethyl acetate fraction bio-oil cured Al plates (samples cured at 160 °C for 4 h). | ||
This effect is further proved by comparing the ATR-IR spectra of the “neutral” and “acidic” ethyl acetate fraction bio-oil with the scrapings of formed polymers after curing (Fig. 13). The polymer of the cured “neutral” ethyl acetate fraction bio-oil shows little structural changes (Fig. 13A), while the polymer of cured “acidic” ethyl acetate fraction bio-oil shows dramatically significant changes after curing (Fig. 13B). Similar to the organic phase bio-oil, the intensity of the carbonyl peak at 1710 cm−1 decreased dramatically due to polymerization on the carbonyl bonds. The intensity of the CC stretching vibration band increased dramatically, and this stretching vibration band slightly shifts to a lower wavenumber (1625 cm−1 to 1617 cm−1). Also, significant structural changes and formation of new chemical bonds can be observed between 1630 cm−1 and 1450 cm−1 , which is characteristic of skeletal vibrations of aromatic rings, suggesting the formation of new aromatic structures during curing. The formation of the significant shoulder peak between 1670 cm−1 and 1530 cm−1, together with the dramatic intensity increase of the CC stretching band at 1617 cm−1, probably implies the formation of conjugated aromatic structures during curing. This is also confirmed by the changes in the alkene/aromatic C–H out of the plane bending region (800 cm−1–900 cm−1). For the “acidic” ethyl acetate fraction bio-oil, after curing between Al plates, the O–H stretching drifts to higher wavenumbers, and the CO stretching peak at 1710 cm−1 slightly drifts to lower wavenumbers, suggesting a reduction of hydrogen bonding.
 | ||
| Fig. 13 ATR-IR spectra of (A) “neutral” ethyl acetate fraction bio-oil and scrapings of cured polymer of this fraction (B) “acidic” ethyl acetate fraction bio-oil and scrapings of cured polymer of this fraction (cured at 160 °C for 4 h). | ||
4. Conclusions
Waste office papers (printed or photocopied) were converted to bio-oil, bio-char and gas products via a microwave-assisted low temperature (<200 °C) fast pyrolysis process. The total liquid yield is about 42%, with 19% for the organic phase and 23% for the aqueous phase bio-oil.The potential application of the organic phase bio-oil as an adhesive for metal-to-metal bonding has been studied. Also, the effects of curing temperatures (120 °C, 140 °C, 160 °C and 180 °C) and curing time (4 and 8 h) have been investigated. A small amount of organic phase bio-oil (70 mg) could glue two aluminium plates with acceptable strengths (>900 N) under all these curing conditions. The highest tensile strengths around 2300 N were obtained when cured at 160 °C for 8 h. Also, the tensile strengths increase with curing time.
To gain an in-depth understanding of the adhesive properties of bio-oil, a liquid–liquid fractionation of the organic phase bio-oil was conducted. Most (hetero-) aromatics were separated into the “acidic” ethyl acetate fraction, while the aqueous fraction mainly contains carbohydrate sugars. From the bonding experiments with different bio-oil fractions, it is revealed that there is a co-operative or synergistic effect between various compounds within the bio-oil. However, the aromatic compounds are significant for good adhesion.
Acknowledgements
Z. Zhang would like to acknowledge the Department of Chemistry, University of York, for a Wild Fund scholarship for PhD study. P. Shuttleworth gratefully acknowledges the Ministerio de Ciencia e Innovación for the concession of a Juan de la Cierva (JCI-2011-10836) contract. We would like to acknowledge Dr. Pedro M. Aguiar and Dr. Helen Parker at Chemistry Department, University of York, for their kind assistance with solid-state 13C CP/MAS NMR and ICP-MS analysis, respectively.Notes and references
- European Recovered Paper Council, Paper Recycling Monitoring Report 2012. http://www.paperforrecycling.eu/uploads/Modules/Publications/WEB_lowres_Monitoring%20report%202012.pdf. Last accessed 17/07/2014.
- H. Merrild, A. Damgaard and T. H. Christensen, Resour., Conserv. Recycl., 2008, 52, 1391–1398 CrossRef PubMed.
- A. Z. Shi, L. P. Koh and H. T. W. Tan, GCB Bioenergy, 2009, 1, 317–320 CrossRef CAS PubMed.
- Y. Ikeda, E. Y. Park and N. Okuda, Bioresour. Technol., 2006, 97, 1030–1035 CrossRef CAS PubMed.
- E. Y. Park, P. N. Anh and N. Okuda, Bioresour. Technol., 2004, 93, 77–83 CrossRef CAS PubMed.
- G. Mtui and Y. Nakamura, Biodegradation, 2005, 16, 493–499 CrossRef CAS PubMed.
- M. Alcalde, M. Ferrer, F. J. Plou and A. Ballesteros, Trends Biotechnol., 2006, 24, 281–287 CrossRef CAS PubMed.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- A. V. Bridgwater and G. V. C. Peacocke, RenewableSustainable Energy Rev., 2000, 4, 1–73 CrossRef CAS.
- A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30, 1479–1493 CrossRef CAS.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- M. Jahirul, M. Rasul, A. Chowdhury and N. Ashwath, Energies, 2012, 5, 4952–5001 CrossRef CAS PubMed.
- S. S. Lam and H. A. Chase, Energies, 2012, 5, 4209–4232 CrossRef CAS PubMed.
- M. F. Demirbas and M. Balat, Energy Convers. Manage., 2006, 47, 2371–2381 CrossRef CAS PubMed.
- F. C. Borges, Z. Du, Q. Xie, J. O. Trierweiler, Y. Cheng, Y. Wan, Y. Liu, R. Zhu, X. Lin, P. Chen and R. Ruan, Bioresour. Technol., 2014, 156, 267–274 CrossRef CAS PubMed.
- D. E. Clark and W. H. Sutton, Ann. Rev. Mater. Sci., 1996, 26, 299–331 CrossRef CAS.
- R. Luque, J. A. Menendez, A. Arenillas and J. Cot, Energy Environ. Sci., 2012, 5, 5481–5488 CAS.
- C. Yin, Bioresour. Technol., 2012, 120, 273–284 CrossRef PubMed.
- J. A. Menéndez, M. Inguanzo and J. J. Pis, Water Res., 2002, 36, 3261–3264 CrossRef.
- A. Domínguez, J. A. Menéndez, M. Inguanzo and J. J. Pis, Fuel Process. Technol., 2005, 86, 1007–1020 CrossRef PubMed.
- A. Domínguez, J. A. Menéndez, M. Inguanzo, P. L. Bernad and J. J. Pis, J. Chromatogr., A, 2003, 1012, 193–206 CrossRef.
- N. Ferrera-Lorenzo, E. Fuente, J. M. Bermúdez, I. Suárez-Ruiz and B. Ruiz, Bioresour. Technol., 2014, 151, 199–206 CrossRef CAS PubMed.
- D. Beneroso, J. M. Bermúdez, A. Arenillas and J. A. Menéndez, Bioresour. Technol., 2013, 144, 240–246 CrossRef CAS PubMed.
- F. C. Borges, Q. Xie, M. Min, L. A. R. Muniz, M. Farenzena, J. O. Trierweiler, P. Chen and R. Ruan, Bioresour. Technol., 2014, 166, 518–526 CrossRef CAS PubMed.
- V. L. Budarin, Y. Zhao, M. J. Gronnow, P. S. Shuttleworth, S. W. Breeden, D. J. Macquarrie and J. H. Clark, Green Chem., 2011, 13, 2330–2333 RSC.
- A. Domínguez, J. A. Menéndez, Y. Fernández, J. J. Pis, J. M. V. Nabais, P. J. M. Carrott and M. M. L. R. Carrott, J. Anal. Appl. Pyrolysis, 2007, 79, 128–135 CrossRef PubMed.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth, S. W. Breeden, A. J. Wilson, D. J. Macquarrie, K. Milkowski, J. Jones, T. Bridgeman and A. Ross, Bioresour. Technol., 2009, 100, 6064–6068 CrossRef CAS PubMed.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth and D. J. Macquarrie, Bioresour. Technol., 2010, 101, 3776–3779 CrossRef CAS PubMed.
- A. Al Shra'ah and R. Helleur, J. Anal. Appl. Pyrolysis, 2014, 105, 91–99 CrossRef PubMed.
- T. J. Appleton, R. I. Colder, S. W. Kingman, I. S. Lowndes and A. G. Read, Appl. Energy, 2005, 81, 85–113 CrossRef CAS PubMed.
- D. Beneroso, J. M. Bermúdez, A. Arenillas and J. A. Menéndez, Fuel, 2014, 132, 20–26 CrossRef CAS PubMed.
- D. Beneroso, J. M. Bermúdez, A. Arenillas and J. A. Menéndez, J. Anal. Appl. Pyrolysis, 2014, 105, 234–240 CrossRef CAS PubMed.
- A. Undri, L. Rosi, M. Frediani and P. Frediani, Fuel, 2014, 116, 662–671 CrossRef CAS PubMed.
- S. S. Lam, A. D. Russell, C. L. Lee, S. K. Lam and H. A. Chase, Int. J. Hydrogen Energy, 2012, 37, 5011–5021 CrossRef CAS PubMed.
- H. B. Goyal, D. Seal and R. C. Saxena, Renewable Sustainable Energy Rev., 2008, 12, 504–517 CrossRef CAS PubMed.
- S. Xiu and A. Shahbazi, RenewableSustainable Energy Rev., 2012, 16, 4406–4414 CrossRef CAS PubMed.
- S. Tsiantzi and E. Athanassiadou, PyNe Newsl., 2000, 10, 10–11 Search PubMed.
- S. Cheng, Z. Yuan, M. Anderson, M. Leitch and C. Xu, J. Appl. Polym. Sci., 2012, 126, E431–E441 CrossRef.
- C. J. Donahue and E. A. Rais, J. Chem. Educ., 2009, 86, 222 CrossRef CAS.
- L. Wang, R. Templer and R. J. Murphy, Appl. Energy, 2012, 99, 23–31 CrossRef CAS PubMed.
- A. Méndez, J. M. Fidalgo, F. Guerrero and G. Gascó, J. Anal. Appl. Pyrolysis, 2009, 86, 66–73 CrossRef PubMed.
- A. M. Prima, R. G. Zhbankov and R. Marupov, J. Struct. Chem., 1965, 5, 783–788 CrossRef.
- S. D. Stefanidis, K. G. Kalogiannis, E. F. Iliopoulou, C. M. Michailof, P. A. Pilavachi and A. A. Lappas, J. Anal. Appl. Pyrolysis, 2014, 105, 143–150 CrossRef CAS PubMed.
- R. J. Evans and T. A. Milne, Energy Fuels, 1987, 1, 123–137 CrossRef CAS.
- G. C. A. Luijkx, F. van Rantwijk and H. van Bekkum, Carbohydr. Res., 1993, 242, 131–139 CrossRef CAS.
- C. A. Mullen, G. D. Strahan and A. A. Boateng, Energy Fuels, 2009, 23, 2707–2718 CrossRef CAS.
- C. Liao and K. Kannan, Environ. Sci. Technol., 2011, 45, 9372–9379 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00768a |
| This journal is © The Royal Society of Chemistry 2015 |