
Cytoprotective effects of fisetin against hypoxia-induced cell death in PC12 cells†
Pei-Yi
Chen‡
a,
Yi-Ru
Ho‡
b,
Ming-Jiuan
Wu
c,
Shun-Ping
Huang
b,
Po-Kong
Chen
b,
Mi-Hsueh
Tai
b,
Chi-Tang
Ho
d and
Jui-Hung
Yen
*b
aCenter of Medical Genetics, Buddhist Tzu Chi General Hospital, Hualien 970, Taiwan. E-mail: pyc571@gmail.com
bDepartment of Molecular Biology and Human Genetics, Tzu Chi University, Hualien 970, Taiwan. E-mail: imyenjh@mail.tcu.edu.tw; Fax: +886-3-856-1422; Tel: +886-3-856-5301 ext 2683
cDepartment of Biotechnology, Chia Nan University of Pharmacy and Science, Tainan 717, Taiwan. E-mail: imwu@mail.chna.edu.tw
dDepartment of Food Science, Rutgers University, 65 Dudley Road, New Brunswick, NJ 08901-8520, USA. E-mail: ho@aesop.rutgers.edu
First published on 14th November 2014
Abstract
Fisetin (3,7,3′,4′-tetrahydroxyflavone), a flavonol compound of flavonoids, exhibits a broad spectrum of biological activities including anti-oxidant, anti-inflammatory, anti-cancer and neuroprotective effects. The aim of this study is to investigate the cytoprotective effect of fisetin and the underlying molecular mechanism against hypoxia-induced cell death in PC12 cells. The results of this study showed that fisetin significantly restored the cell viability of PC12 cells under both cobalt chloride (CoCl2)- and low oxygen-induced hypoxic conditions. Treatment with fisetin successfully reduced the CoCl2-mediated reactive oxygen species (ROS) production, which was accompanied by an increase in the cell viability of PC12 cells. Furthermore, we found that treatment of PC12 cells with fisetin markedly upregulated hypoxia-inducible factor 1α (HIF-1α), its nuclear accumulation and the hypoxia-response element (HRE)-driven transcriptional activation. The fisetin-mediated cytoprotection during CoCl2 exposure was significantly attenuated through the administration of HIF-1α siRNA. Moreover, we demonstrated that MAPK/ERK kinase 1/2 (MEK1/2), p38 MAPK and phosphatidylinositol 3-kinase (PI3 K) inhibitors significantly blocked the increase in cell survival that was induced by fisetin treatment under hypoxic conditions. Consistently, increased phosphorylation of ERK, p38 and Akt proteins was observed in PC12 cells treated with fisetin. However, the fisetin-induced HRE-driven transcription was not affected by inhibition of these kinase signaling pathways. Current results reveal for the first time that fisetin promotes cell survival and protects against hypoxia-induced cell death through ROS scavenging and the activation of HIF1α-, MAPK/ERK-, p38 MAPK- and PI3 K/Akt-dependent signaling pathways in PC12 cells.
1. Introduction
Hypoxia is a pathological state in which the oxygen concentration drops to a level insufficient to support normal metabolism. Neurons are the cells most sensitive to hypoxia, which has been reported to disturb brain function and can lead to a variety of neurological disorders such as motor dysfunction, learning disabilities, epilepsy, seizure and dementia.1–3 Evidence has implicated that prolonged hypoxia causes neuron necrosis and apoptosis in the brain.4 Hypoxia is involved in the development of neurodegenerative diseases. It facilitates the pathogenesis of Alzheimer's disease through Aβ accumulation and tau phosphorylation and by promoting neuron degeneration.5,6Cells have developed a programmed defense response against hypoxia.7 Hypoxia-inducible factor 1 (HIF-1) is a major transcription factor involved in the cell responses to hypoxic stress. HIF-1 consists of a hypoxia inducible subunit HIF-1α and a constitutively expressed subunit HIF-1β. In the presence of oxygen, HIF-1α binds pVHL (Von Hippel–Lindau tumor suppressor) through 2 hydroxylated proline residues. The binding of pVHL leads to the ubiquitination of HIF-1α, which targets it to the proteasome for degradation.8 In contrast, in response to hypoxia, proline hydroxylation is inhibited. pVHL is no longer able to bind and target HIF-1α for proteasomal degradation, which leads to HIF-1α accumulation and translocation to the nucleus. There, HIF-1α dimerizes with HIF-1β and specifically binds to the hypoxia response element (HRE) of the promoters of target genes involved in erythropoiesis, glycolysis, angiogenesis and cell survival.9–11 Furthermore, it has also been shown that HIF-1α plays an essential role in neuroprotection and maintaining the integrity of the brain under hypoxic and ischemic conditions in animal models.12
Flavonoids, the most common group of polyphenolic compounds in the human diet, are abundant in fruits and vegetables.13 They are believed to have beneficial effects against multiple diseases, including cancers, cardiovascular disease and inflammatory and neurodegenerative disorders.14,15 Previous studies have revealed that flavonoid compounds could cross the blood–brain barrier (BBB) and might possess a neuroprotection ability.16 Fisetin (3,7,3′,4′-tetrahydroxyflavone) (ESI Fig. 1†) belongs to the flavonol subgroup of flavonoids found in many vegetables and fruits and is especially rich in apples, strawberries, onions and mangoes. It exhibits antioxidant, anti-inflammatory and anti-carcinogenic activities.17–19 Recently, it was also considered to possess neuroprotective effects against the aging process, cerebral damage and neurodegenerative disorders.20,21 It has been shown to reduce excessive formation of the Aβ protein, which is associated with Alzheimer's disease.22 In addition to neuroprotection, fisetin could promote neuronal differentiation and ERK-dependent long-term potentiation, maintain cognitive function and enhance memory in an animal model.23,24 Taken together, these studies reveal that fisetin is a potential cytoprotective compound for neuronal cells. However, the cytoprotective effect of fisetin on neuronal cells exposed to hypoxic conditions is yet unknown.
It has been reported that cobalt chloride (CoCl2) treatment could mimic some of the hypoxic responses observed in cultured cells, including the generation of reactive oxygen species (ROS) and induction of transcriptional changes of genes, such as HIF-1α, erythropoietin and glycolytic enzymes. In addition, CoCl2-dependent modulation of gene products in hypoxic and DNA damage responses was also observed.25,26 Therefore, CoCl2 has been used as a reagent in both in vitro and in vivo to mimic hypoxic responses.27–29 The PC12 cell line is a rat pheochromocytoma cell line and is a useful model for studying cell survival, cell differentiation and oxygen-sensitive molecular and cellular mechanisms of neuronal cells.30,31 In the present study, we aim to investigate the cytoprotective effect and underlying mechanisms of fisetin against hypoxia-induced cell death in PC12 cells.
2. Materials and methods
2.1 Materials
Fisetin, cobalt chloride (CoCl2), RPMI-1640 medium, non-essential amino acid (NEAA), poly-L-lysine, dimethyl sulfoxide (DMSO), N-acetyl-L-cysteine (NAC), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and other chemicals were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) unless otherwise indicated. Inhibitors U0126, SB203580 and LY294002 were purchased from Promega (Madison, WI, USA). SP600125 was purchased from Enzo Life Sciences (Ann Arbor, MI, USA). The mouse 7S nerve growth factor (NGF) was purchased from Millipore (Billerica, MA, USA).2.2 Cell culture
The PC12 cells were obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). Cells were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated horse serum (HS) (Invitrogen, Carlsbad, CA, USA), 5% fetal bovine serum (FBS) (Biological Industries, Kibbutz Haemek, Israel) and 1% non-essential amino acid (NEAA). The cells were cultured in a 5% CO2 incubator at 37 °C. For experiments using a hypoxia incubator, the cells were cultured in 1% O2, 5% CO2 and 94% N2 at 37 °C. For analysis of differentiated PC12 cells, the cells were seeded on poly-L-lysine-coated 6-well plates and incubated in RPMI medium with 10% HS, 5% FBS and 1% NEAA for 24 h. The medium was replaced with fresh medium with the addition of nerve growth factor (NGF; 50 ng ml−1) for an additional 72 h incubation.2.3 Compound treatments and analysis of cell viability
The PC12 cells (1 × 106 per ml) were pre-treated with the indicated reagent or vehicle (0.1% DMSO) for 1 h before exposure to CoCl2. Cell viability was determined through the reduction of MTT to purple formazan crystals by mitochondria. The cells were incubated with a MTT solution (1 mg ml−1) for 3 h at 37 °C. The crystals were dissolved in DMSO, and the extent of the reduction of MTT was determined as the absorbance at 550 nm.32 For the N-acetyl-L-cysteine (NAC) treatment experiment, the cell viability was measured with a Calcein AM (Invitrogen) assay. PC12 cells were incubated with Calcein AM dye (1 μM) for 30 min at 37 °C. The fluorescence intensity was determined using 485 nm excitation and 530 nm emission.332.4 Analysis of reactive oxygen species (ROS) generation
PC12 cells (1 × 106 per ml) were seeded on poly-L-lysine-coated 6-well plates and incubated in RPMI medium (10% HS, 5% FBS and 1% NEAA) for 24 h. Then, the medium was replaced with fresh serum-free RPMI medium, H2DCFDA (5 μM) was added and the cells were incubated at 37 °C in the dark. After 30 min incubation, the PC12 cells were treated with fisetin (40 μM) for 1 h prior to CoCl2 (200 μM) exposure. After 1 h incubation, the medium was removed and the cells were harvested for ROS analysis. Three independent samples of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were analyzed via flow cytometry using the FL1 emission filter (FACScan, BD Biosciences, San Jose, CA, USA). The data were expressed as the relative percentage of the geometric mean fluorescence intensity.
000 cells were analyzed via flow cytometry using the FL1 emission filter (FACScan, BD Biosciences, San Jose, CA, USA). The data were expressed as the relative percentage of the geometric mean fluorescence intensity.
2.5 Reverse transcription quantitative PCR (RT-Q-PCR) analysis
PC12 cells (1 × 106 per ml) were seeded on 60 mm dishes in RPMI medium (10% HS, 5% FBS and 1% NEAA) and treated with the vehicle (0.1% DMSO) or the indicated compounds for the indicated periods. Total cellular RNA was prepared using the Total RNA Mini Kit (Geneaid, Taipei, Taiwan). The reverse transcription of RNA was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR was performed in a reaction mixture containing cDNA, specific primers [HIF-1α, 5′-CGAGCTGCCTCTTCGACAAG-3′ (forward) and 5′- CCCAGCCGCTGGAGCTA-3′ (reverse); β-actin, 5′-CCTCTGAACCCTAAGGCCAA-3′ (forward) and 5′-AGCCTGGATGGCTACGTACA-3′ (reverse)] and Maxima SYBR Green/ROX qPCR Master Mix (Fermentas, Burlington, CA). The amplification was performed on a Roche Light Cycler 480 sequence detection system. The PCR conditions used are as follows: 50 °C for 2 min, 94 °C for 4 min, 45 cycles at 94 °C for 60 s, 58 °C for 60 s and 72 °C for 60 s. The ΔΔCt method was used for the analysis of HIF-1α mRNA expression, estimated in triplicate samples and normalized to β-actin expression levels.2.6 Western blot analysis
PC12 cells (1 × 106 per ml) were seeded on 100 mm dishes in RPMI medium (10% HS, 5% FBS and 1% NEAA) and subsequently incubated with the indicated agents for the indicated time periods. Total cellular proteins were prepared using RIPA buffer (Thermo Fisher Scientific, Inc., Rockford, IL). For nuclear and cytosolic protein preparation, the cells were harvested using the NE-PER nuclear and cytoplasmic extraction reagent (Thermo Fisher Scientific) according to the manufacturer's instructions.The protein was separated on 10% SDS-PAGE and was subsequently transferred onto a PVDF membrane (PerkinElmer, Boston, MA, USA). The blots were incubated with the following specific antibodies: anti-p44/42 MAPK (Erk1/2), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), anti-p38, anti-phospho-p38 MAPK (Thr180/Tyr182), anti-Akt, anti-phospho-Akt (Ser-147) (Cell Signaling Technology, Inc.), anti-HIF-1α, anti-HDAC2 (GeneTex, Irvine, CA, USA) and monoclonal anti-β-actin (Sigma-Aldrich). The blots were incubated with the appropriate HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h. Proteins were detected using Western Lightning™ Chemiluminescence Reagent Plus (PerkinElmer), and the signal was visualized with Amersham Hyperfilm™ ECL (GE Healthcare, Buckinghamshire, UK).
2.7 Transfection and luciferase reporter assay
PC12 cells (1 × 106 per ml) were seeded on poly-L-lysine-coated 24-well plates in DMEM medium (10% HS and 5% FBS) for 24 h. The cells were transfected with the pGL4.42[Luc2P/HRE/Hygro] plasmid and the Renilla luciferase control vector (Promega) (plasmid ratio, 20:1) using Lipofectamine 2000 (Invitrogen). After 24 h transfection, the cells were treated with the indicated agents for an additional 24 h. The cells were harvested, and the luciferase activities were measured using the Dual-Luciferase Reporter Assay System Kit (Promega). The luciferase intensities were normalized to the activity of the Renilla luciferase.
2.8 Transfection of small interference RNA (siRNA)
PC12 cells (1 × 106 per ml) were seeded onto poly-L-lysine-coated 6-well plates in DMEM medium (10% HS and 5% FBS) for 24 h. Then, the cells were incubated with serum-free OPTI-MEM medium and transfected with the negative control (si-NC) or rat-specific HIF-1α siRNA (si-HIF-1α) duplexes [5′-GAAGGAACCUGAUGCUUUAACUU-3′ (forward) and 5′-AAGUUAAAGCAUCAGGUUCCUUC-3′ (reverse)]34 (GeneDireX Inc., Las Vegas, NV, USA) at a final concentration of 150 pmol ml−1 using Lipofectamine 2000 according to the manufacturer's instructions. After 24 h transfection, the cells were pre-treated with the vehicle or fisetin (40 μM) for 1 h and then exposed to CoCl2 (200 μM) for an additional 24 h. The cell viability was measured with the MTT assay.2.9 Statistical analysis
All experiments were repeated at least three times. All values are expressed as the mean ± SD. The results were analyzed using one-way ANOVA with a Dunnett's post-hoc test, and a p-value of <0.05 was considered statistically significant.3. Results
3.1 Fisetin protects PC12 cells from hypoxia-induced cell death
Cobalt chloride (CoCl2) is widely used as a reagent to mimic hypoxia in PC12 cells. To confirm the cytotoxic effect of CoCl2 on PC12 cells, the cells were treated with CoCl2 (0–250 μM) for 24 h and the cell viability was measured with the MTT assay. Fig. 1A and B confirm that 24 h CoCl2 (0–250 μM) treatment in PC12 cells caused a dose-dependent cytotoxicity. CoCl2 caused cell shrinkage and approximately 55% cell death at a concentration of 200 μM. To examine the protective effect of fisetin in CoCl2-treated PC12 cells, cells were treated with the vehicle (0.1% DMSO) or fisetin (0–60 μM) for 1 h before exposure to 200 μM CoCl2 for 24 h. Fig. 1C shows that fisetin (5–60 μM) significantly increased the cell viability in a dose-dependent manner up to 90.6 ± 8.9% (p < 0.01) from 51.3 ± 0.3% in the CoCl2-treated control group. We further tested whether fisetin could protect PC12 cells cultured in a hypoxia incubator (1% O2, 5% CO2, and 94% N2). The cell viability of the PC12 cells was significantly decreased after 48 h incubation in the hypoxia incubator (ESI Fig. 2†). When cells under hypoxic conditions were treated with fisetin (40 and 60 μM), the cell viability was markedly increased from 51.7 ± 1.0% to 65.0 ± 1.1% and 75.2 ± 3.1%, respectively (Fig. 1D). These data confirm that fisetin restores PC12 cell viability and protects against hypoxia-induced cell death.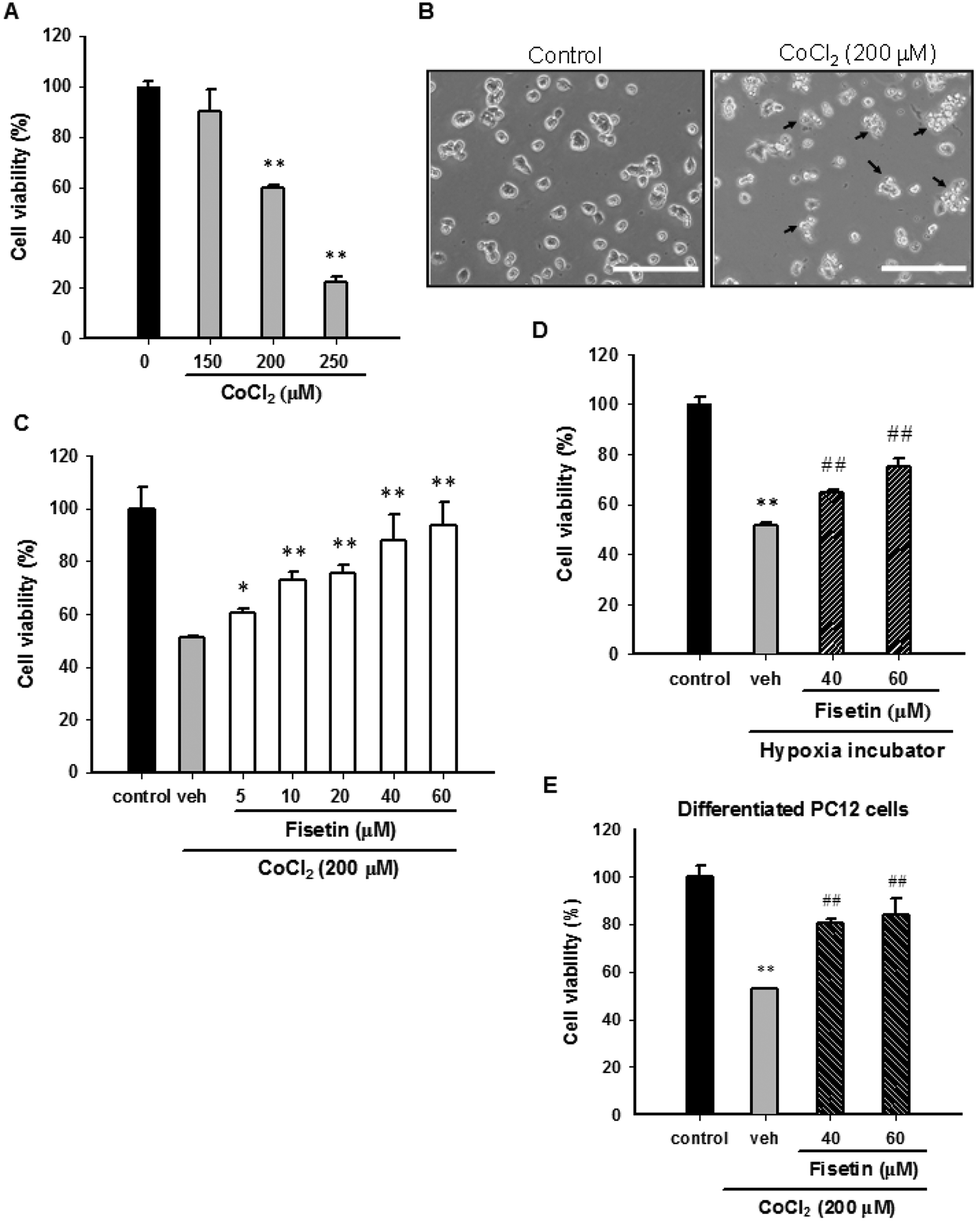 | ||
| Fig. 1 The effect of fisetin on the cell viability in CoCl2-treated PC12 cells. (A) PC12 cells were exposed to CoCl2 (0–250 μM) at 37 °C for 24 h, and the cell viability was measured with the MTT assay. The data represent the mean ± SD of three independent experiments. **p < 0.01 represents significant differences compared with the CoCl2-untreated group. (B) PC12 cells were cultured in poly-L-lysine coated plates and treated with 200 μM CoCl2 at 37 °C for 24 h. Cell morphology was observed using phase-contrast microscopy. Arrowheads indicate the dead cells. Scale bar, 200 μm. (C) PC12 cells were treated with the vehicle (0.1% DMSO) or fisetin (5–60 μM) for 1 h prior to CoCl2 (200 μM) treatment. After 24 h incubation, the cell viability was measured with the MTT assay. The data represent the mean ± SD from three independent experiments. *p < 0.05 and **p < 0.01 represent significant differences compared with the vehicle plus the CoCl2 group. (D) PC12 cells were treated with the vehicle or fisetin and incubated in a hypoxia incubator for 48 h. The cell viability was measured with the MTT assay. The data represent the mean ± SD of three independent experiments. **p < 0.01 represents significant differences compared with cells incubated in a normoxia incubator. ##p < 0.01 represents significant differences compared with the vehicle group. (E) PC12 cells were exposed to NGF (50 ng ml−1) for 72 h and allowed to differentiate. The cells were treated with the vehicle or fisetin for 1 h and then exposed to CoCl2 for an additional 24 h. The cell viability was measured with the MTT assay. The data represent the mean ± SD from three independent experiments. **p < 0.01 represents significant differences compared with the CoCl2-untreated control. ##p < 0.01 represents significant differences compared with the vehicle plus the CoCl2 control. | ||
It has been shown that CoCl2 also causes cytotoxicity in NGF-differentiated PC12 cells.35 We therefore investigated the effect of fisetin on differentiated PC12 cells exposed to CoCl2. The PC12 cells were differentiated by incubating with 50 ng ml−1 nerve growth factor (NGF) for 72 h and then treating with the vehicle (0.1% DMSO) or fisetin (40 and 60 μM) for 1 h followed by exposure to CoCl2 (200 μM) for an additional 24 h. As shown in Fig. 1E, cell viabilities were significantly increased by fisetin (40 and 60 μM) from 52.9 ± 0.2% to 80.4 ± 1.6% and 84.0 ± 6.9%, respectively. These results reveal that fisetin provides cytoprotection against CoCl2 toxicity in both undifferentiated and differentiated PC12 cells.
3.2 Fisetin reduces hypoxia-induced ROS generation and restores cell viability
It has been reported that CoCl2-induced PC12 cell death is mediated by reactive oxygen species (ROS) production.26,36 To examine whether fisetin rescues CoCl2-induced PC12 cell death through scavenging intracellular ROS, the PC12 cells were pre-treated with 40 μM fisetin or 10 mM NAC (as a ROS scavenger control) for 1 h, then 200 μM CoCl2 was added for an additional 1 h incubation, and intracellular ROS was determined as described in the Materials and methods section. Fig. 2A and B illustrate that treatment of PC12 cells with CoCl2 (200 μM) significantly increased the DCF fluorescence to 213.2 ± 7.1% compared to the CoCl2-untreated control. Pre-incubation of cells with fisetin or NAC markedly decreased CoCl2-induced intracellular ROS to 150.6 ± 12.2% or 129.5 ± 7.3% (p < 0.01), respectively. Their effects on cell viability were then analyzed with a Calcein AM assay, instead of the MTT assay because NAC is highly nucleophilic and reduces the MTT even before it enters the cells.37Fig. 2C shows that the cell viability of the cells treated with fisetin (40 μM) and NAC (10 mM) reached 80.1 ± 3.6% and 59.5 ± 7.4% compared with the vehicle (39.9 ± 4.1%) (p < 0.01), respectively. This result indicates that intracellular ROS scavenging is involved in the fisetin-mediated cytoprotection in CoCl2-treated PC12 cells.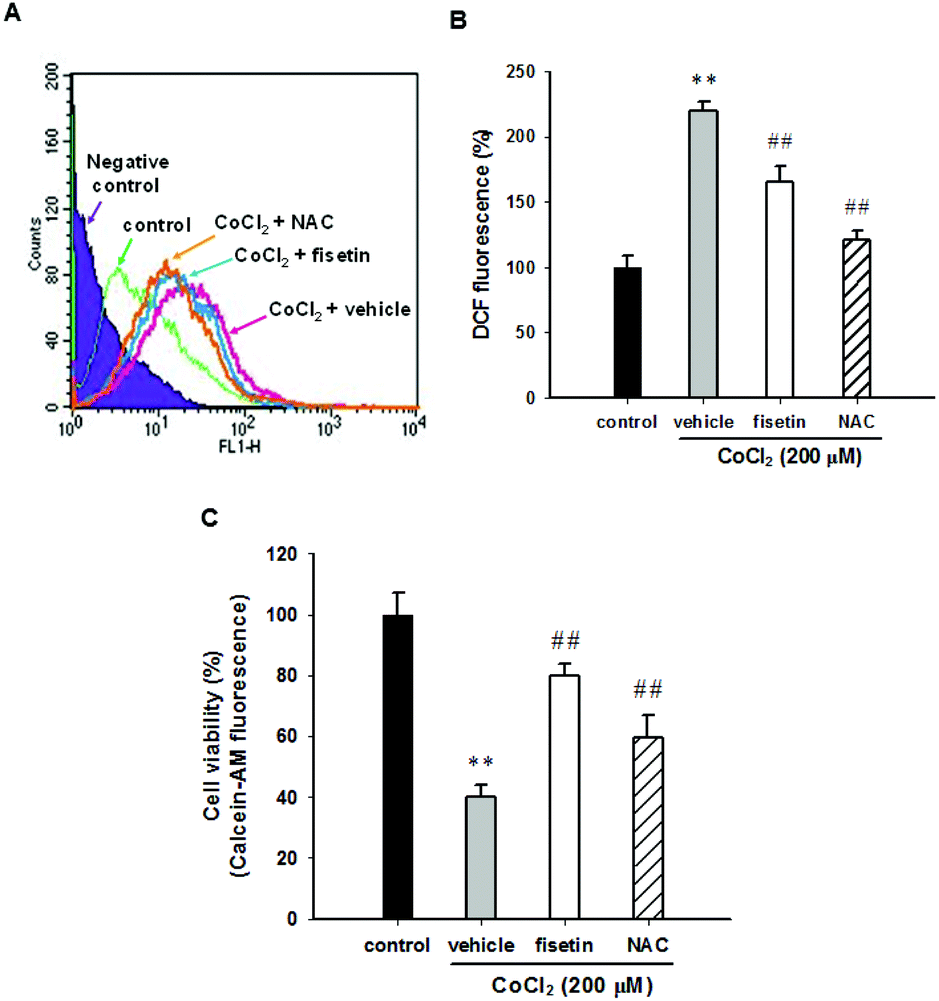 | ||
| Fig. 2 The effect of fisetin on the reactive oxygen species (ROS) production. PC12 cells were seeded in normal serum medium for 24 h, which was replaced with serum free RPMI medium and with the addition of 5 μM H2DCFDA for 30 min incubation. The cells were treated with fisetin (40 μM) or NAC (10 mM) for 1 h followed by exposure to CoCl2 (200 μM) for an additional 1 h. (A) The level of ROS was analyzed using flow cytometry. A representative histogram is shown. (B) The summary of the ROS levels. (C) The cell viability was measured with a Calcein AM assay. The data represent the mean ± SD from three independent experiments. **p < 0.01 represents significant differences compared with the CoCl2-untreated control group. ##p < 0.01 represents significant differences compared with the vehicle plus the CoCl2 group. | ||
3.3 The effect of fisetin on the expression of hypoxia-inducible factor 1α (HIF-1α) in PC12 cells
HIF-1α is a key transcription factor regulating the expression of genes that facilitate cell adaption to hypoxia. HIF-1α protein is stabilized and translocated to the nucleus during hypoxia. Fig. 3A shows that fisetin (40 μM) treatment increased the HIF-1α mRNA expression (approximately 1.5-fold) during 4–8 h compared with the 0 h group (p < 0.01) in PC12 cells. Western blot analysis revealed that the level of cytosolic HIF-1α protein was slightly increased during 2–4 h of fisetin treatment (Fig. 3B). The level of the nuclear HIF-1α protein was markedly elevated approximately 2.2- to 3.1-fold in fisetin-treated PC12 cells (during 4–8 h) (Fig. 3C).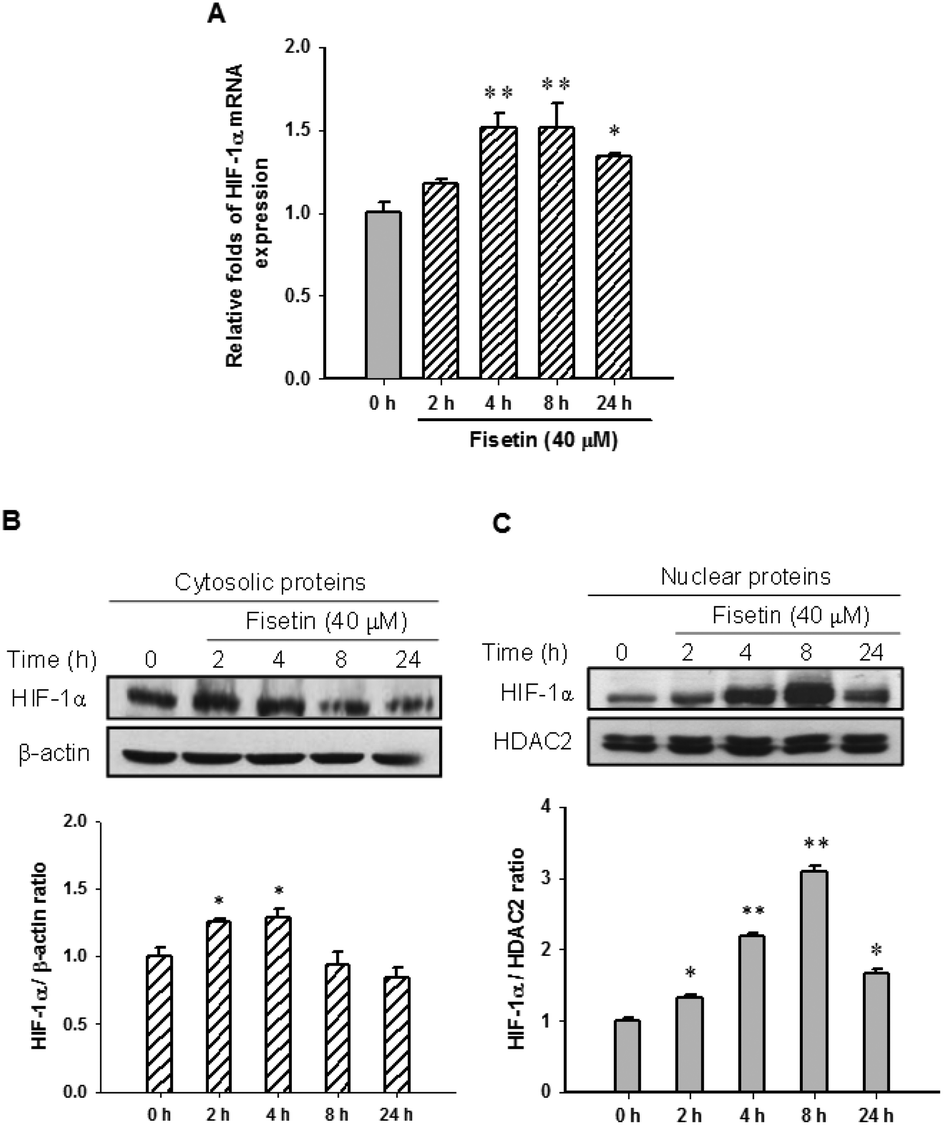 | ||
| Fig. 3 The effect of fisetin on HIF-1α expression and nuclear accumulation. The cells were treated with fisetin (40 μM) for 0–24 h. RNA and proteins were isolated. (A) HIF-1α mRNA expression was analyzed using RT-Q-PCR. The cytosolic HIF-1α and β-actin proteins (B) and the nuclear HIF-1α and HDAC2 proteins (C) were analyzed using western blot analysis. The experiments were replicated at least three times and a representative blot is shown. The normalized intensity of HIF-1α versus β-actin or HDAC2 is presented as the mean ± SD of three independent experiments. *p < 0.05 and **p < 0.01 represent significant differences compared with the 0 h group. | ||
Furthermore, we examined the effect of fisetin on the HIF-1α-mediated transcriptional activation. PC12 cells were co-transfected with hypoxia response element (HRE) reporter plasmids and Renilla control plasmids for 24 h, followed by treatment with fisetin (40 μM) for an additional 24 h. The intensity of luciferase activity was measured as described in the Materials and methods section. As shown in Fig. 4, fisetin significantly increased the HRE-mediated transcriptional activity by approximately 1.6-fold compared with the vehicle group (p < 0.01). While PC12 cells were co-treated with fisetin and CoCl2, the luciferase activity was also significantly elevated compared with the CoCl2 alone group (p < 0.01). These results suggest that fisetin treatment increases HIF-1α gene expression and promotes nuclear translocation of the HIF-1α protein and activation of HIF-1α-mediated transcription in PC12 cells.
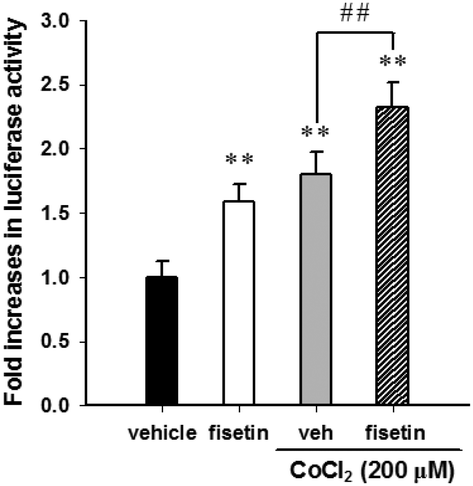 | ||
| Fig. 4 The effect of fisetin on the transactivation of the hypoxia response element (HRE). PC12 cells were transfected with the HRE reporter construct and Renilla luciferase control plasmid. The reporter plasmid-transfected cells were exposed to the vehicle or fisetin (40 μM) for 1 h, then treated with or without CoCl2 (200 μM) for an additional 24 h. The intensity of the luciferase activity was measured, and the data represent the mean ± SD from triplicated experiments. **p < 0.01 represents significant differences compared with the CoCl2-untreated vehicle group. ##p < 0.01 represents significant differences compared with the vehicle plus the CoCl2 group. | ||
The role of HIF-1α in fisetin-mediated cytoprotection in PC12 cells was further investigated using HIF-1α siRNA (si-HIF-1α). Fig. 5A shows that we have successfully achieved an approximately 48% knockdown of the nuclear HIF-1α protein in HIF-1α siRNA-treated cells. When the siRNA-transfected cells were further treated with the vehicle (0.1% DMSO) or fisetin (40 μM) for 1 h and then exposed to CoCl2 (200 μM) for another 24 h, the fisetin-mediated cytoprotection of PC12 cells was significantly attenuated in the HIF-1α knockdown cells (si-HIF-1α) compared with the siRNA negative control cells (si-NC) (p < 0.01) (Fig. 5B). These results suggest that the fisetin-induced nuclear HIF-1α accumulation is involved in the cytoprotective effect against hypoxia-induced cell death.
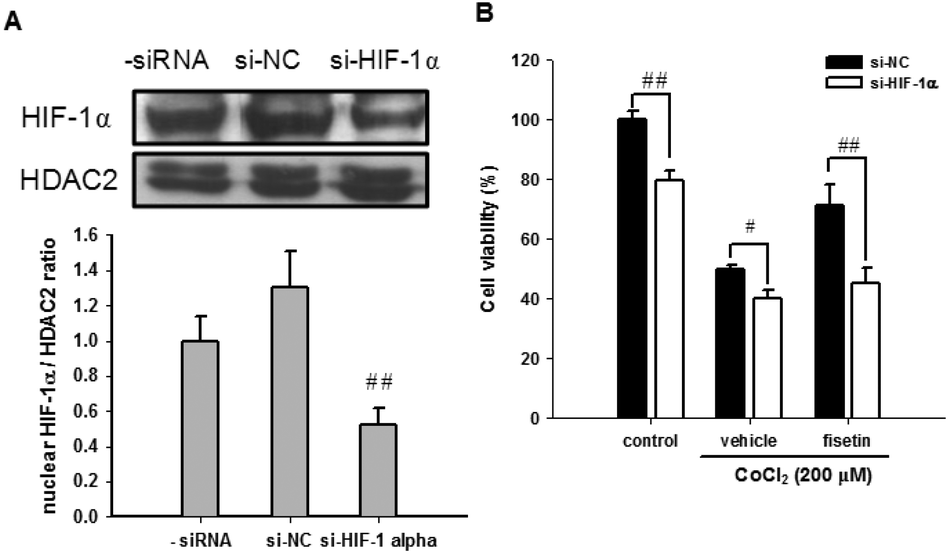 | ||
| Fig. 5 The effect of HIF-1α knockdown on the fisetin-mediated cytoprotection. PC12 cells were transiently transfected with the siRNA negative control (si-NC) or HIF-1α-specific siRNA (si-HIF-1α). (A) The nuclear HIF-1α and HDAC2 proteins were analyzed using western blot analysis. The experiments were replicated at least three times and a representative blot is shown. The normalized intensity of HIF-1α versus HDAC2 is presented as the mean ± SD of three independent experiments. (B) After transfection, PC12 cells were treated with the vehicle or fisetin (40 μM) for 1 h, then exposed to CoCl2 (200 μM) for another 24 h. The cell viability was measured with the MTT assay. The data represent the mean ± SD from triplicated experiments. #p < 0.05 and ##p < 0.01 represent significant differences compared with the siRNA negative control-transfected cell group. | ||
3.4 ERK, p38 MAPK and PI3/Akt pathways are involved in the fisetin-mediated cytoprotection in CoCl2-treated PC12 cells
It has been reported that MAPK and PI3/Akt signaling pathways may contribute to neuronal cell survival.38,39 To investigate whether these signaling pathways are involved in the fisetin-mediated cytoprotection of PC12 cells, the selectively specific kinase inhibitors including MAPK/ERK kinase1/2 (MEK1/2) inhibitor (U0126), JNK inhibitor (SP600125), p38 MAPK inhibitor (SB203580) and PI3 K inhibitor (LY294002) were used. PC12 cells were pre-treated with the inhibitors for 30 min followed by incubation with 40 μM fisetin for 1 h before exposure to 200 μM CoCl2 for another 24 h. As shown in Fig. 6A, the fisetin-mediated cell viability restoration was attenuated by inhibitors U0126, SB203580, and LY294002 compared with inhibitor-untreated cells (p < 0.01). These data suggest that ERK, p38 MAPK and PI3/Akt pathways may be involved in the fisetin-mediated cytoprotection against CoCl2-induced cell death.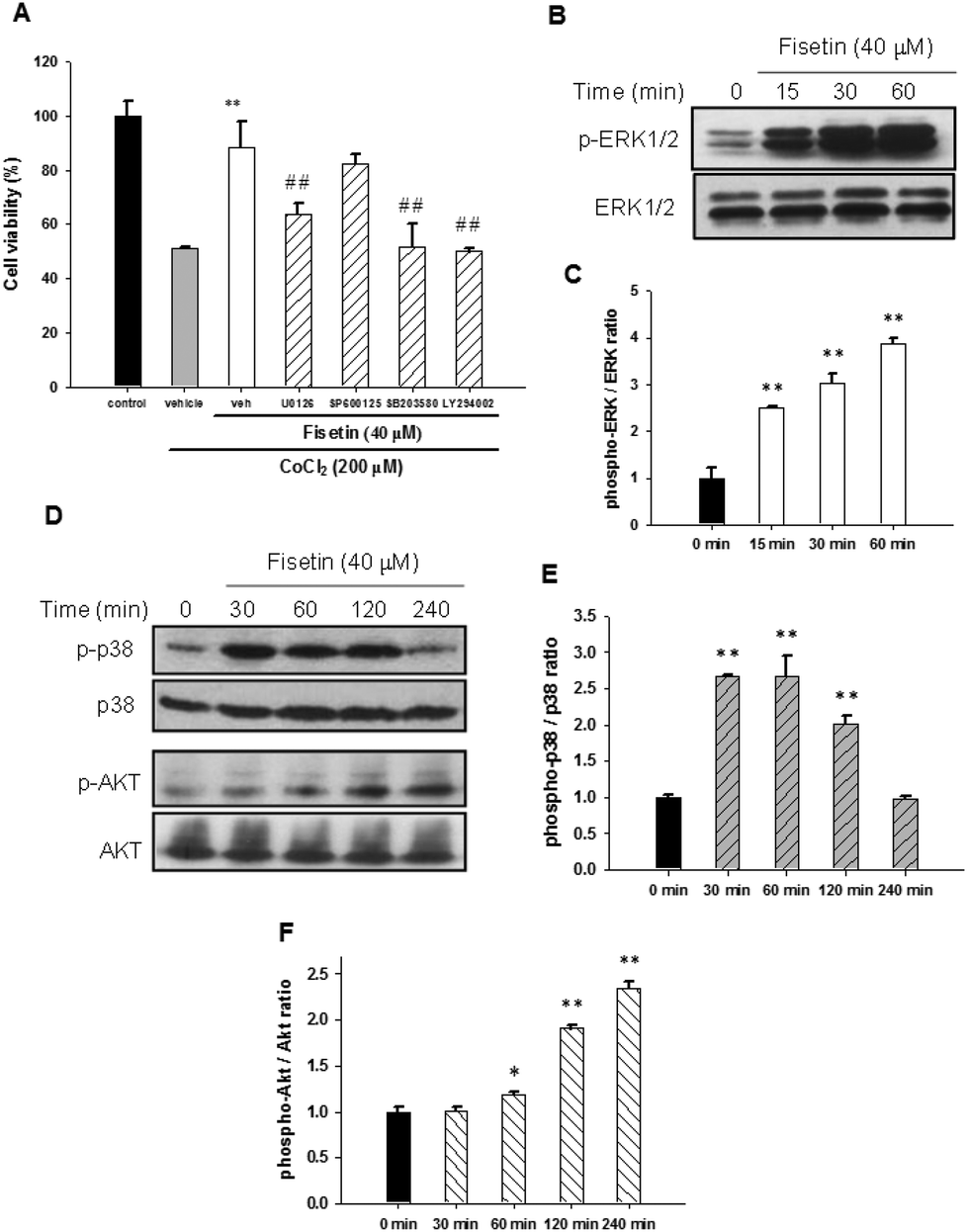 | ||
| Fig. 6 The effect of signaling pathways on the fisetin-mediated cytoprotection. (A) PC12 cells were treated with inhibitors U0126 (10 μM), SP600125 (10 μM), SB203580 (10 μM) and LY294002 (40 μM) for 30 min, followed by fisetin (40 μM) treatment for 1 h prior to CoCl2 (200 μM) exposure. The cell viability was measured with the MTT assay. The data represent the mean ± SD from three independent experiments. **p < 0.01 represents significant differences compared with the vehicle plus the CoCl2 group. ##p < 0.01 represents significant differences compared with the inhibitor-untreated group. (B) The PC12 cells were exposed to fisetin (40 μM) for the indicated period. The phospho-ERK1/2 and ERK1/2 proteins were analyzed using western blot analysis. The experiments were replicated at least three times and a representative blot is shown. (C) The normalized intensity of phospho-ERK1/2 versus ERK1/2 is presented as the mean ± SD of three independent experiments. (D) The phospho-p38 (p-p38), p38, phospho-Akt (p-Akt) and Akt proteins were analyzed using western blot analysis. The experiments were replicated at least three times and a representative blot is shown. The normalized intensity of phospho-p38 versus p38 (E) and phospho-Akt versus Akt (F) are presented as the mean ± SD of three independent experiments. *p < 0.05 and **p < 0.01 represent significant differences compared with the 0 min group. | ||
Furthermore, activation of signaling molecules including ERK, p38 MAPK and PI3/Akt by fisetin was examined using western blot analysis. As shown in Fig. 6B and C, phospho-ERK1/2 was significantly increased in 15 min fisetin treatment and remained at elevated levels for 60 min. Fig. 6D and E show that the level of phospo-p38 peaked during 30–60 min treatment. Moreover, we also found that the level of phospho-Akt was dramatically increased in fisetin-treated PC12 cells during 60–240 min treatment (Fig. 6D and F). Taken together, these data reveal that fisetin may rescue the viability of PC12 cells under hypoxia by activating ERK-, p38 MAPK- and PI3/Akt-dependent signaling pathways.
To examine whether there is crosstalk between the signaling pathways and HIF-1α activation, the PC12 cells were transfected with HRE reporter plasmids and Renilla internal control plasmids for 24 h, treated with the inhibitors U0126, SB203580 and LY294002 for 30 min and subsequently incubated with fisetin (40 μM) alone or followed by the addition of CoCl2 for another 24 h before the luciferase activity was analyzed. The fisetin-induced HIF-1α transcription activity was not affected by treatment with inhibitors U0126, SB203580 and LY294002 under fisetin alone (Fig. 7A) or combination of fisetin and CoCl2 conditions (Fig. 7B). These results indicate that MAPK/ERK-, p38 MAPK- and PI3 K/Akt-dependent pathways are not associated with HIF-1α-mediated transcription in fisetin-treated PC12 cells.
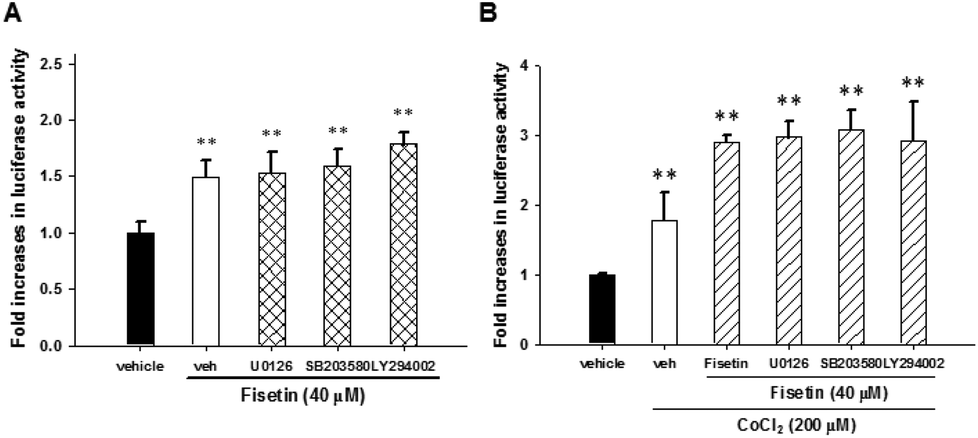 | ||
| Fig. 7 The fisetin-mediated HIF-1α activation was through ERK-, p38 MAPK- and PI3/Akt-independent pathways. The HRE luciferase reporter plasmid transfected cells were pre-treated with U0126 (10 μM), SB203580 (10 μM) and LY294002 (40 μM) for 30 min, followed by fisetin (40 μM) alone treatment for 24 h (A) or fisetin (40 μM) treatment for 1 h prior to CoCl2 (200 μM) exposure (B). Cells were harvested and luciferase activities were determined. The intensity of the luciferase activity was measured and the data represent the mean ± SD from triplicated experiments. **p < 0.01 represents significant differences compared with the CoCl2-untreated vehicle group. | ||
4. Discussion
The consumption of flavonoid-rich foods has been suggested to maintain neuronal function, promote neuroprotection, limit neurodegeneration or reverse deterioration in cognitive performance. Fisetin, a flavonoid distributed in various types of fruits, vegetables and medicinal herbs, has been reported to possess antioxidant, neurotrophic and neuroprotective activities by directly scavenging ROS or affecting signaling pathways in the maintenance of neuronal function and cell survival. However, little information is available on how fisetin affects signaling molecules in response to hypoxia in neuronal cells. In eukaryotic cells, oxygen is important to generate energy for cell survival and functions. Maintenance of neuronal function in the brain depends on a continuous supply of oxygen. The brain is a main consumer of energy and particularly susceptible to hypoxia effects. Hypoxia-induced neuronal cell death is a major concern in several clinical contexts such as stroke, heart failure, neurodegeneration and ischemic disease in the brain. Cobalt chloride (CoCl2) is a hypoxia-mimicking chemical agent that causes cell apoptosis and some hypoxic responses in cultured cells including the generation of ROS, activation of HIF-1α and transcription of a range of hypoxia-responsive HIF-target genes.40 In contrast to the induction of cell death, a small body of literature also shows that CoCl2 stimulated neurite outgrowth in PC12 cells.41,42 In the present study, we demonstrated that chemical CoCl2 (200 μM) treatment significantly induced PC12 cell death, instead of neurite outgrowth (Fig. 1A and B). The fisetin-mediated cytoprotection was observed in PC12 cells cultured under CoCl2 exposure or low oxygen conditions (1% O2) (Fig. 1C and D). We demonstrated that fisetin could restore the cell viability in hypoxia-treated undifferentiated as well as NGF-differentiated PC12 cells (Fig. 1E). These are the first documented findings demonstrating that fisetin provides cytoprotection against cell death in PC12 cells under hypoxic conditions.It has been reported that CoCl2 could induce reactive oxygen species (ROS) generation and is considered to facilitate the PC12 cell apoptosis in hypoxia. Reduction of ROS levels may protect PC12 cells against the hypoxia-induced cell death.36 We further questioned whether fisetin restores the cell viability through reducing ROS production. Both fisetin and N-acetyl-L-cysteine (NAC), a ROS scavenger, can block CoCl2-mediated ROS production, as measured by the reduced DCF fluorescence, and significantly elevate the cell viability. NAC is an aminothiol and synthetic precursor of GSH and is considered an antioxidant for free radical scavenging and increasing intracellular GSH levels. In the current study, we found that depletion of ROS by NAC (10 mM) restored cell viability in CoCl2-treated cells. Fisetin had a higher efficacy in cell viability restoration than NAC in CoCl2-treated cells (Fig. 2C). These findings reveal that fisetin may contribute not only to the intracellular ROS scavenging activity but also to the modulation of other signaling pathways that protect against CoCl2-induced PC12 cell death.
In an attempt to explore possible molecular mechanisms underlying the cytoprotective effects of fisetin, we first focused on HIF-1α expression. The induction of HIF-1α and the consequent activation of HIF-1α target gene expression play a vital role in cellular adaptation and protection against hypoxia. In the brain, HIF-1α expression can be induced by hypoxia in neurons and astrocytes.43 Several reports suggested that neuronal cells with a higher expression of HIF-1α are protective against oxidative stress and more resistant to hypoxia-induced apoptosis.44,45 It is known that the nuclear HIF-1α/HIF-1β dimer binds to p300/CBP and activates the hypoxia response element (HRE) in a broad range of genes such as erythropoietin, glucose transporter 1 and glycolytic enzymes that improve cell survival under low oxygen conditions.46 In the current study, we demonstrated that fisetin significantly induced HIF-1α gene expression, nuclear accumulation and activation of HRE-driven transcription under normoxic or the chemical hypoxic conditions in PC12 cells (Fig. 3 and 4). Furthermore, the siRNA-mediated knockdown of HIF-1α significantly attenuated the fisetin-mediated cytoprotection (Fig. 5). These results reveal that fisetin-induced HIF-1α up-regulation and accumulation of nuclear HIF-1α are associated with the restoration of the cell viability in PC12 cells under hypoxic conditions. However, some reports suggest that HIF-1 signaling might be deleterious instead of adaptive in some circumstances by increasing the expression of p53 and NIP3, leading to cell apoptosis. It was found that the induction of apoptosis in neuron cells depends on the severity and duration of hypoxia. The exact mechanism of HIF-1α involvement in hypoxia-induced apoptosis is unclear. The effect of HIF-1α might be influenced by other proteins or factors that may determine whether HIF-1α acts as a pro- or anti-apoptotic factor.47
Hypoxia occurs in the early stage of disease development, promotes angiogenesis and enhances vascular remodeling. Hypoxia-induced HIF-1α overexpression in tumors is responsible for driving tumor growth and angiogenesis and correlates with poor prognosis.48 In the present study, fisetin transiently and rapidly induced HIF-1α translocation and activation under normoxic and hypoxic conditions, which is correlated with the neuroprotective activity in the neuronal cell model. The molecular basis between anti-ischemic and anti-carcinogenic properties of fisetin under hypoxic conditions remains unclear and needs to be further investigated in vivo. Recently, fisetin has been shown to share antiangiogenic properties and to be potential for anti-cancer therapy. Moreover, it has been reported that fisetin increased nuclear HIF-1α translocation and interacted with p300/CBP, however this protein complex was transcriptionally inactive in lung cancer cells under hypoxic conditions. It suggested that inhibition of hypoxia-induced vascular endothelial growth factor (VEGF) expression was through a HIF-independent pathway.49 The complexity of hypoxia-induced changes including HIF protein expression and its target genes in neuronal function and cancer progression remains to be clarified.
The MAP kinase and PI3 K/Akt pathways are known to be involved in cell survival in several types of cells. Our results show that U0126, SB203580 and LY294002, which are pharmacological inhibitors of ERK, p38 and Akt pathways, respectively, attenuated fisetin-mediated cytoprotection in CoCl2-treated PC12 cells. On the other hand, SP600125, an inhibitor of the JNK pathway, does not participate in the cytoprotection of fisetin (Fig. 6A). This result indicates that activation of MAPK/ERK, p38 and PI3/Akt signaling pathways by fisetin may be involved in the restoration of cell viability in CoCl2-treated cells. Furthermore, we confirmed that fisetin treatment of PC12 cells strongly increased and maintained a sustained rise in ERK phosphorylation (Fig. 6B and C). Fisetin has been reported to facilitate long-term potentiation in hippocampal slices through ERK phosphorylation and that oral administration enhanced memory in mice.23 More recently, ERK activation by fisetin has been shown to be involved in neuroprotection in a Huntington's model, maintaining the neuronal function during aging and improving cognitive function in a transgenic mouse model of Alzheimer's disease.20,21,24 Recently, Jin et al. reported that MAPK/ERK activation is involved in the cytoprotection of cortical neurons under hypoxic conditions.50 Our current results suggest the involvement of the ERK signaling pathway in the fisetin-mediated neuroprotection under hypoxic conditions.
We further examined the effect of fisetin on p38 phosphorylation. It is generally accepted that activation of the p38 MAP kinase pathway is responsible for apoptosis; however, recent studies suggest that transient activation of p38 is also important for cell survival and neuroprotection.51,52 In this study, we found that fisetin markedly induced p38 phosphorylation before 2 h treatment of cells (Fig. 6D and E) and that p38 inhibition in CoCl2-treated cells attenuated fisetin-induced cytoprotection. In addition to MAPK pathways, flavonoids have also been reported to promote neuronal survival through a PI3 K/Akt-dependent pathway.53 Our current results also demonstrated that fisetin treatment led to phosphorylation of Akt in PC12 cells (Fig. 6D and F). Under hypoxic conditions, the addition of the inhibitor LY294002 reduced cell viability in fisetin-treated cells. Activation of Akt signaling is believed to increase the cell viability by inhibiting apoptosis through inactivation of apoptotic factors. Thus, the p38 MAP kinase and Akt signaling pathways may play an important role in fisetin-mediated neuroprotection under hypoxic conditions.
The activation of MAPK- and PI3/Akt-dependent pathways has been reported to involve HIF-1α activation in various cell types.54,55 In the present study, we found that the inhibitors U0126, SB203580 and LY294002 did not change the fisetin-induced HRE-driven transcriptional activity (Fig. 7). This result indicates that cross-talk among the ERK, p38 and Akt pathways and HIF-1α-mediated transactivation in fisetin-treated cells has not been found. The signaling pathways involved in the activation of HIF-1α may differ between cell types. Our current study demonstrates that fisetin up-regulates HIF-1α expression and stimulates HRE-driven transcription in PC12 cells; however, how fisetin affects HIF-1α gene expression and HRE-transactivation is unclear. Whether fisetin modulates HIF-1α at the post-translational level through impairment of its degradation by inhibition of prolyl-hydroxylase remains to be clarified.
Emerging evidence demonstrated that fisetin can transverse the BBB and reach the central nervous system to affect neuronal function. It was found that fisetin showed high brain uptake potential in a cell model for BBB penetration.20 Fisetin administration could reduce the cerebral damage in a rat stroke model, enhance learning and memory and maintenance of neuronal function during aging in mice.23,56,57 Thus, fisetin may serve as a neuroprotective agent for neurodegenerative disorders. However, the bioavailability of fisetin is very low due to its poor water solubility, high lipophilicity and rapid metabolism to hamper its clinical application.58,59 Touil et al. reported that glucuronidated and methoxylated metabolites of fisetin were detected following fisetin administration of a dose of 223 mg kg−1 i.p. in mice. It was found that the maximal concentrations (Cmax) of fisetin could reach 2.5 μg mL−1 (equivalent to 9 μM) at 15 min and have a relatively long terminal half-life in plasma.60 Seguin et al. demonstrated that following administration of fisetin at a dose of 13 mg kg−1 i.v. in mice, the Cmax of fisetin reached 6.0 μg mL−1 (equivalent to 21 μM).61 Shia et al. reported that following oral administration at 50 mg kg−1 in rats, the serum concentration of fisetin declined rapidly and fisetin sulfates/glucuronide metabolites increased. The level of sulfated/glucuronidated fisetin in serum was maintained approximately at 10 μM over 24 h.58 These fisetin metabolites also showed significant bioactivity as compared to the unmodified fisetin. In the present study, we demonstrated the cytoprotective response to fisetin at concentrations from 10 μM to 60 μM, which are widely used in vitro cell systems, although it was much higher than that can be reached in vivo. To improve the fisetin bioavailability and neuroprotective efficacy in vivo, development of novel delivery strategies such as nanoemulsion and liposomal formulation or modulation of the applied route such as injection may increase its absorption toward the concentration employed in this research.59,61,62
5. Conclusions
In the current study, for the first time, we demonstrated that fisetin protects PC12 cells against hypoxia-induced cell death. We found an involvement of ROS scavenging in fisetin-mediated cytoprotection under hypoxic conditions. Furthermore, we demonstrated that fisetin induced the nuclear abundance of HIF-1α in PC12 cells, resulting in the HRE transactivation and restoration of cell viability under hypoxia. Moreover, we found that MAPK/ERK-, p38 MAPK- and PI3/Akt-dependent signaling pathways were involved in the cytoprotection mediated through fisetin in PC12 cells under hypoxic conditions (Fig. 8).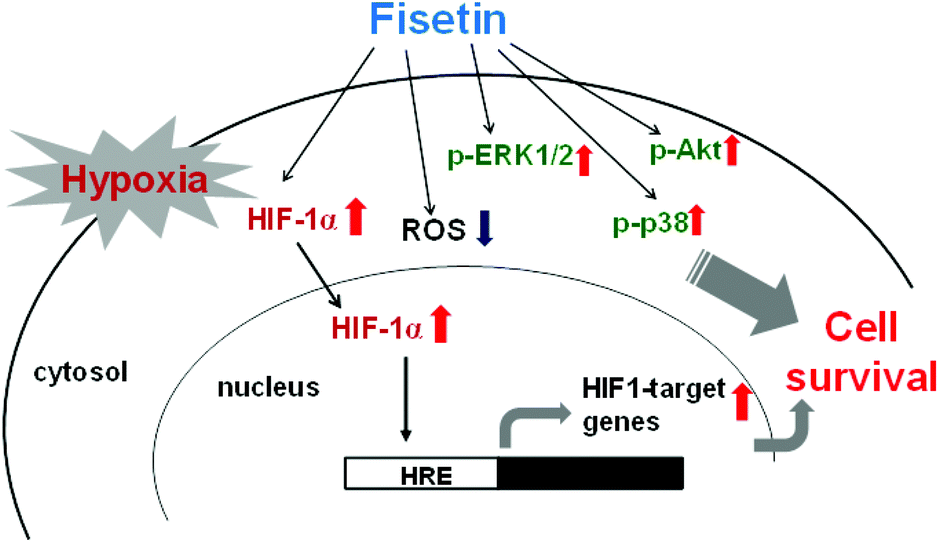 | ||
| Fig. 8 The cytoprotective effects of fisetin on PC12 cells under hypoxic conditions. | ||
Acknowledgements
This study was supported by grant TCRD-103-17 (to P-Y Chen) from Buddhist Tzu Chi General Hospital, and grants NSC 101-2313-B-320-001 and MOST 102-2313-B-320-001-MY3 (to J-H Yen) from the Ministry of Science and Technology, Taiwan.References
- A. Giaccia, B. G. Siim and R. S. Johnson, Nat. Rev. Drug Discovery, 2003, 2, 803–811 CrossRef CAS PubMed.
- R. E. Speer, S. S. Karuppagounder, M. Basso, S. F. Sleiman, A. Kumar, D. Brand, N. Smirnova, I. Gazaryan, S. J. Khim and R. R. Ratan, Free Radicals Biol. Med., 2013, 62, 26–36 CrossRef CAS PubMed.
- B. Lin, Cent. Nerv. Syst. Agents Med. Chem., 2013, 13, 57–70 CrossRef CAS.
- X. Fan, C. J. Heijnen, M. A. van der Kooij, F. Groenendaal and F. van Bel, Brain Res. Rev., 2009, 62, 99–108 CrossRef CAS PubMed.
- L. Li, X. Zhang, D. Yang, G. Luo, S. Chen and W. Le, Neurobiol. Aging, 2009, 30, 1091–1098 CrossRef CAS PubMed.
- G. J. Chen, J. Xu, S. A. Lahousse, N. L. Caggiano and S. M. de la Monte, J. Alzheimers Dis., 2003, 5, 209–228 CAS.
- H. F. Bunn and R. O. Poyton, Physiol. Rev., 1996, 76, 839–885 CAS.
- W. G. Kaelin Jr. and P. J. Ratcliffe, Mol. Cell, 2008, 30, 393–402 CrossRef PubMed.
- J. D. Firth, B. L. Ebert, C. W. Pugh and P. J. Ratcliffe, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6496–6500 CrossRef CAS.
- J. A. Forsythe, B. H. Jiang, N. V. Iyer, F. Agani, S. W. Leung, R. D. Koos and G. L. Semenza, Mol. Cell. Biol., 1996, 16, 4604–4613 CAS.
- P. Carmeliet, Y. Dor, J. M. Herbert, D. Fukumura, K. Brusselmans, M. Dewerchin, M. Neeman, F. Bono, R. Abramovitch, P. Maxwell, C. J. Koch, P. Ratcliffe, L. Moons, R. K. Jain, D. Collen and E. Keshert, Nature, 1998, 394, 485–490 CrossRef CAS PubMed.
- O. Baranova, L. F. Miranda, P. Pichiule, I. Dragatsis, R. S. Johnson and J. C. Chavez, J. Neurosci., 2007, 27, 6320–6332 CrossRef CAS PubMed.
- J. A. Ross and C. M. Kasum, Annu. Rev. Nutr., 2002, 22, 19–34 CrossRef CAS PubMed.
- E. Middleton, Jr., C. Kandaswami and T. C. Theoharides, Pharmacol. Rev., 2000, 52, 673–751 Search PubMed.
- M. C. Kou, S. H. Fu, J. H. Yen, C. Y. Weng, S. Li, C. T. Ho and M. J. Wu, Food Funct., 2013, 4, 602–609 CAS.
- K. A. Youdim, M. Z. Qaiser, D. J. Begley, C. A. Rice-Evans and N. J. Abbott, Free Radicals Biol. Med., 2004, 36, 592–604 CrossRef CAS PubMed.
- Y. Lim do and J. H. Park, Am. J. Physiol. Gastrointest. Liver Physiol., 2009, 296, G1060–G1068 CrossRef CAS PubMed.
- N. Khan, D. N. Syed, N. Ahmad and H. Mukhtar, Antioxid. Redox Signaling, 2013, 19, 151–162 CrossRef CAS PubMed.
- V. M. Adhami, D. N. Syed, N. Khan and H. Mukhtar, Biochem. Pharmacol., 2012, 84, 1277–1281 CrossRef CAS PubMed.
- P. Maher, Genes Nutr., 2009, 4, 297–307 CrossRef CAS PubMed.
- P. Maher, R. Dargusch, L. Bodai, P. E. Gerard, J. M. Purcell and J. L. Marsh, Hum. Mol. Genet., 2011, 20, 261–270 CrossRef CAS PubMed.
- T. Akaishi, T. Morimoto, M. Shibao, S. Watanabe, K. Sakai-Kato, N. Utsunomiya-Tate and K. Abe, Neurosci. Lett., 2008, 444, 280–285 CrossRef CAS PubMed.
- P. Maher, T. Akaishi and K. Abe, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16568–16573 CrossRef CAS PubMed.
- A. Currais, M. Prior, R. Dargusch, A. Armando, J. Ehren, D. Schubert, O. Quehenberger and P. Maher, Aging Cell, 2014, 13, 379–390 CrossRef CAS PubMed.
- N. S. Chandel, E. Maltepe, E. Goldwasser, C. E. Mathieu, M. C. Simon and P. T. Schumacker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11715–11720 CrossRef CAS.
- G. Wang, Nucleic Acids Res., 2000, 28, 2135–2140 CrossRef CAS PubMed.
- L. O. Simonsen, H. Harbak and P. Bennekou, Sci. Total Environ., 2012, 432, 210–215 CrossRef CAS PubMed.
- G. A. Badr, J. Z. Zhang, J. Tang, T. S. Kern and F. Ismail-Beigi, Brain Res. Mol. Brain Res., 1999, 64, 24–33 CrossRef CAS.
- A. Merelli, L. Caltana, P. Girimonti, A. J. Ramos, A. Lazarowski and A. Brusco, Neurotox. Res., 2011, 20, 182–192 CrossRef CAS PubMed.
- L. A. Greene, J. M. Aletta, A. Rukenstein and S. H. Green, Methods Enzymol., 1987, 147, 207–216 CAS.
- Z. Spicer and D. E. Millhorn, Endocr. Pathol., 2003, 14, 277–291 CrossRef CAS PubMed.
- J. Carmichael, W. G. DeGraff, A. F. Gazdar, J. D. Minna and J. B. Mitchell, Cancer Res., 1987, 47, 943–946 CAS.
- L. W. Hu, J. H. Yen, Y. T. Shen, K. Y. Wu and M. J. Wu, PLoS One, 2014, 9, e97880 Search PubMed.
- S. Villanueva, C. Suazo, D. Santapau, F. Perez, M. Quiroz, J. E. Carreno, S. Illanes, S. Lavandero, L. Michea and C. E. Irarrazabal, PLoS One, 2012, 7, e39665 CAS.
- X. L. Tan, X. Y. Huang, W. X. Gao, Y. Zai, Q. Y. Huang, Y. J. Luo and Y. Q. Gao, Neurosci. Lett., 2008, 441, 272–276 CrossRef CAS PubMed.
- W. Zou, M. Yan, W. Xu, H. Huo, L. Sun, Z. Zheng and X. Liu, J. Neurosci. Res., 2001, 64, 646–653 CrossRef CAS.
- R. Bruggisser, K. von Daeniken, G. Jundt, W. Schaffner and H. Tullberg-Reinert, Planta Med., 2002, 68, 445–448 CrossRef CAS PubMed.
- A. Bonni, A. Brunet, A. E. West, S. R. Datta, M. A. Takasu and M. E. Greenberg, Science, 1999, 286, 1358–1362 CrossRef CAS.
- A. Brunet, S. R. Datta and M. E. Greenberg, Curr. Opin. Neurobiol., 2001, 11, 297–305 CrossRef CAS.
- Q. Ke and M. Costa, Mol. Pharmacol., 2006, 70, 1469–1480 CrossRef CAS PubMed.
- E. Kotake-Nara, S. Takizawa, J. Quan, H. Wang and K. Saida, J. Neurosci. Res., 2005, 81, 563–571 CrossRef CAS PubMed.
- Y. Leiser, N. Silverstein, A. Blumenfeld, D. Shilo, A. Haze, E. Rosenfeld, B. Shay, R. Tabakman, S. Lecht, P. Lazarovici and D. Deutsch, J. Cell Physiol., 2011, 226, 165–172 CrossRef CAS PubMed.
- J. C. Chavez, F. Agani, P. Pichiule and J. C. LaManna, J. Appl. Physiol. (1985), 2000, 89, 1937–1942 CAS.
- A. E. Greijer and E. van der Wall, J. Clin. Pathol., 2004, 57, 1009–1014 CrossRef CAS PubMed.
- A. Corcoran and J. J. O'Connor, Acta Physiol., 2013, 208, 298–310 CrossRef CAS PubMed.
- F. R. Sharp and M. Bernaudin, Nat. Rev. Neurosci., 2004, 5, 437–448 CrossRef CAS PubMed.
- J. P. Piret, D. Mottet, M. Raes and C. Michiels, Biochem. Pharmacol., 2002, 64, 889–892 CrossRef CAS.
- J. Brocato, Y. Chervona and M. Costa, Mol. Pharmacol., 2014, 85, 651–657 CrossRef PubMed.
- E. Anso, A. Zuazo, M. Irigoyen, M. C. Urdaci, A. Rouzaut and J. J. Martinez-Irujo, Biochem. Pharmacol., 2010, 79, 1600–1609 CrossRef CAS PubMed.
- K. Jin, X. O. Mao, Y. Zhu and D. A. Greenberg, J. Neurochem., 2002, 80, 119–125 CrossRef CAS.
- A. Matsuzawa and H. Ichijo, Biochim. Biophys. Acta, 2008, 1780, 1325–1336 CrossRef CAS PubMed.
- C. Agca, A. Gubler, G. Traber, C. Beck, C. Imsand, D. Ail, C. Caprara and C. Grimm, Cell Death Dis., 2013, 4, e785 CrossRef CAS PubMed.
- J. P. Spencer, Genes Nutr., 2007, 2, 257–273 CrossRef CAS PubMed.
- E. Minet, T. Arnould, G. Michel, I. Roland, D. Mottet, M. Raes, J. Remacle and C. Michiels, FEBS Lett., 2000, 468, 53–58 CrossRef CAS.
- J. Du, R. Xu, Z. Hu, Y. Tian, Y. Zhu, L. Gu and L. Zhou, PLoS One, 2011, 6, e25213 CAS.
- F. Rivera, J. Urbanavicius, E. Gervaz, A. Morquio and F. Dajas, Neurotox. Res., 2004, 6, 543–553 CrossRef.
- P. Maher, K. F. Salgado, J. A. Zivin and P. A. Lapchak, Brain Res., 2007, 1173, 117–125 CrossRef CAS PubMed.
- C. S. Shia, S. Y. Tsai, S. C. Kuo, Y. C. Hou and P. D. Chao, J. Agric. Food Chem., 2009, 57, 83–89 CrossRef CAS PubMed.
- N. Mignet, J. Seguin, M. Ramos Romano, L. Brulle, Y. S. Touil, D. Scherman, M. Bessodes and G. G. Chabot, Int. J. Pharm., 2012, 423, 69–76 CrossRef CAS PubMed.
- Y. S. Touil, N. Auzeil, F. Boulinguez, H. Saighi, A. Regazzetti, D. Scherman and G. G. Chabot, Biochem. Pharmacol., 2011, 82, 1731–1739 CrossRef CAS PubMed.
- J. Seguin, L. Brulle, R. Boyer, Y. M. Lu, M. Ramos Romano, Y. S. Touil, D. Scherman, M. Bessodes, N. Mignet and G. G. Chabot, Int. J. Pharm., 2013, 444, 146–154 CrossRef CAS PubMed.
- H. Ragelle, S. Crauste-Manciet, J. Seguin, D. Brossard, D. Scherman, P. Arnaud and G. G. Chabot, Int. J. Pharm., 2012, 427, 452–459 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4fo00948g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |