
Ashitaba (Angelica keiskei) extract prevents adiposity in high-fat diet-fed C57BL/6 mice
Tianshun
Zhang
a,
Yoko
Yamashita
a,
Michiko
Yasuda
b,
Norio
Yamamoto
c and
Hitoshi
Ashida
*a
aDepartment of Agrobioscience, Graduate School of Agricultural Science, Kobe University, Kobe 657-8501, Japan. E-mail: ashida@kobe-u.ac.jp; Fax: +81-78-803-5878; Tel: +81-78-803-5878
bOrganization of Advanced Science and Technology, Kobe University, Kobe 657-8501, Japan
cFood Science Research Center, House Wellness Foods Corporation, Imoji 3-20, Itami, Hyogo 664-0011, Japan
First published on 14th October 2014
Abstract
Two main chalcones, 4-hydroxyderricin and xanthoangelol, from Ashitaba, which is a food ingredient and a folk medicine in Asia, have been demonstrated to modulate lipid metabolism in 3T3-L1 and HepG2 cells. In this study, we investigated the effects of Ashitaba extract on adiposity in mice fed a high-fat (HF) diet and its underlying mechanisms based on adipose tissue and hepatic lipid metabolism. C57BL/6 mice were fed a normal or HF diet supplemented with Ashitaba extract (0.01% and 0.1%, w/w) for 16 weeks. Ashitaba extract suppressed the HF diet-induced body weight gain and fat deposition in white adipose tissue, reduced plasma cholesterol, glucose, and insulin levels, increased the adiponectin level, lowered triglyceride and the liver cholesterol content, increased phosphorylation of AMP-activated protein kinase (AMPK) in adipose tissue and liver, inhibited lipogenesis in adipose tissue by down-expression of peroxisome proliferator-activated receptor (PPAR) γ, CCAAT/enhancer-binding protein α and sterol regulatory element-binding protein 1 (SREBP1), inhibited lipogenesis in the liver by down-expression of SREBP1 and its target enzyme fatty acid synthase, and promoted fatty acid oxidation by up-expression of carnitine palmitoyltransferase-1A and PPARα. In conclusion, Ashitaba extract can possibly prevent adiposity through modulating lipid metabolism through phosphorylation of AMPK in adipose tissue and liver.
Introduction
Ashitaba, a Japanese herb, is drunk as a tea and used as a vegetable and is the folk medicine for diuretic, laxative, analeptic and galactagogue purposes. Several attractive compounds have been identified from this plant for health promoting effects including coumarins, flavanones and chalcones.1 Among them, 4-hydroxyderricin (4HD) and xanthoangelol (XAG) are considered to be the major active compounds for various biofunctions including anti-tumor,2,3 anti-inflammatory4 and anti-diabetes5,6 activities. Our previous report7 demonstrated that 4HD and XAG inhibited the differentiation of preadipocytes into adipocytes via down-regulating expression of C/EBPβ, C/EBPα and PPARγ involved in the activation of the AMPK signaling pathway. Moreover, we also found that 4HD and XAG prevented free fatty acid-induced impairment of lipid metabolism through the activation of the liver kinase B1/AMPK pathway in HepG2 cells.8 However, the in vivo effect of Ashitaba extract on a high-fat (HF) diet-induced adiposity is still unclear.Recently, the rates of obesity have increased dramatically.9 Obesity usually results from an energy imbalance:10 excessive energy storage and insufficient energy expenditure induced lipid accumulation in both adipose tissue and liver leading to type II diabetes, cardiovascular diseases, non-alcoholic fatty liver disease and other metabolic disorders.9,11,12 Dietary fat is considered to be one of the most important factors in the pathophysiology of obesity. C57BL/6 mice are an obesity-prone strain and develop obesity, hyperglycemia, and hyperlipidemia when fed HF diet. Thus, C57BL/6 mice are commonly used for research on obesity and obesity-related diseases.13,14
Lipid accumulation and energy metabolism are tightly controlled in the adipose tissue and liver. AMP-activated protein kinase (AMPK) is a key modulator to maintain the cellular as well as whole-body energy balance. There has been increased interest in development of AMPK activators as potential therapies for prevention of amelioration of obesity, diabetes and hepatic steatosis.15,16 Upon activation, AMPK might downregulate an activity of sterol regulatory element binding protein-1 (SREBP1) through protein interaction and/or phosphorylation, and subsequently inhibits target molecules for SREBP1 including fatty acid synthase (FAS) expression, leading to reduced lipogenesis and lipid accumulation.17,18 Moreover, the activation of AMPK also leads to phosphorylation of acetyl-CoA carboxylase (ACC) and inhibits its activity.19 Inactivation of ACC reduces the synthesis of malonyl-CoA, which in turn activates carnitine palmitoyltransferase-1A (CPT-1A) and increases fatty acid oxidation.20 In addition, AMPK also increases fatty acid oxidation by up-regulating expression of peroxisome proliferator-activated receptor (PPAR) α.21 It is also reported that the activation of AMPK inhibits differentiation of adipocytes through down-regulating expression of CCAAT/enhancer-binding protein (C/EBP) α and PPARγ.22
In this study, Ashitaba extract was given to C57BL/6 mice fed the control or HF diet for 16 weeks to examine whether the extract prevents HF diet induced adiposity. We further clarified the underlying molecular mechanisms based on lipid metabolism in the adipose tissue and liver.
Materials and methods
Materials
Ashitaba extract was prepared from Ashitaba Chalcone Powder supplied by Japan Bio Science Laboratory (Osaka, Japan) as previously described.6 The powder (10 g) was extracted with ethyl acetate (100 mL × 3 times) at room temperature, and the obtained extract was dried in vacuo (the yield was 17%). Antibodies for PPARγ, C/EBPα, PPARα, ACC horseradish peroxidase-conjugated anti-rabbit IgG, anti-mouse IgG and anti-goat IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), β-actin, p-AMPK, AMPK and p-ACC were from Cell Signaling Technology (Beverly, MA, USA), SREBP-1 CPT-1A and acyl-CoA carboxylase X1 (ACOX1) were from Abcam (Cambridge, MA, USA) and uncoupling protein-2 (UCP-2) was from BioLegend Inc. (San Diego, CA, USA). All other reagents used were of the highest grade available from the commercial sources.Measurement of 4HD and XAG in Ashitaba extract by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Detection and quantification of 4HD and XAG were performed with a LC-MS/MS (4000 Q TRAP, AB Sciex, Foster City, CA, USA) using electrospray ionization. Chalcone was used as an internal standard compound. HPLC separation was performed with a gradient system using solvent A (0.1% formic acid) and solvent B (acetonitrile) equipped with a L-column-2 ODS (2.1 × 150 mm) column (Chemicals Evaluation and Research Institute, Tokyo, Japan) at a flow rate of 0.2 mL min−1. The column oven was maintained at 40 °C. The gradient program was 0–2 min, 45% A; 2–7 min, linear gradient to 0% A; 7–8 min, 0% A hold; 8–8.1 min, linear gradient to 45% A; and 8.1–15 min, 45% A hold. The chalcones were detected by multiple reaction monitoring as follows: 4HD 339.2/163.1 [M + H]+, XAG 393.2/131.0 [M + H]+, chalcone 209.1/131.0 [M + H]+. For quantification, standard curves of Ashitaba chalcones from 0.05 to 500 fmol per 5 μL injection were generated as described in the previous study.23 Concentrations of 4HD and XAG were corrected by the comparison between the peak area of these compounds and that of the internal standard.Animal treatment
All animal experiments were approved by the Institutional Animal Care and Use Committee (permission #25-04-02) and were carried out according to the guidelines for Animal Experiments at Kobe University. Male C57BL/6 mice (5 weeks old, n = 36) were obtained from Japan SLC (Shizuoka, Japan) and maintained in a temperature-controlled room (22 °C). The mice had free access to tap water and an AIM-93M laboratory purified diet (Oriental Yeast, Tokyo, Japan), and were acclimatized for 7 days before the experiments. They were then randomly divided into six groups of six mice each and fed a control (AIN-93M) or HF diet containing 30% (w/w) lard for 16 weeks. The compositions of the diets and energy densities are shown in Table 1. The diets were supplemented with 0% (C-0 and HF-0 groups), 0.01% (C-0.01 and HF-0.01 groups) or 0.1% (C-0.1 and HF-0.1 groups) Ashitaba extract. Food and water intakes were measured, and the diets were replaced every 2 days. The body weight was measured weekly. After 16 weeks of feeding, the mice were fasted for 18 h and sacrificed under anesthesia with sodium pentobarbital. Blood was collected from cardiac puncture using a heparinized syringe. The liver, white adipose tissues (subcutaneous, epididymal, mesenteric and retroperitoneal adipose tissues) and brown adipose tissue were also collected. Tissue samples were washed with 1.15% (w/v) KCl, weighed, immediately frozen using liquid nitrogen, and kept at −80 °C until use.| Control | HF | |
|---|---|---|
| Ingredients | (g per 100 g diet) | |
| Casein | 14 | 14 |
| L-Cystine | 0.2 | 0.2 |
| Cornstarch | 46.6 | 16.6 |
| Dextrin | 15.5 | 15.5 |
| Sucrose | 10 | 10 |
| Soybean oil | 4 | 4 |
| Cellulose | 5 | 5 |
| Mineral mixture | 3.5 | 3.5 |
| Vitamin mixture | 1 | 1 |
| Choline bitartrate | 0.3 | 0.3 |
| Tertiary butyl hydroxyl quinone | 0.0008 | 0.0014 |
| Lard | 0 | 30 |
| (kcal per 100 g diet) | ||
| Energy density | 348 | 518 |
Measurement of plasma parameters related to lipid and glucose metabolism
Plasma triglyceride, total cholesterol, non-esterified fatty acid (NEFA) and glucose levels were measured using the corresponding commercial assay kit according to the manufacturer's instructions (triglyceride-E test, cholesterol-E test, NEFA-C test, and glucose CII-test, respectively, Wako Pure Chemical). Plasma insulin and adiponectin levels were measured using the commercial ELISA assay kits according to the manufacturer's instructions (mouse insulin ELISA kit and mouse/rat adiponectin ELISA kit, Shibayagi, Shibukawa, Japan). The index of the homeostasis model assessment of insulin resistance (HOMA-IR) was calculated from the values of the plasma glucose and insulin levels according to the following formula:24 HOMA-IR = fasting glucose (mg per 100 mL) × fasting insulin (μU per mL)/405.Measurement of hepatic lipid levels
An aliquot of 100 mg of liver was homogenized with 0.35 mL of distilled water, and the homogenate was extracted three times with 0.7 mL of a chloroform–methanol (2/1, v/v) mixture. The chloroform layer was collected after centrifugation at 1800g for 10 min, and washed with a 1/4 volume of 0.88% (w/v) KCl. The obtained chloroform layer was dried in vacuo, and the weight of the residue as total lipids was measured. After the residue was dissolved in isopropanol containing 10% (v/v) Triton-X, triglyceride and cholesterol levels were measured using the respective commercial kit as described above.Western blotting
Preparation of the cell lysate was performed according to the previous study.25 Proteins in the cell lysate of the adipose tissue and liver were separated using SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. After blocking with commercial Blocking One solution (Nacalai Tesque), the membranes were incubated with primary antibodies for PPARγ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20000), C/EBPα (1:10000), SREBP-1 (1:10000), PPARα (1:20000), CPT-1A (1:5000), ACOX1 (1:20000), UCP-2 (1:20000), β-actin (1:20000), AMPK (1:10000), p-AMPK (1:5000), ACC (1:10000) or p-ACC (1:5000) overnight at 4 °C, followed by the corresponding HRP-conjugated secondary antibody (1:50000) for 1 h at room temperature. Specific immune complexes were detected with the ATTO Light-Capture II Western Blotting Detection System. The density of specific bands was calculated using the ImageJ image analysis software.
:20000), C/EBPα (1:10000), SREBP-1 (1:10000), PPARα (1:20000), CPT-1A (1:5000), ACOX1 (1:20000), UCP-2 (1:20000), β-actin (1:20000), AMPK (1:10000), p-AMPK (1:5000), ACC (1:10000) or p-ACC (1:5000) overnight at 4 °C, followed by the corresponding HRP-conjugated secondary antibody (1:50000) for 1 h at room temperature. Specific immune complexes were detected with the ATTO Light-Capture II Western Blotting Detection System. The density of specific bands was calculated using the ImageJ image analysis software.
Statistical analysis
Data are represented as the means ± SD (n = 6). The statistical significance of experimental observations was determined using the Dunnett's test (Fig. 1) or the Tukey–Kramer multiple comparison test (other tables and figures). The level of significance was set at p < 0.05.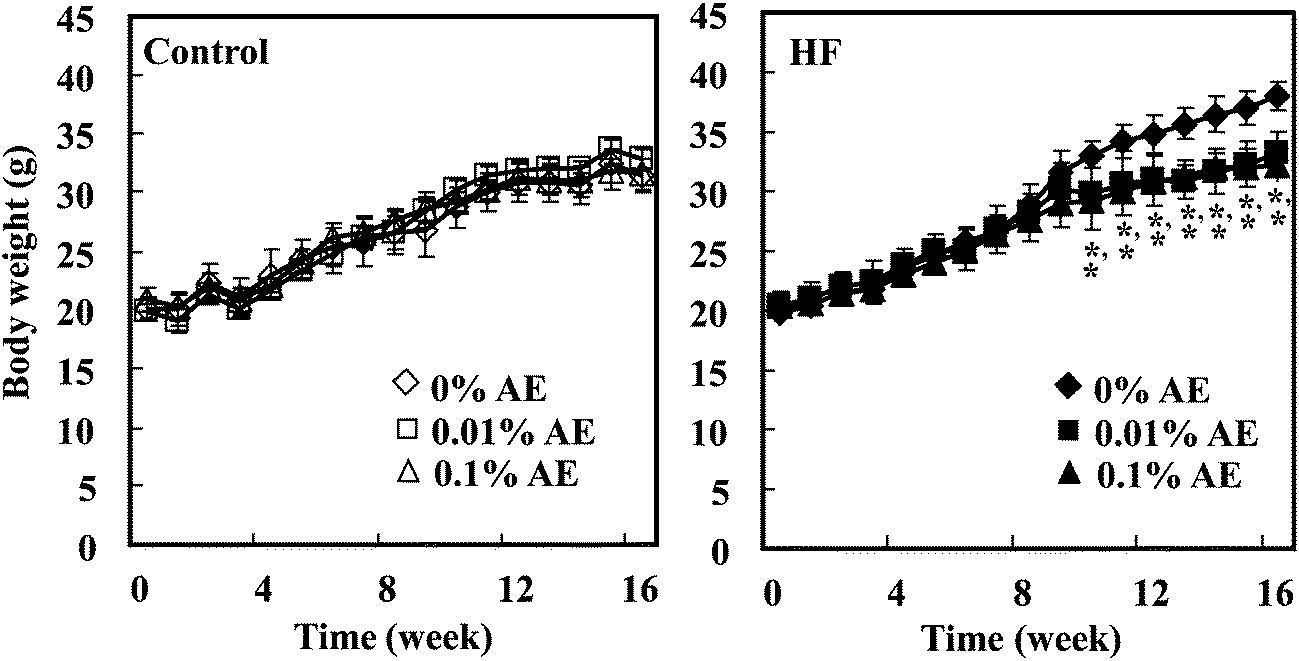 | ||
| Fig. 1 Changes in the body weight of mice fed the control- and HF-diets containing Ashitaba extract (AE) for 16 weeks. Open symbols represent control diet-fed groups, while closed symbols represent HF diet-fed groups. Values are the mean ± SD (n = 6). *p < 0.05 when compared with 0%AE (Dunnett's test). | ||
Results
Effect of Ashitaba extract on the body weight and adipose tissue weight
Ashitaba extract contained 64.89 and 84.86 mg g−1 of 4HD and XAG, respectively, determined by LC-MS/MS analysis. During the feeding period, the body weight of mice was significantly lowered in the HF-0.01 and HF-0.1 groups compared with the HF-0 group from week 10 to 16, respectively (Fig. 1). At the end of the experiment (week 16), we confirmed that the body weight of the mice was significantly higher in the HF-0 group than that in the C-0 group, and that in the HF-0.01 and HF-0.1 groups significantly lowered compared with the HF-0 group (Table 2). The weight of white adipose tissues (epididymal, mesenteric, retroperitoneal, and subcutaneous adipose tissue) was greater in the HF-0 group than that in the C-0 group. Supplementation of Ashitaba extract suppressed the HF-increased adipose tissue weight compared with the HF-0 group. In the groups given the control diet, Ashitaba extract neither affects the body weight nor the adipose tissue weight (Table 2). Ashitaba extract has the potential to reduce the body weight and white adipose weight gain induced by the HF diet.| Group | ||||||
|---|---|---|---|---|---|---|
| Control | HF | |||||
| Ashitaba extract (%) | 0 | 0.01 | 0.1 | 0 | 0.01 | 0.1 |
| a Mice were fed the control or HF diet containing Ashitaba extract for 16 weeks. At the end of the experiment, the body weight and adipose tissue weight were measured after 18 hour fasting. Values are the mean ± SD (n = 6). Values without a common letter in a row differ significantly among groups (p < 0.05) by the Tukey–Kramer multiple comparison test. | ||||||
| Final body weight (g) | 30.8 ± 2.2a | 32.8 ± 1.1ab | 29.2 ± 2.21a | 35.9 ± 3.34b | 31.5 ± 2.11a | 31.5 ± 1.64a |
| Tissue weight (g per 100 g body weight) | ||||||
| Liver | 3.79 ± 0.36a | 3.71 ± 0.33a | 3.88 ± 0.47a | 3.74 ± 0.27a | 3.91 ± 0.32a | 3.62 ± 0.56a |
| White adipose tissue weight | ||||||
| Total | 10.91 ± 4.06a | 13.98 ± 1.18a | 9.75 ± 2.60a | 19.90 ± 4.79b | 11.99 ± 2.8a | 12.25 ± 1.48a |
| Epididymal | 3.22 ± 0.65a | 4.31 ± 0.49ab | 2.94 ± 0.74a | 5.50 ± 1.26b | 3.60 ± 1.26a | 3.85 ± 0.56a |
| Mesenteric | 1.47 ± 0.45ab | 2.10 ± 0.32ab | 1.36 ± 0.24ac | 2.40 ± 0.30b | 1.71 ± 1.03ab | 1.31 ± 0.21ac |
| Retroperitoneal | 1.62 ± 0.61a | 2.22 ± 0.32ab | 1.59 ± 0.42a | 3.10 ± 1.00b | 1.72 ± 0.69a | 2.10 ± 0.73ab |
| Subcutaneous | 4.59 ± 2.47a | 5.53 ± 0.53ab | 3.85 ± 1.61a | 8.90 ± 3.37b | 4.97 ± 1.71a | 4.98 ± 0.87a |
| Brown adipose tissue | 0.73 ± 0.16a | 0.72 ± 0.09a | 0.55 ± 0.12a | 0.47 ± 0.11a | 0.56 ± 0.20a | 0.70 ± 0.19a |
Effects of Ashitaba extract on lipid metabolism
The total plasma cholesterol level was significantly increased in the HF-0 group compared with the C-0 group. HF-0.01 and -0.1 groups significantly lowered the plasma cholesterol level to almost the same level as the control diet-fed groups. On the other hand, Ashitaba extract did not affect plasma triglyceride and NEFA levels in both the control and HF diet-fed groups (Fig. 2). Intake of a HF diet is reported to induce hepatic lipid accumulation, which is involved in systemic insulin resistance.26,27 As shown in Fig. 3, the hepatic total lipid, triglyceride and cholesterol levels in the HF-0 group were significantly higher than those in the C-0 group. The hepatic total lipid, triglyceride and cholesterol levels were significantly lower in HF-0.01 and HF-0.1 groups than those in the HF-0 group. In the control diet-fed mice, Ashitaba extract did not affect hepatic total lipid triglyceride and cholesterol levels. These results indicate that Ashitaba extract has the ability to prevent HF diet-induced lipid accumulation in the liver.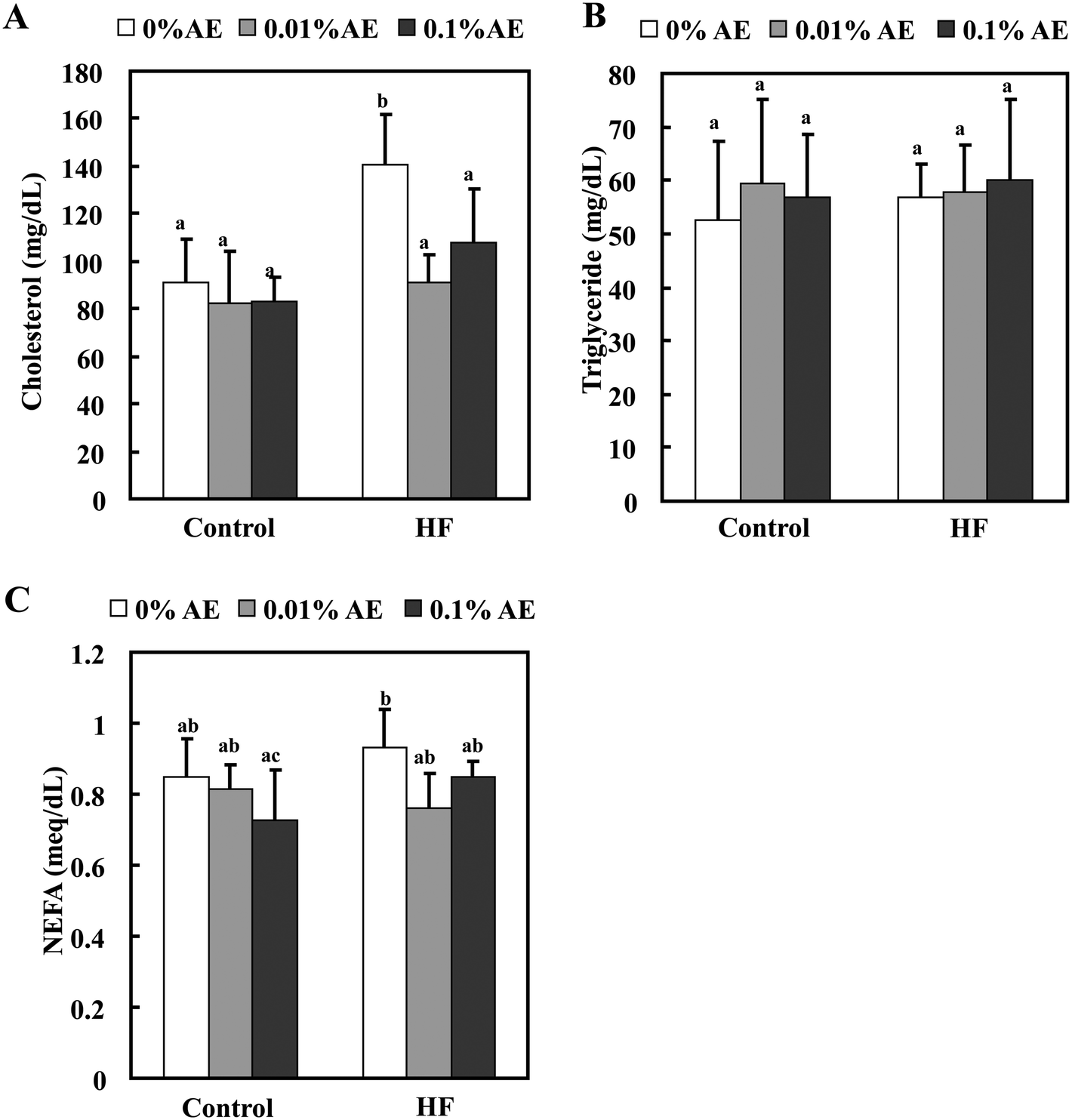 | ||
| Fig. 2 Effect of Ashitaba extract (AE) on the plasma lipid levels. Mice were fed the control- or HF-diet containing Ashitaba extract for 16 weeks. At the end of the experiment, the plasma levels of cholesterol (A), triglyceride (B) and NEFA (C) were measured. Values are the mean ± SD (n = 6). The same letters indicate no significant differences according to the Tukey–Kramer multiple comparison test. p < 0.05 was considered significant. | ||
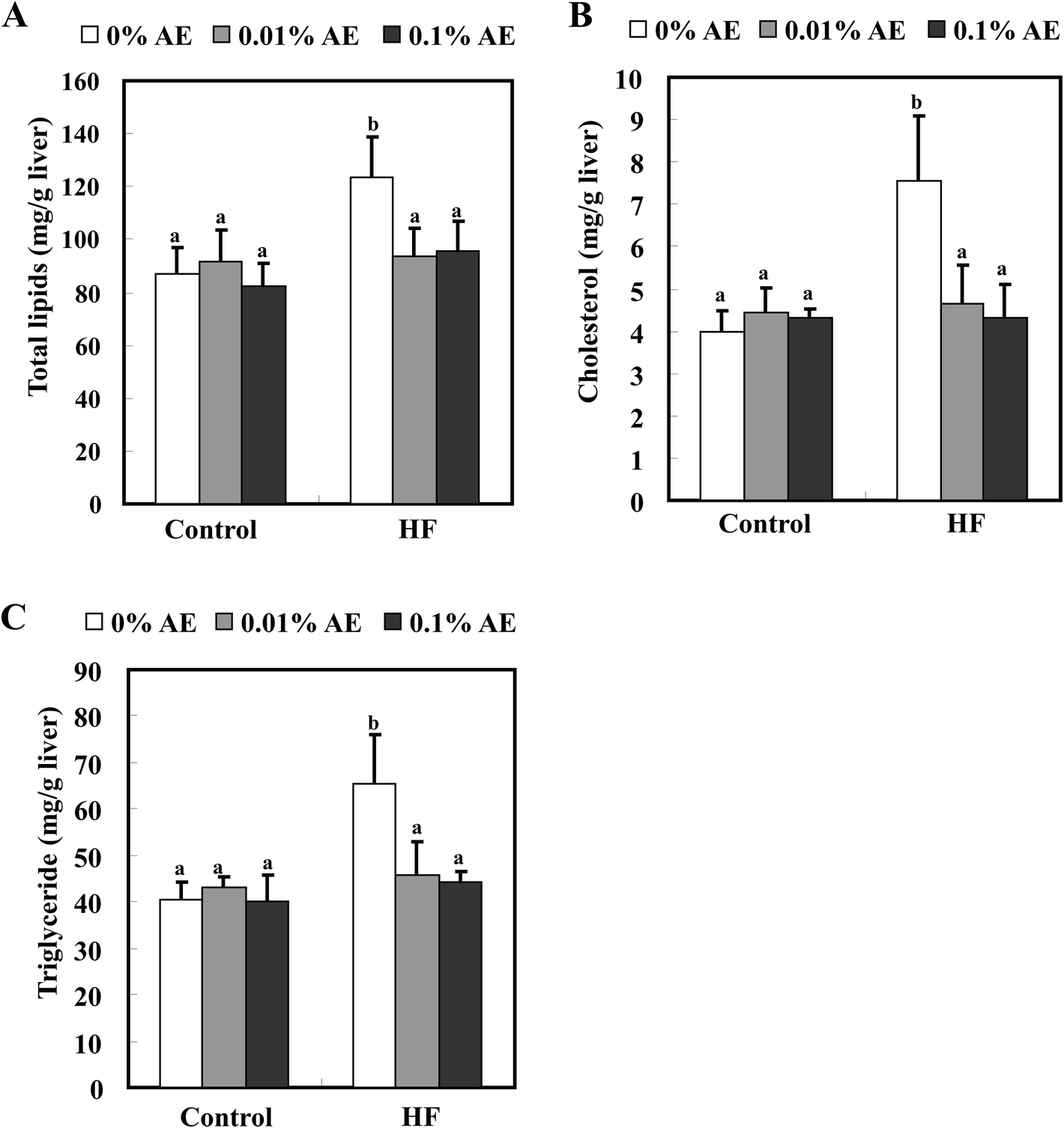 | ||
| Fig. 3 Effects of Ashitaba extract (AE) on the hepatic lipid levels. Mice were fed the control- or HF-diet containing Ashitaba extract for 16 weeks. At the end of the experiment, total lipid (A), cholesterol (B) and triglyceride (C) levels were measured. Values are the mean ± SD (n = 6). The same letters represent no significant differences according to the Tukey–Kramer multiple comparison test. p < 0.05 was considered significant. | ||
Effect of Ashitaba extract on plasma glucose, insulin and adiponectin levels
The plasma glucose level at the end of the experiment was significantly higher in the HF-0 group compared with the C-0 group (Fig. 4A). Supplementation of the HF diet with Ashitaba extract significantly reduced the plasma glucose levels. The plasma insulin level in the HF-0 group was also higher than that in the C-0 group, and supplementation with the Ashitaba extract also reduced the HF-diet increased insulin level to almost the same level as that in the control-diet fed groups (Fig. 4B). Neither glucose nor insulin levels changed among the control diet groups. HOMA-IR, a predictor of total insulin sensitivity, was significantly higher in the HF-0 group than in the C-0 group (Fig. 4C). Supplementation of the HF diet with Ashitaba extract significantly attenuated the HF diet-induced increase in HOMA-IR.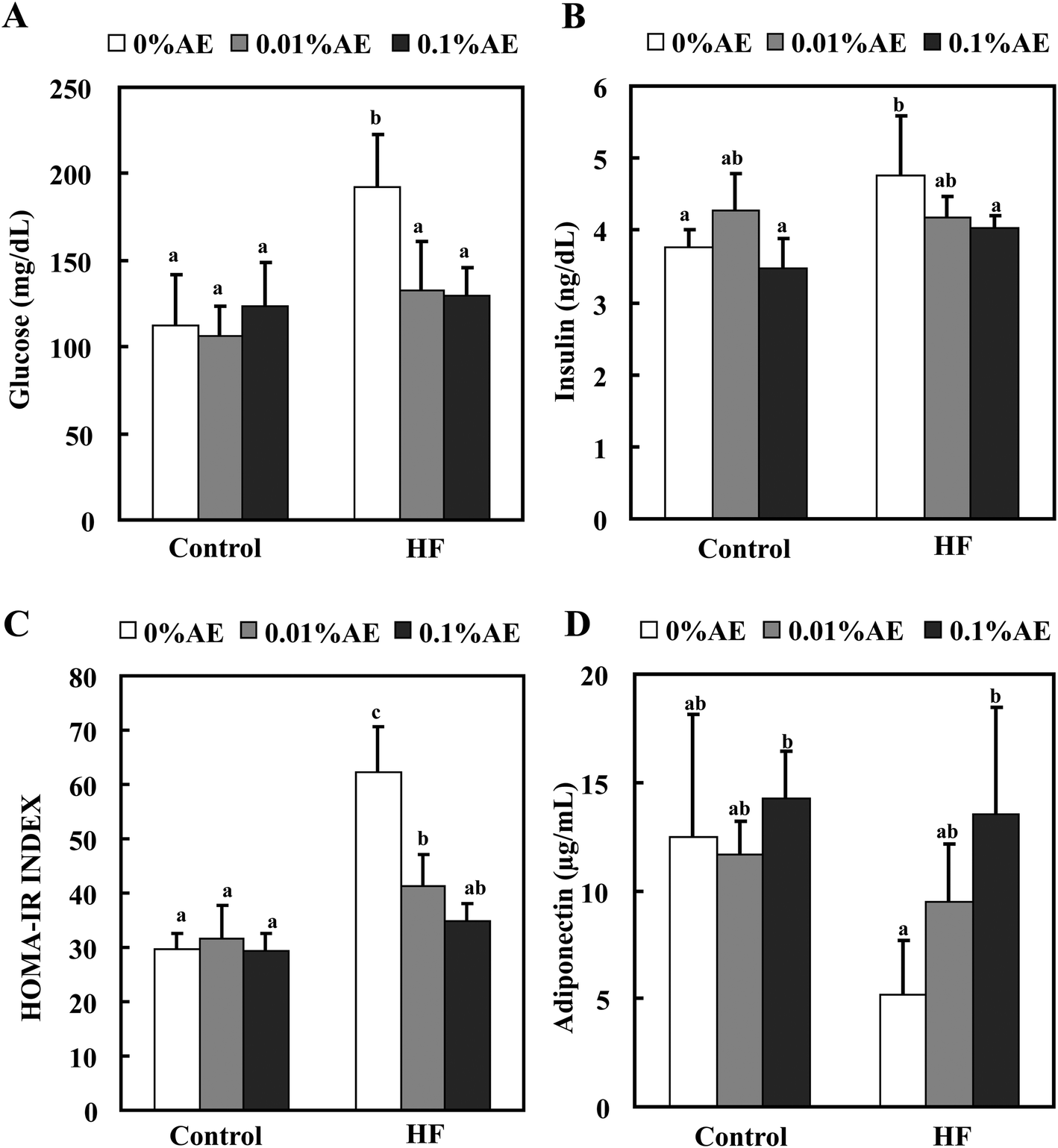 | ||
| Fig. 4 Effects of Ashitaba extract (AE) on plasma glucose, insulin and adiponectin levels. Mice were fed the control- or HF-diet containing Ashitaba extract for 16 weeks. At the end of the experiment, the glucose (A) and insulin (B) levels were measured and the homeostasis model assessment of the insulin resistance index (HOMA-IR) was calculated (C). The plasma adiponectin was also measured (D). Values are the mean ± SD (n = 6). The same letters represent no significant differences according to the Tukey–Kramer multiple comparison test. p < 0.05 was considered significant. | ||
White adipose tissue is a major endocrine tissue that releases various adipocytokines into the bloodstream. Because adiponectin is one of the major adipocytokines associated with maintaining glucose homeostasis,28 we measured the plasma level of adiponectin (Fig. 4D). The plasma adiponectin level was lower in the HF-0 group than that in control-diet fed-groups. Ashitaba extract helped to retain the decreased adiponectin level in the HF diet groups in a dose-dependent manner: the HF-0.1 group revealed a significantly higher adiponectin level compared with the HF-0 group with almost the same level as the control diet groups.
Effect of Ashitaba extract on expression of adipogenesis- and energy metabolism-related proteins in the adipose tissue
AMP-activated protein kinase (AMPK) is a key modulator to maintain the cellular as well as whole-body energy balance.15 It has been known that a HF diet suppresses AMPK activation.29,30 We found that Ashitaba extract supplementation restored AMPK phosphorylation in HF-diet fed-mice (Fig. 5). Since PPARγ, C/EBPα, and SREBP-1 are associated with adipogenesis,31 the expressions of these protein levels in white adipose tissue were determined in Fig. 5; the expressions of PPARγ, C/EBPα and SREBP-1 were significantly increased in the HF-0 group compared with those in the control diet groups. In the HF diet group, supplementation with Ashitaba extract significantly decreased the expression of these adipogenesis-related proteins. In the groups given the control diet, Ashitaba extract did not affect the expression of these proteins. UCP-2, a member of the UCP family, is involved in energy metabolism in adipose tissue and liver.32,33 However, Ashitaba extract did not affect the expression of UCP-2 in both control diet and HF diet groups.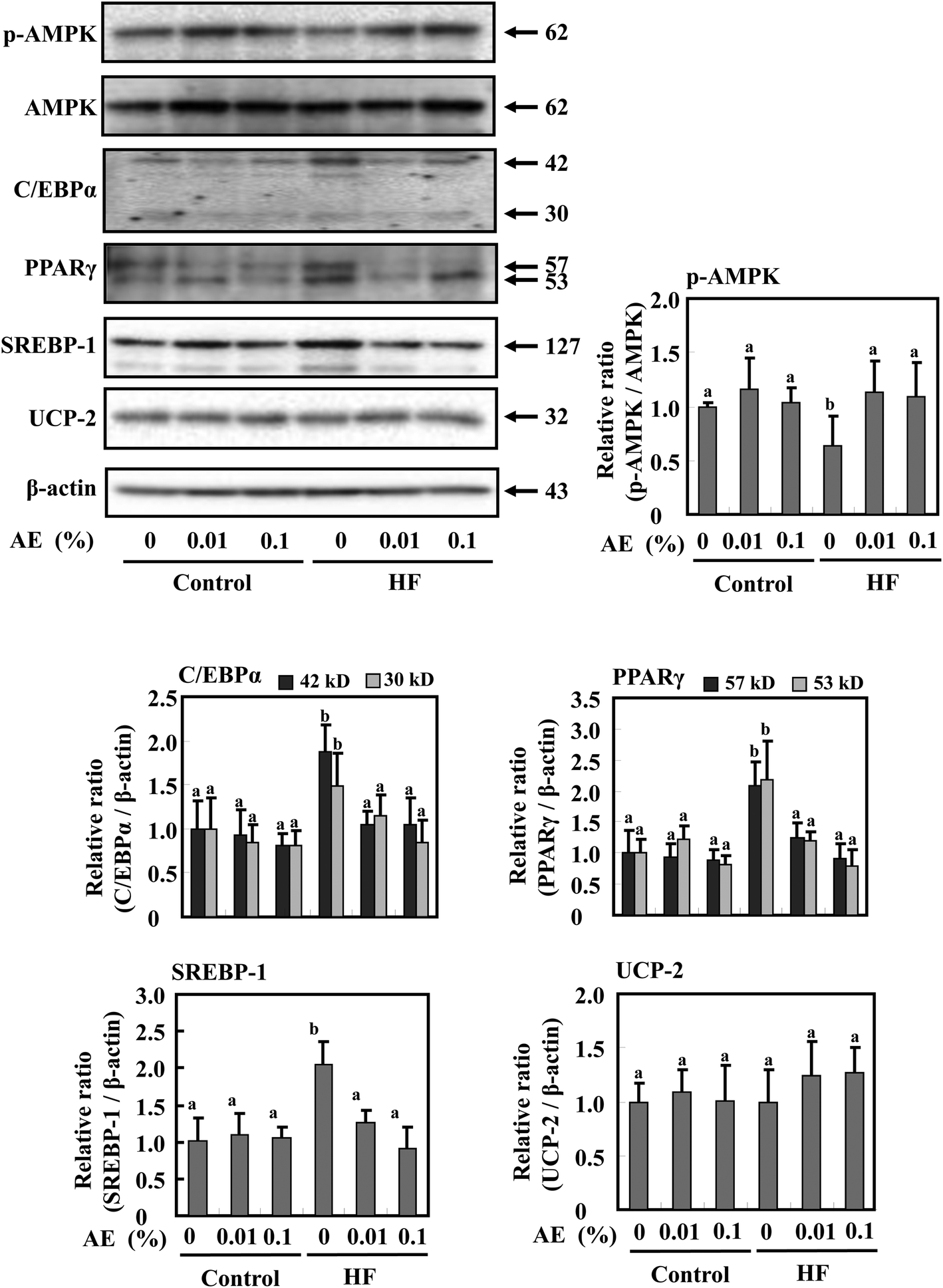 | ||
| Fig. 5 Effects of Ashitaba extract (AE) on expression of adipogenesis- and energy metabolism-related proteins in adipose tissue. Mice were fed the control- or HF-diet containing Ashitaba extract for 16 weeks. The expression of p-AMPK, AMPK, C/EBPα, PPARγ, SREBP-1 and UCP2 in white adipose tissue was evaluated by Western blotting. Densitometric analysis of specific bands for p-AMPK was also shown after normalization by AMPK expression. C/EBPα, PPARγ, SREBP-1 and UCP-2 were shown after normalization by β-actin expression. Values are the mean ± SD (n = 6). The same letters represent no significant differences according to the Tukey–Kramer multiple comparison test. p < 0.05 was considered significant. | ||
Effect of Ashitaba extract on expression of adipogenesis- and energy metabolism-related proteins in the liver
To investigate whether the reduction of fat mass in Ashitaba extract-dosed mice is accompanied by modulation of AMPK activation and lipid metabolism in the liver, Western blotting was performed. As shown in Fig. 6, supplementation of the HF diet groups with Ashitaba extract increased the phosphorylation of AMPK and ACC compared with the HF-0 group. The HF diet significantly increased the expression of SREBP-1 and FAS and supplementation with Ashitaba extract significantly decreased HF diet-induced expression of these proteins, though their expressions in the control-diet fed-mice remained unchanged. Then, we investigated the expression of proteins responsible for fatty acid oxidation and thermogenesis in the liver. The HF diet tended to decrease the expression of CPT1, ACOX1 and PPARα and supplementation with the Ashitaba extract significantly increased HF diet-depressed expression of these proteins. Ashitaba extract did not alter the expression of these proteins in the control diet groups. Similar to the adipose tissue, Ashitaba extract did not affect UCP-2 expression in the liver.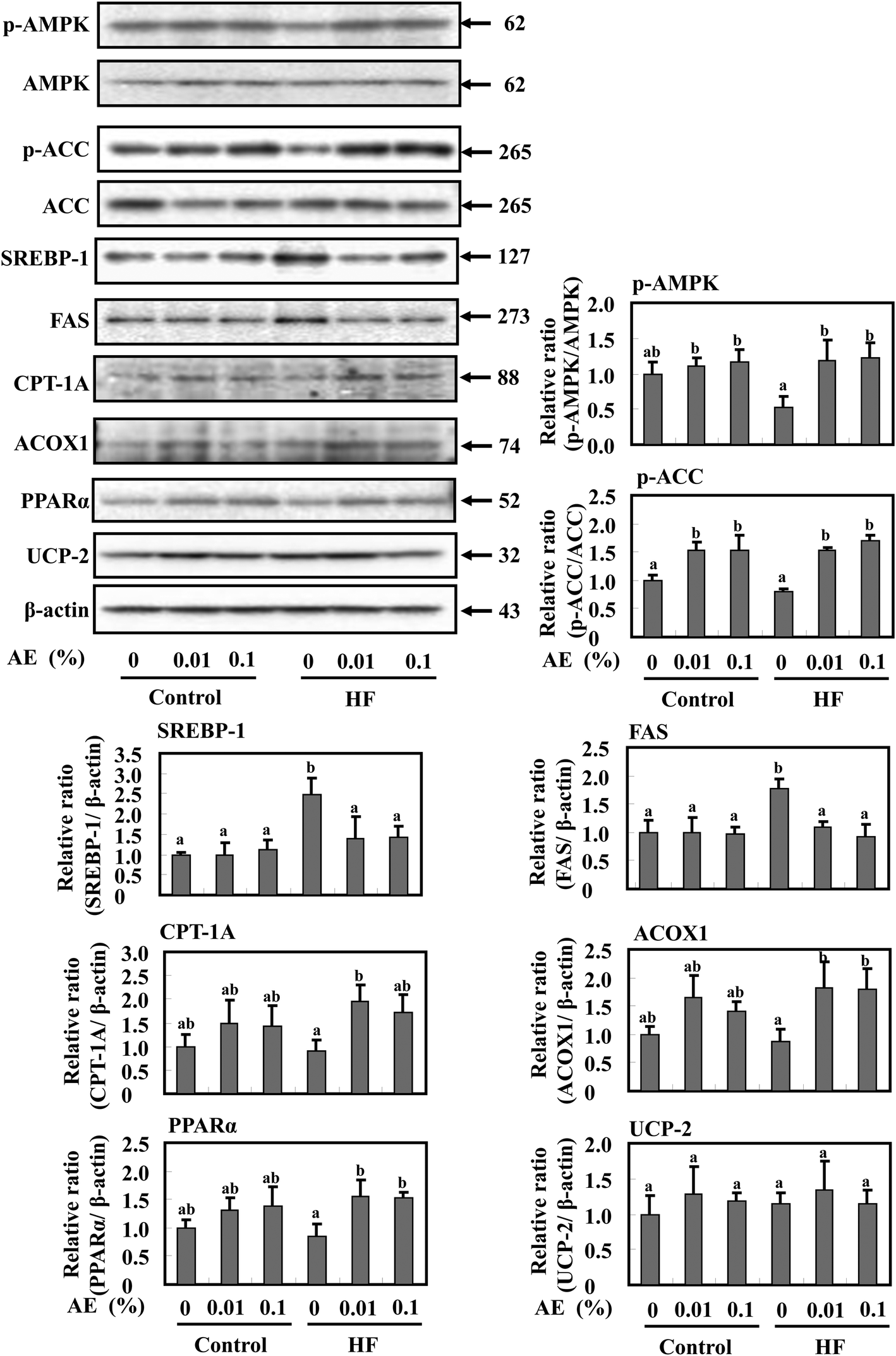 | ||
| Fig. 6 Effects of Ashitaba extract (AE) on expression of adipogenesis- and energy metabolism-related proteins in liver tissue. Mice were fed the control- or HF-diet containing Ashitaba extract for 16 weeks. The expression of p-AMPK, AMPK, p-ACC, ACC, SREBP-1, FAS, CPT-1A, ACOX1, PPARα and UCP-2 in the liver tissue was evaluated by Western blotting. Densitometric analysis of specific bands for p-AMPK and p-ACC was shown after normalization by AMPK and ACC expression, respectively. SREBP-1, FAS, CPT-1A, ACOX1, PPARα and UCP-2 were also shown after normalization by β-actin expression. Values are the mean ± SD (n = 6). The same letters represent no significant differences according to the Tukey–Kramer multiple comparison test. p < 0.05 was considered significant. | ||
Discussion
In this study Ashitaba extract lowered the HF diet-induced body weight and body fat (Table 2 and Fig. 1), accompanied by the prevention of hyperglycemia and hyperlipidemia, as estimated by reducing serum levels of cholesterol, glucose, and insulin and enhancing the adiponectin level (Fig. 2 and 4). Ashitaba extract decreased the hepatic contents of triglyceride and cholesterol, thereby protecting the liver from HF diet-induced dysfunctions (Fig. 3). It was noteworthy that Ashitaba extract restored HF diet-induced inactivation of AMPK in both adipose tissue and liver (Fig. 5 and 6). As the downstream events, Ashitaba extract decreased expression of PPARγ, C/EBPα and SREBP-1, which were involved in adipogenesis in the adipose tissue (Fig. 5), and the extract also decreased the expression of SREBP-1 and FAS, while increased the hepatic expression of CPT-1, ACOX1 and PPARα (Fig. 6).AMPK is a key modulator to maintain the cellular as well as whole-body energy balance. AMPK is activated in response to an increase in the AMP:ATP ratio within the cell and therefore acts as a sensor for cellular energy regulation. Binding of AMP with AMPK allosterically phosphorylates and activates AMPK,34 which in turn shuts down anabolic pathways and supports catabolic pathways through regulating the expression of several proteins of energy metabolism. The activated AMPK interacts with SREBP-1 and inhibits the SREBP-1 target including FAS expression, leading to reduced lipogenesis and lipid accumulation.17,18 Moreover, the activation of AMPK also leads to the phosphorylation and inhibition of ACC activity, resulting in an increase of fatty acid oxidation,19i.e. inactivation of ACC reduces the synthesis of malonyl-CoA, which leads to derepression of CPT-1A and ACOX1, and activates fatty acid oxidation.20 In addition, AMPK also stimulates fatty acid oxidation by up-regulating expression of PPARα.21 The activation of AMPK inhibits the differentiation of adipocytes by down-regulating the expression of C/EBPα and PPARγ.22 It has been known that the HF diet suppresses AMPK activation.29,30 Noticeably, Ashitaba extract treatment restored AMPK phosphorylation in HF-diet fed-mice in both adipose and liver tissues (Fig. 5 and 6). These results indicated that Ashitaba extract regulated lipid metabolism in the adipose tissue and liver through the activation of AMPK.
Ashitaba extract significantly suppressed lipid accumulation in the white adipose tissue, including visceral adipose tissue (Table 2). Furthermore, we found that Ashitaba extract significantly decreased the expression of C/EBPα, PPARγ and SREBP-1 in HF-diet fed-mice (Fig. 5). PPARγ and C/EBPα are considered to be the master regulators or the crucial determinants of the adipocyte fate and play an important role in adipogenesis.31,35 For instance, immortalized fibroblasts lacking PPARγ lose the potential for differentiation into mature adipocytes.36 C/EBPα functions were revealed to be a principal player in adipogenesis from gain-of-function studies in cultured cells37 as well as studies establishing appropriate knockout mice – whole-body C/EBPα-knockout mice, which die shortly after birth owing to liver defects and hypoglycemia because they fail to accumulate lipid in the white or brown adipocytes.38 SREBP-1 regulates lipid metabolism and plays an essential role in the regulation of lipogenesis in fatty acid and triglyceride synthesis.17 Our previous report showed that 4HD and XAG, two main chalcones of Ashitaba, inhibited adipocyte differentiation by down-regulating C/EBP and PPARγ expression.7 The present results indicate that Ashitaba extract suppresses the lipid accumulation in the white adipose tissue by decreasing adipocyte differentiation and lipogenesis.
Ashitaba extract normalized the hepatic lipid content in the HF-diet groups (Fig. 3). Furthermore, we found that Ashitaba extract not only significantly decreased SREBP-1 and FAS expression, but also increased CPT-1A, ACOX1 and PPARα expression in the liver of HF-diet treated mice (Fig. 6). Lipid accumulation in the liver is caused by enhancing de novo lipogenesis and lowering of lipid catabolism. It is known that 25% of liver triglyceride is derived from increased de novo lipogenesis.39De novo lipogenesis is mediated by SREBP-1 that is a key lipogenic transcription factor and nutritionally regulated by glucose and insulin.40,41 SREBP-1 preferentially regulates the lipogenic process by activating genes including FAS, involved in fatty acid and triglyceride synthesis, which contributes to hepatic steatosis. CPT-1, ACOX1 and PPARα were critically associated with the process of fatty acid oxidation.42–45 CPT-1 regulates the transport of fatty acids from the cytoplasm to the mitochondrial matrix across the membrane,42 while ACOX1 is the initial enzyme in the peroxisomal β-oxidation system.43,44 PPARα-mediated responses have been well studied in the liver. It has been reported that the PPARα agonist normalized the fatty liver in HF-diet fed rats45 and markedly improved lipid accumulation in the liver of rats.46 Our previous report showed that 4HD and XAG inhibited the fatty acid-mixture induced lipid accumulation by down-regulating SREBP-1 and up-regulating PPARα expression.8 These results indicated that Ashitaba extract modulated lipid metabolism in the liver by decreasing lipogenesis and increasing fatty acid oxidation.
Visceral adipose tissue is an important predictor of insulin resistance, hyperglycemia and other metabolic risk factors.12,47 Increased adipose tissue weight is accompanied by the induction of inflammatory cytokines involved in insulin resistance.48,49 In addition, visceral adipose tissue has been correlated with the intrahepatic triglyceride content, and an increase in intrahepatic triglycerides is associated with the metabolic abnormalities.26,27,50 The inhibition of fat accumulation in the white adipose tissue and liver by Ashitaba extract may also contribute to prevention of hyperglycemia and insulin resistance. We found that Ashitaba extract decreased plasma glucose and insulin levels and increased the adiponectin level (Fig. 4). Adiponectin is one of the major adipocytokines associated with maintaining glucose homeostasis.28 A previous study showed that Ashitaba extract suppressed acute hyperglycemia in an oral glucose tolerance test of mice, and 4HD and XAG, major polyphenols in Ashitaba extract, stimulate glucose uptake in skeletal muscle cells.6 These results indicate that Ashitaba extract should be an effective material for inhibition of hyperglycemia and insulin resistance.
4HD and XAG are considered to be the major polyphenols in Ashitaba extract. LC-MS/MS results also showed that Ashitaba Chalcone Powder contained 64.89 and 84.86 mg g−1 of 4HD and XAG, respectively. These two compounds modulate lipid metabolism in 3T3-L1 and HepG2 cells.7,8 These results indicate that 4HD and XAG should be the effective compounds for modulating lipid metabolism. The preventive effects of other compounds in Ashitaba extract are not negligible. It has been reported that Ashitaba contains other chalcones, coumarins and flavanones.51 Among them, pteryxin also showed anti-obesity activity.52 However, the preventive effects of other compounds in Ashitaba on adiposity are still not clear. It is necessary to clarify this issue in a future study.
Conclusion
We found that administration of Ashitaba extract reduced the HF diet-induced adiposity, because the extract lowered the body weight gain and serum levels of cholesterol, glucose, and insulin, enhanced the level of adiponectin, and inhibited deposition of lipid in both the adipose tissue and liver. These effects are mainly regulated by the activation of AMPK. Altogether, these findings show that Ashitaba extract should be of benefit to improve the HF diet-induced adiposity.Abbreviations
| ACC | Acetyl CoA carboxylase |
| ACOX1 | Acyl-CoA carboxylase X1 |
| AMPK | AMP-activated protein kinase |
| C/EBP | CCAAT/enhancer-binding protein |
| CPT-1A | Carnitine palmitoyltransferase-1A |
| FAS | Fatty acid synthase |
| 4HD | 4-Hydroxyderricin |
| HF | High-fat |
| PPAR | Peroxisome proliferator-activated receptor |
| SREBP-1 | Sterol regulatory element binding protein-1 |
| UCP-2 | Uncoupling protein-2 |
| XAG | Xanthoangelol |
Acknowledgements
Part of this study was supported by Special Coordination Funds for Promoting Science and Technology, Creation of Innovation Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction Kobe), MEXT, Japan. The authors thank Japan Bio Science Laboratory Co., Ltd for providing the Ashitaba Chalcone Powder. The authors thank Ms Yuki Kimura for her assistance during animal treatment.Notes and references
- M. Kozawa, N. Morita, K. Baba and K. Hata, Chem. Pharm. Bull., 1977, 25, 515–516 CrossRef CAS.
- Y. Kimura, M. Taniguchi and K. Baba, Planta Med., 2004, 70, 211–219 CrossRef CAS PubMed.
- T. Akihisa, H. Tokuda, D. Hasegawa, M. Ukiya, Y. Kimura, F. Enjo, T. Suzuki and H. Nishino, J. Nat. Prod., 2006, 69, 38–42 CrossRef CAS PubMed.
- N. Ohkura, Y. Nakakuki, M. Taniguchi, S. Kanai, A. Nakayama, K. Ohnishi, T. Sakata, T. Nohira, J. Matsuda, K. Baba and G. Atsumi, Biofactors, 2011, 37, 455–461 CrossRef CAS PubMed.
- T. Enoki, H. Ohnogi, K. Nagamine, Y. Kudo, K. Sugiyama, M. Tanabe, E. Kobayashi, H. Sagawa and I. Kato, J. Agric. Food Chem., 2007, 55, 6013–6017 CrossRef CAS PubMed.
- K. Kawabata, K. Sawada, K. Ikeda, I. Fukuda, K. Kawasaki, N. Yamamoto and H. Ashida, Mol. Nutr. Food Res., 2011, 55, 467–475 CAS.
- T. Zhang, K. Sawada, N. Yamamoto and H. Ashida, Mol. Nutr. Food Res., 2013, 57, 1729–1740 CAS.
- T. Zhang, N. Yamamoto and H. Ashida, Food Funct., 2014, 5, 1134–1141 CAS.
- J. M. Friedman, Nature, 2000, 404, 632–634 CAS.
- M. Takahashi, S. Ikemoto and O. Ezaki, J. Nutr. Sci. Vitaminol., 1999, 45, 583–593 CrossRef CAS.
- K. Lois, J. Young and S. Kumar, Int. J. Clin. Pract., 2008, 62, 932–938 CrossRef CAS PubMed.
- J. P. Després, I. Lemieux, J. Bergeron, P. Pibarot, P. Mathieu, E. Larose, J. Rodés-Cabau, O. F. Bertrand and P. Poirier, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 1039–1049 CrossRef PubMed.
- S. Nishikawa, A. Yasoshima, K. Doi, H. Nakayama and K. Uetsuka, Exp. Anim., 2007, 56, 263–272 CrossRef CAS.
- R. S. Surwit, M. N. Feinglos, J. Rodin, A. Sutherland, A. E. Petro, E. C. Opara, C. M. Kuhn and M. Rebuffe-Scrive, Metabolism, 1995, 44, 645–651 CrossRef CAS.
- D. Carling, Trends Biochem. Sci., 2004, 29, 18–24 CrossRef CAS PubMed.
- H. M. O'Neill, G. P. Holloway and G. R. Steinberg, Mol. Cell. Endocrinol., 2013, 366, 135–151 CrossRef PubMed.
- M. S. Brown and J. L. Goldstein, Cell, 1997, 89, 331–340 CrossRef CAS.
- A. J. Sanyal, Nat. Clin. Pract. Gastroenterol. Hepatol., 2005, 2, 46–53 CrossRef CAS PubMed.
- A. Ejaz, D. Wu, P. Kwan and M. Meydani, J. Nutr., 2009, 139, 919–925 CrossRef CAS PubMed.
- J. D. McGarry and N. F. Brown, Eur. J. Biochem., 1997, 244, 1–14 CAS.
- W. J. Lee, Biophys. Res. Commun., 2006, 340, 281–295 Search PubMed.
- Z. Cao, R. M. Umek and S. L. McKnight, Genes Dev., 1991, 5, 1538–1552 CrossRef CAS.
- T. Nakamura, T. Tokushima, K. Kawabata, N. Yamamoto, M. Miyamoto and H. Ashida, Arch. Biochem. Biophys., 2012, 521, 71–76 CrossRef CAS PubMed.
- K. K. Trout, C. Homko and N. C. Tkacs, Biol. Res. Nurs., 2007, 8, 305–318 CrossRef CAS PubMed.
- S. Nishiumi, N. Yamamoto, R. Kodoi, I. Fukuda, K. Yoshida and H. Ashida, Arch. Biochem. Biophys., 2008, 470, 187–199 CrossRef CAS PubMed.
- K. M. Korenblat, E. Fabbrini, B. S. Mohammed and S. Klein, Gastroenterology, 2008, 134, 1369–1375 CrossRef CAS PubMed.
- J. H. Hwang, D. T. Stein, N. Barzilai, M. H. Cui, J. Tonelli, P. Kishore and M. Hawkins, Am. J. Physiol.: Endocrinol. Metab., 2007, 293, 1663–1669 CrossRef PubMed.
- H. Tilg and A. R. Moschen, Mol. Med., 2008, 14, 222 CAS.
- J. Pang, Y. Choi and T. Park, Arch. Biochem. Biophys., 2008, 476, 178–185 CrossRef CAS PubMed.
- S. J. Kim, J. Y. Jung, H. W. Kim and T. Park, Biol. Pharm. Bull., 2008, 31, 1415–1421 CAS.
- M. I. Lefterova and M. A. Lazar, Trends Endocrinol. Metab., 2009, 20, 107–114 CrossRef CAS PubMed.
- O. Boss, P. Muzzin and J. P. Giacobiono, Eur. J. Endocrinol., 1998, 139, 1–9 CrossRef CAS PubMed.
- D. F. Ricquier and F. Bouillaud, Biochem. J., 2000, 345, 161–179 CrossRef CAS.
- N. B. Ruderman, H. Park, V. K. Kaushik, D. Dean, S. Constant, M. Prentki and A. K. Saha, Acta Physiol. Scand., 2003, 178, 435–442 CrossRef CAS PubMed.
- P. Tontonoz, E. Hu, R. A. Graves, A. I. Budavari and B. M. Spiegelman, Genes Dev., 1994, 8, 1224–1234 CrossRef CAS.
- E. D. Rosen, C. H. Hsu, X. Wang, S. Sakai, M. W. Freeman, F. J. Gonzalez and B. M. Spiegelman, Genes Dev., 2002, 16, 22–26 CrossRef CAS PubMed.
- S. O. Freytag, D. L. Paielli and J. D. Gilbert, Genes Dev., 1994, 8, 1654–1663 CrossRef CAS.
- N. D. Wang, M. J. Finegold, A. Bradley, C. N. Ou, S. V. Abdelsayed, M. D. Wilde, L. R. Taylor, D. R. Wilson and G. J. Darlington, Science, 1995, 269, 1108–1112 CrossRef CAS.
- K. L. Donnelly, C. I. Smith, S. J. Schwarzenberg, J. Jessurun, M. D. Boldt and E. J. Parks, J. Clin. Invest., 2005, 115, 1343–1351 CrossRef CAS PubMed.
- J. L. Goldstein and M. S. Brown, J. Clin. Invest., 2008, 118, 1220–1222 CrossRef CAS PubMed.
- G. Zhou, R. Myers, Y. Li, Y. Chen, X. Shen, J. Fenyk-Melody, M. Wu, J. Centre, T. Doebber, N. Fujii, N. Musi, M. F. Hirshman, L. J. Goodyear and D. E. Moller, J. Clin. Invest., 2001, 108, 1167–1674 CrossRef CAS PubMed.
- J. P. Bonnefont, F. Djouadi, C. Prip-Buus, S. Gobin, A. Munnich and J. Bastin, Mol. Aspects Med., 2004, 25, 495–520 CrossRef CAS PubMed.
- S. Miyazawa, H. Hayashi, M. Hijikata, N. Ishii, S. Furuta, H. Kagamiyama, T. Osumi and T. Hashimoto, J. Biol. Chem., 1987, 262, 8131–8137 CAS.
- R. J. A. Wanders, S. Ferdinandusse, G. A. Jansen, P. Vreken, H. R. Waterham, C. W. T. Van Roermund and E. G. Van Grunsven, Biochem. Soc. Trans., 2001, 29, 250–266 CrossRef CAS.
- O. Braissant and W. Wahli, Endocrinology, 1998, 139, 2748–2754 CAS.
- J. M. Ye, M. A. Iglesias, D. G. Watson, B. Ellis, L. Wood, P. B. Jensen, R. V. Sorensen, P. J. Larsen, G. J. Cooney, K. Wassermann and E. W. Kraegen, Am. J. Physiol. Endocrinol. Metab., 2003, 284, 531–540 Search PubMed.
- A. Gastaldelli, Y. Miyazaki, M. Pettiti, M. Matsuda, S. Mahankali, E. Santini, R. A. DeFronzo and E. Ferrannini, J. Clin. Endocrinol. Metab., 2002, 87, 5098–5103 CrossRef CAS PubMed.
- G. S. Hotamisligil, Nature, 2006, 444, 860–867 CrossRef CAS PubMed.
- T. You, R. Yang, M. F. Lyles, D. Gong and B. J. Nicklas, Am. J. Physiol. Endocrinol. Metab., 2005, 288, 741–747 CrossRef PubMed.
- E. Fabbrini, F. Magkos, B. S. Mohammed, T. Pietka, N. A. Abumrad, B. W. Patterson, A. Okunade and S. Klein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15430–15435 CrossRef CAS PubMed.
- T. Akihisa, H. Tokuda, M. Ukiya, M. Iizuka, S. Schneider, K. Ogasawara, M. Teruo, I. Kenji, S. Takashi and H. Nishino, Cancer Lett., 2003, 201, 133–137 CrossRef CAS.
- R. N. Nugara, M. Inafuku, K. Takara, H. Iwasaki and H. Oku, Nutrition, 2014, 30, 1177–1184 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |