
Ex vivo digestion of carp muscle tissue – ACE inhibitory and antioxidant activities of the obtained hydrolysates
J.
Borawska†
*a,
M.
Darewicz†
a,
G. E.
Vegarud
b,
A.
Iwaniak
a and
P.
Minkiewicz
a
aDepartment of Food Biochemistry, Faculty of Food Science, University of Warmia and Mazury in Olsztyn, Cieszyński 1, 10-726 Olsztyn, Poland. E-mail: justyna.borawska@uwm.edu.pl
bDepartment of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, 1432 Ås, Norway
First published on 16th October 2014
Abstract
In the digestive tract of humans, bioactive peptides, i.e. protein fragments impacting the physiological activity of the body, may be released during the digestion of food proteins, including those of fish. The aim of the study was to establish the method of human ex vivo digestion of carp muscle tissue and evaluate the angiotensin I-converting enzyme inhibitory and antioxidant activities of hydrolysates obtained after digestion. It was found that the hydrolysates of carp muscle tissue obtained with the three-stage method of simulated ex vivo digestion showed ACE inhibitory as well as antioxidative activities. It was demonstrated that the degree of hydrolysis depended on the duration of individual stages and the degree of comminution of the examined material. Although the applied gastric juices initiated the process of hydrolysis of carp muscle tissue, the duodenal juices caused a rapid increase in the amount of hydrolysed polypeptide bonds. The antihypertensive and antioxidative activities of the hydrolysates of carp muscle tissue increased together with progressive protein degradation. However, the high degree of protein hydrolysis does not favour an increase in the activity of free radical scavenging. The presented results are an example of the first preliminary screening of the potential health-promoting biological activity of carp muscle tissue in an ex vivo study.
Introduction
Fish have been used in human nutrition for over 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years. Fish and fish products are valuable nutritional foodstuffs and carp is among them, the most commonly bred fish in the world.1 In 2009, nearly 97% of European carp production came from 6 countries, mainly Poland, the Czech Republic, Germany, Hungary, France and Lithuania. Poland is one of the biggest producers of carp (Cyprinus carpio), a common freshwater fish. In 2010–2011 carp production in Poland exceeded 15 thousand tons.2 Carp meat contains 15–18% proteins and 2–8% lipids, of which 13–20% are PUFA.3
000 years. Fish and fish products are valuable nutritional foodstuffs and carp is among them, the most commonly bred fish in the world.1 In 2009, nearly 97% of European carp production came from 6 countries, mainly Poland, the Czech Republic, Germany, Hungary, France and Lithuania. Poland is one of the biggest producers of carp (Cyprinus carpio), a common freshwater fish. In 2010–2011 carp production in Poland exceeded 15 thousand tons.2 Carp meat contains 15–18% proteins and 2–8% lipids, of which 13–20% are PUFA.3
In recent years, there has been increased interest in analysing the digestion of food and its components in the digestive tract in humans. Digestion begins in the mouth and is completed in the small intestine.4 From the perspective of result reliability, it is most desirable to conduct studies on humans or, alternatively, on animals. However, high cost, time-consuming nature and ethical considerations have prompted a search for alternative methods. In practice, after implementing in vitro methods and, due to the complexity of digestion, there may be discrepancies between the results of in vivo and in vitro studies.5 The aspects that must be taken into account during development of an in vitro or ex vivo method of digestion are related to, among others, the type of food matrix (liquid/solid) and the form of sample comminution (e.g. food particles, a homogenate or an extract of the tested substances), the number of digestion stages (the mouth, stomach, duodenum, small intestine and large intestine), the specificity of added enzymes and their activities, pH and duration.5–7
During the digestion of food proteins (including fish proteins), bioactive peptides, i.e. protein fragments that impact the physiological activity of the body, may be released in the human digestive tract.8 Angiotensin I-converting enzyme (ACE) [EC 3.4.15.1] is an important enzyme for controlling blood pressure. Antihypertensive peptides with an ACE inhibitory activity are the best-known group of food protein-derived bioactive peptides.9 It has been reported that hydrolysates obtained in vitro from fish display ACE inhibitory activity.9 Numerous in vivo studies of antihypertensive peptides derived from marine resources have shown potent ACE inhibitory activity in spontaneously hypertensive rats (SHR).10
Fragments possessing antioxidative activity that may reduce the rate of enzymatic and non-enzymatic oxidation are another group of peptides which are interesting in prophylaxis of diet-related diseases. To reduce the risk of diet-related diseases, the removal of reactive oxygen molecules is recommended. The reviews of the production and biological activities of marine-derived biologically active peptides, including ACE inhibitory and antioxidant activities from fish sources, were reported by Ryan et al.11 These peptides may be used in the prophylaxis of diet-related diseases as components of functional food.
Enzymes derived from the human digestive tract (ex vivo digestion) have been used in only a few experimental studies on the enzymatic release of biologically active peptides from food proteins. These studies have mimicked or approximated the physiological conditions of the human body.12,13 The results of studies with human digestive enzymes (ex vivo digestion) are closer to the results from in vivo rather than in vitro experiments.5,14
The carp production plays an important role in the fishery in Poland and other European countries. Moreover, this fish has not been studied so far. Thus, we focused our present research on carp potential to generate bioactive peptides during digestion. Since its potential as a substrate for the production of bioactive peptides via human digestive juices has not been reported earlier, the aim of the present study was to establish the method of human ex vivo digestion of carp muscle tissue and evaluate the angiotensin I-converting enzyme inhibitory and antioxidant activities of hydrolysates obtained after digestion.
Experimental
Materials
Fresh carp (Cyprinus carpio) was purchased directly from a fish farm near Olsztyn (Poland) and transported immediately to the laboratory on ice. Fish samples, averaged from fragmented 3 kg of skinless fillets, were washed, packed and stored in a freezer at −70 °C. Sodium dodecyl sulphate (SDS), ACE from rabbit lungs, hippuryl-L-histidyl-L-leucine (HHL), pyridine, benzene sulphonyl chloride (BSC), DPPH (1,1-diphenyl-2-picrylhydrazyl), ABTS (2,2-azinobis(3-ethyl-benzothiazoline-6-sulphonic acid)), phosphate buffered saline (PBS), and Trolox were purchased from Sigma-Aldrich (USA), an LMW-SDS Calibration Kit from GE Healthcare Life Sciences (UK), Protein Assay Dye Reagent Concentrate and bovine serum albumin (BSA) from Bio-Rad (USA). All other chemicals used in the experiments were of analytical grade. Highly purified water was prepared using a Milli-Q PLUS (Millipore Corp. NY, USA) and used for the preparation of all buffers and solutions.Methods
Ex vivo digestion
The ex vivo digestion method was performed according to Almaas et al.12,14 with modifications. Human gastric juice (HGJ) and human duodenal juice (HDJ) were collected according to Ulleberg et al.15 All gastric and duodenal enzymes used in the study were a batch obtained from six healthy adults. The aspiration was approved by the Norwegian Ethics Committee, and all volunteers had given a written consent to participate in the study. The pH values of individual juices varied from 1.0 to 4.0 (gastric juice) and from 5.9 to 7.0 (duodenal juice). The pepsin activity of HGJ was 36.85 U mL−1, and the ‘total proteolytic activity’ in duodenal juice was 12.35 U mL−1.Digestion was carried out in three steps: (1) “chewing” – 3 min, (2) “stomach” with a gradual lowering of pH from ∼6.2 to 2.5 that continued for 1–2 hours, (3) “duodenal” lasting for 1 hour, at pH = 7.0, as is presented in Fig. 1.
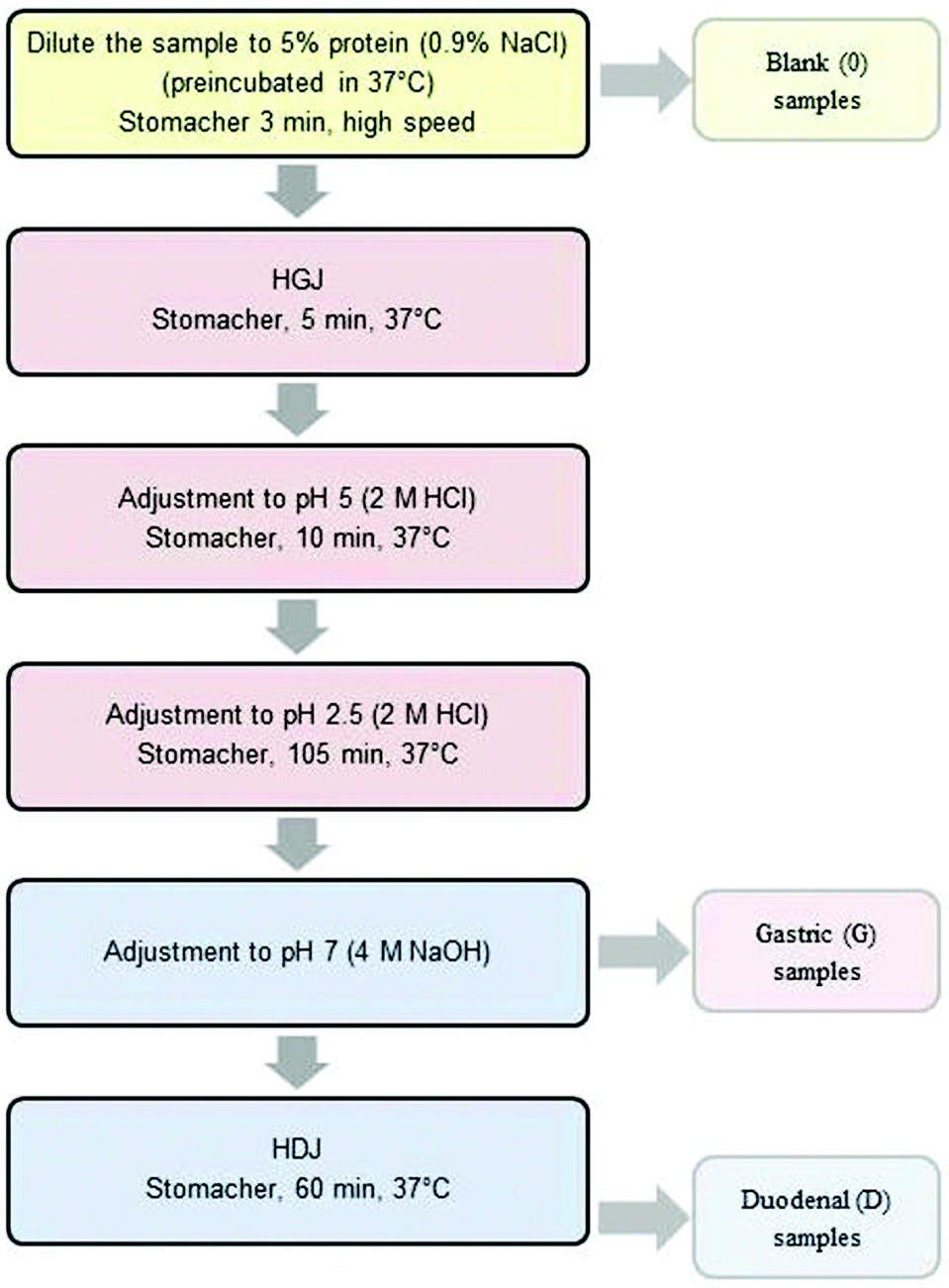 | ||
| Fig. 1 The scheme of ex vivo digestion. | ||
In order to investigate the impact of the degree of comminution on the range of carp protein hydrolysis, three degrees of comminution were used: homogenization (homog.), cutting into cubes with a side length of about 1 cm (cubes) and cutting and crushing the tissue with a knife (middle).
The comminuted muscle tissue of carp fillets was diluted up to the final protein content of 5% (w/v) with a 0.9% NaCl (w/v) solution at 37 °C. These samples were mixed in a Stomacher 400 (Seward, UK) for 3 minutes at 37 °C and a “blank” (“0”) sample was collected. Subsequently, HGJ was added at 15 U per gram of protein. After mixing for 5 minutes in a Stomacher 400 (37 °C), the pH of the samples was reduced to 5.0 (2 M HCl) and after 10 additional minutes to 2.5 and the samples were then incubated for 45 or 105 minutes. Thereafter, pH was increased to 7.0 with a 4 M NaOH solution. At this stage, the “gastric” samples (“G”) were collected. Next, HDJ was added at 31.2 U per gram of protein. The digested carp tissue was mixed in a Stomacher 400 (37 °C) for 60 minutes. “Duodenal” (D) samples were taken every 15 minutes.
The samples collected at each stage of the experiment were cooled in ice and centrifuged (9000g, 20 min, 4 °C). Supernatants were collected, frozen and then freeze-dried and stored at −18 °C. The ex vivo digestion procedure was performed in triplicate.
Protein content
The protein samples were measured with a microprotein assay according to Bradford.16 A microprotein assay using the diluted Protein Assay Dye Reagent Concentrate and BSA was used to determine the standard curve. Samples or BSA (0.02 mL) were mixed with Bradford Dye Reagent (1 mL). The absorbance was measured at 595 nm after 5–10 min against a reagent blank.Based on the results, a standard curve of known concentrations of BSA was used to calculate the protein concentration in the tested samples. The measurements were taken in triplicates and the results were presented as average values.
The remaining protein content of samples was calculated as the ratio of the protein content of the sample after digestion to the protein content of the “0” sample. The obtained results were expressed in percentages.
SDS-PAGE method
SDS–PAGE was carried out to evaluate the protein profile after each digestion step (“Mini-PROTEAN”, Bio-Rad, USA). The assay was performed according to standard protocols,17 using 12% and 15% separating acrylamide gels. The molecular mass markers used were the Low MW standard kit (97000–14400 Da, GE Healthcare Life Sciences, UK). Staining was performed according to the standard Coomassie procedure (Bio-Rad, USA).
ACE inhibition assay
ACE inhibitory activity was assayed by measuring the release of HA from the substrate HHL according to Jimsheena and Gowda.18 The assay mixture contained 0.125 mL of a 0.05 M sodium borate buffer (pH 8.2), containing 0.3 M NaCl, 0.05 mL of 5 mM HHL and 0.025 mL of ACE (2.5 mU), which was pre-incubated with different sample concentrations. The reaction was stopped after incubation at 37 °C for 30 min by the addition of 0.2 mL of 1 M HCl. Pyridine (0.4 mL) was added followed by a 0.2 mL of BSC (the order of addition of reagents is critical), mixed by inversion for 1 min and cooled on ice. The absorbance was measured at 410 nm.The degree of ACE inhibition (%) was calculated with the following equation19:
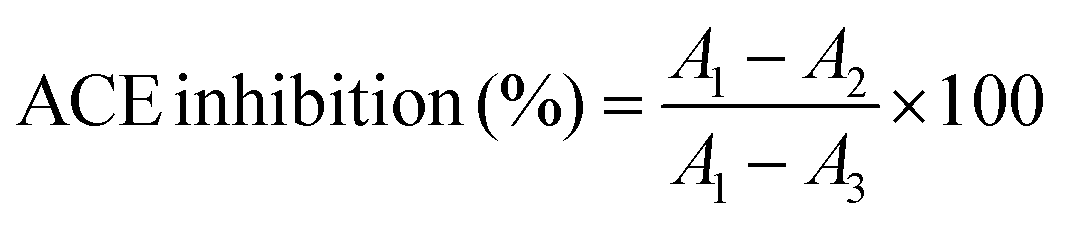 | (1) |
The values presented in the paper are the mean of triplicate analyses.
The IC50 value is defined as the concentration required to decrease the ACE activity by 50%. The percent inhibition curves were plotted using a minimum of five measurements for each sample concentration and the mean IC50 values were obtained using Graph Pad Prism® v. 5.02 for Windows (GraphPad Software, USA).
DPPH radical scavenging activity assay
DPPH radical scavenging activity was determined by the method according to Wu et al.20 with modification. All of the freeze-dried digests were dissolved in distilled water at 10 mg mL−1. A 0.5 mL of sample was then mixed with the same volume of 0.15 mM DPPH that was dissolved in 95% ethanol. The mixture was then shaken vigorously using a mixer and kept in the dark for 30 min. The absorbance of the resulting solution was recorded at 517 nm. The scavenging activity was calculated using the following equation: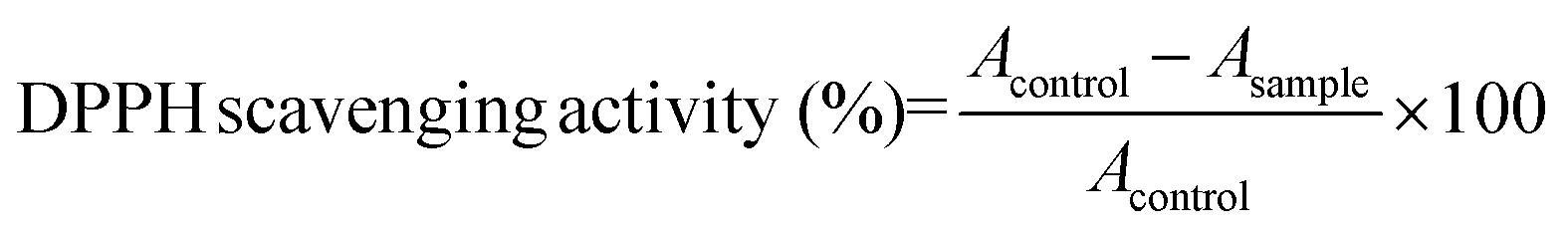 | (2) |
In addition, a standard curve based on known concentrations of Trolox was obtained and the results were presented as the number of μmol of Trolox corresponding to the activity of 1 g of hydrolysate. The curve was generated with SigmaPlot v. 12 (Systat Software Inc., USA). The values presented in the paper are the mean of triplicate analyses.
ABTS radical scavenging activity assay
ABTS˙+ radical scavenging activity was determined as described by Re et al.21 with a modification. The ABTS˙+ solution was prepared with final concentrations of 7 mM ABTS˙+ and 2.45 mM potassium persulphate. The mixture was left in the dark at room temperature for 12–16 h before use. Prior to the assay, the ABTS˙+ solution was diluted with 10 mM PBS (pH 7.4) to an absorbance of 0.70 ± 0.02 at 734 nm. 10 μL of samples (with a concentration of the hydrolysate 10.0 mg mL−1) were then added to 1 mL of diluted ABTS˙+ solution. The mixture was shaken vigorously for 30 s and left in the dark for 6 min. An equivalent volume of distilled water instead of the sample was used as a blank. The absorbance of the resulting solution was measured at 734 nm. The scavenging activity was calculated using the following equation22: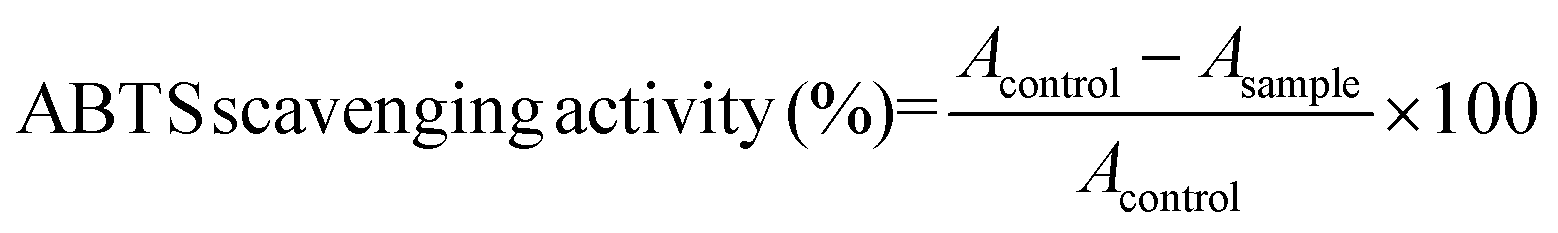 | (3) |
A standard curve was also prepared with 10 μL of Trolox and 1 mL of diluted ABTS˙+ solution. The degree of ABTS˙+ radical scavenging activity of carp digests was calculated on the basis of the Trolox standard curve and was expressed as the Trolox equivalent antioxidant capacity (TEAC, μm g−1). The presented values are the mean of triplicate analyses.
Statistical analysis
The results of the analyses are presented as means ± standard deviation. Statistical analyses were conducted with Statistica v. 10 (StatSoft, Poland) using an ANOVA Kruskal–Wallis test with a significance level of P < 0.05.Results and discussion
Development of a carp muscle ex vivo digestion method
The ex vivo digestion method was adapted from the procedure described by Almaas et al.12,14 The above-mentioned method was previously developed to digest milk, a liquid food, thus we had to modify a comminution type and duration of digestion stages to mimic digestion of fish meat tissue, which is an example of solid food.The impact of comminution type and digestion time of the individual stages of simulated digestion of carp muscle tissue on the protein hydrolysis was monitored with the SDS-PAGE method (Fig. 2). The results indicated that the degree of fish tissue comminution impacted the protein hydrolysis. We examined three degrees of comminution: homogenization (homog.), which is the most convenient under the laboratory conditions, cutting into cubes with a side length of about 1 cm (cubes), and cutting and crushing the tissue with a knife (middle) – a method closest to the physiological conditions, but less repeatable than homogenization and on the other hand more repeatable than cutting. The less the fish samples were comminuted, the slower the process of hydrolysis was, especially for proteins with a molecular weight (MW) >30 kDa in the “gastric” stage. The same trend was observed based on the protein content measurement (data not shown; for description of the method see the section: Protein content). Cutting the carp tissue into cubes resulted in a higher number of bands in the samples after the “gastric” stage; these bands were between 30 and 66 kDa and were not observed in the homogenized samples. The “duodenal” stage was visualized by a significant reduction in bands with MW >30 kDa, especially for the homogenized samples. Similar trends were observed by other researchers for hydrolysates prepared from salmon,23 catfish24 and red tilapia.25 Since the studies attempted to imitate the process that occurs in the digestive tract in humans, it was decided to use an intermediate method of comminution the samples are exposed to ex vivo digestion, i.e. cutting and crushing the tissue with a knife (middle), although that homogenization would facilitate working with biological materials.
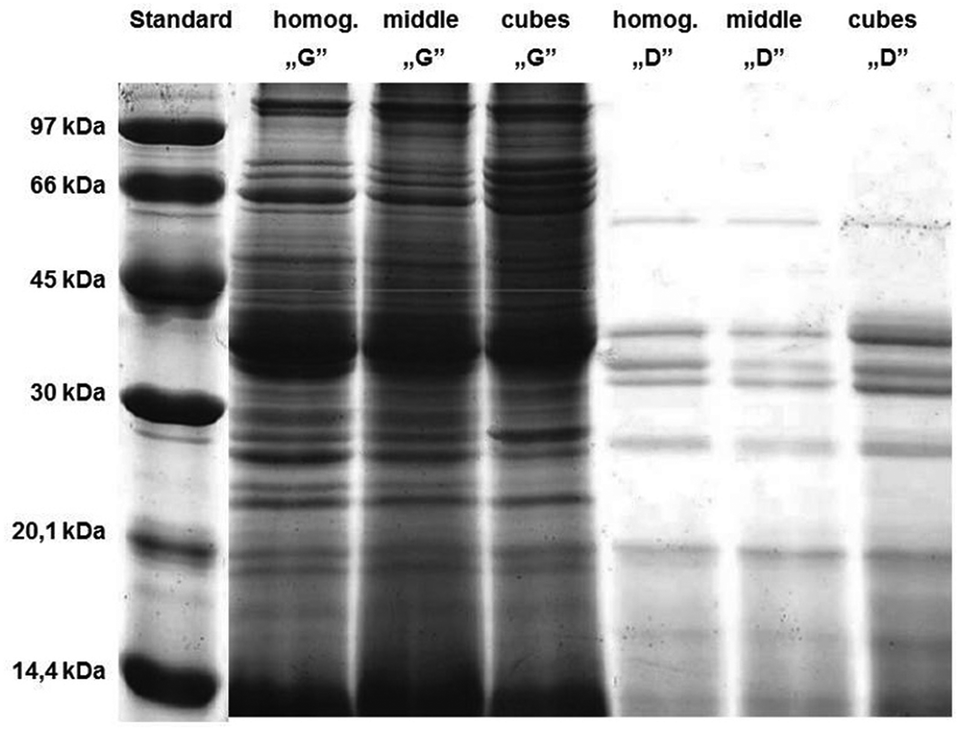 | ||
| Fig. 2 Electrophoretic separation (SDS-PAGE, 15%) of the hydrolysates of carp muscle tissue with a different degree of comminution: after homogenization (homog.); after cutting into cubes with 1 cm side length (cubes); and after cutting and crushing with a knife (middle). Standard, mass marker (97–14.4 kDa); G, samples after the two-hour “gastric” stage; D, samples after the one-hour “duodenal” stage. For details see the Experimental section. | ||
The effect of the duration of digestion on the protein hydrolysis was the next parameter to be analysed. Based on the data available in the literature, it was assumed that “gastric” digestion should last for about 1–2 hours (with gradual pH reduction from 5.0 to ca. 2.5), whereas the “duodenal” stage should last about one hour (pH approximately 7).7,26,27 The value of pH in the stomach usually increases to ca. 5 after food intake, because of the buffering capacity of food components. The secretion of HCl lowers the pH to a value observed while fasting.
After one and two hours of “gastric” digestion (Fig. 3), the protein profile showed a reduction in bands with MW between 45 kDa and 60 kDa and a reduction in the intensity of other bands, while more visible and distinct bands appeared in the low molecular weight region below 14 kDa. Since the analysis of electrophoregrams revealed that digestion with human gastric juices sampled from the stomach (HGJ) was much slower than the “duodenal” stage, it was decided to assume a two-hour “gastric” stage and a one-hour “duodenal” stage.
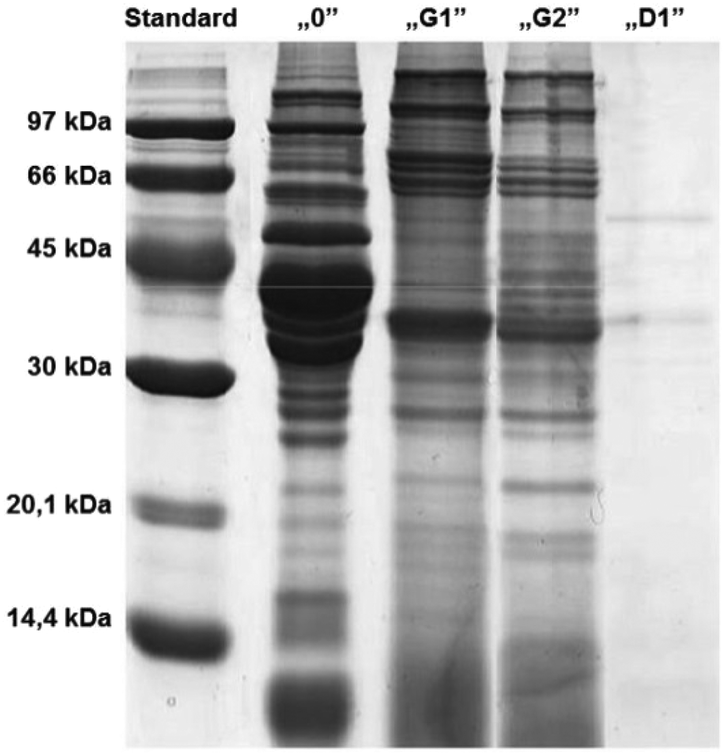 | ||
| Fig. 3 Electrophoretic separation (SDS-PAGE, 15%) of the hydrolysates of carp muscle tissue after cutting and crushing with a knife (middle). Hydrolysates obtained after one- (G1) and two-hour (G2) “stomach” digestion and a one-hour “duodenal” digestion (D1). Standard, mass marker (97–14.4 kDa); “0”, samples after the “chewing” stage. For details see section Experimental. | ||
It seemed that the carp muscle tissue was hydrolysed very rapidly during the duodenal step and therefore the samples were redrawn at 15 minute intervals (Fig. 4). The results confirmed a very rapid protein degradation. After 60 minutes of “duodenal” digestion, no intact bands were observed. This could be related to the degree of hydrolysis, which increased with the time of hydrolysis. Similar observations were reported by Shamloo et al.25
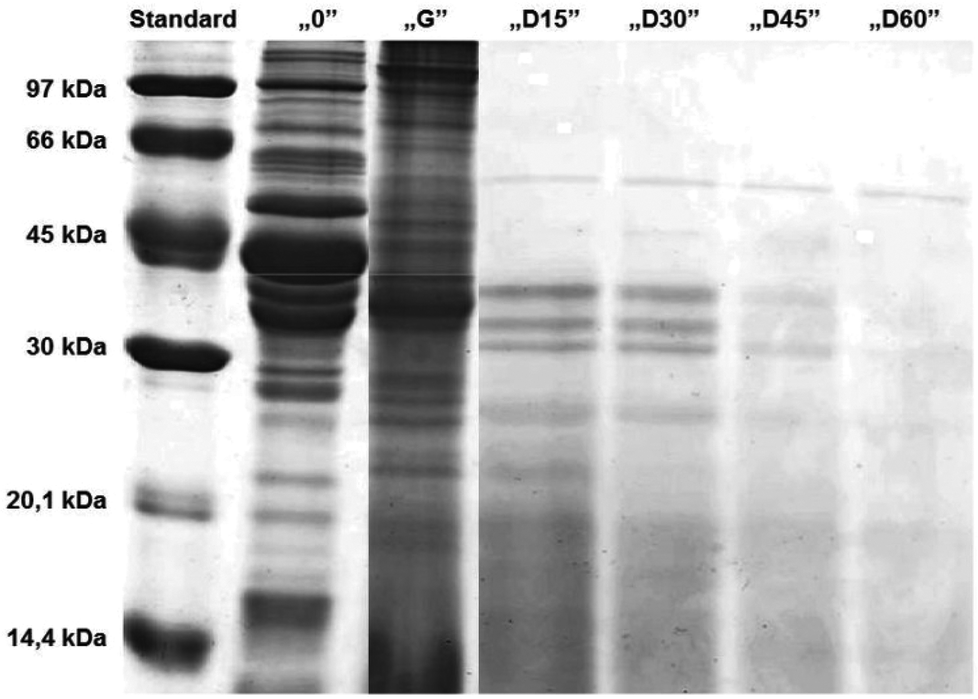 | ||
| Fig. 4 Electrophoretic separation (SDS-PAGE, 15%) of the hydrolysates of carp muscle tissue after cutting and crushing with a knife (middle). Hydrolysates obtained after a two-hour “stomach” digestion (G) and the one-hour “duodenal” digestion, where samples were taken every 15 minutes (D15, D30, D45, D60). Standard, mass marker (97–14.4 kDa); “0”, samples after the “chewing” stage. For details see section Experimental. | ||
Protein degradation
The content of proteins remaining intact in the fish muscle hydrolysates was used as an indirect parameter to monitor the changes in the degree of carp muscle tissue hydrolysis (Table 1). The obtained results confirmed the observations based on the SDS-PAGE findings. The pepsin in the gastric juice initiated hydrolysis of the carp muscle tissue proteins, whereas all the proteolytic enzymes of duodenal juice generated a more rapid increase in the amount of hydrolysed polypeptide bonds. After 15 minutes of duodenal digestion, the protein content in a sample decreased from approx. 72% (“gastric” stage) to 7.33%. An increase in time during the “duodenal” phase from 15 minutes to 60 minutes caused only a minor reduction in the amount of the intact protein. Similar observations were reported by Nakajima et al.28 with in vitro studies on salmon (Salmo salar and Onchorhynchus kisutch) protein hydrolysis. Pancreatin hydrolysates of fish muscles showed a significantly higher degree of hydrolysis than pepsin hydrolysates. Similar observations of the digestion dynamics of whey protein in goat milk were recorded by Eriksen et al.29| Stage | Protein remaining intact (%) |
|---|---|
| Mean ± SD | |
| G | 71.96 ± 2.87abcd |
| D15 | 7.33 ± 0.12aefg |
| D30 | 5.19 ± 0.06beh |
| D45 | 4.46 ± 0.16cfh |
| D60 | 4.55 ± 0.46dg |
Lipids in the muscle tissue in the carp seem to hinder the activity of proteolytic enzymes. Devle et al.6 observed that the presence of lipids in full fat bovine milk inhibited the degradation of β-lactoglobulin. Human digestive juices are a complex mixture of proteases, amylases and lipases occuring in different isoforms in combination with inhibitors, activators, bile salts, bilirubin and other compounds that may impact the hydrolysis of proteins.6 Bile found in HDJ emulsifies lipids, thereby influencing the rate of protein digestion in this segment of the digestive tract.
The ex vivo method used in this study does not include a membrane where the water-soluble digestion products are continuously removed. However, when compared with other static in vitro digestion methods using a commercial enzyme preparation, the ex vivo method seemed to be similar to the human digestion process.6
ACE inhibitory activity
There are several methods to determine the ACE inhibitory activity which can be used to select foods with potential hypotensive activity in vivo.9 The spectrophotometric method of Jimsheena and Gowda18 is based on the commonly used method introduced by Cushman and Cheung.30 In these studies, the results are expressed as the percentage of ACE inhibition by hydrolysates with the final concentration in the reaction tube of 3 mg mL−1. As shown in Fig. 5, samples after chewing inhibited ACE by 30%. The results of ACE inhibitory activity in the carp muscle tissue that was not exposed to hydrolysis may suggest the presence of compounds that act as inhibitors of ACE. In general, non-hydrolysed proteins do not show any inhibitory activity against ACE or demonstrate a low degree of this activity.31 It is supposed that in the muscle tissue of the carp there may be some endogenous or autolysis-generated peptides-ACE inhibitors or other substrates that are hydrolysed by ACE instead of HHL. Due to the complexity of the muscle tissue matrix, the presence of other enzymes hydrolysing HHL to HA or compounds, other than HA, interfering at 410 nm wavelength cannot be excluded.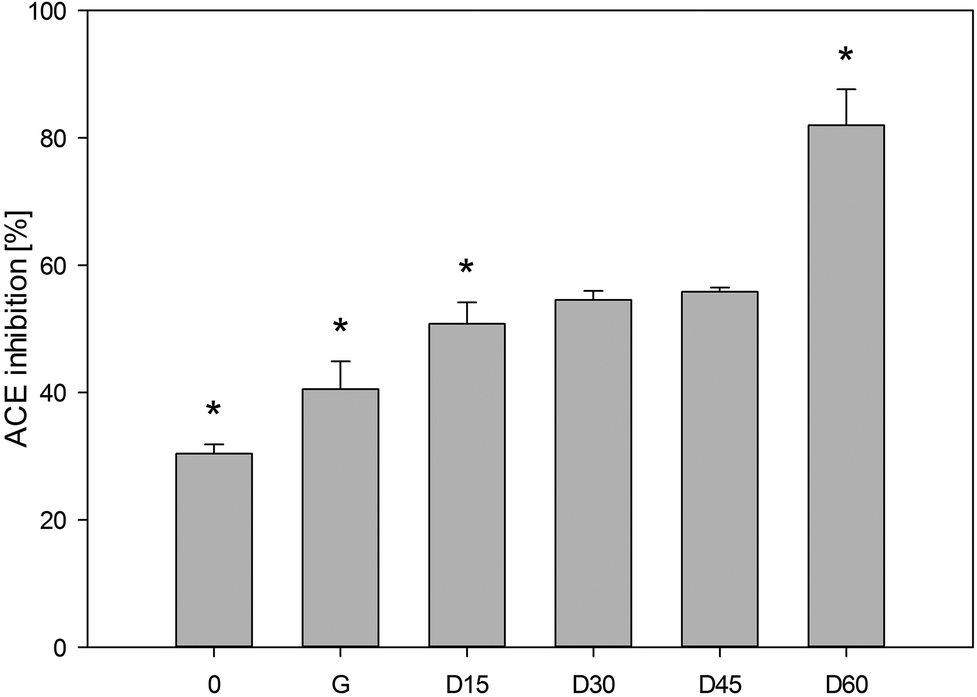 | ||
| Fig. 5 Degree of ACE inhibition by the hydrolysates of carp muscle tissue (3 mg ml−1). 0, a sample after the “chewing” stage; G, a sample after the two-hour “gastric” stage; D, samples after the “duodenal” stage collected after 15, 30, 45 and 60 minutes in the course of this stage. The data is presented as means (with SD). The pillars denoted with “*” differ statistically at P < 0.05. | ||
It has been observed that the ACE inhibitory activity increases along with the duration of ex vivo digestion. After 2 hours, gastric samples inhibited ACE by 41% and the duodenal digested samples after 15 minutes inhibited ACE by 51%. A similar tendency was observed by Nakajima et al.28 when pepsin (2 h) and pancreatin (3 h) hydrolysed samples of Atlantic salmon, Coho salmon, Alaska pollack and southern blue whiting inhibited ACE by 69.4–77%. Theodore and Kristinsson32 showed that the channel catfish protein hydrolysate with a protein concentration of 1.5 mg mL−1 inhibited ACE by approximately 60%. The ACE inhibitory activity determined after the two-hour “gastric” stage and one-hour “duodenal” digestion was approx. 2.7 times higher than in the samples collected after the “chewing” stage. A significant difference was demonstrated between the values of ACE inhibitory activity in the samples after the “gastric” stage and after 15 and 60 minutes of the “duodenal” phase. Pepsin, the main component of human gastric juices, has broad specificity to cleave the peptide bonds. It was used in the past for the production of ACE inhibitory peptides.33 In our study, peptides derived from carp proteins by HGJ were slightly resistant to digestion in the stomach.
The results of IC50 of the hydrolysates of carp muscle tissue demonstrated that they might inhibit ACE. The highest activity (IC50 = 1.90 mg mL−1) was detected in the hydrolysate generated after two-hour “gastric” digestion and one-hour “duodenal” stages. The hydrolysates after two-hour “gastric” digestion were less active, which was confirmed from IC50 = 9.26 mg mL−1. The results recorded after simulated digestion of pea and whey proteins showed that the duration of “gastric” and “duodenal” phases significantly impacted the ACE inhibitory activity of hydrolysates.31
An increase in the ACE inhibitory activity during the digestion time was also recorded for proteins in grass carp tissues subjected to hydrolysis with Alcalase or neutral protease AS1398, and Atlantic salmon hydrolysed with Alcalase and papain.34–36 In this study, a 60 min carp muscle duodenal hydrolysate showed a stronger ACE inhibition than that reported by Nakajima et al.28 The pepsin and pancreatin hydrolysates of Atlantic salmon, Coho salmon, Alaska pollack, and southern blue whiting showed IC50 values of 5.00, 3.70, 2.90 and 3.60 mg mL−1, respectively. Extracts from the pickled mackerel, fermented mackerel, sardine muscle hydrolysate and hard clam extract were reported to have IC50 values of 0.1–0.4, 0.06–0.20, 0.25–0.62 and 1.090–0.036 mg mL−1, respectively.28 In comparison with these reports, the hydrolysate of carp muscle obtained from gastric and duodenal human juices showed either lower or similar ACE inhibitory activity. Most inhibitory peptides derived from marine proteins were reported to be short-chain and obtained from hydrolysates with a high degradation level.9 Since these mostly antihypertensive di- or tripeptides are too small to be substrates of digestive proteases, they should have high resistance to gastrointestinal digestion. One should bear in mind that to exert an antihypertensive effect, the ACE inhibitory peptides released after gastrointestinal digestion have to be absorbed from the intestine in an active form and have to reach the cardiovascular system as reported by Iwaniak et al.9
Antioxidative activity
Due to the diversity of oxidation processes and antioxidant activity of carp muscle hydrolysates, the use of a single method to evaluate the antioxidant activity cannot provide a clear view of their real antioxidant potential. The products of ex vivo digestion of the carp muscle tissue were characterized based on their antioxidative activity, which was expressed as the capacity to scavenge DPPH˙ and ABTS+˙ free radicals (Table 2). While analysing the results of the antioxidative activity of the tested hydrolysates, it should be taken into account that the tests used to measure the antioxidative activity serve to determine the total antioxidative capacity of a given sample, but not the impact of individual components.37| Stage | Antioxidative activity determined by DPPH method | Antioxidative activity determined by ABTS method | ||||||
|---|---|---|---|---|---|---|---|---|
| DPPH˙ scavenging activity (%) | Trolox (μM g−1) | ABTS+˙ scavenging activity (%) | Trolox (μM g−1) | |||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 0 | 62.2abcde | 2.9 | 141abcde | 20 | 16.3abcde | 0.9 | 37abcde | 86 |
| G | 8.3afg | 0.4 | 61afgh | 3 | 57.9afghi | 6.2 | 191afghi | 24 |
| D15 | 5.4b | 0.1 | 40bf | 1 | 73.5bf | 2.0 | 246bf | 8 |
| D30 | 5.7c | 0.2 | 42c | 1 | 82.3cg | 3.2 | 277cg | 12 |
| D45 | 4.8df | 0.2 | 35dg | 1 | 79.1dh | 4.4 | 266dh | 17 |
| D60 | 3.5eg | 0.4 | 25eh | 3 | 79.7ei | 2.9 | 268ei | 11 |
DPPH˙ is often used as a substrate to evaluate the antioxidative activity since, as a free radical, it receives an electron or hydrogen ion, forming a stable molecule.38 All samples generated after the “chewing” stage had a capacity to scavenge free radicals. The results may be related to the presence of peptides naturally found in meat, for example glutathione (gamma-Glu-Cys-Gly), carnosine (beta-alanyl-L-histidine), anserine (beta-alanyl-L-1-methylhistidine) and ophidine (beta-alanyl-L-3-methylhistidine).39 Nakajima et al.28 demonstrated that, for instance, the content of anserine in 100 g of muscle tissue in Atlantic salmon is 712 ± 147 mg. In addition, the muscle tissue of the carp may be a source of numerous compounds with reductive properties, enzymes, hormones, free amino acids, vitamins C and E, coenzyme Q, carotenoids and manganese, zinc, selenium and copper.40 It has been proven that carotenoids are found in complexes with actomyosin molecules (e.g. astaxantin with α-actinin) in the muscle tissue in salmons.41 The hydrolysates of carp muscle tissue showed an activity against DPPH˙ radicals, but a higher capacity to scavenge free radicals was demonstrated by the samples from the “gastric” stage, i.e. the samples with a higher content of the non-hydrolysed protein. A similar relationship was reported by Bougatef et al.42 during their studies on sardinelle (Sardinella aurita) proteins. These authors found that an increase in the degree of hydrolysis of sardinelle proteins generated products with lower activity against DPPH˙ radicals. Moreover, Zhong et al.43 found that pepsin hydrolysates of silver carp proteins showed a higher capacity to scavenge DPPH˙ radicals than trypsin hydrolysates.
Similar to the test with DPPH, the ABTS method allowed to observe the antioxidative capacity of the samples obtained after the “chewing” stage. An increase in the antioxidative activity of hydrolysates was recorded. A statistically significant increase in this activity was observed after two hours of the “gastric” stage as well as after a one-hour “duodenal” stage. Similar relationships of the increased activity of ABTS+˙ radical scavenging were seen during hydrolysis of fish-derived gelatin with papain44 and protein hydrolysates prepared from ornate threadfin bream (Nemipterus hexodon) muscle, using skipjack tuna pepsin.45 Generally, all hydrolysates contained peptides which were hydrogen donors and could react with the radicals to convert them to a more stable product, thereby terminating the radical chain reaction.46
By comparing the results of the antioxidative activities of carp muscle tissue hydrolysates produced with DPPH (in an ethanol solution) and ABTS (in an aqueous solution), it is supposed that over the course of hydrolysis, an increase in the solubility of peptides and amino acids in an aqueous solution favoured higher results in the ABTS test. The differences in the results obtained with the methods of DPPH and ABTS free radical scavenging may be explained not only with differences in the solubility of samples but also with the different stereoselectivity of radicals and varied stoichiometry of reactions between the antioxidants and radicals.47 Furthermore, the obtained results indicate that the hydrolysates contain peptides and amino acids that may act as electron donors and may react with free radicals both in a polar and non-polar environment to form stable products and terminate the chain of free-radical reactions.
Conclusions
Until now there are many reports in the literature on digestion of food proteins using commercial digestive enzymes of non-human origin with further identification of generated molecules during in vitro digestion. However, relatively few studies on digestion of foods using human gastrointestinal enzymes have been performed. Thus, our strategy to simulate the gastrointestinal digestion of carp muscle tissue to obtain the hydrolysates possessing biological activity seems to be an innovative approach.The present study shows new data on carp muscle tissue degradation by ex vivo digestion with HGJ and HDJ. The carp muscle hydrolysates obtained via a proposed digestion method showed high ACE inhibitory activity and ABTS radical scavenging activity ex vivo, which was more effective than the DPPH radical scavenging activity.
This is a preliminary screening of the potential health-promoting biological activity of carp muscle tissue proteins in an ex vivo study. Further work is being carried out to analyse the ACE inhibitory and antioxidative activity of certain carp and other fish muscle proteins and identify peptide sequences that may have an ex vivo physiological relevance in the regulation of blood pressure and oxidation processes. Much attention should be paid to the fish proteins as food components being the valuable agents in the prevention of some diet-related diseases.
Acknowledgements
The authors would like to thank Irene Comi for the aspiration of human digestive juices in cooperation with the Oestfold Hospital Trust, Norway, and for analysing all enzyme activities. J.B. is grateful to Prof. G.E. Vegarud from the Norwegian University of Life Sciences, Norway, for her supervision during the stay in Norway.This research was supported by grants from the National Science Centre in Poland (project no. N N312 465240), University of Warmia and Mazury (528-0712-0882, 528-0712-0809) and co-financed by technical assistance funds of the EEA Financial Mechanism and the Norwegian Financial Mechanism within the framework of the Scholarship and Training Fund. J.B. was supported by the European Union within the European Social Fund.
Notes and references
- FAO, Fishery and Aquaculture Statistics 2010 (2012). Yearbook. http://ftp//ftp.fao.org/FI/CDrom/CD_yearbook_2010/navigation/index_intro_e.htm (accessed 08/03/2014).
- K. Turkowski and A. Lirski, Acta Ichthyol. Piscat., 2010, 40, 137–144 CrossRef.
- Z. Usydus, J. Szlinder-Richert and M. Adamczyk, Food Chem., 2009, 112, 139–145 CrossRef CAS PubMed.
- I. Sensoy, Crit. Rev. Food Sci. Nutr., 2014, 54, 902–909 CrossRef CAS PubMed.
- S. J. Hur, B. O. Lim, E. A. Decker and D. J. McClements, Food Chem., 2011, 125, 1–12 CrossRef CAS PubMed.
- H. Devle, E. K. Ulleberg, C. F. Naess-Andresen, E.-O. Rukke, G. Vegarud and D. Ekeberg, Int. Dairy J., 2014, 36, 6–13 CrossRef CAS PubMed.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carrière, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Ménard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. J. Wickham, W. Weitschies and A. Brodkorb, Food Funct., 2014, 1113–1124 CAS.
- C. C. Udenigwe and R. E. Aluko, J. Food Sci., 2012, 77, R11–R24 CrossRef CAS PubMed.
- A. Iwaniak, P. Minkiewicz and M. Darewicz, Compr. Rev. Food Sci. Food Saf., 2014, 13, 114–134 CrossRef CAS.
- Y. Zhao, B. Li, S. Dong, Z. Liu, X. Zhao, J. Wang and M. Zeng, Peptides, 2009, 30, 1028–1033 CrossRef CAS PubMed.
- J. T. Ryan, R. P. Ross, D. Bolton, G. F. Fitzgerald and C. Stanton, Nutrients, 2011, 3, 765–791 CrossRef CAS PubMed.
- H. Almaas, H. Holm, T. Langsrud, R. Flengsrud and G. E. Vegarud, Br. J. Nutr., 2006, 96, 562–569 CAS.
- T. M. Qureshi, G. E. Vegarud, R. K. Abrahamsen and S. Skeie, J. Dairy Sci., 2013, 96, 838–853 CrossRef CAS PubMed.
- H. Almaas, A.-L. Cases, T. G. Devold, H. Holm, T. Langsrud, L. Aabakken, T. Aadnoey and G. E. Vegarud, Int. Dairy J., 2006, 16, 961–968 CrossRef CAS PubMed.
- E. K. Ulleberg, I. Comi, H. Holm, E. B. Herud, M. Jacobsen and G. E. Vegarud, Food Dig., 2011, 2, 52–61 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- V. K. Jimsheena and L. R. Gowda, Anal. Chem., 2009, 81, 9388–9394 CrossRef CAS PubMed.
- Y. Nakamura, N. Yamamoto, K. Sakai, A. Okubo, S. Yamazaki and T. Takano, J. Dairy Sci., 1995, 78, 777–783 CrossRef CAS.
- H.-C. Wu, H.-M. Chen and C.-Y. Shiau, Food Res. Int., 2003, 36, 949–957 CrossRef CAS.
- R. Re, N. Pellegrimi, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radicals Biol. Med., 1999, 26, 1231–1237 CrossRef CAS.
- T. K. Mohamed, A. Issoufou and H. Zhou, Int. Food Res. J., 2012, 19, 207–213 CAS.
- H. G. Kristinsson and B. A. Rasco, J. Agric. Food Chem., 2000, 48, 657–666 CrossRef CAS PubMed.
- A. E. Theodore, S. Raghavan and H. G. Kristinsson, J. Agric. Food Chem., 2008, 56, 7459–7466 CrossRef CAS PubMed.
- M. Shamloo, J. Bakar, D. Mat Hashim and A. Khatib, Int. Food Res. J., 2012, 19, 183–188 CAS.
- E. Escudero, M. A. Sentandreu and F. Toldrá, J. Agric. Food Chem., 2010, 58, 5160–5165 CrossRef CAS PubMed.
- M. Wickham, R. Faulks and C. Mills, Mol. Nutr. Food Res., 2009, 53, 952–958 CAS.
- K. Nakajima, Y. Yoshie-Stark and M. Ogushi, Food Chem., 2009, 114, 844–851 CrossRef CAS PubMed.
- E. K. Eriksen, H. Holm, E. Jensen, R. Aaboe, T. G. Devold, M. Jacobsen and G. E. Vegarud, Br. J. Nutr., 2010, 104, 374–381 CrossRef CAS PubMed.
- D. W. Cushman and H. S. Cheung, Biochem. Pharmacol., 1971, 20, 1637–1648 CrossRef CAS.
- A. Jakubczyk, M. Karaś, B. Baraniak and M. Pietrzak, Food Chem., 2013, 141, 3774–3780 CrossRef CAS PubMed.
- A. E. Theodore and H. G. Kristinsson, J. Sci. Food Agric., 2007, 87, 2353–2357 CrossRef CAS.
- Y. Yu, J. Hu, X. Bai, Y. Du and B. Lin, Process Biochem., 2006, 41, 1589–1593 CrossRef CAS PubMed.
- J. Chen, Y. Wang, Q. Zhong, Y. Wu and W. Xia, Peptides, 2012, 33, 52–58 CrossRef CAS PubMed.
- F. Zhang, Z. Wang and S. Xu, Food Chem., 2009, 117, 387–392 CrossRef CAS PubMed.
- R.-Z. Gu, C.-Y. Li, W.-Y. Liu, W.-X. Yi and M.-Y. Cai, Food Res. Int., 2011, 44, 1536–1540 CrossRef CAS PubMed.
- A. Pihlanto, Int. Dairy J., 2006, 16, 1306–1314 CrossRef CAS PubMed.
- G. Marinova and V. Batchvarov, Bulg. J. Agric. Sci., 2011, 17, 11–24 Search PubMed.
- A. G. P. Samaranayaka and E. C. Y. Li-Chan, J. Funct. Foods, 2011, 4, 229–254 CrossRef PubMed.
- I. Medina, J. M. Gallardo and S. P. Aubourg, Int. J. Food Sci. Technol., 2009, 44, 1467–1479 CrossRef CAS PubMed.
- S. M. Ojagh, R. Núñez-Flores, M. E. López-Caballero, M. P. Montero and M. C. Gómez-Guillén, Food Chem., 2011, 125, 595–606 CrossRef CAS PubMed.
- A. Bougatef, N. Nedjar-Arroume, L. Manni, R. Ravallec, A. Barkia, D. Guillochon and M. Nasri, Food Chem., 2010, 118, 559–565 CrossRef CAS PubMed.
- S. Zhong, C. Ma, Y. C. Lin and Y. Luo, Food Chem., 2011, 126, 1636–1642 CrossRef CAS PubMed.
- L. You, J. M. Regenstein and R. H. Liu, J. Food Sci., 2010, 75, C582–C587 CrossRef PubMed.
- S. Nalinanon, S. Benjakul, H. Kishimura and F. Shahidi, Food Chem., 2011, 124, 1354–1362 CrossRef CAS PubMed.
- W. Binsan, S. Benjakul, W. Visessanguan, S. Roytrakul, M. Tanaka and H. Kishimura, Food Chem., 2008, 106, 185–193 CrossRef CAS PubMed.
- A. P. F. Corrêa, D. J. Daroit, J. Coelho, S. M. M. Meira, F. C. Lopes, J. Segalin, P. H. Risso and A. Brandelli, J. Sci. Food Agric., 2011, 91, 2247–2254 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |