
Induction of Nrf2-mediated genes by Antrodia salmonea inhibits ROS generation and inflammatory effects in lipopolysaccharide-stimulated RAW264.7 macrophages
Hsin-Ling
Yang†
a,
Shu-Wei
Lin
a,
Chuan-Chen
Lee†
b,
Kai-Yuan
Lin
c,
Chun-Huei
Liao
a,
Ting-Yu
Yang
a,
Hui-Min
Wang
d,
Hui-Chi
Huang
e,
Chi-Rei
Wu
e and
You-Cheng
Hseu
*bf
aInstitute of Nutrition, China Medical University, Taichung 40402, Taiwan
bDepartment of Health and Nutrition Biotechnology, Asia University, Taichung 41354, Taiwan
cDepartment of Medical Research, Chi-Mei Medical Center, Tainan 710, Taiwan
dDepartment of Fragrance and Cosmetic Science, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
eSchool of Chinese Pharmaceutical Sciences and Chinese Medicine Resources, College of Pharmacy, China Medical University, Taichung 40402, Taiwan
fDepartment of Cosmeceutics, College of Pharmacy, China Medical University, Taichung 40402, Taiwan. E-mail: ychseu@mail.cmu.edu.tw; Fax: +886 4 22078083; Tel: +886 4 22053366 x5308
First published on 17th October 2014
Abstract
Antrodia salmonea (AS), a well-known medicinal mushroom in Taiwan, has been reported to exhibit anti-oxidant, anti-angiogenic, anti-atherogenic, and anti-inflammatory effects. In the present study, we investigated the activation of Nrf2-mediated antioxidant genes in RAW264.7 macrophages by the fermented culture broth of AS, studied the resulting protection against lipopolysaccharide (LPS)-stimulated inflammation, and revealed the molecular mechanisms underlying these protective effects. We found that non-cytotoxic concentrations of AS (25–100 μg mL−1) protected macrophages from LPS-induced cell death and ROS generation in a dose-dependent manner. The antioxidant potential of AS was directly correlated with the increased expression of the antioxidant genes HO-1, NQO-1, and γ-GCLC, as well as the level of intracellular GSH followed by an increase in the nuclear translocation and transcriptional activation of the Nrf2–ARE pathway. Furthermore, Nrf2 knockdown diminished the protective effects of AS, as evidenced by the increased production of pro-inflammatory cytokines and chemokines, including PGE2, NO, TNF-α, and IL-1β, in LPS-stimulated macrophages. Notably, AS treatment significantly inhibited LPS-induced ICAM-1 expression in macrophages. Our data suggest that the anti-inflammatory potential of Antrodia salmonea is mediated by the activation of Nrf2-dependent antioxidant defense mechanisms. Results support the traditional usage of this beneficial mushroom for the treatment of free radical-related diseases and inflammation.
Introduction
Macrophages play a pivotal role in the innate immune response and serve as the first line of defense against invading pathogens by facilitating cytoprotection and repair processes.1 Despite its beneficial role in host defence, aberrant macrophage activation by endotoxins induces a number of major cellular responses that play roles in the pathogenesis and progression of inflammatory responses.2 Lipopolysaccharide (LPS), which is a component of the cell walls of Gram-negative bacteria, can activate a number of signals within macrophages during inflammation and infection.3 Activated macrophages produce a variety of pro-inflammatory molecules, including nitric oxide (NO), prostaglandin E2 (PGE2), tumor necrosis factor-α (TNF-α), and interleukins, which are involved in the development and progression of inflammatory diseases and cancer.4,5 Macrophage activation by LPS can initiate an increase in oxygen uptake that gives rise to a variety of reactive oxygen species (ROS), which are the major factors driving the oxidative stress-induced inflammation in immune cells.6A number of lines of evidence suggest that nuclear factor E2-related factor-2 (Nrf2) plays an important role in protecting macrophages from LPS-stimulated inflammation.7 Nrf2 belongs to the basic-leucine zipper (bZIP) family of transcriptional activator proteins and can be activated by endogenous products of oxidative stress.8 The expression levels of many cytoprotective enzymes that are induced in response to oxidative stress are regulated primarily at the transcriptional level. The activation of the cis-acting anti-oxidant response element (ARE) is dependent on the transcription factor Nrf2. Nrf2 activates several antioxidant genes that are important for the elimination of oxidative stress in cellular systems: hemeoxygenase-1 (HO-1), NAD(P)H-quinone oxidoreductase-1 (NQO-1), γ-glutamylcysteine synthetase (γ-GCLC), glutathione (GSH), and glutathione S-transferase A2 (GST-A2).9,10 These enzymes exert cytoprotective, anti-oxidant, and anti-inflammatory effects in endotoxin-induced macrophages.11
The medicinal mushroom Antrodia salmonea (or Taiwanofungus salmoneus), a newly identified medicinal fungus species, belongs to the genus Taiwanofungus. The fruiting body of Antrodia salmonea has long been used in Taiwanese folk medicine for the treatment of diarrhea, abdominal pain, hypertension, itchy skin, and liver cancer. It has also been used as a food and drug detoxicant.12 In our previous study, Antrodia salmonea in submerged culture exhibited antioxidant activities in vitro and protected human erythrocytes and low-density lipoproteins from oxidative modification.13 Moreover, the safety levels and nontoxic characteristics of the fermented culture broth of Antrodia salmonea were evaluated using acute toxicity studies in a mouse model.14 To date, several novel compounds have been isolated from the basidiomata of Antrodia salmonea, and in vitro studies have shown that these compounds display anti-oxidative and anti-inflammatory activities.15–17 However, the possible mechanisms underlying the protective activity of Antrodia salmonea have not yet been fully elucidated. Recently, several naturally occurring compounds and crude extracts of medicinal herbs have been reported to induce the expression of phase II detoxifying enzymes in different cell types; in some of these studies, Nrf2-mediated genes were shown to exert anti-oxidant and anti-inflammatory activities.18 Therefore, the present study was designed to investigate whether the anti-oxidant and anti-inflammatory potential of Antrodia salmonea is mediated by the activation of Nrf2-stimulated defense mechanisms. This study may help us to further evaluate the clinical therapeutic food detoxifying potential of Antrodia salmonea with respect to its anti-oxidant and anti-inflammatory properties.
Materials and methods
Chemicals
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), glutamine, and penicillin–streptomycin were purchased from GIBCO BRL (Grand Island, NY). LPS (from Escherichia coli 055:B5), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and 2′,7′-dihydrofluorescein-diacetate (DCFH2-DA) were purchased from Sigma-Aldrich (St Louis, MO). Mouse monoclonal antibodies against TNF-α and HO-1 were purchased from Abcam (Cambridge, UK). Nrf2, NQO-1, and IL-1β antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-γ-GCLC antibody was obtained from GeneTex Inc. (Irvine, CA). Antibodies against p-JNK1/2, p-ERK1/2, p-p38 MAPK, and p-PI3K were obtained from Cell Signaling Technology Inc. (Danvers, MA). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from Calbiochem (La Jolla, CA). All other chemicals were of the highest grade commercially available and were supplied by either Merck (Darmstadt, Germany) or Sigma.Antrodia salmonea from submerged cultures
The strain of the fungus Antrodia salmonea was collected in Nantou County in middle Taiwan. The plant was identified by Dr Shy-Yuan Hwang of Endemic Species Research Institute, Nantou, Taiwan. Voucher specimens (no. AS001) have been deposited in China Medical University, Taichung, Taiwan. The hyphae of Antrodia salmonea were separated from the fruiting bodies. The whole colony was cut and placed in a flask with 50 mL of sterile water. After homogenization, the fragmented mycelial suspension was then inoculated into a culture medium comprising 2.0% glucose, 0.1% wheat powder, and 0.1% peptone in distilled water. The medium was adjusted to an initial pH of 5.0. Each culture was grown in a 2 L Erlenmeyer flask (containing 1 L of the medium) and incubated at 25 °C for 10 days with shaking at 120 rpm. Thereafter, 3.5 L of the culture was inoculated into a 500 L fermenting tank containing 300 L of culture medium. The resulting mixtures were then cultured at 25 °C for 30 days. The fermentation conditions were the same as those used for the seed fermentation but with an aeration rate of 0.075 vvm, to produce a mucilaginous medium containing the mycelia. The experiments were performed using 2–4 different batches of whole fermented culture of Antrodia salmonea.Sample preparation
The fermentation product was poured through a non-woven fabric on a 20 mesh sieve to separate the deep yellow fermented culture into the filtrate and mycelia and subsequently concentrated under vacuum and freeze dried. The dry matter yield of the fermented culture was approximately 15 g L−1. The freeze-dried samples were ground, shaken with distilled water, and then centrifuged at 3000g for 5 min, followed by passage through a 0.2 μm filter. The aqueous extracts were concentrated under vacuum and freeze dried to form a powder. The yield of the fermented culture broth (AS) (1 g) was approximately 0.375 g. To prepare the stock solution, the powdered AS samples were dissolved in 10 mM sodium phosphate buffer (pH 7.4) containing 0.15 M NaCl (PBS) at 25 °C. The solution was stored at −20 °C before its anti-oxidant and anti-inflammatory potential were analyzed.Cell culture
The murine macrophage (RAW264.7) cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured in DMEM containing 2 mM glutamine, 1% penicillin–streptomycin, and 10% heat-inactivated FBS at 37 °C under a humidified atmosphere containing 5% CO2. For all experiments, the supernatant was removed 1 h after AS supplementation, the cells were washed with PBS, and the culture medium was replaced with a new medium with or without LPS (1 μg mL−1) for the indicated time. The LPS was dissolved in PBS (pH 7.2).MTT assay
The effect of AS on cell viability was monitored by the MTT colorimetric assay. RAW264.7 cells (4 × 105 cells per well) were grown to confluence on 12-well cell culture plates, incubated with AS (25–100 μg mL−1) in the absence or presence of LPS (1 μg mL−1), and allowed to proliferate for 24 h. After treatment, the cells were incubated with 400 μL of 0.5 mg mL−1 MTT in PBS for 2 h. The culture supernatant was removed, the remaining MTT formazan was dissolved in 400 μL of isopropanol, and the absorbance was measured at 570 nm using an ELISA reader (Bio-Tek Instruments, Winooski, VT). The effect of AS on cell viability was assessed as the percentage of viable cells compared with the vehicle-treated control cells, which were arbitrarily designated as 100%. The assay was performed in triplicate at each concentration.Measurement of intracellular ROS generation
The production of intracellular ROS was detected by fluorescence microscopy using DCFH2-DA as described previously.19 Briefly, RAW264.7 cells at a density of 4 × 105 cells per well in a 12-well plate were incubated with AS (25–100 μg mL−1) in the presence or absence of LPS (1 μg mL−1) for 30 min. Then, 10 μM DCFH2-DA was added to the culture medium, and the cells were incubated at 37 °C for further 30 min. After incubation, the cells were washed with warm PBS, and the ROS production was measured by changes in fluorescence due to the intracellular production of DCF caused by the oxidation of DCFH2. The DCF fluorescence was measured via fluorescence microscopy (200× magnification) (Olympus, Center Valley, PA). The fold increase of intracellular ROS compared with the control was quantified by measuring the DCF fluorescence intensity in the microscope field.Preparation of cell extracts and Western blot analysis
RAW264.7 cells (4 × 106 cells per dish in a 10 cm dish) were incubated with AS (50 μg mL−1), after which they were incubated with LPS for the indicated time. After treatment, the cells were detached, washed once in cold PBS, and suspended in 100 μL lysis buffer (10 mM Tris-HCl [pH 8.0], 0.32 M sucrose, 1% Triton X-100, 5 mM EDTA, 2 mM DTT, and 1 mM phenylmethyl sulfonyl fluoride). The suspension was kept on ice for 20 min and then centrifuged at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C. The total protein content was determined using a Bio-Rad protein assay reagent, with bovine serum albumin (BSA) as the standard. Protein extracts were reconstituted in sample buffer (0.062 M Tris-HCl [pH 6.8], 2% SDS, 10% glycerol and 5% β-mercaptoethanol), and the mixture was boiled for 5 min. Equal amounts (50 μg) of denatured protein were loaded into each lane and separated on 8–15% SDS-PAGE, followed by transfer onto PVDF membranes overnight. The membranes were blocked with 0.1% Tween-20 in Tris-buffered saline containing 5% non-fat dry milk for 30 min at room temperature and then reacted with primary antibodies for 2 h. They were then incubated with a horseradish peroxidase-conjugated goat anti-rabbit or an anti-mouse antibody for 2 h and developed using the enhanced chemiluminescence substrate (Pierce Biotechnology, Rockford, IL). A densitometric graph of band intensities was generated using commercially available software (AlphaEase, Genetic Technology Inc., Miami, FL), with the control representing 100% or 1.0-fold, as shown below the gel data.
000g for 20 min at 4 °C. The total protein content was determined using a Bio-Rad protein assay reagent, with bovine serum albumin (BSA) as the standard. Protein extracts were reconstituted in sample buffer (0.062 M Tris-HCl [pH 6.8], 2% SDS, 10% glycerol and 5% β-mercaptoethanol), and the mixture was boiled for 5 min. Equal amounts (50 μg) of denatured protein were loaded into each lane and separated on 8–15% SDS-PAGE, followed by transfer onto PVDF membranes overnight. The membranes were blocked with 0.1% Tween-20 in Tris-buffered saline containing 5% non-fat dry milk for 30 min at room temperature and then reacted with primary antibodies for 2 h. They were then incubated with a horseradish peroxidase-conjugated goat anti-rabbit or an anti-mouse antibody for 2 h and developed using the enhanced chemiluminescence substrate (Pierce Biotechnology, Rockford, IL). A densitometric graph of band intensities was generated using commercially available software (AlphaEase, Genetic Technology Inc., Miami, FL), with the control representing 100% or 1.0-fold, as shown below the gel data.
Immunofluorescence staining
RAW264.7 cells at a density of 1 × 104 cells per well in 8-well plates were cultured in DMEM containing 10% FBS in an eight-well glass Tek chamber and treated with AS (50 μg mL−1 for 1 h). The cells were then fixed in 2% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, washed, blocked with 10% FBS in PBS, and then incubated with an anti-Nrf2 primary antibody in 1.5% FBS for 2 h, followed by incubation with a FITC (488 nm)-conjugated secondary antibody for 1 h in 6% BSA. Cells were stained with 1 μg mL−1 of DAPI for 5 min. The stained cells were washed with PBS and visualized using a confocal microscope at 630× magnification.RNA extraction and RT-PCR analysis
RAW264.7 cells were seeded at a density of 4 × 106 cells per dish in a 10 cm dish. After reaching 90% confluence, the cells were incubated with AS (50 μg mL−1) for various times (1–12 h). Total RNA from cultured cells was prepared using the TRIzol reagent (Invitrogen, Carlsbad, CA). A 1 μg sample of total RNA was subjected to RT-PCR using a Bio-Rad iCycler PCR instrument (Bio-Rad, Hercules, CA) and the SuperScript-III® One-Step RT-PCR Platinum taq® Kit (Invitrogen); amplification was performed in 30–38 cycles of 94 °C for 45 s (denaturing), 60–65 °C for 45 s (annealing), and 72 °C for 1 min (primer extension). The sequences of the primers used were as follows: HO-1 forward, 5′-TTACCTTCCCGAACATCGAC-3′, reverse: 5′-GCATAAATTCCCACTGCCAC-3′; NQO-1 forward, 5′-CAGATCCTGGAAGGATGGAA-3′, reverse, 5′-AAGTTAGTCCCTCGGCCATT-3′; and Nrf2 forward, 5′-AGCAGGACATGGAGCAAGTT-3′, reverse, 5′-TTCTTTTTCCAGCGAGGAGA-3′. The PCR products were electrophoresed in a 1% agarose gel and stained with ethidium bromide.ARE promoter activity
To examine the promoter activity, we used a dual-luciferase reporter assay system (Promega, Madison, WI). Briefly, RAW264.7 cells were cultured in 24-well plates until reaching ∼80% confluence and then incubated for 5 h in serum-free DMEM that did not contain antibiotics. The cells were then transfected with the pcDNA vector or the ARE plasmid with β-gal and GFP using Lipofectamine 2000 (Invitrogen). After plasmid transfection, cells were treated with AS (50 μg mL−1) for 24 h. After incubation, the cells were lysed, and the luciferase activity was measured using a luminometer (Bio-Tek Instruments Inc., Winooski, VT). The luciferase activity was normalized to the β-galactosidase activity in the cell lysates, and data were expressed as the averages of three independent experiments.Determination of intracellular GSH
RAW264.7 cells were seeded at a density of 4 × 106 cells per dish in a 10 cm dish. After reaching 90% confluence, the cells were incubated with AS (50 μg mL−1) for various times (1–12 h). The total GSH content of macrophages was determined using a Cayman's GSH assay kit (Cayman Chemical Co., Ann Arbor, MI). The kit uses an enzymatic recycling method involving glutathione reductase to quantify glutathione (GSH). The sulfhydryl group of GSH reacts with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) and Ellman's reagent to produce a yellow-colored compound, 5-thio-2-nitrobenzoic acid (TNB). The rate of TNB production is directly proportional to the rate of this recycling reaction, which is in turn directly proportional to the concentration of GSH in the sample. Thus, the net absorbance at 405 nm yields an accurate estimate of GSH in the sample. The GSH values were extrapolated from the absorbance values based on the standard curve provided with the assay kit.Transient transfection of siRNA targeting Nrf2
RAW264.7 cells were transfected with Nrf2 siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. For the transfections, RAW264.7 cells were grown in DMEM containing 10% FBS and plated in 6-well plates to 60% confluence at the time of transfection. On the next day, the culture medium was replaced with 500 μL of Opti-MEM (Invitrogen), and the cells were transfected using the RNAiMAX transfection reagent (Invitrogen). For each transfection, 5 μL RNAiMAX was mixed with 250 μL of Opti-MEM and incubated for 5 min at room temperature. In a separate tube, siRNA (100 pM, for a final concentration of 100 nM in 1 mL of Opti-MEM) was added to 250 μL of Opti-MEM, and the siRNA solution was added to the diluted RNAiMAX reagent. The resulting siRNA/RNAiMAX mixture (500 μL) was incubated for an additional 25 min at room temperature to allow complex formation. Subsequently, the solution was added to the cells in the 6-well plates for a final transfection volume of 1 mL. After incubation for 6 h, the transfection medium was replaced with 2 mL of standard growth medium, and the cells were cultured at 37 °C. Then, the cells were co-incubated with or without AS (50 and 100 μg mL−1) in the presence or absence of LPS (1 μg mL−1) for 1–24 h. The protein expression levels of Nrf2 and β-actin and the production of NO, PGE2, TNF-α, and IL-1β were quantified.Determination of nitric oxide production
The concentration of NO in the culture supernatant was determined based on the accumulation of nitrite, a major stable product of NO, using a Griess reagent-based colorimetric assay. RAW264.7 cells were seeded at a density of 4 × 105 cells per well in 12-well plates. After transfection with Nrf2 siRNA, cells were pre-treated with or without AS (50 and 100 μg mL−1) prior to incubation with LPS (1 μg mL−1) for 18 h. Culture supernatants (100 μL) were mixed with an equivalent volume of Griess reagent, and the absorbance of the mixture at 540 nm was measured using an ELISA microplate reader. A standard curve was constructed using known concentrations of sodium nitrate.Determination of PGE2 production
The PGE2 concentration in the culture media was determined using an EIA kit (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol. RAW264.7 cells were seeded at a density of 4 × 105 cells per well in 12-well plates. After Nrf2 siRNA transfection, cells were pre-treated with or without AS (50 and 100 μg mL−1) prior to incubation with LPS (1 μg mL−1) for 18 h. The culture supernatants (100 μL) were collected, and the PGE2 concentrations were determined using an ELISA microplate reader.Quantification of TNF-α and IL-1β
The TNF-α and IL-1β concentrations in the cell culture media were determined using ELISA kits (R&D Systems), as described previously.20 Briefly, Nrf2 siRNA-transfected cells were seeded at a density of 4 × 105 cells per well in 12-well plates and incubated with or without AS (50 and 100 μg mL−1) in the presence or absence of LPS (1 μg mL−1) for 18 h. Culture media were collected and transferred into 96-well plates that were coated with anti-mouse TNF-α or IL-β monoclonal antibodies, and the plates were incubated at room temperature for 2 h. After five washes, biotinylated anti-mouse TNF-α or IL-β monoclonal antibodies and avidin–horseradish peroxidase were added, and the plates were incubated for 2 h at room temperature. After a final wash, the chromogenic substrate (tetramethylbenzidine and hydrogen peroxide) was added to each well, and the plates were incubated for 30 min at room temperature in the dark. The absorbance at 405 nm was read on an ELISA plate reader after the addition of a stop solution.Statistical analysis
The experimental results are presented as the mean ± standard deviation (mean ± SD). All the study data were analyzed using an analysis of variance (ANOVA), followed by a Dunnett's test for pairwise comparisons. Statistical significance was defined as p < 0.05 for all tests.Results
The effects of AS on cell viability in RAW264.7 macrophages
Prior to the in vitro anti-oxidant and anti-inflammatory study, we examined whether AS (25–100 μg mL−1) affects RAW264.7 cell viability using an MTT assay. The macrophages were treated with various concentrations of AS for 24 h, and the cell viability was unaffected by AS at concentrations of up to 100 μg mL−1 (Fig. 1A). As shown in Fig. 1B, treatment with LPS (1 μg mL−1) slightly reduced the cell viability to 89 ± 3%, and this LPS-induced reduction of cell viability was significantly prevented by AS at 50 or 100 μg mL−1 (p < 0.05). Based on these data, we used the non-cytotoxic concentrations of AS (i.e., <100 μg mL−1) to evaluate its antioxidant/anti-inflammatory effects in RAW264.7 cells.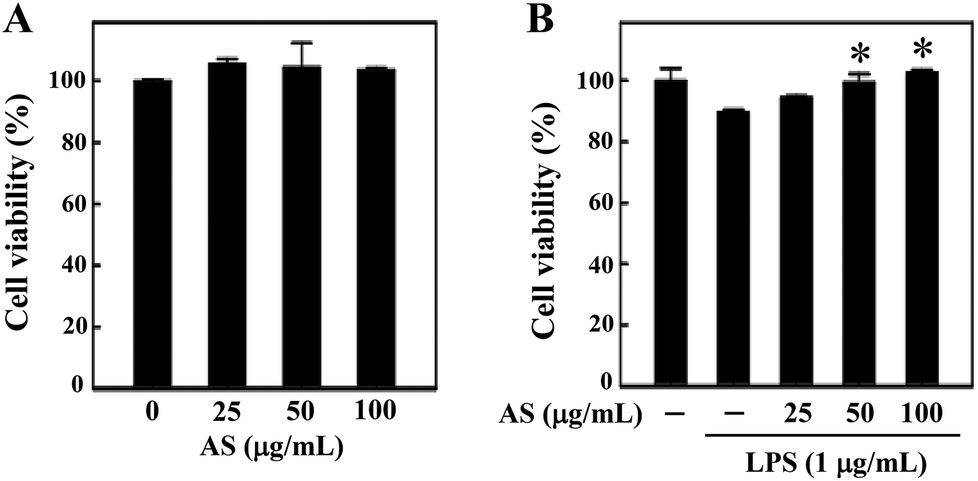 | ||
| Fig. 1 The effects of the fermented culture broth of Antrodia salmonea (AS) on murine macrophage cell viability. RAW264.7 cells were treated with various concentrations of AS (25, 50, and 100 μg mL−1) in the absence (A) or presence (B) of LPS (1 μg mL−1) for 24 h. After treatment, cells were incubated with MTT for 2 h. The culture supernatant was removed and re-suspended with isopropanol to dissolve the MTT formazan, and the absorbance was measured at 570 nm using an ELISA microplate reader. The results are the mean ± SD of three assays. *Indicates a significant difference from the LPS-activated group (p < 0.05). | ||
AS inhibits LPS-induced ROS generation in LPS-stimulated macrophages
LPS-induced intracellular ROS accumulation was monitored within cells in vitro using a DCFH2-DA fluorescence microscopic analysis. As shown in Fig. 2A and B, the incubation of RAW264.7 cells with LPS (1 μg mL−1) caused a significant increase in the intracellular ROS in RAW264.7 cells (24-fold), whereas co-treatment with AS (25–100 μg mL−1) resulted in a significant (p < 0.05) dose-dependent reduction (<10-fold) of ROS accumulation in LPS-induced RAW264.7 cells. In addition, when the cells were incubated with AS alone for the same length of time, the concentration of ROS was maintained at a background level similar to that in unstimulated cells (data not shown). Therefore, we concluded that AS could suppress LPS-induced ROS generation in macrophages.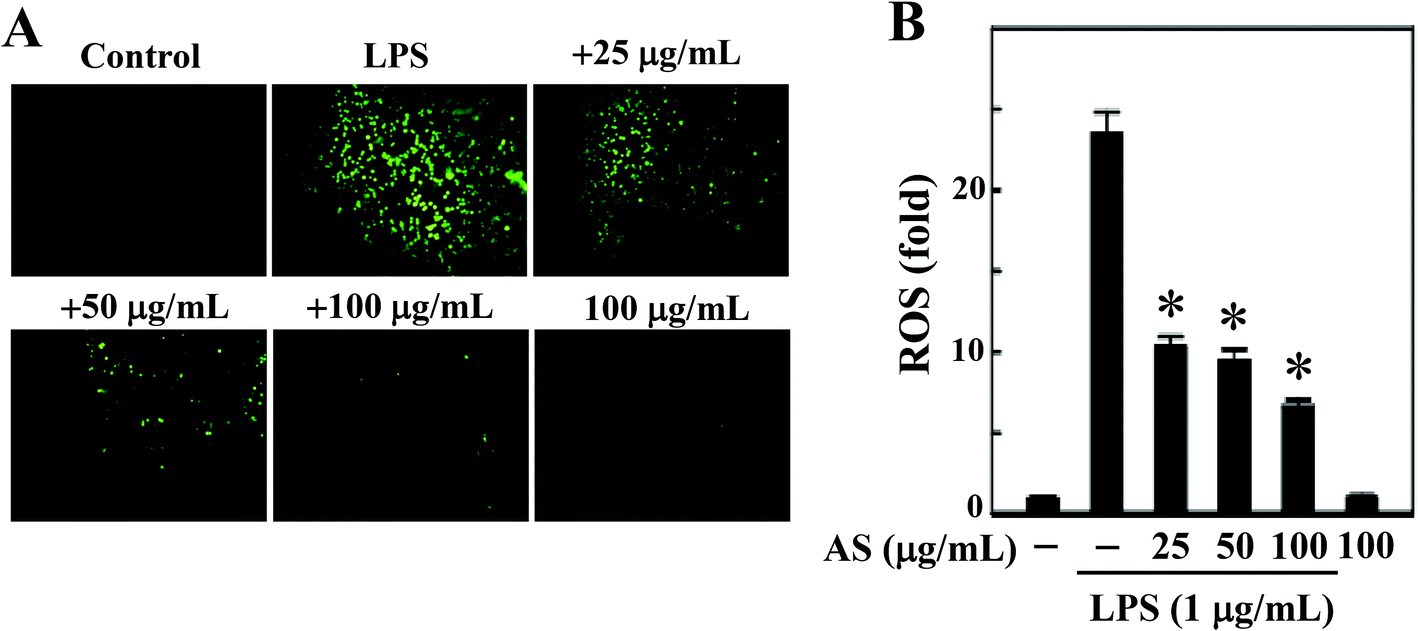 | ||
| Fig. 2 AS suppresses ROS generation in LPS-stimulated macrophages. (A) RAW264.7 cells were treated with various concentrations of AS (25, 50, and 100 μg mL−1) in the presence or absence of LPS (1 μg mL−1) for 30 min. The non-fluorescent cell membrane-permeable probe DCFH2-DA was added to the culture medium at a final concentration of 10 μM. DCFH2 was reacted with cellular ROS and metabolized to fluorescent DCF. DCF fluorescence was imaged under a fluorescence microscope (200× magnification). (B) The fold increase of intracellular ROS, compared to the control (1-fold), was quantified by measuring the DCF fluorescence intensity in the microscopic field. The results shown are the mean ± SD of three assays. *Indicates a significant difference from the LPS-activated group (p < 0.05). | ||
AS activates the antioxidant gene Nrf2 in macrophages
Nrf2 is a transcription factor that serves as a sensor for oxidative stress and coordinates the expression of antioxidant stress response genes upon exposure to oxidative stimulation. We hypothesized that the protective effects of AS against LPS-induced oxidative stress (intracellular ROS accumulation) were mediated by the induction of antioxidant genes through Nrf2. As we predicted, Western blotting showed that AS (50 μg mL−1) significantly increased the nuclear translocation of Nrf2, as evidenced by the accumulation of Nrf2 in the nucleus. AS treatment also significantly increased the total Nrf2 expression in LPS-induced macrophages (Fig. 3A). Aberrant Nrf2 activation by AS was observed within 2 h, whereas the increase in Nrf2 expression was gradually decreased when AS was applied at later time points (Fig. 3A). To further confirm this protective effect, we monitored the nuclear translocation of Nrf2 using an immunofluorescence assay. As shown in Fig. 3B, the nuclear accumulation of Nrf2 was significantly increased by AS compared with the control cells. More precisely, Nrf2 expression in control cells was observed in the cytoplasm, whereas Nrf2 accumulation in the nucleus was observed after 1 h of treatment with AS (Fig. 3B). These results strongly suggest that AS promotes the activation and nuclear translocation of Nrf2 in macrophages.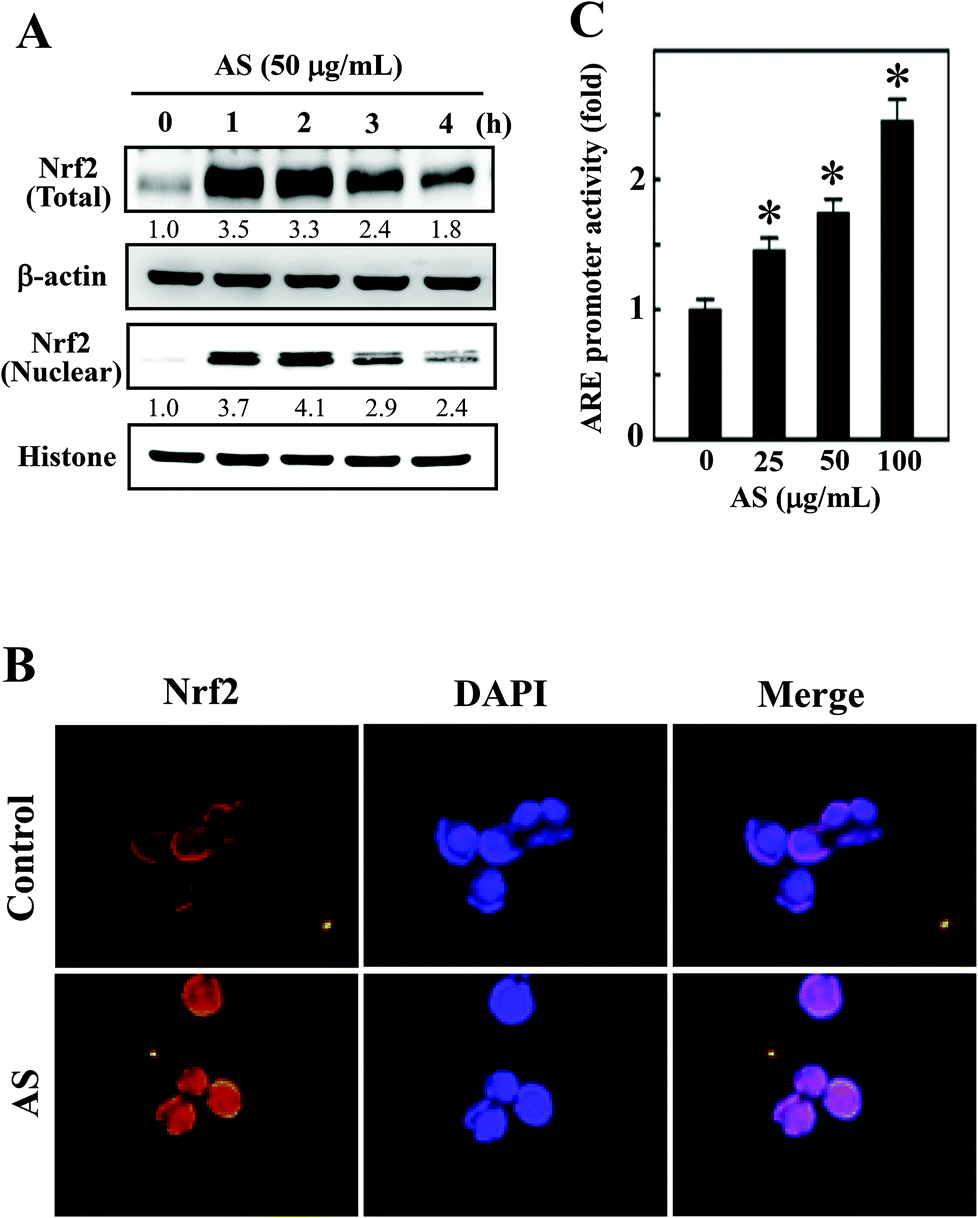 | ||
| Fig. 3 AS upregulates Nrf2 transcriptional activation in macrophages. (A) RAW264.7 cells were treated with AS (50 μg mL−1) for 1–4 h. Western blotting was performed to analyze the overall and nuclear Nrf2 expression using specific whole-cell and nuclear extracts. Equal amounts (50 μg) of the cell lysate from each sample were resolved by 8–15% SDS-PAGE. β-Actin and histone served as internal controls for the total and nuclear fractions, respectively. The relative changes in the intensities of the protein bands were measured by densitometry. (B) Cells grown in chamber slides were pre-treated with 50 μg mL−1 AS for 1 h, fixed, and permeabilized. Cells were incubated with an anti-Nrf2 antibody, followed by incubation with a FITC-labeled secondary antibody. The subcellular and nuclear localization of Nrf2 was visualized using a confocal microscope at 40× magnification. (C) ARE-luciferase activity was measured after transient transfected RAW264.7 cells were incubated with various concentrations of AS (25–100 μg mL−1) for 6 h. The results are the mean ± SD of three assays. *Indicates significant difference in comparison with the LPS-activated group (p < 0.05). | ||
AS activates ARE activity in macrophages
To further test the hypothesis that AS may promote the transcriptional activation of Nrf2 in RAW264.7 cells, the promoter activity of Nrf2 (ARE) was measured using a luciferase reporter assay. An Nrf2 promoter construct in a pcDNA vector was transiently transfected into RAW264.7 cells, and the induced luciferase activity was measured after treatment with various concentrations of AS (25–100 μg mL−1) for 1 h. The luciferase activity was normalized to the β-galactosidase activity in the cell lysate and was considered the basal level (1-fold). The luciferase activity derived from the ARE promoter was consistently increased after treatment with AS in a dose-dependent manner from 1-fold to 1.5-, 1.7-, and 2.5-fold by 25, 50, and 100 μg mL−1, respectively (Fig. 3C). The luciferase activity of blank plasmid pcDNA in RAW264.7 cells was not affected by AS treatment (data not shown). This result confirms that AS treatment not only induces the nuclear translocation of Nrf2 but also promotes ARE-driven transcriptional activity in RAW264.7 cells.AS upregulates HO-1 and NQO-1 expression in macrophages
The activated Nrf2 might induce various proteins with antioxidant and anti-inflammatory activities. HO-1 and NQO-1 are the anti-oxidant/anti-inflammatory molecules that are most strongly upregulated by Nrf2 under various stress conditions or in response to oxidants. Therefore, we hypothesized that the nuclear translocation and transcriptional activation of Nrf2 by AS might induce HO-1 and NQO-1 gene expression. As shown in Fig. 4A, compared with untreated control cells, AS (50 μg mL−1) significantly increased the HO-1 mRNA expression in a time-dependent manner. In particular, higher HO-1 expression was observed at 9 h after AS treatment in RAW264.7 cells (245 ± 4%). Furthermore, the mRNA expression of NQO-1 was also significantly increased by AS (50 μg mL−1) in a time-dependent manner; the highest expression was observed after 2 h (228 ± 6%), after which the NQO-1 level gradually declined (Fig. 4A). Concomitant with the mRNA expression, we also found that the protein levels of HO-1 were significantly increased by AS in a time-dependent manner, and the highest expression was observed after 8 h (210 ± 3%) (Fig. 4B). Similarly, the NQO-1 protein expression was significantly increased by AS. The highest level of NQO-1 expression was observed after 2–4 h treatment (243 ± 5%), after which the expression gradually declined (Fig. 4B). These data support our hypothesis that AS-induced Nrf2 activation promotes the transcription of antioxidant genes, including HO-1 and NQO-1.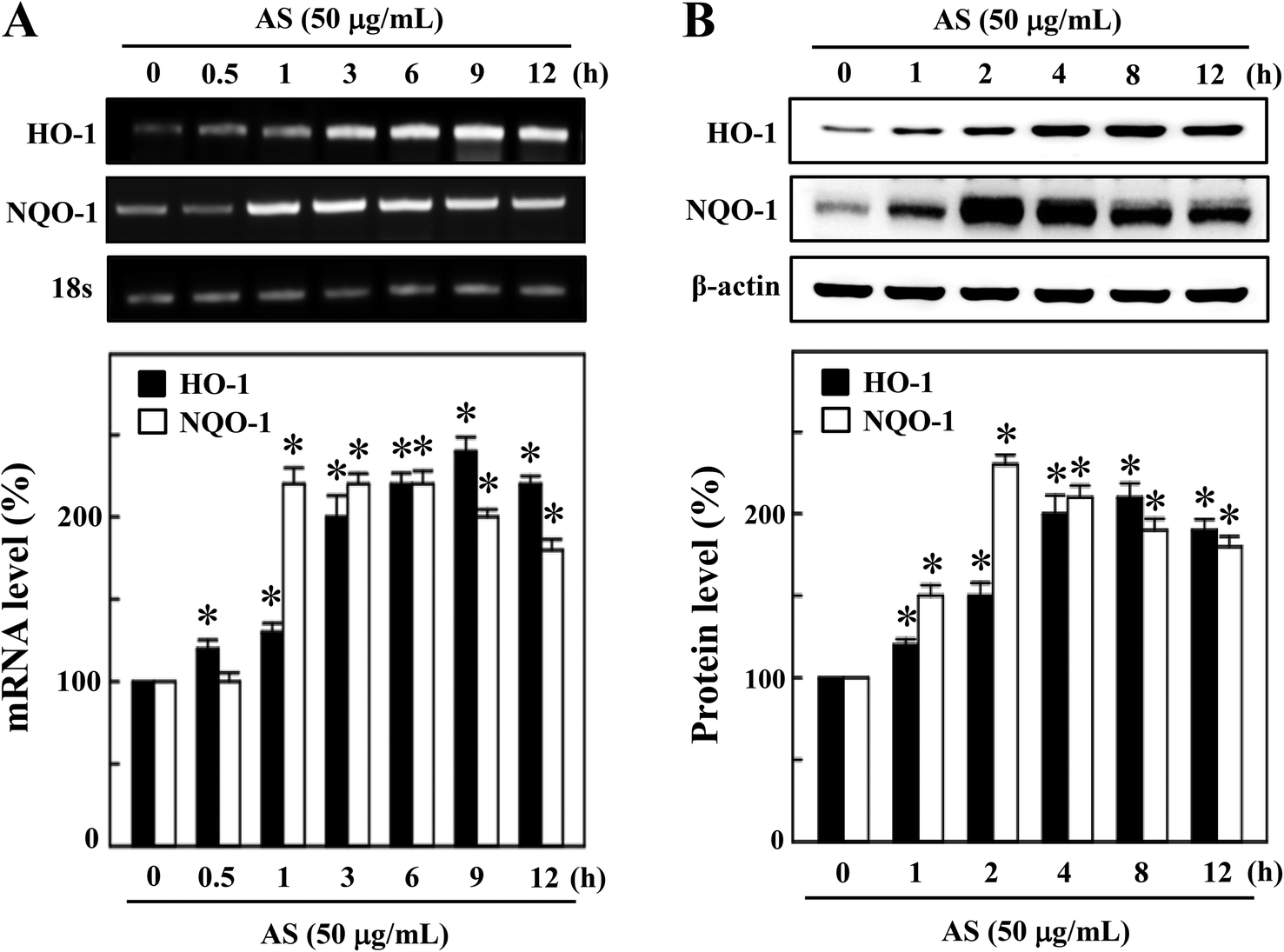 | ||
| Fig. 4 AS induces the expression of the antioxidant genes HO-1 and NQO-1 in macrophages. (A) RAW264.7 cells were incubated with or without 50 μg mL−1 AS for 0.5–12 h, and total RNA was subjected to RT-PCR using a one-step RT-PCR master mix. The RT-PCR products were separated by 1% agarose gel electrophoresis. GAPDH served as an internal control. Relative changes in mRNA bands were measured by densitometry. (B) Cells were incubated with or without 50 μg mL−1 AS for 1–12 h, and total cell lysate was subjected to Western blotting to monitor the HO-1 and NQO-1 protein expression using specific antibodies. Equal amounts (50 μg) of total lysate from each sample were resolved by 8–15% SDS-PAGE, with β-actin as a control. The relative changes in the intensities of the protein bands were measured by densitometry. The results are the mean ± SD of three assays. *Indicates significant difference from the LPS-activated group (p < 0.05). | ||
AS increased the intracellular GSH and γ-GCLC expression in macrophages
GSH is the predominant low-molecular weight thiol and the most important non-enzyme antioxidant in mammalian cells. GSH effectively protects cells from various oxidative stresses caused by scavenging free radicals, suppressing lipid peroxidation and removing hydrogen peroxides in the cellular system.21 Therefore, we hypothesized that the antioxidant potential of AS may be mediated by the induction of intracellular GSH, and the levels of intracellular GSH were determined. As shown in Fig. 5A, the untreated control cells showed a basal level of 280 ± 18 μM GSH, and AS (50 μg mL−1) treatment consistently increased intracellular GSH to 343 ± 35 μM, 368 ± 14 μM, and 423 ± 7 μM after 4, 8, and 12 h, respectively. To further confirm this protective effect, the level of γ-GCLC protein expression was monitored by Western blotting. GSH is sequentially synthesized from glutamate, cysteine, and glycine, and this synthesis is controlled by the rate limiting enzymes of these reactions, including γ-GCLC. We found that AS (50 μg mL−1) treatment significantly increased γ-GCLC expression in a time-dependent manner. Compared to the untreated control cells (100%), the AS-treated macrophages exhibited a more than 2-fold (200 ± 20%) increase in γ-GCLC expression (Fig. 5B). These data may suggest that AS eliminates LPS-induced oxidative stress by inducing the expression of γ-GCLC, leading to the elevation of intracellular GSH.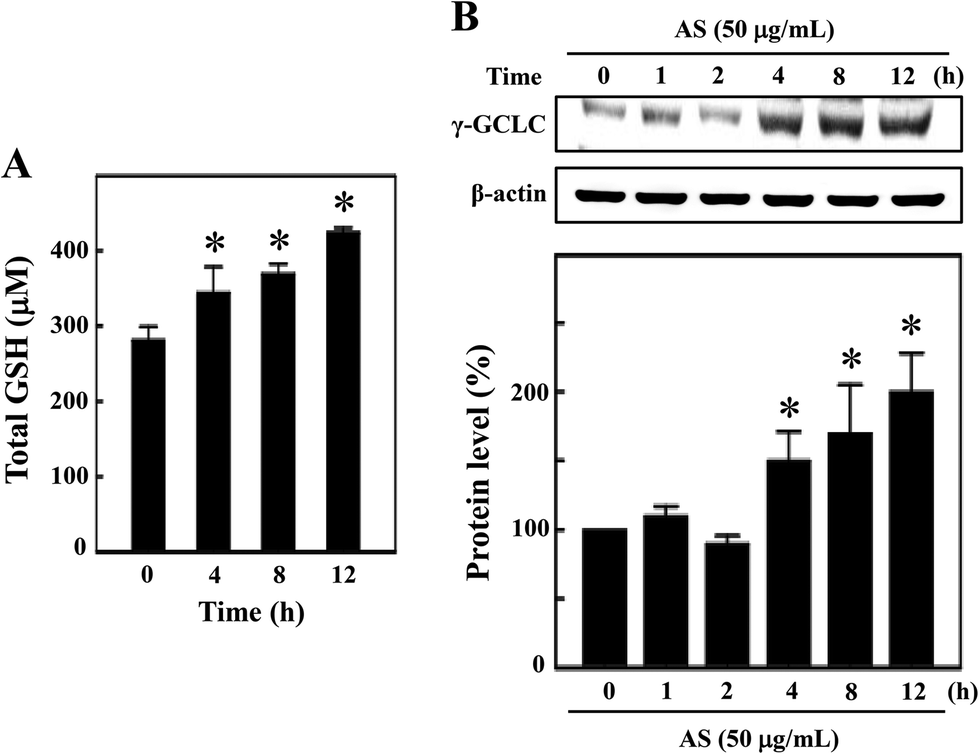 | ||
| Fig. 5 AS induces GSH formation and γ-GCLC protein expression in macrophages. (A) Intracellular GSH was measured using a commercially available ELISA kit, as described in the Materials and methods section. RAW264.7 cells were incubated with 50 μg mL−1 AS for 4–12 h. (B) Total cell lysate was subjected to Western blotting using specific antibodies to monitor γ-GCLC protein expression. Cells were incubated with or without AS (50 μg mL−1) for 1–12 h. Equal amounts (50 μg) of total lysate from each sample were resolved by 8–15% SDS-PAGE with β-actin as a control. The relative changes in the intensities of the protein bands were measured by densitometry. The results are the mean ± SD of three assays. *Indicates significant difference from the LPS-activated group (p < 0.05). | ||
Nrf2 knockdown diminishes the protective effect of AS in LPS-stimulated macrophages
To demonstrate the importance of Nrf2 regulation, we developed an Nrf2 knockdown model in RAW264.7 cells using siRNA transfection. The efficacy of the Nrf2 gene knockdown was confirmed by Western blot analysis, which showed that the transfection of siNrf2 (100 pM) led to a reduction in the Nrf2 protein level (Fig. 6A). However, the siRNA-induced reduction in Nrf2 was not altered by AS treatment (Fig. 6A) even after 18 h of AS treatment; that is, the transfection with siNrf2 abrogated the protective effect of AS on LPS-induced production of NO, PGE2, TNF-α, and IL-1β in RAW264.7 cells (Fig. 6B–E). This finding is evidence that the augmentation of Nrf2 is directly involved in AS-mediated anti-inflammatory effects in macrophages.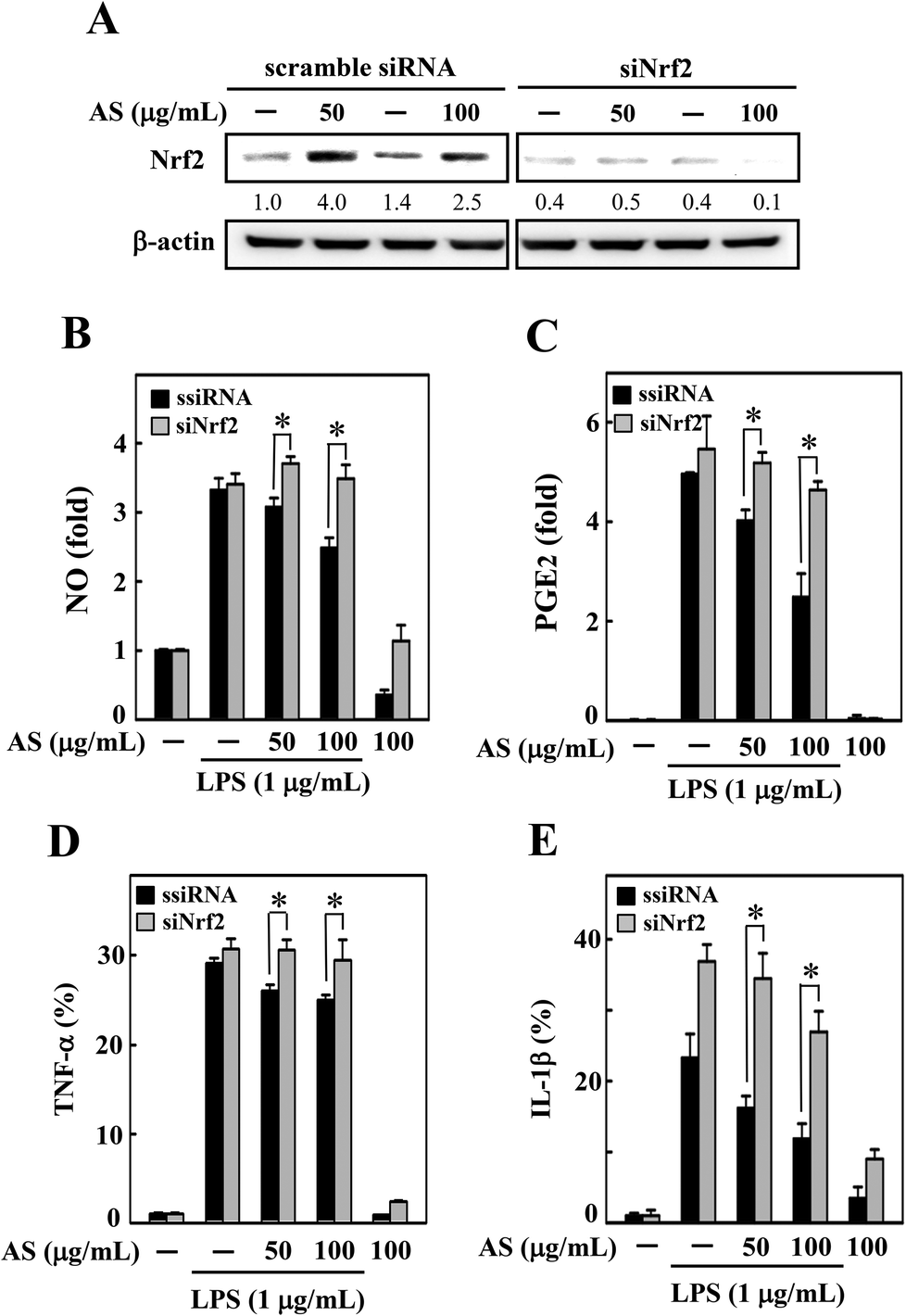 | ||
| Fig. 6 Nrf2 knockdown attenuates the protective effects of AS in LPS-stimulated macrophages. (A) RAW264.7 cells were transfected with siRNA targeting Nrf2 or a non-silencing control. Following transfection for 24 h, the cells were incubated with or without AS (50 or 100 μg mL−1) for 18 h. The Nrf2 knockdown was confirmed by Western blotting. (B–E) The control and Nrf2 siRNA-treated macrophages were incubated with or without AS (50 or 100 μg mL−1) for 18 h in the presence or absence of LPS (1 μg mL−1). (B) NO production was determined by measuring the formation of nitrite, the stable end-metabolite of NO in the culture media using the Griess assay. The levels of PGE2 (C), TNF-α (D), and IL-1β (E) in the culture media were measured using a commercial ELISA kit, as described in the Materials and methods section. The results are the mean ± SD of three assays. *Indicates a significant difference from the LPS-activated group (p < 0.05). | ||
AS upregulates the AKT and MAPK signaling pathways in macrophages
The role of PI3K/AKT activation in the transcriptional regulation of Nrf2 has been studied in detail.11 Based on these reports, we used Western blot analysis to monitor whether AS could induce PI3K activation in macrophages. As shown in Fig. 7, AS treatment caused a time-dependent increase in PI3K phosphorylation in macrophages. The phosphorylation levels peaked within 45 min and were sustained for 60 min.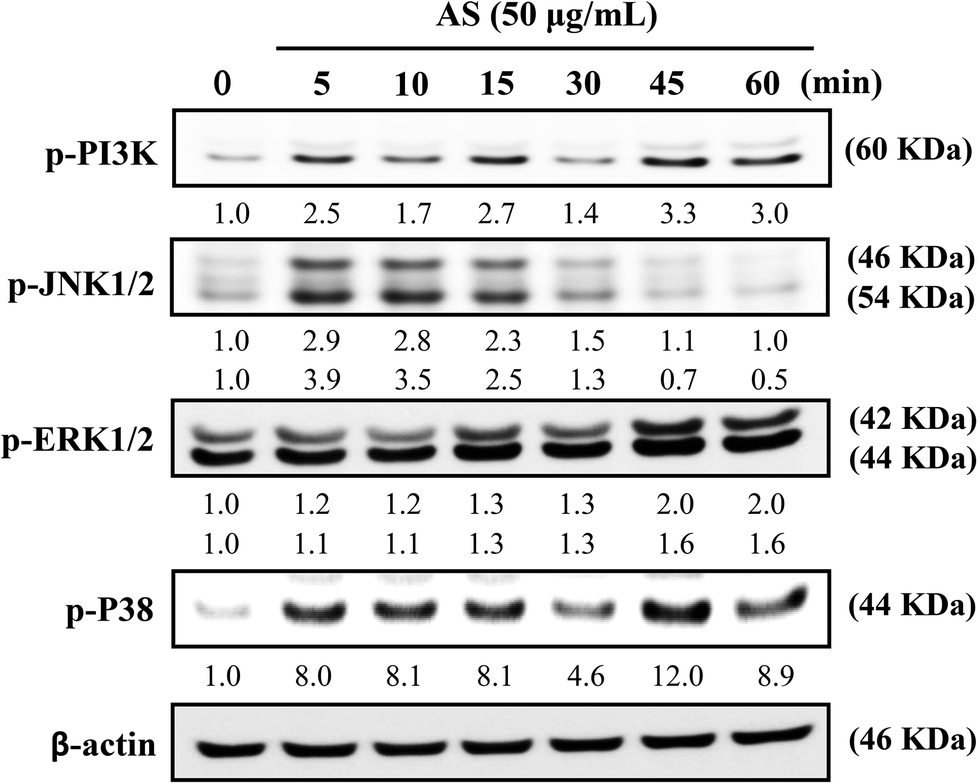 | ||
| Fig. 7 AS activates the PI3K and MAPK signaling pathways in macrophages. RAW264.7 cells were incubated with 50 μg mL−1 AS for 5–60 min, and the levels of phosphorylated PI3K, JNK1/2, ERK1/2, and p38 MAPK were determined using Western blot analysis. Equal amounts of total cell lysate (50 mg) were resolved by 8–12% SDS-PAGE, and Western blotting was performed. β-Actin was used as an internal control. The relative changes in the intensities of the protein bands were determined by densitometry. | ||
MAPK family proteins, such as JNK1/2, ERK1/2, and p38 MAPK, have been shown to play important roles in the activation of Nrf2.11 Therefore, the effects of AS on MAPK activation were examined. Macrophages were treated with AS (50 μg mL−1) for 5–60 min, and the resulting phosphorylation levels of JNK1/2, ERK1/2, and p38 MAPK were assessed by immunoblotting. As shown in Fig. 7, AS treatment caused a marked increase in JNK1/2 phosphorylation after 5–10 min, after which the phosphorylation gradually declined to the basal level. AS treatment also increased the phosphorylation of ERK1/2 in a time-dependent manner. The highest level of ERK1/2 phosphorylation was observed after 45 min and was sustained thereafter (Fig. 7). Furthermore, the phosphorylation of p38 MAPK was observed after 5 min of AS treatment and peaked after 45 min of treatment (Fig. 7). Taken together, these results suggest that AS-mediated induction of Nrf2 is driven by the activation of upstream kinases including PI3K/AKT, JNK1/2, ERK1/2, and p38 MAPK.
AS inhibits LPS-induced ICAM-1 expression in macrophages
Moreover, during inflammation, macrophage adhesion to endothelial cells is highly influenced by ICAM-1. Also, expression of ICAM-1 in macrophage cells is induced by various cytokines including IL-1β, TNF-α, and LPS.22 Therefore, next we sought to investigate the effect of AS on LPS-induced ICAM-1 expression in macrophage cells. AS shown in Fig. 8, compared with unstimulated control cells, LPS treatment significantly increased ICAM-1 expression (7.1-fold), whereas AS significantly reduced LPS-induced ICAM-1 expression.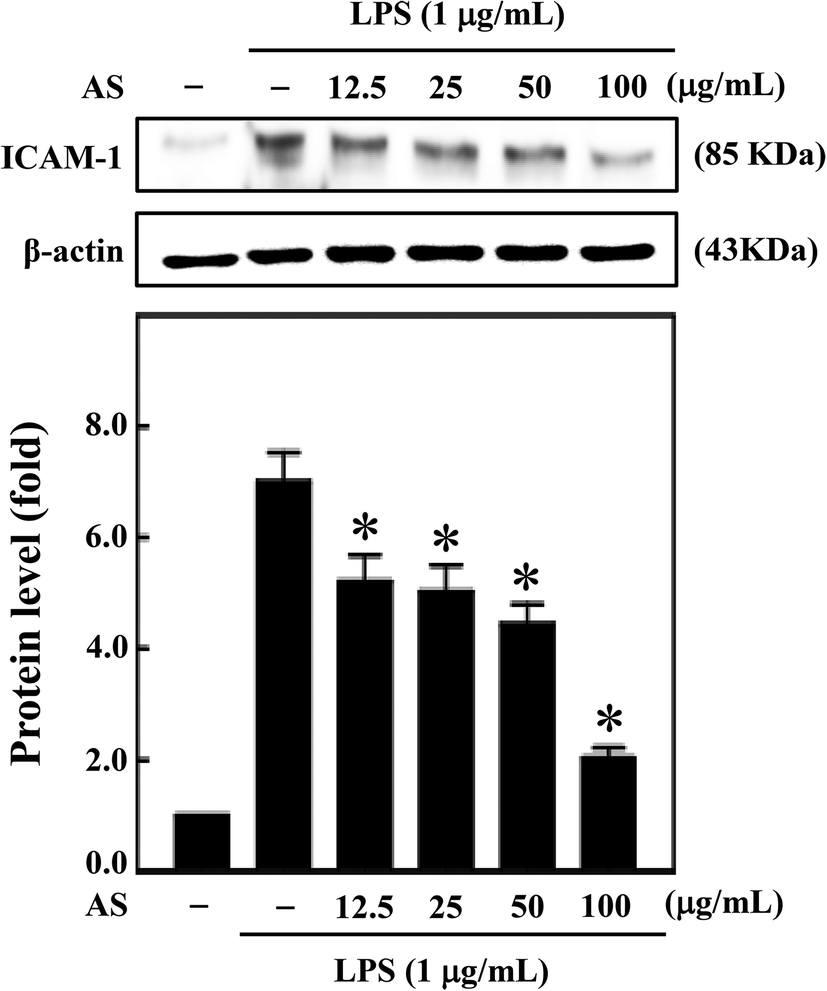 | ||
| Fig. 8 AS inhibits LPS-induced ICAM-1 expression in macrophage cells. RAW264.7 cells were treated with AS (12.5–100 μg mL−1) in the absence or presence of LPS (1 μg mL−1) for 24 h. Equal amounts (50 μg) of total lysate from each sample were resolved by 8–15% SDS-PAGE with β-actin as a control. The relative changes in the intensities of the protein bands were measured by densitometry. The results are the mean ± SD of three assays. *Indicates significant difference from the LPS-activated group (p < 0.05). | ||
Discussion
The results of this study clearly indicate that the antioxidant potential of AS was directly correlated with the increased expressions of the antioxidant genes HO-1, NQO-1, and γ-GCLC and the level of intracellular GSH, which was followed by an augmentation of nuclear translocation and the transcriptional activation of Nrf2-mediated antioxidant gene pathways. Nrf2 knockdown diminished the protective effects of AS, as evidenced by the increased production of pro-inflammatory cytokines and chemokines including PGE2, NO, TNF-α, and IL-1β in LPS-stimulated RAW264.7 cells. These results suggest that the activation of Nrf2-mediated antioxidant defense mechanisms by Antrodia salmonea protects macrophages from LPS-stimulated inflammation.ROS are created by a variety of cellular processes as part of cellular signaling events. A number of lines of evidence suggest that ROS participate in inflammation, and LPS-induced ROS generation has been widely studied in various in vitro and in vivo systems.23,24 In particular, ROS are major oxidative products that are mainly released by the mitochondria, cytochrome p450 metabolism, peroxisomes, and inflammatory cell activation by endotoxins in macrophages.25 Moreover, ROS production is an important component of the initiation and enhancement of cell death via apoptosis or autophagy.26 In this study, we observed that LPS treatment enhanced intracellular ROS accumulation in RAW264.7 macrophages and that the elevated ROS generation was significantly inhibited by AS.
It has been reported that Nrf2/ARE-mediated antioxidant genes are induced by various external stimuli and plant-derived natural products.11,27,28 Activated Nrf2 translocates from the cytoplasm to the nucleus and binds DNA at ARE-binding motifs to activate the transcription of various detoxifying (phase II) enzymes including HO-1, NQO-1, and γ-GCLC.5 This induction of these anti-oxidant genes has been assumed to be the mechanism through which Nrf2 inhibits LPS-induced inflammation. Several studies revealed that increases in the expressions of antioxidant enzymes (HO-1, NQO-1, and γ-GCLC) can reduce ROS levels in cells, eventually creating a reducing intracellular environment and maintaining Nrf2 in its augmented configuration.29 In this study, we found that the nuclear translocation of Nrf2 in macrophages significantly increased in the presence of AS, suggesting that the elimination of intercellular ROS may be due to the transcriptional activation of Nrf2 by AS. This is consistent with previous reports that the extracts of many plants contain anti-inflammatory agents that inhibit ROS formation via the activation of Nrf2 cascades in macrophages.11
The discovery of RNA interference has revolutionized the study of specific gene functions and pathways.30 Importantly, siRNA-directed transcriptional silencing is conserved in mammalian cells, providing a means of inhibiting the functions of specific mammalian genes.30 Our data show that siRNA-mediated knockdown of Nrf2 expression enhances the production of NO, PGE2, TNF-α, and IL-β in macrophages. AS failed to protect Nrf2-knockdown macrophages from LPS-induced NO, PGE2, TNF-α, and IL-β production. These data demonstrate that the protective effect of AS is mediated mainly by Nrf2 activation.
HO-1 is a well-known antioxidant enzyme that plays an important role in the defense against LPS-induced ROS generation and inflammation in macrophages.31 Several lines of evidence have demonstrated that HO-1 expression driven by Nrf2 and the products of HO-1 activity are likely to inhibit NO production.32 The genes regulated by Nrf2 include phase II enzymes such as NQO-1, which detoxify endogenous and exogenous chemicals through reduction and conjugation reactions.33 A previous study showed that NQO-1 knockdown led to the increased expression of LPS-induced inflammatory cytokines including TNF-α and IL-1β in cultured monocytes.34 The regulation of GSH/GCLC by Nrf2 also protects cells from LPS-induced inflammatory responses.35 These antioxidant enzymes are vital for protecting cells from oxidative stress and electrophile toxicity as well as preventing carcinogenesis. Thus, AS protects the macrophages from LPS-induced inflammation mainly by elevating the intracellular anti-oxidative enzymes by enhancing the accumulation of Nrf2, a transcription factor for antioxidant genes, and thus dramatically inducing the expression of the antioxidant genes HO-1, NQO-1, and γ-GCLC in response to LPS stimulation.
Moreover, cross-talk between the transcription factors NF-κB and Nrf2 is involved in inflammatory gene expression.28 The over-expression of Nrf2 suppressed the expression of pro-inflammatory genes in human arotic endothelial cells.36 Paur et al. (2010) reported that extracts of oregano, coffee, thyme, clove, and walnut inhibited LPS-induced NF-κB activation via the upregulation of Nrf2-mediated antioxidant genes in cultured monocytes.37 Furthermore, pre-treatment of peritoneal macrophage cells with the Nrf2 inducer sulforaphane appeared to inhibit LPS-induced induction of iNOS, COX-2, TNF-α, and IL-1β expressions.38 Thus, it is reasonable to assume that there are at least two possible explanations for the mechanism underlying the anti-inflammatory effect of AS: the inactivation of NF-κB and activation of Nrf2, which stimulates anti-inflammatory HO-1 gene expression. In this regard, controlling the macrophage activation may have potential therapeutic implications for the treatment of various inflammatory diseases.
The PI3K/AKT and MAPK signaling pathways, including the JNK, ERK, and MAPK p38 pathways, and their downstream transcription factors play critical roles in cell fate determination. In addition, previous studies have demonstrated that the transduction of oxidative stress signals to ARE-mediated gene expression involves three main pathways, i.e., the activation of the PI3K/AKT and MAPK cascades.11 Therefore, to further identify the upstream regulatory mechanisms involved in AS-induced Nrf2 signaling, the PI3K/AKT and MAPK pathways were examined in the present study. We found that AS significantly induced the phosphorylation of PI3K, JNK, ERK, and p38 MAPK in LPS-challenged RAW264.7 cells. One possible explanation for this observation is that the activation of the Nrf2/ARE pathway may be associated with the activation of the PI3K/AKT and MAPK cascades.
Recent studies have shown that certain bioactive compounds, such as ergostanes, lanostanes, and naphthoquinones, are present in AS and exhibit potential anti-oxidative and anti-inflammatory effects in vitro.15,17 Three new ergostanes exhibited anti-oxidative effects by inhibiting NADPH oxidase activity but did not exhibit free radical scavenging activity. In addition, the ergostanes strongly inhibited nitric oxide production in activated neutrophils and microglial cells. A previous study also showed that ethanolic extracts of AS significantly reduced the serum levels of NO and TNF-α as well as the expression of the iNOS protein, both in vitro and in vivo, which might be mediated through the pharmacological effect of adenosine.16 Huang et al. (2012) also reported that two bioactive compounds, adenosine and zhankuic acid A, have been identified in the ethanolic extract of the fruiting body of AS.16 During the inflammation, adenosine levels rise and modulate inflammatory responses by interacting with their receptors.39 In other experimental studies it was also observed that Antrodia salmonea contains an enormous amount of polyphenols, which are believed to be the molecules responsible for its anti-oxidative effects.15,17 Moreover, our recent study with HPLC analysis showed that AS was composed ∼11.6% of 2,4-dimethoxy-6-methylbenzene-1,3-diol.13 Taken together, these studies demonstrate that different extraction conditions produced compounds with different biological activities. Our previous studies revealed that fermented culture broths of Antrodia camphorata, a similar medicinal fungus, exhibited various biological activities.40–42 Therefore, we believe that the anti-oxidant and anti-inflammatory effects of AS may be attributed to the pharmacological activities of its major bioactive compounds. However, further studies of these bioactive compounds are highly warranted.
Our finding that Antrodia salmonea can induce the expression of antioxidant genes through the activation of the Nrf2 signaling pathway is novel and important. The delayed and increased induction of HO-1, NQO-1, γ-GCLC and the elevation of GSH likely tilt the balance in favor of an anti-oxidative and anti-inflammatory environment. Based on the above reported bioactivities, it is clear that Antrodia salmonea exerted an anti-inflammatory effect on LPS-stimulated macrophages through the upregulation of Nrf2-mediated antioxidant defense mechanisms. This antioxidant and anti-inflammatory potential may therefore indicate the presence of a novel mechanism of action underlying the apparent efficacy of this beneficial mushroom.
Abbreviations
| AS | Antrodia salmonea |
| LPS | Lipopolysaccharide |
| ROS | Reactive oxygen species |
| Nrf2 | NF-E2-related factor-2 |
| ARE | Antioxidant response element |
| HO-1 | Heme oxygenase-1 |
| NQO-1 | NAD(P)H:quinone acceptor oxidoreductase 1 |
| γ-GCLC | γ-glutamate-cysteine ligase catalytic subunit |
| GSH | Glutathione |
| NO | Nitric oxide |
| PGE2 | Prostaglandin E2 |
| TNF-α | Tumor necrosis factor-α |
| IL-1β | Interleukin-1β |
| PI3K | Phosphatidylinositol 3-kinase |
| JNK | c-JUN N-terminal kinase |
| ERK | Extracellular signal-regulated kinase |
| ICAM-1 | Intercellular adhesion molecule-1 |
Acknowledgements
This work was supported by grants NSC-101-2320-B-039-050-MY3, NSC-103-2622-B-039-001-CC2, CMU 100-ASIA-13, and CMU 100-ASIA-14 from the National Science Council, China Medical University, and Asia University, Taiwan.References
- A. Linde, D. Mosier, F. Blecha and T. Melgarejo, Innate immunity and inflammation—New frontiers in comparative cardiovascular pathology, Cardiovasc. Res., 2007, 73, 26–36 CrossRef CAS PubMed.
- Y. Kharraz, J. Guerra, C. J. Mann, A. L. Serrano and P. Munoz-Canoves, Macrophage plasticity and the role of inflammation in skeletal muscle repair, Mediators Inflamm., 2013, 2013, 491497 CrossRef PubMed.
- T. R. Martin, Recognition of bacterial endotoxin in the lungs, Am. J. Respir. Cell Mol. Biol., 2000, 23, 128–132 CrossRef CAS PubMed.
- J. W. Jeong, W. S. Lee, S. C. Shin, G. Y. Kim, B. T. Choi and Y. H. Choi, Anthocyanins downregulate lipopolysaccharide-induced inflammatory responses in BV2 microglial cells by suppressing the NF-κB and Akt/MAPKs signaling pathways, Int. J. Mol. Sci., 2013, 14, 1502–1515 CrossRef CAS PubMed.
- J. K. Kundu and Y. J. Surh, Breaking the relay in deregulated cellular signal transduction as a rationale for chemoprevention with anti-inflammatory phytochemicals, Mutat. Res., 2005, 591, 123–146 CrossRef CAS PubMed.
- M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Free radicals, metals and antioxidants in oxidative stress-induced cancer, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS PubMed.
- M. J. Alcaraz, A. M. Vicente, A. Araico, J. N. Dominguez, M. C. Terencio and M. L. Ferrandiz, Role of nuclear factor-kappaB and heme oxygenase-1 in the mechanism of action of an anti-inflammatory chalcone derivative in RAW 264.7 cells, Br. J. Pharmacol., 2004, 142, 1191–1199 CrossRef CAS PubMed.
- M. Kobayashi and M. Yamamoto, Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species, Adv. Enzyme Regul., 2006, 46, 113–140 CrossRef CAS PubMed.
- N. Li, J. Alam, M. I. Venkatesan, A. Eiguren-Fernandez, D. Schmitz, E. Di Stefano, N. Slaughter, E. Killeen, X. Wang, A. Huang, M. Wang, A. H. Miguel, A. Cho, C. Sioutas and A. E. Nel, Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals, J. Immunol., 2004, 173, 3467–3481 CrossRef CAS.
- T. Nguyen, P. Nioi and C. B. Pickett, The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress, J. Biol. Chem., 2009, 284, 13291–13295 CrossRef CAS PubMed.
- Y. J. Surh, Cancer chemoprevention with dietary phytochemicals, Nat. Rev. Cancer, 2003, 3, 768–780 CrossRef CAS PubMed.
- C. C. Shen, C. F. Lin, Y. L. Huang, S. T. Wan, C. C. Chen, S. J. Sheu, Y. C. Lin and C. C. Chen, Bioactive components from the mycelium of Antrodia salmonea, J. Chin. Chem. Soc., 2008, 55, 854–857 CAS.
- Y. C. Hseu, C. C. Lee, Y. C. Chen, K. J. Senthil Kumar, C. S. Chen, C. T. Tsai, H. C. Huang, H. M. Wang and H. L. Yang, Antrodia salmonea in submerged culture exhibits antioxidant activities in vitro and protects human erythrocytes and low-density lipoproteins from oxidative modification, Food Chem. Toxicol., 2014, 66, 150–157 CrossRef CAS PubMed.
- S. J. Sheu, Y. L. Chen, C. Y. Yang, C. C. Chen, K. Xia, C. M. Chiu and H. F. Chou, Studies on acute toxicity of Antrodia salmonea fermented product, 4th General Assembly of Health Food Society of Taiwan, 2005 Search PubMed.
- C. C. Shen, Y. C. Shen, Y. H. Wang, L. C. Lin, M. J. Don, K. T. Liou, W. Y. Wang, Y. C. Hou and T. T. Chang, New lanostanes and naphthoquinones isolated from Antrodia salmonea and their antioxidative burst activity in human leukocytes, Planta Med., 2006, 72, 199–203 CAS.
- G. J. Huang, C. H. Pan, F. C. Liu, T. S. Wu and C. H. Wu, Anti-inflammatory effects of ethanolic extract of Antrodia salmonea in the lipopolysaccharide-stimulated RAW246.7 macrophages and the lambda-carrageenan-induced paw edema model, Food Chem. Toxicol., 2012, 50, 1485–1493 CrossRef CAS PubMed.
- C. C. Shen, Y. H. Wang, T. T. Chang, L. C. Lin, M. J. Don, Y. C. Hou, K. T. Liou, S. Chang, W. Y. Wang, H. C. Ko and Y. C. Shen, Anti-inflammatory ergostanes from the basidiomata of Antrodia salmonea, Planta Med., 2007, 73, 1208–1213 CrossRef CAS PubMed.
- H. O. Pae, G. S. Jeong, H. S. Kim, W. H. Woo, H. Y. Rhew, H. S. Kim, D. H. Sohn, Y. C. Kim and H. T. Chung, Costunolide inhibits production of tumor necrosis factor-alpha and interleukin-6 by inducing heme oxygenase-1 in RAW264.7 macrophages, Inflamm. Res., 2007, 56, 520–526 CrossRef CAS PubMed.
- Y. C. Hseu, C. W. Chou, K. J. Senthil Kumar, K. T. Fu, H. M. Wang, L. S. Hsu, Y. H. Kuo, C. R. Wu, S. C. Chen and H. L. Yang, Ellagic acid protects human keratinocyte HaCaT cells against UVA-induced oxidative stress and apoptosis through the upregulation of the HO-1 and Nrf-2 antioxidant genes, Food Chem. Toxicol., 2012, 50, 1245–1255 CrossRef CAS PubMed.
- Y. C. Hseu, H. C. Huang and C. Y. Hsiang, Antrodia camphorata suppresses lipopolysaccharide-induced nuclear factor-kappaB activation in transgenic mice evaluated by bioluminescence imaging, Food Chem. Toxicol., 2010, 48, 2319–2325 CrossRef CAS PubMed.
- D. Han, N. Hanawa, B. Saberi and N. Kaplowitz, Mechanisms of liver injury. III. Role of glutathione redox status in liver injury, Am. J. Physiol. Gastrointest Liver Physiol., 2006, 291, G1–G7 CrossRef CAS PubMed.
- P. G. Frank and M. P. Lisanti, ICAM-1: role in inflammation and in the regulation of vascular permiability, Am. J. Physiol. Heart Circ. Physiol., 2008, 295, H-926–H-927 CrossRef CAS PubMed.
- S. Cuzzocrea, D. P. Riley, A. P. Caputi and D. Salvemini, Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury, Pharmacol. Rev., 2001, 53, 135–159 CAS.
- K. Rahman, Studies on free radicals, antioxidants, and co-factors, Clin. Interv. Aging, 2007, 2, 219–236 CAS.
- M. Mohsenzadegan and A. Mirshafiey, The immunopathogenic role of reactive oxygen species in Alzheimer disease, Iran. J. Allergy Asthma Immunol., 2012, 11, 203–216 CAS.
- G. Manda, M. T. Nechifor and T. M. Neagu, Reactive oxygen species, cancer and anti-cancer therapies, Curr. Chem. Biol., 2009, 3, 342–366 CrossRef CAS.
- G. Scapagnini, S. Vasto, N. G. Abraham, C. Caruso, D. Zella and G. Fabio, Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders, Mol. Neurobiol., 2011, 44, 192–201 CrossRef CAS PubMed.
- Y. J. Surh and H. K. Na, NF-kappaB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals, Genes Nutr., 2008, 2, 313–317 CrossRef CAS PubMed.
- W. Droge, Free radicals in the physiological control of cell function, Physiol. Rev., 2002, 82, 47–95 CAS.
- B. A. Narayanan, N. K. Narayanan, L. Davis and D. Nargi, RNA interference-mediated cyclooxygenase-2 inhibition prevents prostate cancer cell growth and induces differentiation: modulation of neuronal protein synaptophysin, cyclin D1, and androgen receptor, Mol. Cancer Ther., 2006, 5, 1117–1125 CrossRef CAS PubMed.
- J. P. Roach, E. E. Moore, D. A. Partrick, S. S. Damle, C. C. Silliman, R. C. McIntyre Jr. and A. Banerjee, Heme oxygenase-1 induction in macrophages by a hemoglobin-based oxygen carrier reduces endotoxin-stimulated cytokine secretion, Shock, 2009, 31, 251–257 CrossRef CAS PubMed.
- F. A. Wagener, H. D. Volk, D. Willis, N. G. Abraham, M. P. Soares, G. J. Adema and C. G. Figdor, Different faces of the heme-heme oxygenase system in inflammation, Pharmacol. Rev., 2003, 55, 551–571 CrossRef CAS PubMed.
- R. K. Thimmulappa, K. H. Mai, S. Srisuma, T. W. Kensler, M. Yamamoto and S. Biswal, Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray, Cancer Res., 2002, 62, 5196–5203 CAS.
- S. A. Rushworth, D. J. MacEwan and M. A. O'Connell, Lipopolysaccharide-induced expression of NADPH:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes, J. Immunol., 2008, 181, 6730–6737 CrossRef CAS.
- R. K. Thimmulappa, C. Scollick, K. Traore, M. Yates, M. A. Trush, K. T. Liby, M. B. Sporn, M. Yamamoto, T. W. Kensler and S. Biswal, Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide, Biochem. Biophys. Res. Commun., 2006, 351, 883–889 CrossRef CAS PubMed.
- X. L. Chen, G. Dodd, S. Thomas, X. Zhang, M. A. Wasserman, B. H. Rovin and C. Kunsch, Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression, Am. J. Physiol. Heart Circ. Physiol., 2006, 290, H1862–H1870 CrossRef CAS PubMed.
- I. Paur, T. R. Balstad, M. Kolberg, M. K. Pedersen, L. M. Austenaa, D. R. Jacobs Jr. and R. Blomhoff, Extract of oregano, coffee, thyme, clove, and walnuts inhibits NF-kappaB in monocytes and in transgenic reporter mice, Cancer Prev. Res., 2010, 3, 653–663 CrossRef PubMed.
- W. Lin, R. T. Wu, T. Wu, T. O. Khor, H. Wang and A. N. Kong, Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway, Biochem. Pharmacol., 2008, 76, 967–973 CrossRef CAS PubMed.
- B. N. Cronstein, Adenosine, an endogenous anti-inflammatory agent, J. Appl. Physiol., 1994, 76, 5–13 CAS.
- Y. C. Hseu, W. C. Chang, Y. T. Hseu, C. Y. Lee, Y. J. Yech, P. C. Chen and H. L. Yang, Protection of oxidative damage by water extract from Antrodia camphorata mycelia in normal human red blood cells, Life Sci., 2002, 71, 469–482 CrossRef CAS.
- H. L. Yang, C. S. Chen, W. H. Chang, F. J. Lu, Y. C. Lai, C. C. Chen, T. H. Hseu, C. T. Kuo and Y. C. Hseu, Growth inhibition and induction of apoptosis in MCF-7 breast cancer cells by Antrodia camphorata, Cancer Lett., 2006, 231, 215–227 CrossRef CAS PubMed.
- H. L. Yang, K. J. Senthil Kumar and Y. C. Hseu, Multiple molecular targets of Antrodia camphorata: A suitable candidate for ovarian cancer chemoprevention, in Targeting New Pathways and Cell Death in Breast Cancer, ed. R. Aft, Intech Press, Rijeka, Croatia, 2012, pp. 157–180 Search PubMed.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |