
Biocatalytic perchlorate reduction: kinetics and effects of groundwater characteristics
Justin M.
Hutchison
 and
Julie L.
Zilles†
*
and
Julie L.
Zilles†
*
Department of Civil and Environmental Engineering, University of Illinois, Urbana, Illinois 61801, USA. E-mail: jzilles@illinois.edu; Fax: +(217) 333 9464; Tel: +(217) 244 2925
First published on 3rd September 2015
Abstract
Biocatalytic reduction of perchlorate can minimize the effects of competitive electron acceptors and completely reduce perchlorate into chloride and oxygen, but to date has only been demonstrated under idealized laboratory conditions. This work investigated biocatalytic perchlorate reduction in two groundwater drinking water sources, under a range of conditions and with a variety of electron donors. The biocatalysts, perchlorate reductase and chlorite dismutase from Azospira oryzae, had a maximum activity of 162.5 ± 8.4 U (μg Mo)−1 in buffered solution and retained 82–94% of their activity in groundwater samples. The half saturation concentration for perchlorate was 92.0 μM. Perchlorate reduction rates were higher than nitrate reduction rates, with nitrate as the sole electron acceptor having reduction rates 7.5 to 9.7% of the maximum perchlorate reduction rates in groundwater. Activity was consistent from pH 6.5 to 9.0. The temperature dependence of biocatalytic perchlorate reduction was well defined by the Arrhenius equation. No significant difference in biocatalytic activity was observed with calcium and magnesium concentrations over the tested range of 0 to 400 mg L−1 or with natural organic matter up to 6 mg L−1. Ascorbic acid with addition of an electron shuttle resulted in reduction of more than 99% of perchlorate in less than 6 hours, an order of magnitude loss in activity compared to methyl viologen. These results suggest the potential of the biocatalysts for treating perchlorate over a range of concentrations and conditions representative of industrial and groundwater perchlorate contamination.
Water impactPerchlorate, an endocrine disrupter, is toxic to sensitive populations at low concentrations. This manuscript investigated biocatalytic removal of perchlorate for use in drinking water treatment. Robust biocatalytic activity was observed in groundwater samples over a range of conditions. Measured perchlorate removal rates in groundwater samples provide a basis for reactor design. The results support the potential for biocatalytic perchlorate removal. |
1. Introduction
Widespread perchlorate contamination of drinking water has been found in over 20 U.S. states, resulting in advisory or regulatory limits in several states and a pending regulatory limit of 15 μg L−1 from the United States Environmental Protection Agency.1–3 These regulations are intended to prevent developmental defects in fetuses and young children arising from preferential uptake of perchlorate in the thyroid.4–6To remove perchlorate from drinking water, municipalities primarily use non-selective or selective ion exchange.1 Whole-cell biological processes have also been shown to reduce perchlorate.1 However, these technologies have significant drawbacks. Non-selective ion exchange produces brine waste with elevated perchlorate concentrations and is less effective for perchlorate removal in the presence of high concentrations of competing anions such as nitrate and sulfate.7 Specialized, bi-functional resins target perchlorate more specifically, reducing the impact of competing anions.8 The disadvantage is that these specialized resins are not easily regenerated and are generally incinerated after saturation,8 increasing costs and environmental impacts. Whole cell biological reduction has been explored in a number of configurations including fixed beds,9,10 bioelectrochemical reduction,11 and membrane biofilm reactors.12 However, biological reduction of perchlorate also performs poorly in the presence of co-contaminating nitrate, sulfate, and oxygen, since these are preferred electron acceptors for many microorganisms.13–15 Other challenges associated with biological perchlorate removal include the potential for hydrogen sulfide production, the possible growth of pathogenic organisms, and public perception.
Because co-contaminating nitrate and sulfate are common in drinking water sources,16 these problems necessitate advances in perchlorate treatment. To that end, a wide variety of approaches are being investigated, including chemical17 and biological18 processes to treat perchlorate in waste brines, direct biological regeneration of perchlorate-selective resins,19,20 ion exchange membrane bioreactors,21 and two stage membrane biofilm reactors that minimize sulfate reduction.22 We recently proposed a system that selectively reduces perchlorate into innocuous chloride and oxygen using cell-free biocatalysts, specifically perchlorate reductase (PR) and chlorite dismutase (CD) from Azospira oryzae, and provided proof of concept for this approach in buffered, laboratory solutions.23 PR is a soluble, periplasmic protein with similarity to nitrate reductases.24 CD is also a soluble protein and catalyzes an intramolecular electron transfer to form the final products of chloride and oxygen.25
Biocatalytic treatment is generally attractive due to high substrate affinities and reaction rates, specificity, and optimal activities under ambient conditions of temperature, pressure, and pH. However, to date biocatalysts have largely been used only for high value products such as pharmaceuticals.26 A prominent exception is the extracellular biocatalyst laccase, which has been used to treat phenolic compounds in industrial wastewater (e.g. forest products industry27 and textile and dye-making industry28). Laccase has also been proposed for oxidation of phenolic compounds such as pharmaceuticals in wastewater effluent.29
For treatment of perchlorate and other chlorine oxyanions, a biocatalytic approach shares with biological treatment processes the advantage of completely degrading the contaminant, but avoids some of the challenges associated with whole-cell reduction of perchlorate. Specifically, unlike whole cells, which preferentially use nitrate, the biocatalysts target perchlorate even in the presence of excess nitrate and have no activity with sulfate, reducing the amount of electron donor that would be required to treat perchlorate in the presence of competing anions.23 Furthermore, because the biocatalysts are non-living, the process can operate under nutrient limited conditions, avoiding formation of hydrogen sulfide, mitigating any hazard posed by pathogenic organisms, and minimizing the formation of biofilms. In addition, these biocatalysts showed good stability, maintaining perchlorate reduction up to 23 days.23 While promising, this initial proof-of-concept study did not determine kinetic parameters for perchlorate reduction. It was further limited by its exclusive use of buffered laboratory solutions and ideal electron donors.
To better understand the potential advantages and limitations of the biocatalytic system for perchlorate removal, this work measured the biocatalysts' kinetic activities in two real world groundwater samples and laboratory buffered conditions. The effects of temperature, pH, natural organic matter (NOM), calcium, and magnesium were specifically investigated, and a variety of potential electron donors were tested. The results provide a basis for evaluating the practical potential for biocatalytic removal of perchlorate during drinking water treatment.
2. Materials and methods
2.1 Biocatalyst preparation, media, and chemicals
Biocatalysts were obtained from the perchlorate-reducing A. oryzae strain PS (ATCC number BAA-33). The anaerobic growth media was as previously described,23 with 14.7 mM acetate as electron donor and 7 mM perchlorate as electron acceptor. Preparation of A. oryzae soluble protein fraction containing PR and CD was also as previously described, including addition of glycerol to a final concentration of 10% before storage.23To normalize activity across different preparations, two measurements were used: molybdenum content, as an indirect measure of PR concentration, and total protein. To determine molybdenum content, aliquots of each soluble protein fraction were taken prior to addition of glycerol and dialyzed to remove salts and free molybdenum using 3000 Dalton molecular weight cut off dialysis cassettes (Thermo Scientific) with three 50 mM phosphate buffer exchanges. Original volume of sample was maintained. Samples were analyzed with inductively coupled plasma-optical emission spectrometry (ICP-OES) (PerkinElmer Optima 2000DV, Waltham, MA). To facilitate comparison to the literature, activity was also normalized to total protein concentrations in the soluble protein fractions, as determined using the Bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Soluble protein fractions produced in this work contained an average of 21.22 ± 1.76 mg mL−1 protein and 357 ± 39 μg L−1 molybdenum.
All solutions were prepared with Nanopure water (18 MΩ cm), produced from deionized water in an EMD Millipore Milli-Q (Model Number: Z00QSV0US) System (Billerica, MA). Unless otherwise specified, chemicals were purchased from Fisher Scientific (Pittsburgh, PA). Anaerobic solutions were prepared by degassing with N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 for 30 minutes, and headspace was degassed with the same mixture for 5 minutes. The ratio of N2:CO2 was varied in the range of 80:20 to 100:0 to maintain the desired pH.
:CO2 for 30 minutes, and headspace was degassed with the same mixture for 5 minutes. The ratio of N2:CO2 was varied in the range of 80:20 to 100:0 to maintain the desired pH.
2.2 Groundwater sampling and characterization
Groundwater was collected from two sources. The Illinois groundwater (Illinois GW) was harvested from a depth of 46.3 meters in the Illinoisian Formation above the Mahomet Aquifer. This water was known to have high amounts of iron and manganese and was therefore pretreated in a manganese greensand filter. Water was also collected in Eastern Iowa (Iowa GW), from a depth of 151 meters in the Silurian-Devonian Aquifer, without pretreatment. Five gallon samples were collected and stored in polypropylene jerricans in the dark at 4 °C.The groundwater samples were characterized after equilibration with the atmosphere and, for the Illinois sample, after pretreatment, corresponding to the expected placement of the biocatalysts in the treatment train for drinking water prior to disinfection. Oxygen concentration was measured using a rugged dissolved oxygen (RDO) probe (Thermo Scientific, 087020MD). pH was measured using a Thermo Orion 8172 BN ROSS Sure-Flow pH electrode. Bicarbonate concentration was estimated from alkalinity pH titration measurement using 0.1, 0.01 and 0.001 M HCl. Hardness was tested by titration (Hach Total Hardness Kit, HA-71A). Selected metals were analyzed with inductively coupled plasma-optical emission spectrometry (ICP-OES) (PerkinElmer Optima 2000DV). Total ammonia (NH3/NH4 mg L−1 NH–N) was analyzed by colorimetric analysis (Hach salicylate kit). Halides were measured using Thermo Scientific Ion Selective Electrodes. Perchlorate was quantified using ion chromatography (IC) with conductivity detection (IC-CD; Dionex ICS-2000) on an Ion Pac AG-16 and AS-16,30 and nitrate, sulfate, chlorate and chlorite were analyzed on an Ion Pac AG-18 and AS-18 hydroxide-selective anion exchange column as previously described.23
2.3 Colorimetric biocatalytic activity assays
Biocatalytic activities were analyzed using a standard colorimetric assay for perchlorate reduction, which uses methyl viologen (MV) as an electron donor.24 As previously described,23 the assays were performed in stoppered anaerobic cuvettes (Absorption Cells 117.104, Hellma USA, Inc., Plainview, NY) at room temperature. In brief, the methyl viologen was first reduced with sodium dithionite, twenty μL soluble protein fraction was added and monitored until the absorbance (578 nm) slope stabilized, and then electron acceptor (perchlorate, chlorate, nitrate, sulfate, or anion combinations as specified) was added and the reaction was followed by absorbance measurements. The background activity of the soluble protein fraction (measured without electron acceptor) was subtracted. An extinction coefficient of 13.1 mM−1 cm−1 was used.31 Units (U) represent one μmole of MV oxidized per minute. Because MV donates electrons for perchlorate reduction and can also react with the oxygen that is produced, up to eight moles of MV could be oxidized per mole of perchlorate reduced to chloride. Activity measurements were performed in triplicate from independent growths (biological replicates) and reported with standard deviation. Data was fit to the single substrate Michaelis–Menten kinetic equation:| v = Vmax[S]/(Km + [S]) |
Using the MV assay, biocatalytic activity was tested over a range of conditions. The pH was varied from 6–9 in increments of 0.5 in assays conducted with 1 mM perchlorate and Iowa GW. Iowa GW pH was adjusted with hydrochloric acid or sodium hydroxide prior to degassing. pH was maintained during the degassing process by determined ratios of carbon dioxide and nitrogen.
Biocatalytic activity was also determined over a temperature range from 5 °C to 30 °C in increments of 5 °C, again using the MV assay, 1 mM perchlorate, and Iowa GW. Temperature was controlled by putting the spectrophotometer (Thermo Scientific Genesys 20) in an incubator (Thermo Scientific MaxQ 6000). Solutions were allowed to equilibrate to the specified temperature before measurement. The data was fit to the Arrhenius' equation shown:
| k = Ae−Ea/(RT) |
The effects of calcium, magnesium, and NOM were determined in 50 mM Tris Cl− (pH 7.5) buffered conditions. Calcium chloride and magnesium chloride were tested individually at concentrations up to 400 mg L−1. Suwanee River NOM (IHSS, St. Paul, MN) was tested from 1 to 6 mg L−1.
2.4 Alternative electron donors
To characterize the range of electron donors that can be used by the biocatalysts, a variety of organic electron donors were tested in buffered solutions. Based on previous work with NADH,32 each of these potential donors was tested with and without the electron shuttle 5-methylphenazinium methyl sulfate (PMS) (Acros Organics, New Jersey). Twenty μL of soluble protein fraction were incubated in 10 mL samples containing 5 mM electron donor, 0 or 100 μM PMS, and 1 mM perchlorate. Initial assays were incubated on the benchtop (approximately 22 °C) for 24 hours and then frozen at −80 °C to halt enzyme activity. Perchlorate removal was monitored by IC as detailed in section 2.2. Controls included no soluble protein fraction and no perchlorate samples for each reaction mixture. The initial reaction rate was quantified for one promising candidate, ascorbic acid, by scaling the reaction up to 100 mL with 1 mL of soluble protein fraction in stoppered anaerobic media bottles and withdrawing 3 mL samples hourly for perchlorate measurements.2.5 Preliminary design calculations
Initial calculations for perchlorate treatment in a batch reactor system were determined using an influent perchlorate concentration of 100 μg L−1 and an effluent concentration of 10 μg L−1. The reactor was modeled using an integrated form of the Michaelis–Menten equation and a hydraulic retention time (HRT) of two hours. For initial calculations, nitrate was not included as an inhibitory effect.2.6 Statistical analysis
The assumption of equal variance was tested using F-test. Statistical analysis was performed using the independent-samples t-test with equal variance. Samples were considered significantly different with an alpha of less than 0.05.3. Results
3.1 Characterization of groundwater
The two groundwater samples were similar in composition (Table 1). The hardness and alkalinity measurements are characteristic of very hard water in the United States.33 The alkalinity of the samples nevertheless represents a decrease in buffering capacity as compared to the laboratory buffered system. Other than hardness, the groundwater characteristics were within typical ranges (Table 1). No perchlorate, chlorate or nitrate were detected, and the levels of sulfate were below the EPA regulatory and advisory limits.34| Component | Units | Illinois GW | Iowa GW | MCLa or NSDWRb | Typical valuesc |
|---|---|---|---|---|---|
| a Maximum Contaminant Level.34 b National Secondary Drinking Water Regulations.34 c Ref. 33. d ND – Not detected, detection limits NO3− (10 ppb), ClO3− (10 ppb), ClO4− (5 ppb). e Temperatures were determined for near surface groundwater from mean annual temperature. | |||||
| pH | 7.34 | 7.19 | 6.5–8.5b | 6.0–8.5 | |
| Alkalinity | mg L−1 as HCO3− | 393.0 | 378.9 | — | — |
| Hardness | mg L−1 of CaCO3 | 342.0 | 376.2 | — | 121–180 |
| Ca | ppm | 66.8 | 70.5 | — | >15 |
| Fe | ppb | 0.5 | 480 | 300b | <10000 |
| K | ppm | 1.62 | 0.78 | — | <10 |
| Mg | ppm | 26.9 | 28.5 | — | <300 |
| Mn | ppb | 68 | 37 | 50b | <200 |
| Mo | ppm | 0 | 0 | 0.03–1 | — |
| Na | ppm | 26.2 | 12.08 | 0.2 | <1000 |
| P | ppm | 0.16 | 0.13 | — | — |
| S | ppm | 1.24 | 1.03 | — | — |
| Ammonia | mg L−1 NH3–N | <0.4 | <0.4 | ||
| Fluoride | ppm | 0 | 0 | 4a/2b | <10 |
| Chloride | ppm | 61 | 52 | 250b | <10 |
| Bromide | ppm | 2 | 0 | — | — |
| Iodide | ppm | 0 | 0 | — | — |
| Perchlorate | ppm | NDd | ND | — | — |
| Chlorate | ppm | ND | ND | — | — |
| Chlorite | ppm | ND | ND | 1.0 | — |
| Nitrate | ppm | ND | ND | 44a | <50 |
| Sulfate | ppm | 9.5 | 9.7 | 250b | <1000 |
| TOC | ppm | 1.74 | 1.22 | — | 0.1–6 |
| Temperature | °C | — | 2.78–25e | ||
| DO | mg L−1 | 9.66 | 10.06 | — | — |
3.2 Biocatalytic activity in groundwater
To determine activities of the biocatalysts at realistic perchlorate concentrations and in groundwater, the soluble protein fractions were assayed in real groundwater over a range of perchlorate concentrations. Although the biocatalysts were not purified, throughout this work the measured activity is attributed to PR and CD. This assumption is supported by the high expression of PR in A. oryzae cells grown on perchlorate24 and by the unique activity of CD. It is however possible that some of the measured activity was due to a nitrate reductase, which can also show activity for perchlorate.24 To account for variation in biocatalyst content across different preparations, activities were normalized to molybdenum concentration, because subunit A of PR has one molecule of molybdenum.24 The biocatalysts showed good activity in groundwater (Fig. 1), maintaining 82% (Illinois GW) and 94% (Iowa GW) of their activity in laboratory solutions. To facilitate comparison to previously published results,23,24 the biocatalysts' activity was also normalized to total protein content. The activity values were 2.49 ± 0.22 U mg total protein−1 in buffer, 2.22 ± 0.38 U mg total protein−1 in the Illinois GW, and 2.28 ± 0.12 U mg total protein−1 in Iowa GW. The background activity in groundwater was less than 0.1% of the maximum perchlorate reducing rates. The maximum reaction rates (Vmax) and half saturation constants (Km) of the soluble protein fractions were calculated using the single substrate form of Michaelis Menten kinetic equation (Table 2). Kinetic values for Illinois GW and Iowa GW were not statistically different from buffer.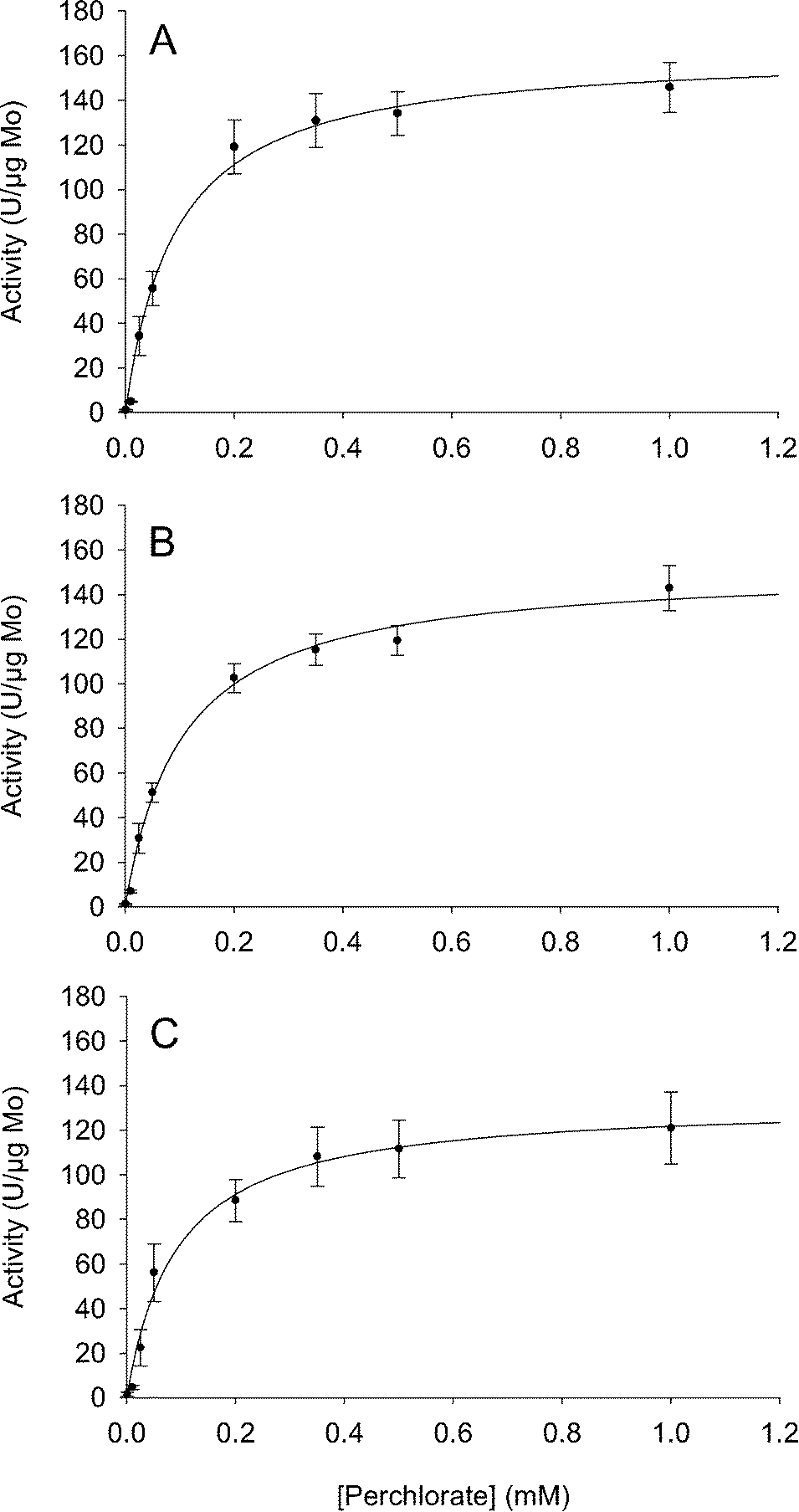 | ||
| Fig. 1 Biocatalytic perchlorate reduction in buffered systems (A) and groundwater (B, Iowa GW; C, Illinois GW). Results of MV activity assays conducted on triplicate independent soluble protein fractions at perchlorate concentrations from 0.0005–1 mM. Activity is given in Units (U), defined as 1 μmol MV oxidized per minute, and are normalized to molybdenum as an indirect measure of PR concentration. Solid line represents Michaelis–Menten kinetics model. Error bars represent standard error. | ||
| Component | Illinois GW | Iowa GW | Buffer system |
|---|---|---|---|
| a Average ± standard error. b Units (U) are defined as 1 μmol MV oxidized per minute and are normalized to molybdenum as an indirect measure of PR concentration. | |||
| Maximum velocitya (Vmax) (U (μg Mo)−1)b | 132.9 ± 9.8 | 152.4 ± 6.3 | 162.5 ± 8.4 |
| Half saturation constanta (Km) (mM) | 0.091 ± 0.026 | 0.105 ± 0.016 | 0.092 ± 0.019 |
When chlorate was supplied instead of perchlorate, the maximum activity was higher (658.3 ± 36.8 U (μg Mo)−1versus 152.4 ± 6.3 U (μg Mo)−1), and the half saturation constant was lower (50 ± 12 μM versus 105 ± 16 μM) in Iowa GW. This suggests the system will also be effective for chlorate remediation and that chlorate will not accumulate during perchlorate removal. The activity with chlorite could not be tested in this assay because it reacts with MV.
Because a key advantage of the biocatalytic system is the specificity it exhibits for perchlorate,23 the specificity was confirmed in groundwater. Assays with 1 mM nitrate as the sole electron acceptor showed slow reduction, with rates only 9.7 ± 0.4% of perchlorate reduction rates in Iowa GW and 7.5 ± 2.3% in Illinois GW. These results are slightly better than previous results in a buffered system, where nitrate had 24.9 ± 3.6% of perchlorate activity.23 Nitrate activity could be due to the presence of a putative nitrate reductase35 in the soluble protein fractions or to the similarity between PR and nitrate reductase.36 Simultaneous addition of 1 mM nitrate and 1 mM perchlorate lowered the observed reduction rates to 72.1 ± 1.1% of perchlorate reduction rates in Iowa GW and 71.8 ± 3.2% in Illinois GW. This rate is difficult to interpret, since the colorimetric response could come from either electron acceptor. However, by quantifying perchlorate in endpoint assays, prior work demonstrated that the biocatalysts showed good perchlorate removal even in the presence of 100-fold excess nitrate.23 There was no observed sulfate activity in either groundwater or in previous work.23
3.3. Effects of groundwater characteristics on activity
In addition to rapid and selective perchlorate reduction in real world waters, application of biocatalysts requires an understanding of their response to common variables. Several important factors: pH, temperature, calcium, magnesium, and NOM, were tested here for their impact on the perchlorate reducing activities of the biocatalysts. pH and temperature were tested in Iowa GW, while calcium, magnesium, and NOM were tested in buffered conditions.Over the pH range tested here (6.0–9.0), the biocatalysts showed robust perchlorate reduction (Fig. 2). Activity decreased only at pH 6.0, with a 48% loss of activity, but even at pH 6.0, the values were not significantly different (P = 0.10) from pH 7.0. A stronger response to temperature was observed, with a gradual decrease in activity as temperature decreased, culminating in a 68% decrease in activity when comparing activity at 10 °C to 25 °C. Using the Arrhenius equation, the activation energy of the biocatalysts was 45.6 kJ mole−1, and the pre-exponential factor was ln(21.6) s−1. Data fit the equation with a coefficient of determination of .970 (Fig. 3).
 | ||
| Fig. 2 Effect of pH on biocatalytic perchlorate reduction. Results of MV assays conducted in Iowa GW with 1 mM perchlorate. Activity is given in Units (U), defined as 1 μmol MV oxidized per minute, and are normalized to molybdenum as an indirect measure of PR concentration. Average and standard deviation of triplicate independent soluble protein fractions are presented. | ||
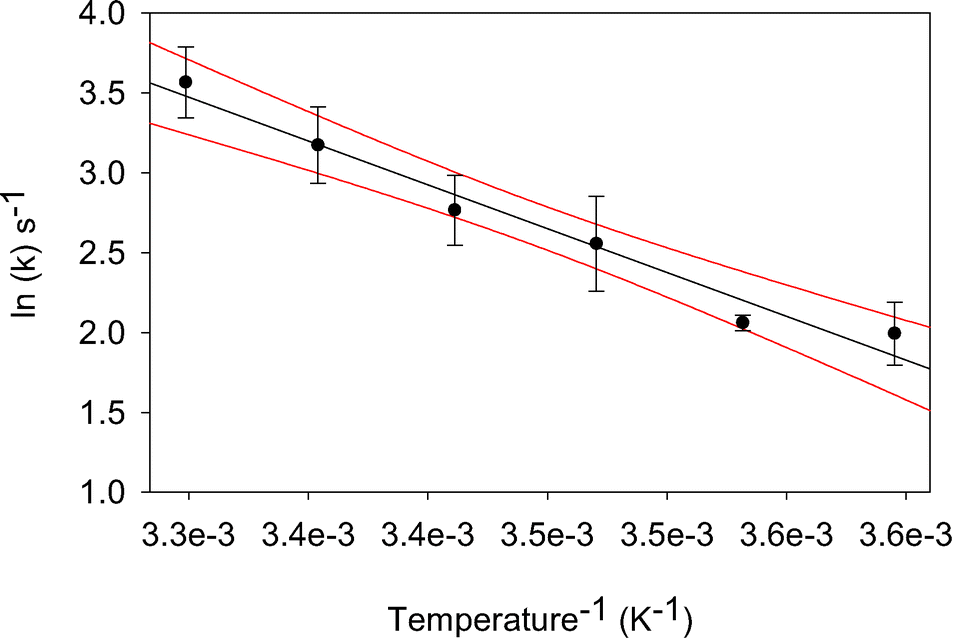 | ||
| Fig. 3 Effect of temperature on biocatalytic perchlorate reduction. Results of MV assays conducted in Iowa GW with 1 mM perchlorate at specified temperature. Average and standard deviation of triplicate independent soluble protein fractions are presented. Black line indicates Arrhenius equation fit, and red lines indicate 95% confidence interval. | ||
No statistically significant differences in biocatalyst activity were observed over calcium and magnesium concentrations from 0 to 400 m L−1, although a slight decreasing trend might be occurring for calcium (Fig. 4), culminating with a drop in activity of 24.2%. Suwanee River NOM also had no statistically significant impact on perchlorate reducing activity over the range from 1–6 mg L−1 of NOM tested (Fig. 4). Slight differences in the zero-point reference activities are due to fluctuations in room temperature.
 | ||
| Fig. 4 Effect of calcium and magnesium (A) and NOM (B) on biocatalytic perchlorate reduction. Results of MV assays conducted in buffered system with 1 mM perchlorate. Activity is given in Units (U), defined as 1 μmol MV oxidized per minute, and are normalized to molybdenum as an indirect measure of PR concentration. Average and standard deviation of triplicate independent soluble protein fractions are presented. | ||
3.4. Alternative electron donors
Previous studies on perchlorate reduction have used either MV or NADH/PMS as electron donors (e.g.ref. 23, 24, 32). However, these compounds are relatively expensive. MV and PMS are also oxygen-sensitive. We therefore tested a variety of alternative organic electron donors: sodium acetate, ascorbic acid, citric acid, ethanol, formic acid, and sodium pyruvate. The electron donors tested in this study are common metabolites. In conditions without an additional electron shuttle, there was no detectable perchlorate removal.However, with the addition of 100 μM PMS as an electron shuttle, the results were more promising. Ascorbic acid showed the most potential to act as an electron donor for the perchlorate reducing enzymes, achieving a statistically significant 32.0 ± 15.7% decrease as compared to the initial concentration of perchlorate (P = .028). With formic acid 14.3 ± 9.7% of the perchlorate was reduced as compared to the initial perchlorate concentration; however, the results were not statistically significant (P = 0.086). Citric acid and pyruvate also were able to reduce perchlorate; however, these results were inconsistent across replicates, perhaps due to the involvement of an additional component from the soluble protein fraction. No perchlorate reduction was detected with acetate or ethanol. No perchlorate reduction was observed in controls without biocatalysts.
As ascorbic acid with PMS demonstrated the most promise for perchlorate reduction, the rate of perchlorate reduction was tested for this system. Robust perchlorate reduction was observed, with 52.3 ± 8.4% perchlorate reduced within the first hour (Fig. 5). This initial perchlorate reduction rate corresponds to 2.2 μmol perchlorate reduced per min per μg of molybdenum. For comparison to MV assay results, assuming the methyl viologen reaction consumes 8 electrons for each perchlorate molecule, the perchlorate reduction rate for methyl viologen at 1 mM perchlorate would be 19.6 μmol min−1 μg−1. Using ascorbic acid with PMS as an electron donor system therefore results in approximately an order of magnitude loss in activity. After six hours, over 99% of the perchlorate had been reduced.
 | ||
| Fig. 5 Biocatalytic perchlorate reduction with ascorbic acid and PMS. Reactions were conducted in buffer with an initial concentration of 1 mM perchlorate, and reaction progress was monitored by quantification of perchlorate. Average and standard deviation of triplicate independent soluble protein fractions are presented. | ||
4. Discussion
This work demonstrates the activity of the perchlorate-reducing biocatalysts under application-appropriate conditions. Activities in real groundwater were only slightly lower than in laboratory buffered solutions, comparing to buffered values measured here and in previous reports.23,32 Considering a broader range of typical groundwater conditions, the activity was relatively insensitive to pH, hardness, and NOM, and showed a gradual decrease with decreasing temperature. Substitution of ascorbic acid and electron shuttle PMS for MV resulted in approximately an order of magnitude drop in activity. Here we discuss these findings in the context of the literature and describe their implications for process design.The half saturation constants measured here (91–105 μM) were within the range of reported values for PR and perchlorate, which span a bacterial consortium reported at 0.28 μM (ref. 13) to 4700 μM for Dechlorosoma sp KJ with the electron donor acetate.37 They are higher than that published for purified PR from another A. oryzae strain, GR-1, which was 27 μM Km for perchlorate.24 This discrepancy could reflect differences in the PR encoded by these two strains, or it could be due to our use of soluble protein fractions rather than purified protein. Another component in the soluble protein fractions could cause some type of interference or competition that raises the apparent Km. If the affinity for perchlorate becomes a limiting factor for technology development, it should be possible to improve it by removing interfering factors and/or using a higher-affinity homolog.
The effects of groundwater characteristics reported here are generally consistent with the limited information available in prior publications. Our soluble protein fraction has shown perchlorate reducing activity as low as 5 °C, with the highest activity at the highest temperature tested, 30 °C. Purified PR from strain perc1ace has perchlorate reducing activity in the range of 20 to 40 °C with optimal activity at 25 to 35 °C.38 To our knowledge, no prior reports of activation energy for PR or CD exist, but a related enzyme, nitrate reductase, has activation energies of 41–42 kJ mol−1,39,40 very similar to the value of 45.6 kJ mol−1 reported here for perchlorate reduction. Work with strain perc1ace showed consistent PR activity over a pH range from 7.0 to 9.0, in agreement with our results.38 For strain GR-1, optimal CD activity is achieved at a pH of 6.0 and drops precipitously below 6.0.25 A direct comparison of our results with these CD results is, however, not possible, because the colorimetric assay used in our experiments measures the combined effects of PR and CD. To our knowledge, the effects of magnesium, calcium, and NOM on perchlorate reduction have not been previously studied.
The electron donors tested in this study were selected based on their occurrence in bacterial metabolism, which was anticipated to increase the likelihood of successful interaction with PR. However, these common electron donors were unable to donate electrons directly for perchlorate reduction; the addition of a shuttle was required. While the biocatalysts are able to reduce perchlorate in the presence of oxygen, at this point, the known options for supplying reducing power (MV or a shuttle with NADH or ascorbic acid) for biocatalytic perchlorate reduction all involve an oxygen-sensitive component. Biocatalytic treatment of perchlorate for drinking water therefore would require anaerobic operation of the system. Alternatively, development of an oxygen-stable electron donor or shuttle would provide a broader range of potential operational conditions.
In comparison to ligand-enhanced rhenium complex/palladium catalysts under development for perchlorate reduction,41 the biocatalysts show much higher activity. To correspond as nearly as possible to the chemical convention of normalizing to active site, we used the Mo-normalized values for the biocatalysts. Comparing perchlorate reduction rates at 1 mM initial perchlorate, these chemical catalysts reduced 0.317 mmol perchlorate min−1 (mmol Rh)−1, compared to the biocatalysts' rate of 1900 mmol min−1 (mmol Mo)−1. This represents a 6000 fold higher activity for the biocatalysts. Another option is to compare the kcat for the biocatalysts (1716 min−1) to these chemical catalysts' kobs value of 0.0415 min−1 value: a 41000 fold larger turnover number for the biocatalysts. For large-scale application, the biocatalysts' activity at neutral pH is also a significant advantage, as the chemical catalysts' activities were reported at pH 3, where they are most active.
Considering the implications of the kinetics reported here for practical application of the biocatalysts, the best available basis for comparison is a recent life cycle analysis (LCA) of perchlorate treatment options. For traditional rhenium/palladium catalysts, this LCA projected that a 20-fold increase in activity was required for the technology to be competitive versus ion exchange and biological reduction.42,43 Considering that the ligand-enhanced catalysts used as a comparison here already represent an approximately 140-fold increase in activity over the values used in the LCA,41 and that the biocatalytic activities reported in this work are orders of magnitude higher than the enhanced catalysts, suggests that a biocatalytic process will be competitive with existing technologies. However, a comprehensive evaluation of the costs and environmental impacts of the biocatalysts is needed to guide continued progress towards application. The results presented here provide a solid basis for conducting such an evaluation.
This work also provides a basis for preliminary design calculations. For treatment of groundwater-sourced drinking water, in their current soluble form, the biocatalysts could be applied in batch reactors. Based on the kinetic results, a batch reactor operating at 25 °C and an average HRT of 2 hours would require a dosing rate of 0.1 μg molybdenum equivalence of biocatalyst for each liter of water treated. This dosage corresponds to 280 μL of biocatalysts for each liter of water treated. If the pH were at or below pH 6.5, the dosage would be 0.15 μg molybdenum equivalence of biocatalyst each liter of water treated. Operating at 5 °C would require dosing rates of 0.38 μg molybdenum equivalence of biocatalyst each liter of water treated. From prior applications of enzymes industrially, there are also a variety of methods for immobilizing enzymes, which could be applied to PR and CD to reduce the dosage, costs and environmental impacts. Finally, it is important to note that while molybdenum is an essential trace element, it can also contribute to copper deficiency and cause toxic effects at high levels of consumption. However, even if all of the molybdenum was released from PR, these dosage values are approximately two orders of magnitude lower than the reference dose limits for molybdenum of 5 μg kg−1 day−1 recommended by the US EPA.44
5. Conclusions
This work demonstrates the potential of biocatalysts for perchlorate reduction in drinking water treatment. The biocatalysts showed effective perchlorate reduction over a perchlorate range from 0.5 μM to 1 mM, representing perchlorate contamination found in municipal drinking water to industrial/military industrial sites, in real groundwater and under typical ranges of groundwater characteristics. Preliminary design calculations suggest that perchlorate could be removed to concentrations less than the likely EPA regulation limit of 15 ppb with hydraulic retention times of 2 hours, supporting its practical potential, although a detailed economic and environmental assessment is still needed.The biocatalysts have advantages compared to traditional treatment technologies. As compared to the industry standard of ion exchange, the biocatalysts completely reduce perchlorate to innocuous chloride and oxygen and show minimal interference from competing anions nitrate and sulfate. In comparison to whole-cell based biological perchlorate remediation, the biocatalysts have a lower demand for electron donor, because they show specificity for perchlorate over nitrate. Because the biocatalysts are non-living, they should pose lower risk and be more widely accepted for use in drinking water treatment.
Acknowledgements
We thank Brittany Webb for assisting with kinetic experiments, Kellen Mobilia for helpful discussion, and anonymous reviewers for their constructive comments. Support for this research was provided by the U.S. National Science Foundation (CBET 1336620 and DGE 1144245), ARCS® (Achievement Rewards for College Scientists) Foundation, Inc.'s Scholar Illinois Chapter (2014, 2015), and The University of Illinois Clean Energy Education Fellowship.References
- Perchlorate Treatment Technology Update, U.S. Environmental Protection Agency, 542-R-05-015, Washington, D.C., 2005 Search PubMed.
- Drinking Water: Preliminary Regulatory Determination on Perchlorate, US Environmental Protection Agency, Federal Register 73 FR 60262, 2008, 60262-60282 Search PubMed.
- EPA Seeks Advice on Perchlorate in Drinking Water - Agency Issues Interim Health Advisory, US Environmental Protection Agency, EPA-HQ-OW-2009-0297-0675, Washington, D.C., 2009 Search PubMed.
- R. J. Brechner, G. D. Parkhurst, W. O. Humble, M. B. Brown and W. H. Herman, J. Occup. Environ. Med., 2000, 42, 777–782 CrossRef CAS.
- M. A. Greer, G. Goodman, R. C. Pleus and S. E. Greer, Environ. Health Perspect., 2002, 110, 927–937 CrossRef CAS.
- T. Zewdie, C. M. Smith, M. Hutcheson and C. R. West, Environ. Health Perspect., 2010, 118, 42–48 CAS.
- A. R. Tripp and D. A. Clifford, J. - Am. Water Works Assoc., 2006, 98, 105–114 CAS.
- B. H. Gu, G. M. Brown and C. C. Chiang, Environ. Sci. Technol., 2007, 41, 6277–6282 CrossRef CAS.
- J. C. Brown, R. D. Anderson, J. H. Min, L. Boulos, D. Prasifka and G. J. G. Juby, J. - Am. Water Works Assoc., 2005, 97, 70–81 CAS.
- S. K. de Long, X. Li, S. Bae, J. C. Brown, L. Raskin, K. A. Kinney and M. J. Kirisits, J. Appl. Microbiol., 2012, 112, 579–592 CrossRef CAS PubMed.
- C. S. Butler, P. Clauwaert, S. J. Green, W. Verstraete and R. Nerenberg, Environ. Sci. Technol., 2010, 44, 4685–4691 CrossRef CAS PubMed.
- K. J. Martin and R. Nerenberg, Bioresour. Technol., 2012, 122, 83–94 CrossRef CAS PubMed.
- M. R. London, S. K. de Long, M. D. Strahota, L. E. Katz and G. E. Speitel Jr, Water Res., 2011, 45, 6593–6601 CrossRef CAS PubMed.
- J. C. Brown, V. L. Snoeyink, L. Raskin and R. Lin, Water Res., 2003, 37, 206–214 CrossRef CAS.
- S. G. Lehman, M. Badruzzaman, S. Adham, D. J. Roberts and D. A. Clifford, Water Res., 2008, 42, 969–976 CrossRef CAS PubMed.
- E. T. Urbansky, Perchlorate in the Environment, Kluwer Academic/Plenum Publishers, New York, 2000 Search PubMed.
- J. Liu, J. K. Choe, Z. Sasnow, C. J. Werth and T. J. Strathmann, Water Res., 2013, 47, 91–101 CrossRef CAS PubMed.
- B. E. Logan, J. Wu and R. F. Unz, Water Res., 2001, 35, 3034–3038 CrossRef CAS.
- A. K. Venkatesan and J. R. Batista, Biorem. J., 2011, 15, 1–11 CrossRef CAS PubMed.
- M. Sharbatmaleki and J. R. Batista, Water Res., 2012, 46, 21–32 CrossRef CAS PubMed.
- C. T. Matos, S. Velizarov, J. G. Crespo and M. A. Reis, Water Res., 2006, 40, 231–240 CrossRef CAS PubMed.
- A. Ontiveros-Valencia, Y. Tang, R. Krajmalnik-Brown and B. E. Rittmann, Water Res., 2014, 55, 215–224 CrossRef CAS PubMed.
- J. M. Hutchison, S. K. Poust, M. Kumar, D. M. Cropek, I. E. MacAllister, C. M. Arnett and J. L. Zilles, Environ. Sci. Technol., 2013, 47, 9934–9941 CrossRef CAS PubMed.
- S. W. Kengen, G. B. Rikken, W. R. Hagen, C. G. van Ginkel and A. J. Stams, J. Bacteriol., 1999, 181, 6706–6711 CAS.
- C. G. van Ginkel, G. B. Rikken, A. G. M. Kroon and S. W. M. Kengen, Arch. Microbiol., 1996, 166, 321–326 CrossRef CAS.
- A. J. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13, 548–556 CrossRef CAS.
- P. Widsten and A. Kandelbauer, Enzyme Microb. Technol., 2008, 42, 293–307 CrossRef CAS PubMed.
- D. T. D'Souza, R. Tiwari, A. K. Sah and C. Raghukumar, Enzyme Microb. Technol., 2006, 38, 504–511 CrossRef PubMed.
- S. Ba, A. Arsenault, T. Hassani, J. P. Jones and H. Cabana, Crit. Rev. Biotechnol., 2013, 33, 404–418 CrossRef CAS PubMed.
- D. Shuai, B. P. Chaplin, J. R. Shapley, N. P. Menendez, D. C. McCalman, W. F. Schneider and C. J. Werth, Environ. Sci. Technol., 2010, 44, 1773–1779 CrossRef CAS PubMed.
- R. N. F. Thorneley, Biochim. Biophys. Acta, Bioenerg., 1974, 333, 487–496 CrossRef CAS.
- M. Heinnickel, S. C. Smith, J. Koo, S. M. O'Connor and J. D. Coates, Environ. Sci. Technol., 2011, 45, 2958–2964 CrossRef CAS PubMed.
- American Water Works Association, Groundwater, American Water Works Association, Denver, CO, 3rd edn, 2003 Search PubMed.
- Drinking Water Contaminants, US Environmental Protection Agency, EPA 819-F-09-0004, 2009 Search PubMed.
- K. G. Byrne-Bailey and J. D. Coates, J. Bacteriol., 2012, 194, 2767–2768 CrossRef PubMed.
- M. J. Oosterkamp, F. Mehboob, G. Schraa, C. M. Plugge and A. J. Stams, Biochem. Soc. Trans., 2011, 39, 230–235 CrossRef CAS PubMed.
- B. E. Logan, H. Zhang, P. Mulvaney, M. G. Milner, I. M. Head and R. F. Unz, Appl. Environ. Microbiol., 2001, 67, 2499–2506 CrossRef CAS PubMed.
- B. C. Okeke and W. T. Frankenberger Jr., Microbiol. Res., 2003, 158, 337–344 CrossRef CAS PubMed.
- J. R. Mahan, T. D. Sherman and E. A. Funkhouser, Plant Cell Physiol., 1988, 29, 735–737 CAS.
- B. Lledo, R. M. Martinez-Espinosa, F. C. Marhuenda-Egea and M. J. Bonete, Biochim. Biophys. Acta, 2004, 1674, 50–59 CrossRef CAS PubMed.
- J. Liu, J. K. Choe, Y. Wang, J. R. Shapley, C. J. Werth and T. J. Strathmann, ACS Catal., 2015, 5, 511–522 CrossRef CAS.
- J. K. Choe, M. H. Mehnert, J. S. Guest, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2013, 47, 4644–4652 CrossRef CAS PubMed.
- K. D. Hurley, Y. Zhang and J. R. Shapley, J. Am. Chem. Soc., 2009, 131, 14172–14173 CrossRef CAS PubMed.
- US Environmental Protection Agency, IRIS - Molybdenum, http://www.epa.gov/iris/subst/0425.htm#oralrfd, (accessed March, 2015).
Footnote |
| † Mailing address: 3230C Newmark Civil Engineering Laboratory MC250, 205 North Mathews Avenue, Urbana, IL 61801, USA. |
| This journal is © The Royal Society of Chemistry 2015 |