
Effects of ultraviolet light on silver nanoparticle mobility and dissolution†
Anjuliee M.
Mittelman
a,
John D.
Fortner
b and
Kurt D.
Pennell
*a
aDepartment of Civil and Environmental Engineering, Tufts University, 200 College Ave, Medford, Massachusetts 02155, USA. E-mail: kurt.pennell@tufts.edu; Fax: +1 (617) 627 3994; Tel: +1 (617) 627 3099
bDepartment of Energy, Environmental, and Chemical Engineering, Washington University at St. Louis, 1 Brookings Drive, St. Louis, Missouri 63130, USA
First published on 2nd September 2015
Abstract
Nanomaterials are subject to various physical, chemical, and biological transformations, necessitating a better understanding of the impact of “aging” processes on nanoparticle fate and transport in engineered and natural porous media. The objective of this study was to evaluate the mobility and dissolution of citrate-coated silver nanoparticles (nAg, 11 nm diameter) in water-saturated sand following ultraviolet (UV) irradiation with UVA (320–400 nm) or UVB (280–320 nm) light. A 3-day UV exposure resulted in up to a 5-fold increase in mean diameter, a 10 to 15 mV increase in zeta potential (i.e., less negative), red shifts in surface plasmon resonance, and up to a 25-fold increase in Ag+ release. The addition of a reactive oxygen species (OH˙) scavenger, tert-butyl alcohol, reduced aggregation and dissolution of nAg exposed to UV light up to 50%, indicating that free radical activity plays a central role in aging. Transport experiments conducted in columns packed with 40–50 mesh Ottawa sand revealed that 25 to 50% more UVA- and UVB-aged nAg were retained compared to freshly prepared (un-aged) nAg. Additionally, 35 to 50% of the applied UV-aged nAg mass eluted as Ag+, compared to less than 1% in experiments with fresh nAg. UVB exposure resulted in up to 4-fold greater Ag+ release and greater nAg retention compared with UVA exposure, consistent with the less negative zeta potential of UVB- compared to UVA-aged nAg (−31 vs. −37 mV). These findings demonstrate that exposure to UV light significantly enhances nAg retention and dissolution in porous media, and thus, oxidative aging of nAg is likely to enhance Ag+ release.
Nano impactNanomaterials will experience surface transformations as a result of chemical, physical, and biological processes in natural and engineered systems. The potential impacts of “aging” processes on nanomaterial fate and transport behavior in porous media are not typically considered in nanoparticle transport studies. In this work, silver nanoparticles (nAg) were exposed to ultraviolet (UV) light and monitored for changes in morphology, surface chemistry, and dissolution kinetics. UV exposure resulted in aggregation, reduced stability, and increased Ag+ release. In sand columns, aged nAg exhibited 50% greater retention and dissolved more rapidly compared with fresh nAg. These findings demonstrate that aged nanomaterials are likely to exhibit reduced mobility in natural and engineered porous media, and may undergo more rapid release of core materials. |
1. Introduction
Over the course of their life cycle, engineered nanomaterials may be subjected to various transformation processes, including photodegradation, passivation by natural organic matter, biodegradation, and oxidation.1 These processes are likely to modify or “age” nanomaterials, resulting in changes to nanoparticle surface properties, which may in turn alter their fate and transport in natural and engineered systems. Most nanoparticles are coated with inorganic or organic compounds to protect the core material and to increase their stability in aqueous suspensions.2 For example, silver nanoparticles are typically coated with citrate or polyvinylpyrrolidone,3 while titanium dioxide nanoparticles are often coated with alumina or silica to control reactivity and provide an amphiphilic polymer layer to improve dispersability.3 The transport of nanomaterials and nanomaterial residues is of interest due to both the potential for water contamination and the need to better understand the performance of filters and membranes used in water and wastewater treatment processes.4 Additionally, the breakdown of the protective surface coating and dissolution of the nanoparticle core could lead to release of dissolved metal ions and/or catalyzing reactive (oxygen) species.5,6Previous studies have evaluated the aging of nanoparticles exposed to natural waters,7–9 swimming pool water,10 organic acids,11 clay and dissolved organic matter,12,13 and ultra-violet (UV) light.2,4,7,14–17 Exposure to sunlight is a particularly interesting aging mechanism, as UV light can catalyze radical reactions, which in turn can cause surface degradation and the formation of potentially toxic byproducts.4 For example, UV light was shown to accelerate degradation of the Al(OH)3–polydimethylsiloxane (PDMS) coating of nano-TiO2 used in cosmetics, resulting in an increase in dissolved concentrations of Si and Al.4 Hou and Jafvert15 found that aqueous nC60 clusters, when exposed to sunlight in water, were transformed via oxidation to soluble derivatives. UV light has also been shown to accelerate the oxidative aging of citrate-coated silver nanoparticles (nAg). When nAg were irradiated with UVB light, Gorham14 found that the rate of surface plasmon resonance (SPR) loss in UV-vis spectra corresponded to an increase in free citrate concentrations, release of silver ions (Ag+), and the formation of a core–shell structure (Ag0 core/Ag2O shell).
Several studies have also examined the role that aging plays in nanomaterial toxicity in environmental and biological systems.16–21 Following aging of quantum dots (QDs) under acidic or alkaline conditions, Mahendra18 observed a significant increase in microbial toxicity due to loss of the organic coating and release of Cd and Se ions from the metal core. Lee16 reported that the toxicity of QDs against the freshwater macroinvertebrate Daphnia magna increased in the presence of UV light, which was attributed to the decreasing size of the particles due to coating degradation and dissolution, release of core ions, and the generation of reactive oxygen species. In contrast, Cheng17 reported that polymer-coated nAg were less toxic following sunlight exposure due to the formation of larger aggregates.
While previous research has demonstrated that nanomaterials exhibit changes in size, surface chemistry, stability, and toxicity following exposure to environmental or biological media, the potential effects of aging are typically not accounted for in nanoparticle transport experiments. With the exception of one study by Kim,22 prior transport studies have utilized freshly prepared suspensions and did not consider aging processes that may occur in the environment. Thus, the objective of this study was to evaluate the transport behavior of citrate-coated nAg in water-saturated sand following exposure to either UVA (320–400 nm) or UVB (280–320 nm) light in a photo-reactor. Over a 7-day exposure period, the particle size of nAg suspensions was characterized using dynamic light scattering (DLS) and transmission electron microscopy (TEM), surface charge was determined by laser Doppler velocimetry, changes in surface chemistry were evaluated using UV-vis spectroscopy, and total and dissolved silver ion content were monitored. The role of free radicals in surface aging was evaluated through batch experiments with a reactive oxygen species (ROS) scavenger, tert-butyl alcohol (tBOH). Transport experiments were conducted with 3-day UV-aged nAg in columns packed with water-saturated 40–50 mesh Ottawa sand at two ionic strengths (10 and 20 mM NaNO3) and at three flow rates (0.5, 1.0, and 1.5 mL min−1). An ionic strength of 10 mM is representative of a groundwater, while the higher ionic strength of 20 mM was selected to assess the effects increasing salinity on nAg aging and transport. Column influent and effluent samples were monitored for total and dissolved silver content and mean particle size, while the column was destructively sampled at the conclusion of each experiment to obtain nAg retention profiles.
2. Materials and methods
2.1. Materials
Silver nanoparticles were prepared using previously described methods23,24 with sodium borohydride (Alfa Aesar, Ward Hill, MA) as the reducing agent and silver nitrate (Sigma Aldrich, St. Louis, MO) as the precursor. The sodium borohydride reduction method produces a ca. 50 mg L−1 suspension of nAg with a mean diameter of 11 ± 2 nm containing <0.02 mg L−1 of Ag+. The pH and zeta potential of the nAg stock as prepared were ca. 8.2 and −50 mV, respectively. The free citrate concentration in freshly prepared nAg suspensions was determined to be 7.5 mg L−1 using ion chromatography (Dionex, ICS-3000).Ottawa sand (F-75, 30–270 mesh) was obtained from U. S. Silica (Frederick, MD) and sieved to obtain the 40–50 mesh size fraction (d50 = 355 μm). Prior to use in column experiments, the sand was treated using a series of acid washes, rinses with deionized water, and ultrasonication to remove organic and metal oxide impurities.25
2.2. Batch experiments
UV light exposure studies were conducted in a fan-cooled photo-reactor (LZC-4X, Luzchem Research, Gloucester, ON). The photo-reactor was operated at an intensity of ca. 2.5 mW cm−2, typical of sea level solar radiation exposure in Boston, MA (42.3 °N), using combinations of top and side irradiation from eight–30.5 cm lamps. UVA and UVB lamps had spectral bands centered at 350 nm and 300 nm, respectively, and were rated at 8 Watts each. Fused quartz 50 mL Erlenmeyer flasks (Technical Glass Products, Painesville, OH) were used in place of borosilicate glassware to achieve maximum UV light transmittance (>90%). Flasks containing a total Ag concentration of 3 mg L−1 were prepared in triplicate by combining 2.7 mL of nAg stock with 42.3 mL DI water, and adjusting the pH to 7.00 ± 0.05 with dilute NaOH or HNO3. The flasks were placed on a rotary platform shaker (DOS-20S, ELMI, San Diego, CA) operated at 80 rpm to ensure uniform mixing and light exposure. Dark control flasks containing “fresh” nAg were prepared under identical conditions and wrapped in aluminum foil in the photo-reactor. Batches were sampled daily and characterized by DLS and UV-vis spectroscopy, and analyzed for total and dissolved silver content. Shifts in UV-vis spectra were monitored over time to qualitatively assess changes in nAg surface chemistry.A series of batch reactors was also prepared with tBOH, a free radical scavenger, in order to evaluate the potential contribution of ROS, such as the hydroxyl radical (OH˙), to nanoparticle surface aging.26–28 Flasks with a total Ag concentration of 3 mg L−1 were prepared as described previously with 15 mM tBOH (Fisher Scientific, NH) and exposed to UVA or UVB light for 7 days.
Additional studies were conducted to monitor the size distribution, zeta potential, and dissolution kinetics of nAg suspensions that had been aged for 3 days. Batch reactors were prepared in triplicate with 10 mM and 20 mM NaNO3 solutions in 100 mL borosilicate glass flasks (Kimble Chase, Vineland, NJ). Suspensions were prepared by adding NaNO3 to 3 day-aged nAg stock (3 mg L−1) and adjusting pH to 7.00 ± 0.05. The reactors were sampled every 30 min for 180 min, to characterize the size distribution using DLS, and to analyze for total and dissolved silver. These batch studies were designed to simulate the initial conditions and residence time of the nanoparticle suspensions in the column studies.
2.3. Column experiments
A total of nine transport experiments were conducted in borosilicate glass columns (12.5 cm length × 2.5 cm inside diameter) packed with 40–50 mesh Ottawa sand and saturated with a background electrolyte solution following the methods of Wang et al.25 A non-reactive tracer test (1000 mg L−1 NaBr, pH 7) was conducted for each column experiment prior to nanoparticle introduction using methods described previously.25 Tracer effluent samples were collected continuously (5 samples per pore volume) using a fraction collector (CF-2, Spectrum Laboratories) and analyzed for bromide concentration. Effluent breakthrough curves obtained for pulse injections of bromide were fit to a one-dimensional form of the advective–dispersive transport (ADR) equation using the CXTFIT program (ver. 2.0)29 to obtain the hydrodynamic dispersion coefficient for each column.Following the non-reactive tracer test, a 3 pore volume (PV) pulse of nanoparticle suspension (ca. 3 mg L−1) was introduced into each column, followed by 3 PV of nanoparticle-free solution with identical chemical conditions to the injection suspension. The nanoparticle suspensions were prepared by combining either fresh or aged nAg stock with 10 or 20 mM NaNO3 and adjusting the pH to 7.00 ± 0.05 using dilute HNO3 or NaOH. A syringe pump was used to deliver both nAg suspensions and background electrolyte solutions to the column in upflow mode at a rate of 0.5, 1, or 1.5 mL min−1. These flow rates corresponded to pore-water velocities and column residence times of 3.8 m d−1 and 52 min, 7.6 m d−1 and 26 min, and 11.5 m d−1 and 17 min, respectively. The influent solution was sampled at the beginning and conclusion of the pulse injection (ca. 75 min) and analyzed for total Ag, Ag+, mean particle diameter, and zeta potential. Effluent samples were collected continuously at a resolution of 5 samples per PV, and analyzed for total Ag and Ag+ to generate effluent breakthrough curves. At the conclusion of each column experiment, the columns were dissected into 1.25 cm increments. Each increment was homogenized by vortexing for 30 seconds, after which a 5 g sample was digested and analyzed for Ag+.
2.4. Analytical methods
The hydrodynamic diameter and electrophoretic mobility of nAg suspensions were measured with a Malvern ZetaSizer (Malvern, Worcestershire, UK) using dynamic light scattering (DLS) and laser Doppler velocimetry, respectively. These measurements were carried out in triplicate at 25 °C using a measurement angle of 173° (backscatter). A refractive index and viscosity of 1.330 and 0.8872 cP, respectively, were used for the dispersant (water). Electrophoretic mobility measurements were converted into zeta potential values using the Smoluchowski approximation.30 UV-vis spectra of nAg suspensions were monitored to assess changes in surface chemistry during UV light exposure.24,31 An aliquot (1 mL) of nAg suspension was transferred to a glass cuvette (Shimadzu, Kyoto, Japan) and absorbance spectra (200–600 nm) were recorded using a spectrophotometer (UV-3100, Shimadzu). Transmission electron micrographs were collected using a JEOL-2100 (JEOL USA, Peabody, MA). TEM samples were prepared on ultra-thin 400 mesh carbon-coated copper grids (Ted Pella, Redding, CA).Bromide concentrations were measured using an ion specific probe (Cole Parmer). Total silver content (nAg + Ag+) in all samples was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES, 7300 DV, Perkin Elmer). Graphite furnace-atomic absorption spectroscopy (GF-AAS, iCE 3300, Thermo Scientific) was used to quantify Ag+. The detection limits of the ICP-OES and GF-AAS methods for Ag+ were 9 μg L−1 and 0.8 μg L−1, respectively.32 Dissolved silver was separated from nAg by centrifugation at 2500 × g in ultrafiltration units (Amicon 3k, Millipore). Less than 3% of Ag+ was retained on the membranes following ultrafiltration based on results from control tests with 0.01–3.0 mg L−1 AgNO3 (data not shown). Solid-phase samples were digested in 10 mL of concentrated nitric acid (Fisher, 15.9 M) at 190 °C using a microwave digester unit (SP-D Discover, CEM).24 The digested solution was diluted (4×) in DI water and analyzed for silver content using ICP-OES.
3 Results and discussion
3.1. Exposure of nAg to UVA and UVB light
A series of batch experiments was conducted to investigate the effects of UVA and UVB light exposure on citrate-stabilized nAg (pH 7, zero ionic strength). Over the 7-day exposure period, the mean diameter and zeta potential of fresh nAg (i.e., un-aged control) remained constant at 10.2 ± 0.6 nm and −44.8 ± 1.1 mV, respectively (Fig. 1). The mean diameter of UVA-aged nAg also remained relatively stable, ranging from 9 to 12 nm, with a slight increase (to 14 nm) observed on day 7. In contrast, exposure to UVB light produced a much greater effect on the mean diameter of nAg, which increased from 10.3 ± 0.4 nm to 45.4 ± 0.6 nm after 48 h, and then gradually increased to 53.8 ± 1.8 nm by day 7 (Fig. 1a). These data were supported by TEM images (Fig. 2), which show the formation of nAg aggregates following exposure to UVB light (additional images provided in ESI†). The zeta potential of UVA- and UVB-aged nAg became less negative over the 7-day exposure period, approaching values of −10.0 ± 4.1 mV and −26.7 ± 1.2 mV, respectively, at the conclusion of the experiment (Fig. 1b). Thus, exposure to both UVA and UVB light resulted in substantial changes in the zeta potential of nAg, while only UVB light yielded a significant (P = 0.05) increase in the mean diameter of nAg. | ||
| Fig. 1 Change in (A) size and (B) zeta potential of nAg exposed to UVA and UVB light for 7 days in a photoreactor. Diameter and zeta potential at time 0 were 9.7 ± 2.1 nm and −44.6 ± 1.4 mV, respectively. | ||
 | ||
| Fig. 2 Transmission electron micrographs of (A) fresh and (B) 3-day UVB-aged nAg. | ||
Aggregation kinetic studies were conducted with fresh nAg and UV-aged (1, 3 and 7 day exposure) nAg suspensions containing NaNO3 at concentrations ranging from 5 to 300 mM (pH 7.00 ± 0.05). The critical coagulation concentration (CCC) for each system was determined by accounting for reaction- and diffusion-limited aggregation based on the method of Chen and Elimelech33 (see ESI†). Plots of the attachment efficiency (α) versus NaNO3 concentration (Fig. S2†) yielded two distinct aggregation regimes, and the extrapolated intersection yielded the CCC value. The transition to a diffusion-limited regime (α = 1) occurred more rapidly in aged nAg suspensions compared with fresh nAg. The resulting CCC values for fresh, 3 day UVA- and 3 day UVB-aged nAg were 161.4, 37.8, and 21.4 mM, respectively, indicating that UV exposure greatly reduced nAg stability. CCC values declined substantially after 1 day, and did not change appreciably over the subsequent 7-day exposure period (Table S1†), indicating that the effects of UV exposure on nAg suspension stability occurred at early time.
UV-vis spectroscopy studies revealed evidence of chemical and morphological changes as a result of UV light exposure (see ESI,† Fig. S3). UV-vis spectra for fresh and aged nAg displayed characteristic peaks in absorbance at ca. 400 nm, consistent with results from previous nAg batch studies.14,31 UVA and UVB light exposure caused broadening in the spectra, a decrease in surface plasmon resonance (SPR), and a 5 to 6 nm red shift in the peak maximum. Changes in the absorbance spectra of UV-aged nAg are indicative of oxidative processes and the formation of oxide layers (Ag2O) on the particle surface, consistent with previous studies.7,17,31,34–36 A decrease in SPR is also characteristic of aggregation5,35 and the decrease in SPR over 7 days observed for UVB-aged suspensions (ca. 0.2 absorbance units) coincided with the observed increase in mean particle diameter (ca. 40 nm).
The dissolution kinetics of fresh and aged nAg over the 7-day UVA and UVB exposure period are shown in Fig. 3. Fresh nAg suspensions exhibited minimal dissolution, with dissolved silver levels only reaching a maximum value of 0.06 mg L−1 after 7 days. In comparison, Ag+ concentrations in UVA-aged suspensions increased to 0.6 mg L−1 after 7 days, while the concentration of Ag+ in UVB-exposed nAg suspensions steadily increased to a maximum value of 1.25 mg L−1 after 7 days. The greater rate of nAg dissolution observed for UVB exposure compared to UVA exposure (equivalent intensity of 2.5 mW cm−2) is consistent with the higher energy of shorter UVB wavelengths. Silver ion concentrations in UVA-aged nAg suspensions were variable over the first four days of exposure, with cyclical decreases followed by increases in Ag+ release. Li and Lenhart7 observed a similar initial decrease in Ag+ concentrations, which was attributed to re-adsorption of released silver onto particle surfaces. In prior studies, the initial decrease in dissolution was attributed to sorption of silver ions onto the citrate coating and complexation of Ag+ in solution by free citrate.20,23 It is also possible that some photoreduction of Ag+ to Ag(0) occurred in the presence of citrate, a process that has been widely used to synthesize nAg and manipulate the size and shape of prepared particles.37,38 These reactions may also have occurred in UVB-aged suspensions, but were likely masked by the much greater rates of dissolution (2-fold higher compared with UVA) during the first few days of aging.
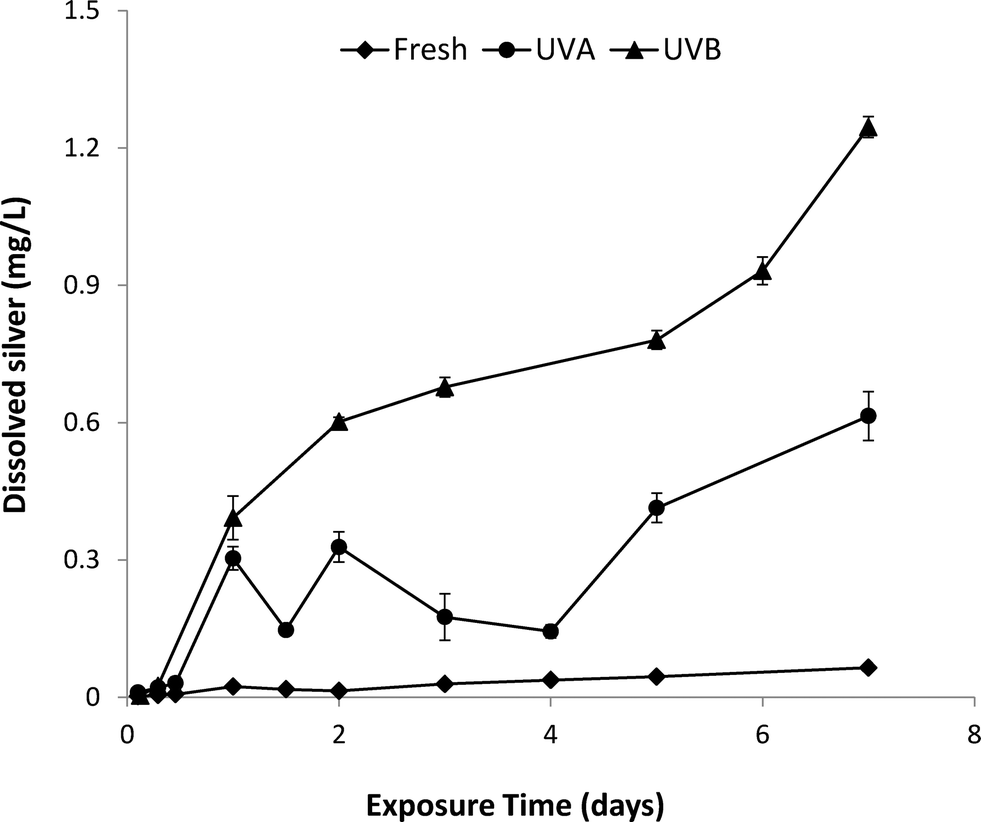 | ||
| Fig. 3 Ag+ release from nAg exposed to UVA or UVB light for 7 days in a photo-reactor. Error bars represent standard deviation of mean Ag+ release from 3 replicate batch reactors. | ||
Based on these findings, there are two likely mechanisms responsible for UV aging of nAg: direct action of UV light on the particle surface and free radical activity. The UVA and UVB wavelengths used to age nAg overlap with the UV-vis adsorption band measured for nAg, and it is probable that UV irradiation disturbed electrons at the nAg surface, in turn driving dipole–dipole interactions and the formation of aggregates.7,39,40 These same surface energy phenomena also disturb the equilibrium of silver in the oxide layer, leading to enhanced silver dissolution.7 The potential contribution of free radical activity to nAg aging was assessed through an additional set of batch experiments with tBOH which can be catalyzed by UV light in water and is a known OH˙ scavenger.27,28 In the presence of tBOH, aggregation of nAg was reduced by up to 50% and the net change in zeta potential decreased by ca. 60% during UV light exposure (ESI,† Fig. S6–S8). For example, the mean diameter and zeta potential of 7-day UVB-aged nAg with and without tBOH were ca. 53 nm and 25 nm and −26 mV and −35 mV, respectively. In addition, the presence of tBOH reduced nAg dissolution as indicated by a 3–6 fold decrease in Ag+ release over the 7-day exposure period (ESI,† Fig. S8).
3.2. Behavior of UV-aged nAg in electrolyte solutions
Prior to conducting column transport experiments, the mean diameter, zeta potential, and dissolution of 3-day UV-aged nAg were monitored for 180 min in 10 mM and 20 mM NaNO3 solutions (see ESI†). These conditions were representative of the initial conditions and residence time, respectively, of the nanoparticle suspensions used in column studies. The size and zeta potential of fresh (un-aged) nAg in 10 mM and 20 mM NaNO3 solutions did not change significantly (P < 0.05), remaining stable at 10.7 ± 0.7 nm and 11.3 ± 1.2 nm, and −45.6 ± 0.7 mV and −40.9 ± 0.8 mV, respectively, over the 180 min study (Fig. S4†). In contrast, the addition of NaNO3 to aqueous suspensions of 3-day UV-aged nAg resulted in 8–30 fold increases in the mean diameter of nAg. UVA-aged nAg aggregated upon addition of 10 mM and 20 mM NaNO3, reaching mean diameters of 86.2 ± 2.1 nm and 298 ± 5 nm respectively, after 180 min. Similarly, the size of UVB-aged nAg increased rapidly, reaching mean diameters of 245 ± 6 nm and 270 ± 6 nm in 10 mM and 20 mM NaNO3, respectively (Fig. S4A†). The zeta potential of UVA-aged nAg in NaNO3 solutions ranged from ca. −37 to −33 mV, while the zeta potential UVB-aged nAg became less negative after NaNO3 addition, steadily decreasing from an initial value of −31.3 mV to ca. −29 mV and −25 mV at 10 mM and 20 mM, respectively (Fig. S4B†). These batch results are consistent with those of El Badawy41 and Li,35 who reported an increase in aggregation and net increase in zeta potential (i.e., less negative) of nAg with increasing NaNO3 concentration.The addition of NaNO3 had a negligible effect on the dissolution of fresh and UVA-aged nAg over 180 min, but accelerated dissolution of UVB-aged nAg (Fig. S5†). Fresh nAg displayed very minimal dissolution (<0.04 mg L−1) in 10 or 20 mM NaNO3 solutions (pH 7), while Ag+ concentrations in UVA-aged nAg suspensions remained stable at ca. 0.32 ± 0.03 mg L−1 (P < 0.05) 180 min after NaNO3 addition. Dissolution in UVB-aged solutions containing 10 mM or 20 mM NaNO3 increased from ca. 0.7 mg L−1 to 1.1 and 1.4 mg L−1, respectively, within 30 min of NaNO3 addition, and then remained constant for the next 150 min. Enhanced dissolution of nAg at higher electrolyte concentrations (i.e., 10 mM versus 20 mM NaNO3) has been reported previously,3,35 and was attributed to destabilization of the electrical double layer surrounding particles and competitive exchange of Ag+ for Na+ or other cations in solution.35
3.3. Transport and dissolution of UV-aged nAg
A series of column experiments was performed to quantify the effects of UV light exposure (3 day) on coupled transport and dissolution of nAg in water-saturated Ottawa sand. Model fits to non-reactive tracer tests, which were conducted immediately prior to the injection of nAg suspensions, yielded a retardation factor (RF) of 0.99 to 1.02, consistent with ideal transport of a tracer, and a Peclet number (Pe) of 209 to 213, which corresponds to a hydrodynamic dispersivity of ca. 0.06 cm. These results are consistent with prior tracer tests conducted in columns packed with 40–50 mesh Ottawa sand.42 Total and nAg breakthrough curves, expressed as normalized effluent concentration (C/C0) versus dimensionless pore volumes (PV), obtained from column experiments conducted at 10 mM NaNO3 are shown in Fig. 4A. Concentrations of Ag+ in effluent samples are presented in Fig. 4B. Breakthrough of fresh nAg in the column effluent began at ca. 1 PV, gradually climbed to a maximum C/C0 value of 0.9 and then decreased sharply 1 PV after introduction of nanoparticle-free 10 mM NaNO3 solution (Fig. 4A). The shape of the fresh nAg breakthrough curve was asymmetrical, suggesting the existence of a limited retention capacity, which is consistent with behavior observed in previous nAg transport studies.24,43 The diameter of fresh nAg remained stable at ca. 10 nm in the influent chamber and in effluent samples (Table 1). Minimal dissolution occurred under these experimental conditions (pH 7.0, 10 mM NaNO3), with effluent samples containing less than 0.02 mg L−1 of Ag+ (Fig. 4B).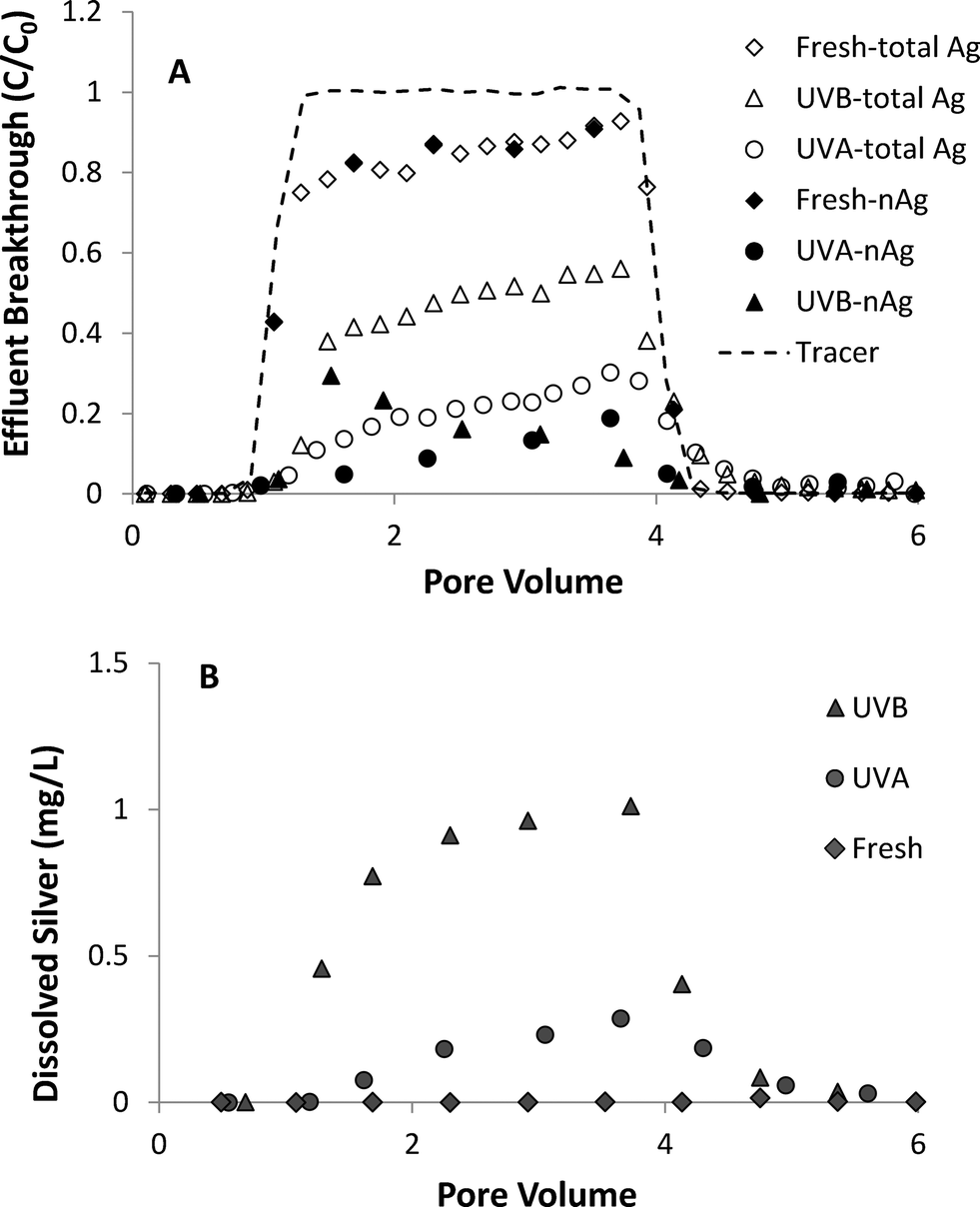 | ||
| Fig. 4 Effluent breakthrough of (A) total silver and nAg and (B) dissolved silver from column experiments (1.0 mL min−1) with fresh and UV-aged nAg in 10 mM NaNO3 solutions. | ||
| Preparation | Flow rate (mL min−1) | Ionic strength (mM) | Size-start/enda (nm) | Zeta potential (mV) | Ag+ start/endab (mg L−1) | % retained | Mass balance |
|---|---|---|---|---|---|---|---|
| a Influent chamber sampled at beginning and end of injection period (0 and 75 minutes). b D.L. = detection limit (0.008 mg L−1). | |||||||
| Fresh | 1.0 | 10 | 9.21/10.13 | −42.7 | <D.L./0.02 | 15.0 | 97.5 |
| UVA-aged | 0.5 | 10 | 12.28/199.0 | −29.8 | 0.19/0.31 | 80.5 | 97.2 |
| UVA-aged | 1.0 | 10 | 11.18/170.2 | −33.5 | 0.15/0.28 | 67.2 | 94.6 |
| UVA-aged | 1.5 | 10 | 12.17/130.7 | −35.3 | 0.23/0.43 | 39.8 | 103.0 |
| UVB-aged | 1.0 | 10 | 96.53/214.8 | −33.1 | 0.58/0.73 | 58.1 | 101.5 |
| Fresh | 1.0 | 20 | 10.17/9.98 | −39.8 | <D.L./0.04 | 28.6 | 98.9 |
| UVA-aged | 1.0 | 20 | 11.01/288.3 | −27.8 | 0.25/0.38 | 73.0 | 93.9 |
| UVB-aged | 1.0 | 20 | 137.5/280.4 | −22.4 | 0.48/0.91 | 50.3 | 98.1 |
| Fresh | 10.17/9.98 | −39.8 | <D.L./0.04 | 28.6 | 98.9 | ||
| UVA-aged | 11.01/288.3 | −27.8 | 0.25/0.38 | 73.0 | 93.9 | ||
| UVB-aged | 137.5/280.4 | −22.4 | 0.48/0.91 | 50.3 | 98.1 | ||
UVA- and UVB-aged (3 days) nAg were considerably less mobile compared to fresh nAg, with 45–50% less nAg mass recovered in the column effluent. UVA-aged nAg displayed asymmetrical breakthrough behavior at 10 mM NaNO3, and were strongly retained in the Ottawa sand, reaching a maximum C/C0 value of only 0.19 (Fig. 4A). The observed reduction in mobility was consistent with the less negative zeta potential of UVA-aged nAg (−36.5 mV) compared to fresh nAg (−45.0 mV). UVB-aged nAg exhibited a downward-sloping effluent concentration plateau, first reaching a maximum relative concentration of 0.29 at 1.5 PV and then gradually declining to 0.05 after ca. 4 PV. This type of decrease in effluent concentrations has been attributed to physical straining44 or aggregation.45–47 Rapid aggregation of UVB-aged nAg occurred in the influent chamber, with the mean diameter increasing from 96.5 to 214.8 nm over the course of the 75 min injection period. Experiments conducted by Xu48 suggest that straining can occur at particle diameter to sand grain diameter ratios as low as 8 × 10−3. However, the ratio of diameters (215 nm) to 40–50 mesh Ottawa sand (355 μm) was 6.1 × 10−4, which is more than one order of magnitude smaller than the proposed threshold value for straining. Therefore, the down-ward sloping breakthrough curve plateau observed with UVB-aged nAg was attributed to particle aggregation, a phenomenon that was previously observed in transport experiments conducted with unstable, aggregating zinc oxide45 and titanium dioxide46,47 suspensions.
The percentages of fresh, UVA- and UVB-aged nAg mass that were retained in sand columns at 10 mM NaNO3 was 15.0, 67.2 and 58.1%, respectively (Table 1). UVB-aged nAg displayed a less negative zeta potential (−32.1 mV) in 10 mM NaNO3 compared to UVA-aged nAg (−36.5 mV), suggesting that UVB-aged nAg should be more strongly retained on negatively-charge Ottawa sand.25 However, a substantial amount of UVB-aged nAg eluted from the column as dissolved silver, with Ag+ effluent concentrations reaching 0.98 mg L−1. Approximately 30% and 65% of effluent silver was in the form of Ag+ in columns experiments with UVA- and UVB-aged nAg, respectively. These observations are consistent with nAg behavior observed in batch reactor experiments (Fig. S5†), where 3-day UVB-aged nAg dissolved more readily compared to UVA-aged nAg. Although no nanoparticles were detected in effluent samples after ca. 4 PV, tailing was observed in total Ag effluent breakthrough curves for columns with aged nAg, indicating that retained nAg was dissolving over time (Fig. 4A). A reduction in dissolution of retained particles is likely due to oxidation of the particle surface and the resulting slower dissolution of the Ag2O layer relative to faster oxidation of the bare Ag core.7
The effect of ionic strength on UV-aged nAg transport and dissolution was evaluated by comparing results from column experiments conducted at 10 and 20 mM NaNO3. Effluent breakthrough curves for total Ag and nAg for 20 mM NaNO3 experiments using 3-day aged nAg are shown in Fig. 5A, and concentrations of Ag+ in effluent samples are shown in Fig. 5B. Similar to results obtained at 10 mM, the breakthrough curve obtained for fresh nAg at 20 mM NaNO3 was asymmetrical in shape with minimal dissolution (<0.04 mg L−1) (Fig. 5A) and no observed changes in particle diameter in influent or effluent samples. Both UVA- and UVB-aged nAg exhibited a gradual decrease in plateau effluent concentrations at 20 mM NaNO3 (Fig. 5A), consistent with increased aggregation measured in the influent chambers. The size of 3-day UVA- and UVB-aged nAg increased from 11.01 to 288.3 nm and from 137.5 to 280.4 nm, respectively, during the injection period. The amount of nAg detected in effluent samples at 20 mM NaNO3 was considerably lower compared to 10 mM NaNO3, with breakthrough of UVA- and UVB-aged nAg reaching maximum C/C0 values of 0.16 and 0.14, respectively. This behavior was consistent with the less negative zeta potential of UVA- (−27.8 mV) and UVB-aged nAg (−22.4 mV) in 20 mM NaNO3 solutions (Table 1). The percentage of applied fresh, UVA- and UVB-aged nAg mass that was retained in sand columns at 20 mM NaNO3 was 28.6, 73.0 and 50.3%, respectively. Effluent silver breakthrough at 20 mM NaNO3 was primarily in the form of dissolved ion, with approximately 50% and 80% eluting as Ag+ in columns with UVA- and UVB-aged nAg, respectively. Maximum Ag+ concentrations of 0.34 and 1.3 mg L−1 were observed in effluent samples. These levels are 15 to 20% higher compared to values obtained from 10 mM NaNO3 columns, which is consistent with increased dissolution of nAg observed in batch experiments at 20 mM compared to 10 mM (Fig. S5†).
 | ||
| Fig. 5 Effluent breakthrough of (A) total silver and nAg and (B) dissolved silver from column experiments (1.0 mL min−1) with fresh and UV-aged nAg in 20 mM NaNO3 solutions. | ||
The effect of flow rate on the transport and dissolution UVA-aged nAg in sand columns (10 mM NaNO3, pH 7.0) is shown in Fig. 6. Results from column studies at three different flow rates show that effluent breakthrough of nAg decreased as flow rate was decreased, with maximum C/C0 values of 0.05, 0.11, and 0.56 measured at flow rates of 0.5, 1.0 and 1.5 mL min−1, respectively. Increasing retention with decreasing flow rate is consistent with findings from prior nanoparticle transport studies and with behavior predicted by filtration theory.49 Dissolution of nAg and release of Ag+ from the columns was strongly dependent upon flow rate, with ca. 75%, 40%, and 20% of silver mass eluting as dissolved ion at flow rates of 0.5, 1.0, and 1.5 mL min−1, respectively. Increased Ag+ release at lower flow rates was attributed to the increased contact time (greater column residence time) between aqueous phase and the attached particles, which resulted in greater nAg dissolution. The percentage of applied nAg retained in the sand with increasing flow rate was 67.2, 80.5, and 39.8%, respectively. Although the lowest amount of nAg in column effluent was observed at the slowest flow rate (0.5 mL min−1), increased dissolution resulted in lower overall silver mass retention compared with the 1.0 mL min−1 column. Tailing in total Ag and Ag+ breakthrough curves was observed at all three flow rates tested, and was most pronounced at the lowest flow rate (0.5 mL min−1). After the pulse injection was terminated, Ag+ continued to elute from the 0.5 mL min−1 column at relatively high levels, increasing from ca. 0.5 mg L−1 at 4 PV to 0.7 mg L−1 at 6 PV.
 | ||
| Fig. 6 Effect of flow rate on breakthrough of (A) total silver and nAg and (B) dissolved silver in column experiments with UVA-aged nAg in 10 mM NaNO3. | ||
Silver retention profiles obtained at the conclusion of each column experiment are shown in Fig. S9.† The majority of retained mass was in the form of nAg, since dissolved silver concentrations were relatively low (<0.06 mg L−1 at 6 PV) at the conclusion of most experiments, and the resident solution was drained from the columns prior to solid phase sampling. Although UVB-aged nAg were retained more strongly compared with UVA-aged nAg at the different ionic strengths tested, they also dissolved more readily resulting in lower overall silver retention. Retention profiles obtained from all fresh and UV-aged columns were hyper-exponential in shape, with the highest attachment measured near the column inlet. This retention behavior is consistent with hyper-exponential profiles reported in prior transport studies performed with nAg43 and other nanoparticle systems.45,50,51
4 Conclusions
The experimental results presented here provide a detailed evaluation of the effects of UV light surface aging on nAg fate and transport in porous media. Batch studies indicate that exposure to both UVA and UVB light significantly alters the size, surface charge, surface chemistry, and dissolution rate(s) of nAg. Free radical activity (OH˙) was identified as a primary mechanism responsible for surface oxidation of nAg during UV exposure based on the results of experiments conducted in the presence of tBOH, a free radical scavenger. Subsequent column studies demonstrated that up to 50% more UV-aged nAg was retained in water-saturated sand compared to freshly prepared, unreacted nAg suspensions. While UV-aged nAg are less mobile than fresh (un-aged) nAg, column results indicate that attached aged particles more readily dissolve in porous media, releasing Ag+ at levels as high as 1.2 mg L−1. Thus, exposure to UV light resulted in substantially greater attachment of nAg on quartz sand, however, UV-aged nAg that were retained in the column readily dissolved, releasing Ag+ into effluent at relatively high concentrations. Irradiation with higher energy UVB wavelengths was shown to induce greater surface aging relative to UVA, in turn resulting in relatively lower mobility and more rapid dissolution of UVB-aged nAg in porous media. The oxidation of nAg and subsequent dissolution of the Ag2O layer to form Ag+ have been shown to play a key role in observed toxicity to different organisms.52,53 These findings demonstrate that surface aging can substantially alter the transport and dissolution behavior of nAg in porous media, and should be considered when evaluating the fate and antimicrobial effects of nAg in natural and engineered porous media systems.Acknowledgements
The authors wish to thank Wenlu Li at Washington University for his advice regarding photoreactor experiments. Support for this research was provided by a grant from the National Science Foundation (NSF), Award no. CBET-1236653. TEM images were collected at the University Harvard Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported under NSF award no. ECS-0335765. The work has not been subject to NSF review, and therefore, does not necessarily reflect the views of the organization and no official endorsement should be inferred.References
- D. Lin, X. Tian, F. Wu and B. Xing, J. Environ. Qual., 2010, 39, 1896 CrossRef.
- J. Labille, J. Feng, C. Botta, D. Borschneck, M. Sammut, M. Cabie, M. Auffan, J. Rose and J.-Y. Bottero, Environ. Pollut., 2010, 158, 3482–3489 CrossRef CAS PubMed.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- M. Auffan, M. Pedeutour, J. Rose, A. Masion, F. Ziarelli, D. Borschneck, C. Chaneac, C. Botta, P. Chaurand, J. Labille and J.-Y. Bottero, Environ. Sci. Technol., 2010, 44, 2689–2694 CrossRef CAS PubMed.
- J. Lee, J. D. Fortner and J. Kim, Environ. Sci. Technol., 2007, 41, 2529–2535 CrossRef CAS.
- J. Lee, K. Ji, J. Kim, C. Park, K. H. Lim, T. H. Yoon and K. Choi, Environ. Toxicol., 2009, 593–600 CAS.
- X. Li and J. J. Lenhart, Environ. Sci. Technol., 2012, 46, 5378–5386 CrossRef CAS PubMed.
- V. Sarathy, P. G. Tratnyek, J. T. Nurmi, D. R. Baer, J. E. Amonette, C. L. Chun, R. L. Penn and E. J. Reardon, J. Phys. Chem. C, 2008, 112, 2286–2293 CAS.
- B. C. Reinsch, B. Forsberg, R. L. Penn, C. S. Kim and G. V. Lowry, Environ. Sci. Technol., 2010, 44, 3455–3461 CrossRef CAS PubMed.
- J. Virkutyte, S. R. Al-Abed and D. D. Dionysiou, Chem. Eng. J., 2012, 191, 95–103 CrossRef CAS PubMed.
- I. A. Mudunkotuwa, J. M. Pettibone and V. H. Grassian, Environ. Sci. Technol., 2011, 46, 7001–7010 CrossRef PubMed.
- C. Coutris, E. J. Joner and D. H. Oughton, Sci. Total Environ., 2012, 420, 327–333 CrossRef CAS PubMed.
- K. G. Scheckel, T. P. Luxton, A. M. El Badawy, C. A. Impellitteri and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1307–1312 CrossRef CAS PubMed.
- J. M. Gorham, R. I. MacCuspie, K. L. Klein, D. H. Fairbrother and R. D. Holbrook, J. Nanopart. Res., 2012, 14, 1139 CrossRef.
- W. Hou and C. T. Jafvert, Environ. Sci. Technol., 2009, 43, 362–367 CrossRef CAS.
- J. Lee, M. I. N. Cho, J. D. Fortner and J. B. Hughes, Environ. Sci. Technol., 2009, 43, 4878–4883 CrossRef CAS.
- Y. Cheng, L. Yin, S. Lin, M. Wiesner, E. Bernhardt and J. Liu, J. Phys. Chem. C, 2011, 115, 4425–4432 CAS.
- S. Mahendra, H. Zhu, V. L. Colvin and P. J. Alvarez, Environ. Sci. Technol., 2008, 42, 9424–9430 CrossRef CAS.
- T. Phenrat, T. C. Long, G. V. Lowry and B. Veronesi, Environ. Sci. Technol., 2009, 43, 195–200 CrossRef CAS.
- X. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-kim and J. N. Meyer, Environ. Sci. Technol., 2011, 46, 1119–1127 CrossRef PubMed.
- W. Song, J. Zhang, J. Guo, J. Zhang, F. Ding, L. Li and Z. Sun, Toxicol. Lett., 2010, 199, 389–3897 CrossRef CAS PubMed.
- H.-J. Kim, T. Phenrat, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2009, 43, 3824–3830 CrossRef CAS.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- A. M. Mittelman, A. Taghavy, Y. Wang, L. M. Abriola and K. D. Pennell, J. Nanopart. Res., 2013, 15, 1765 CrossRef.
- Y. Wang, Y. Li, J. D. Fortner, J. B. Hughes, L. M. Abriola and K. D. Pennell, Environ. Sci. Technol., 2008, 42, 3588–3594 CrossRef CAS.
- R. Joshi, S. Adhikari, B. S. Patro, S. Chattopadhyay and T. Mukherjee, Free Radical Biol. Med., 2001, 30, 1390–1399 CrossRef CAS.
- J. Staehelld and J. Holgne, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef PubMed.
- T. Garoma and M. D. Gurol, Environ. Sci. Technol., 2004, 38, 5246–5252 CrossRef CAS.
- N. Toride, F. J. Leij and M. Th. van Genuchten, The CXTFIT Code for Estimating Transport Parameters from Laboratory or Field Tracer Experiments; Ver. 2.0, Research report No. 137, U.S. Department of Agriculture, Salinity Laboratory, Riverside, CA, 1995 Search PubMed.
- R. J. Hunter, Zeta potential in colloid science, Academic Press, London, 1981 Search PubMed.
- M. Chen, L.-Y. Wang, J.-T. Han, J.-Y. Zhang, Z.-Y. Li and D.-J. Qian, J. Phys. Chem. B, 2006, 110, 11224–11231 CrossRef CAS PubMed.
- A. Hubaux and G. Vos, Anal. Chem., 1970, 42, 849–855 CrossRef CAS.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 117, 10994–11001 CrossRef PubMed.
- A. Henglein, Anal. Chem., 1998, 2, 444–450 Search PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26, 16690–16698 CrossRef CAS PubMed.
- M. Schmidt, A. Masson and C. Bréchignac, Phys. Rev. Lett., 2003, 91, 1–4 Search PubMed.
- M. Maillard, P. Huang and L. Brus, Nano Lett., 2003, 3, 1611–1615 CrossRef CAS.
- A. M. Ahern and R. L. Garrell, Anal. Chem., 1987, 2813–2816 CrossRef CAS.
- X. Lu, M. Rycenga, S. E. Skrabalak, B. Wiley and Y. Xia, Annu. Rev. Phys. Chem., 2009, 60, 167–927 CrossRef CAS PubMed.
- Z. Tang, N. A. Kotov and M. Giersig, Science, 2002, 297, 237–2340 CrossRef CAS PubMed.
- A. M. El Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS PubMed.
- M. D. Becker, Y. Wang, K. D. Pennell and L. M. Abriola, Environ. Sci.: Nano, 2015, 2, 155–166 RSC.
- Y. Liang, S. A. Bradford, J. Simunek, H. Vereecken and E. Klumpp, Water Res., 2013, 47, 2572–2582 CrossRef CAS PubMed.
- S. A. Bradford, S. Torkzaban and S. L. Walker, Water Res., 2007, 41, 3012–3024 CrossRef CAS PubMed.
- X. Jiang, M. Tong, H. Li and K. Yang, J. Colloid Interface Sci., 2010, 350, 427–434 CrossRef CAS PubMed.
- G. Chen, X. Liu and C. Su, Environ. Sci. Technol., 2012, 46, 7142–7150 CrossRef CAS PubMed.
- I. G. Godinez, C. J. G. Darnault, A. P. Khodadoust and D. Bogdan, Environ. Pollut., 2013, 174, 106–113 CrossRef CAS PubMed.
- S. Xu, B. Gao and J. E. Saiers, Water Resour. Res., 2006, 42 DOI:10.1029/2006WR004948.
- Y. Li, Y. Wang, K. D. Pennell and L. M. Abriola, Environ. Sci. Technol., 2008, 42, 7174–7180 CrossRef CAS.
- I. Chowdhury, Y. Hong, R. J. Honda and S. L. Walker, J. Colloid Interface Sci., 2011, 360, 548–555 CrossRef CAS PubMed.
- D. Kasel, S. A. Bradford, J. Šimůnek, M. Heggen, H. Vereecken and E. Klumpp, Water Res., 2013, 47, 933–944 CrossRef CAS PubMed.
- C. Lok, C. Ho, R. Chen, Q. He, W. Yu, H. Sun, P. K. Tam, J. Chiu and C. Che, J. Proteome Res., 2006, 916–924 CrossRef CAS PubMed.
- C. Gunawan, W. Y. Teoh, C. P. Marquis, J. Lifia and R. Amal, Small, 2009, 5, 341–344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details regarding the attachment efficiency calculations, UV-vis spectra of UVA- and UVB-aged nAg, free radical activity, aggregation and dissolution kinetics of 3 day-aged nAg in electrolyte solutions, and retention profiles. See DOI: 10.1039/c5en00145e |
| This journal is © The Royal Society of Chemistry 2015 |