
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nanosphere adsorbents for removal of arsenate and selenate from water
Man
Li
,
Chengwei
Wang
,
Michael J.
O'Connell
and
Candace K.
Chan
*
Materials Science and Engineering, School for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, AZ, USA. E-mail: candace.chan@asu.edu; Fax: +(480) 727 9321; Tel: +(480) 727 8614
First published on 16th March 2015
Abstract
Porous carbon nanospheres prepared using spray pyrolysis were evaluated as adsorbents for removal of arsenate and selenate in de-ionized (DI), canal, and well waters. The carbon nanospheres displayed good binding to both metals in DI water and outperformed commercial activated carbons for arsenate removal in pH > 8, likely due to the presence of basic surface functional groups, high surface-to-volume ratio, and suitable micropores formed during the synthesis.
Nano impactCarbon nanomaterials have attracted attention as sorbent materials for remediation of pollutants and harmful metals from the environment due to their higher surface-to-volume ratios compared to conventional activated carbons. However, carbon nanotubes and graphene are ineffective for removing oxoanions from water due to acidic surface functional groups. Here we show that carbon nanospheres prepared using a facile spray pyrolysis method display good adsorption to arsenate and selenate in DI water solutions. A key characteristic of the carbon nanospheres is the presence of micropores originating from the removal of metal oxide nanoparticles, as well as the presence of basic surface groups. The carbon nanospheres had superior removal of arsenate in canal and well waters with slightly alkaline pH compared to commercial activated carbon. These results highlight the potential for carbon nanospheres to be used as adsorbents for toxic metal treatment at neutral to alkaline pH. |
New sorbent materials are greatly needed in order to remove harmful contaminants from drinking and industrial waste water that can cause negative health effects and adverse consequences to the environment. Activated carbons are commonly used adsorbents for water treatment applications and are a mature technology for the removal of harmful organic compounds1 and metals2,3 such as chromium, lead, and mercury. Not only do activated carbons need to have suitable surface functional groups for adsorption of species, but some studies have also shown that a microporous structure can improve the removal of inorganic oxoanions.4,5 This can be challenging to control due to the wide range of preparation conditions for activated carbon, which can give different structures, porosity, surface chemistry, and surface area.3
With the development of carbon nanotechnology, there has been interest in exploiting the high surface-to-volume ratios of these nanomaterials for water treatment. Recently, carbon nanotube and graphene-based sorbents have been demonstrated for removal of metals such as mercury,6 arsenic,7–9 chromium,10–12 and selenium13 with promising results. However, such carbon nanostructures may have cost prohibitive synthesis methods and also cytotoxicity issues.14–16 For instance, a recent report found that graphene oxide actually amplified the phytotoxicity of arsenate in wheat plants and affected the plants' natural detoxification processes.17 Recently, we developed a facile spray pyrolysis method for synthesizing highly porous carbon nanospheres that displayed excellent properties for dye adsorption and electrochemical double-layer energy storage.18 Unlike carbon nanotubes and graphene, spherical shaped carbon nanostructures have been shown to have good biocompatibility.19–21 Therefore, carbon nanospheres may be promising materials for environmental remediation applications such as the removal of harmful metals from water.
Here we evaluate the adsorption properties of carbon nanospheres (CNS) for removal of arsenate (As(V)) and selenate (Se(VI)). The toxic and carcinogenic properties of arsenic22 are well known. Although arsenate is less toxic than arsenite (As(III)), it is the predominate form of arsenic in oxygen rich and oxidizing environments such as drinking and surface waters.23 While selenium is an essential element, excessive levels can lead to toxicity in humans and wildlife, particularly in aquatic environments where bioaccumulation can be quite rapid. For example, only 2–5 ppb of waterborne selenium species can cause reproductive failure in fish.24 Selenate is more difficult to remove compared to the lower oxidation state species and there are few effective adsorbents available.25
CNS were prepared using a spray pyrolysis method described in our previous work.18 Unlike most other synthesis techniques for CNS that required templates such as silica26,27 or polymer nanospheres,28,29 our solution-based, spray pyrolysis method is able to create porous CNS directly without the use of any templates and can be easily scaled. Briefly, a metal salt is added to the precursor solution and decomposed with heating to form metal oxide nanoparticles that act as nucleation sites for carbonization of a carbohydrate source. Post-synthesis etching removes the metal oxide nanoparticles and leaves highly porous CNS (Fig. 1A). The metal oxide nanoparticle size, and hence pore diameter in the resulting CNS, can be controlled by tuning the ratio of the precursors.18 Therefore, this synthesis method can be used to prepare carbon with the desired micropores for metal oxoanion adsorption. Furthermore, the nanoscale size of the CNS creates a high outer surface-to-volume ratio which can reduce the diffusion distance for metals to the binding sites down to the nanometer scale. In comparison, although conventional activated carbons have similar specific surface area as the CNS, much of it originates from inner surfaces. For activated carbons composed of micron-sized particles, the diffusion distance of metals to binding sites will be much larger than in the CNS.
 | ||
| Fig. 1 (A) Schematic of formation mechanism for synthesis of microporous carbon nanospheres. (B) SEM images of synthesized carbon nanospheres. TEM images of carbon nanospheres (C) after carbonization as a composite with metal oxide nanoparticles (noted with arrows) and (D) after acid etching to dissolve the metal nanoparticles. Scale bar = 50 nm. | ||
In a typical synthesis, 1 g sucrose and 1 g Mn(NO3)2 were dissolved in 100 mL DI water and sprayed using N2 carrier gas into a tube furnace heated at 1000 °C. High temperature annealing at 1200 °C for 2 hours under Ar was performed, followed by etching with concentrated HCl and washing in DI water to form the final product consisting of CNS with a median diameter of ~70 nm, with the largest particle size <1 micron.18Fig. 1B shows typical scanning electron microscopy (SEM) images of the CNS. Transmission electron microscopy (TEM) of the material after carbonization showed that the CNS contained manganese oxide nanoparticles 2–10 nm in diameter (Fig. 1C). After etching, the nanoparticles were dissolved to reveal empty micropores (Fig. 1D). Based on the X-ray diffraction (XRD, Fig. 2A) and Raman spectroscopy (Fig. 2B) analysis, the CNS adopted a disordered amorphous structure with predominately carbon sp3 bonding.18
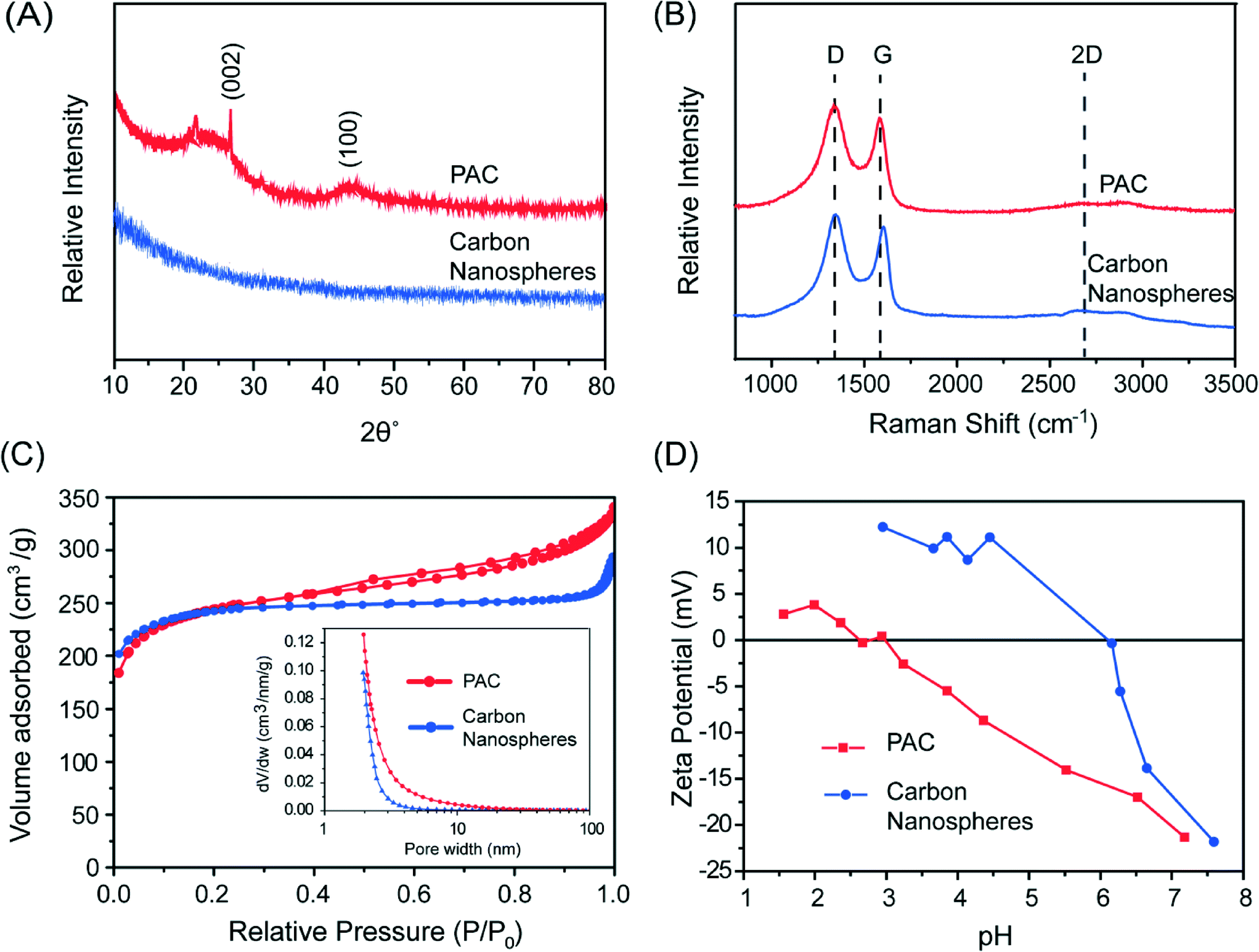 | ||
| Fig. 2 (A) XRD pattern, (B) Raman spectra, (C) N2-sorption curve with pore size distribution the inset, and (D) zeta potential measurement for PAC and carbon nanospheres. | ||
Cabot Norit® 20BF powdered activated carbon (PAC, 325 mesh) and GAC-820 granular activated carbon (GAC, 8 × 20 mesh) were obtained and used without further treatment as comparison sorbent materials to the CNS. Both of these activated carbons are prepared from bituminous coal. The Raman spectrum for PAC showed a similar disordered structure as that in the CNS (Fig. 2B) but the (002) and (100) planes associated with graphitic carbon can be discerned in the XRD pattern (Fig. 2A). Thus, the structure of PAC is likely a mixture of disordered carbon with some regions of graphitic, sp2 carbon.
The specific surface area for the CNS using the Brunauer–Emmett–Teller (BET) method at 77 K in nitrogen (Micromeritics TriStar II 3020) was around 1000 m2 g−1 with a pore volume around 0.28 cm3 g−1. Gas sorption measurements on PAC determined a BET surface area of 864 m2 g−1 and pore volume of 0.22 cm3 g−1. GAC has been reported with a BET surface area of 908 m2 g−1 and pore volume of 0.5 cm3 g−1.30 The nitrogen-sorption isotherm for the CNS and PAC are shown in Fig. 2C. While PAC shows a type IV isotherm with hysteresis, indicating some mesoporosity,31 the CNS had a type I isotherm with no hysteresis. This indicates that the CNS contained mostly micropores <2 nm and no significant pore volume associated with mesopores (2–50 nm) or macropores (>50 nm), as shown by the Barrett–Joiner–Halenda (BJH) derivative pore distribution plot (Fig. 2C, inset). This is consistent with the formation mechanism of the porous CNS after the removal of the metal oxide nanoparticles with etching (Fig. 1A). Similar nanoporous structure is observed in carbide-derived carbon, which is formed by etching metal from a metal carbide such as TiC,32 although the pore widths are larger for our CNS due to the larger size of the metal oxide nanoparticles.
Zeta potential measurements were performed on the PAC and CNS (ZetaPALS, Brookhaven Instruments). As shown in Fig. 2D, the isoelectric point (pH where the zeta potential is zero) for PAC was around pH 2.5–3, which is typical for coal-derived activated carbons with acidic surface groups (L-type carbon).33 In contrast, the isoelectric point for the CNS was around 6.16. The higher isoelectric point in the CNS is a result of the high temperature annealing temperatures used in the synthesis, which can remove the oxidised acidic surface groups.34 Basic carbons with anionic exchange properties (H-type carbons) can be obtained when heating >950 °C in vacuum or inert atmosphere.33,35
Batch adsorption experiments were performed using the carbon materials at a concentration of 0.44 g L−1 in water spiked with 1 ppm Na2SeO4 and 1 ppm Na2HAsO4·7H2O. The sorbents were added to the spiked solutions and stirred at a constant speed with sampling at different time periods. The sorbents were then removed with filtration and the filtrate was analysed with inductively coupled plasma atomic emission spectroscopy (ICP-OES). Ultrapure DI water (18.3 MΩ cm, pH 5.5) was used for synthetic water solutions. The predominant species at this pH are SeO42− and H2AsO4−.23,36 Water samples were also obtained from the service canal and B well that serve as makeup water for the boiler and cooling towers at the Salt River Project Santan Generating Facility in Gilbert, AZ. The canal and well waters were spiked with 1 ppm selenate or arsenate in the same manner. The pH of the canal and well waters were 8.54 and 8.30, respectively. In this pH range, the dominant Se(VI) species is still SeO42−, but the As(V) is found as the doubly charged anion, HAsO42−.23,36
The arsenate and selenate removal over time using carbon sorbents is shown in Fig. 3 in the different water matrices. The CNS showed good binding to both metal species. For removing arsenate in DI water, 53% was removed in 2 hours, with 100% removal observed by 22 hours (Fig. 3A). In the canal (Fig. 3C) and well waters (Fig. 3D), the arsenate removal rates were slower, with only about 3% removed after 2 hours. However, by 22 hours, >89% of the arsenate was removed, with the removal efficacy in the canal water very similar as in the DI water. The slightly lower removal efficacy for arsenate in the canal and well waters compared to DI water can be explained by their higher pH. The anionic adsorption capability of carbons is typically attributed to surface functional groups such as –COOH, –OH2+, –COO−, –OH, –O−, which become protonated and/or positively charged when dispersed into aqueous solutions.2,11,37 The arsenate adsorption capacities of activated carbons reported in the literature typically reach a maximum at pH 2–5,38,39 where the carbon surface has a more positive charge. Similarly, carbon nanotubes with oxygen-containing surface functional groups showed low arsenate binding capacities due to negative zeta-potentials from pH 3–10.40 For this reason, many carbon-based adsorbents rely on modification with iron, which can form inner-sphere complexes with arsenic.41–43 Here, we see that our CNS display good arsenate adsorption at pH > 8 without requiring this modification due to their higher isoelectric point. The canal and well waters also contain other competing anion species such as nitrate (typically 60–130 ppm) and sulphate (700–1000 ppm), which did not appear to have a large effect on the arsenate binding.
 | ||
| Fig. 3 Percentage removed of 1 ppm As(V) and 1 ppm Se(VI) on carbon nanospheres compared to powdered activated carbon (PAC) and granular activated carbon (GAC) at dosage of 0.44 g L−1 in (A) DI water, pH 5.5, (B) DI water, pH 8.3, (C) canal water, and (D) well water. | ||
In DI water, the CNS could remove 56% of the starting selenate concentration in 2 hours (Fig. 3A). After 22 hours of exposure, 97% of the selenate was removed. However, at 26 hours, the selenate concentration in the water increased, suggesting some desorption of selenate from the CNS surface. Similar desorption behaviour has been observed on inorganic sorbents in high ionic strength electrolytes.44 Since selenate is a weak binding anion and adsorbs through outer-sphere complexes,45 it can easily become displaced by competing anions. In DI water, which is slightly acidic, the adsorption of protons onto the basic surface groups of the CNS will cause the solution pH to increase. For instance, the DI water solution containing 1 ppm arsenate and selenate had an initial pH of 5.5, which increased to 7.35 after the CNS were added and stirred for 2 hours. The increase of pH in the solution until the equilibrium is reached could result in desorption of some of the selenate.
Nonetheless, these results are much better than what was previously observed on other nanocarbon sorbents. For example, graphene oxide evaluated in a similar water matrix (1 ppm selenate in DI water, pH 6) but at a higher dose of 1 g L−1 could only remove 30% of the selenate after 24 hours exposure time.13 This could be due to a number of reasons, including low effective surface area or differences in surface functional groups. Although dilute graphene oxide suspensions have surface area as high as 736 m2 g−1, this value decreases due to agglomeration starting at concentrations of 50 mg L−1.46 The surface chemistry of graphene oxide is complex and heterogenous, but is generally accepted to consist of predominately epoxides and tertiary alcohols in the basal plane and ketones, carboxylic acids, ethers, and enols on the edges.47 Studies have found that strong hydrogen bonding between water molecules and functional groups in the basal plane play a key role in maintaining the layer stacking of graphene oxide,48 which may further inhibit the ability for selenate to adsorb, since it may have to intercalate in between the layers or compete with water for binding sites. Furthermore, the graphene oxide surface is acidic in character49 and would have a low number of suitably charged binding sites for adsorption of selenate at pH 6.
When tested in the canal (Fig. 3C) and well waters (Fig. 3D), the selenate removal efficacy of the CNS was very low, about 2–3%, due to the presence of completing anions. Sulphate has been found to compete with selenate binding on various sorbents due to its similar anion structure and adsorption behavior.50,51 However, one way to potentially address this problem is to use a barium salt to precipitate out the sulphate from the water prior to its exposure to the sorbent.45
In spiked DI water, PAC could adsorb arsenate and selenate faster than the CNS, with 93% arsenate and 77% selenate removed in 45 minutes. Similar to the CNS, all of the arsenate and selenate could be removed at longer exposure times. On the other hand, GAC removed only 35% of the arsenate and 62% of the selenate after 22 hours. Due to the poor performance in DI water, GAC was not tested further.
Despite the good adsorption behaviour in DI water, PAC could not remove arsenate and selenate from the canal and well waters. To further investigate whether the low removal efficacy of PAC in the canal and well waters was due to the higher pH or the presence of competing ions, the pH of the DI water was adjusted to 8.3 by adding NaOH. The results for these tests are shown in Fig. 3B. Comparing these results to those obtained in DI water without pH adjustment (Fig. 3A), both arsenate and selenate removal efficiencies by PAC decreased by about half, which means the higher pH of the solution does have a negative effect on the adsorption properties. This suggests that the worse performance of PAC in the canal and well waters is due to a decrease in positively charged surface binding sites as a result of the higher pH.
This also shows that the basic surface properties of the CNS allows for good arsenate adsorption in the canal and well waters. The exact nature of the basic sites will require further detailed study, as it is a controversial topic in carbon science. Some contributors to basicity have been proposed as: (1) the electron donating character of π-electrons on graphitic basal planes, (2) oxygen surface functionalities such as chromene, diketon, quinone, and pyrone groups, (3) nitrogen-containing functionalities, and (4) inorganic impurities.52 The contribution of (1) seems to be less likely in this case, since the CNS have little graphitic structure and moreover have very similar disordered carbon structure as PAC, based on Raman spectroscopy and X-ray diffraction. Due to the lack of nitrogen functional groups in the sucrose precursor used to make the CNS, (3) is also less likely. Instead, the annealing procedures used to prepare the CNS may create basic oxygen-containing functional groups. The last contribution cannot be ruled out since the manganese salt is an important component of the synthesis. However, any manganese compounds should be removed from the CNS after the post-synthesis HCl etching. Also, previous studies have found that manganese oxide species can successfully remove arsenate from water only at pH < 5 due to their low point-of-zero charge.53,54
Conclusion
In summary, we have found that carbon nanospheres prepared using a facile spray pyrolysis method can display good activity for arsenate and selenate adsorption in synthetic DI water solutions. In water solutions composed of canal and well water at pH > 8, the carbon nanospheres could outperform PAC likely due to the presence of basic functional groups, higher surface area, and suitable microporous structure as a result of the formation mechanism arising from the synthesis method. However, competing anions in these waters completely inhibited selenate adsorption on the carbon nanospheres, whereas the arsenate binding kinetics were only slightly decreased. As conventional activated carbons and nanostructured carbons such as carbon nanotubes and graphene typically show good adsorption properties in acidic pH, these results highlight the potential for carbon nanospheres to be used as adsorbents for toxic metal treatment at neutral to alkaline pH. Future work will elucidate the nature of the surface functional groups on the carbon nanospheres and focus on obtaining more understanding on the mechanism of adsorption.Acknowledgements
The work was funded through the Arizona State University/Salt River Project Joint Research Program, under the project “Photocatalytic Treatment of Selenium and Arsenic in SRP Waters”. We would like to thank D. Pelley, F. Fuller, and R. Woods from Salt River Project for their support of this research. We thank P. Westerhoff for assistance with the zeta potential measurements. We also acknowledge the use of facilities within the LeRoy Eyring Center for Solid State Science and Goldwater Environmental Laboratory at ASU.Notes and references
- J. M. Montgomery, Water treatment principles and design, John Wiley & Sons, New York, 1985 Search PubMed.
- C. P. Huang and M. H. Wu, Water Res., 1977, 11, 673–679 CrossRef CAS.
- J. Rivera-Utrilla, M. Sánchez-Polo, V. Gómez-Serrano, P. M. Alvarez, M. C. M. Alvim-Ferraz and J. M. Dias, J. Hazard. Mater., 2011, 187, 1–23 CrossRef CAS PubMed.
- M. Asadullah, I. Jahan, M. B. Ahmed, P. Adawiyah, N. H. Malek and M. S. Rahman, J. Ind. Eng. Chem., 2014, 20, 887–896 CrossRef CAS PubMed.
- Z. Hu, L. Lei, Y. Li and Y. Ni, Sep. Purif. Technol., 2003, 31, 13–18 CrossRef CAS.
- V. Chandra and K. S. Kim, Chem. Commun., 2011, 47, 3942–3944, 10.1039/C1CC00005E.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- A. K. Mishra and S. Ramaprabhu, Desalination, 2011, 282, 39–45 CrossRef CAS PubMed.
- S. Vadahanambi, S. H. Lee, W. J. Kim and I. K. Oh, Environ. Sci. Technol., 2013, 47, 10510–10517 CAS.
- Z. C. Di, J. Ding, X. J. Peng, Y. H. Li, Z. K. Luan and J. Liang, Chemosphere, 2006, 62, 861–865 CrossRef CAS PubMed.
- J. Hu, C. Chen, X. Zhu and X. Wang, J. Hazard. Mater., 2009, 162, 1542–1550 CrossRef CAS PubMed.
- H. Jabeen, V. Chandra, S. Jung, J. W. Lee, K. S. Kim and S. B. Kim, Nanoscale, 2011, 3, 3583–3585 RSC.
- Y. Fu, J. Wang, Q. Liu and H. Zeng, Carbon, 2014, 77, 710–721, DOI:10.1016/j.carbon.2014.05.076.
- G. Jia, H. Wang, L. Yan, X. Wang, R. Pei, T. Yan, Y. Zhao and X. Guo, Environ. Sci. Technol., 2005, 39, 1378–1383 CrossRef CAS.
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300, DOI:10.1073/pnas.1222276110.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Durán, Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef CAS PubMed.
- X. Hu, J. Kang, K. Lu, R. Zhou, L. Mu and Q. Zhou, Sci. Rep., 2014, 4, 6122 CrossRef CAS PubMed.
- C. Wang, Y. Wang, J. Graser, R. Zhao, F. Gao and M. J. O'Connell, ACS Nano, 2013, 7, 11156–11165 CrossRef CAS PubMed.
- Q. L. Zhao, Z. L. Zhang, B. H. Huang, J. Peng and M. Zhang, Chem. Commun., 2008, 5116–5118 RSC.
- S. T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J. H. Liu, Y. Liu, M. Chen, Y. Huang and Y. P. Sun, J. Phys. Chem. C, 2009, 113, 18110–18114 CAS.
- R. Zhang, Y. B. Liu and S. Q. Sun, J. Nanopart. Res., 2013, 15, 1–12 Search PubMed.
- P. B. Tchounwou, A. K. Patlolla and J. A. Centeno, Toxicol. Pathol., 2003, 31, 575–588 CAS.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- A. D. Lemly, Ecotoxicol. Environ. Saf., 2004, 59, 44–56 CrossRef CAS.
- K. Hagelstein and T. I. Mudder, TMS 2009 138th Annual Meeting & Exhibition: Supplemental Proceedings, 2009, vol. 3, pp. 77–88 Search PubMed.
- J. E. Hampsey, Q. Hu, Z. Wu, L. Rice, J. Pang and Y. Lu, Carbon, 2005, 43, 2977–2982 CrossRef CAS PubMed.
- M.-M. Titirici, A. Thomas and M. Antonietti, Adv. Funct. Mater., 2007, 17, 1010–1018 CrossRef CAS.
- Y. Yan, F. Zhang, Y. Meng, B. Tu and D. Zhao, Chem. Commun., 2007, 2867–2869 RSC.
- J.-S. Lee, S.-I. Kim, J.-C. Yoon and J.-H. Jang, ACS Nano, 2013, 7, 6047–6055 CrossRef CAS PubMed.
- D. Hanigan, J. Zhang, P. Herckes, S. W. Krasner, C. Chen and P. Westerhoff, Environ. Sci. Technol., 2012, 46, 12630–12639 CrossRef CAS PubMed.
- K. S. W. Sing and D. H. Everett, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- R. Dash, J. Chmiola, G. Yushin, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer and S. Kucheyev, Carbon, 2006, 44, 2489–2497 CrossRef CAS PubMed.
- M. O. Corapcioglu and C. P. Huang, Carbon, 1987, 25, 569–578 CrossRef CAS.
- P. Chingombe, B. Saha and R. J. Wakeman, Carbon, 2005, 43, 3132–3143 CrossRef CAS PubMed.
- H. P. Boehm, Carbon, 2002, 40, 145–149, DOI:10.1016/S0008-6223(01)00165-8.
- J. Torres, V. Pintos, S. Domínguez, C. Kremer and E. Kremer, J. Solution Chem., 2010, 39, 1–10 CrossRef CAS.
- N. V. Mandich, S. B. Lalvani, T. Wiltkowski and L. S. Lalvani, Met. Finish., 1998, 96, 39–44 CrossRef CAS.
- H. E. Eguez and E. H. Cho, JOM, 1987, 39, 38–41 CrossRef CAS.
- L. Lorenzen, J. S. J. van Deventer and W. M. Landi, Miner. Eng., 1995, 8, 557–569 CrossRef CAS.
- X. Peng, Z. Luan, J. Ding, Z. Di, Y. Li and B. Tian, Mater. Lett., 2005, 59, 399–403 CrossRef CAS PubMed.
- Z. Gu, J. Fang and B. Deng, Environ. Sci. Technol., 2005, 39, 3833–3843 CrossRef CAS.
- K. D. Hristovski, H. Nguyen and P. K. Westerhoff, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 354–361 CrossRef CAS PubMed.
- Q. Chang, W. Lin and W. Ying, J. Hazard. Mater., 2010, 184, 515–522 CrossRef CAS PubMed.
- N. Jordan, H. Foerstendorf, S. Weiß, K. Heim, D. Schild and V. Brendler, Geochim. Cosmochim. Acta, 2011, 75, 1519–1530 CrossRef CAS PubMed.
- G. Jegadeesan, K. Mondal and S. B. Lalvani, Environ. Technol., 2005, 26, 1181–1188 CrossRef CAS PubMed.
- P. Montes-Navajas, N. G. Asenjo, R. Santamaría, R. Menendez, A. Corma and H. García, Langmuir, 2013, 29, 13443–13448 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. E. Johns and M. C. Hersam, Acc. Chem. Res., 2013, 46, 77–86 CrossRef CAS PubMed.
- T. Szabo, E. Tombacz, E. Illes and I. Dekany, Carbon, 2006, 44, 537–545 CrossRef CAS PubMed.
- K. H. Goh and T. T. Lim, Chemosphere, 2004, 55, 849–859 CrossRef CAS PubMed.
- C. M. Gonzalez, J. Hernandez, J. G. Parsons and J. L. Gardea-Torresdey, Microchem. J., 2010, 96, 324–329 CrossRef CAS PubMed.
- M. A. Montes-Moran, D. Suarez, J. A. Menéndez and E. Fuente, Carbon, 2004, 42, 1219–1225 CrossRef CAS PubMed.
- H. S. Posselt, F. J. Anderson and W. J. Weber, Environ. Sci. Technol., 1968, 2, 1087–1093 CrossRef CAS.
- A. Hanson, J. Bates, D. Heil and A. Bristol, Water Treatment Technology Report, 1999, vol. 41 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |