
Exploring controls on the fate of PVP-capped silver nanoparticles in primary wastewater treatment†
Stephen M.
King
*a,
Helen P.
Jarvie
b,
Michael J.
Bowes
b,
Emma
Gozzard
b,
Alan J.
Lawlor
c and
M. Jayne
Lawrence
d
aISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Oxford, Didcot, OX11 0QX, UK. E-mail: stephen.king@stfc.ac.uk; Tel: +44 (0)1235 446437
bNERC Centre for Ecology & Hydrology, Mclean Building, Benson Lane, Crowmarsh Gifford, Wallingford, OX10 8BB, UK
cNERC Centre for Ecology & Hydrology, Lancaster Environment Centre, Lancaster LA1 4AP, UK
dPharmaceutical Science Division, King's College London, Franklin-Wilkins Building, Stamford Street, London, SE1 9NH, UK
First published on 21st January 2015
Abstract
Small-angle neutron scattering has been used to examine the settling behaviour of partially-passivated silver nanoparticles (AgNP), capped with a polyvinylpyrolidone (PVP) stabiliser, in water and domestic wastewater, in a primary clarification ‘microcosm’ as a function of time. The impact of two flocculants routinely used in the wastewater treatment process has also been studied. The settling velocity is found to be time-dependent, but always exceeds 100 mm h−1 during the first hour at the point of input. Particle removal by settling is almost three times greater in wastewater than it is in pure water. The results are rationalised in terms of a generic, but synergistic, interaction between non-ionic capping agents and anionic components of wastewater, and we show how this may afford an explanation for some of the diversity of behaviour previously reported in studies of several different NPs in wastewater treatment. We conclude that AgNPs entering primary clarification with non-ionic surface coatings, whether present by design or environmental transformation, pose no threat to the viability of the biofilms in secondary wastewater treatment.
Nano impactThe sources, transport, and fate of silver nanoparticles (AgNPs) are of concern due to the chemical toxicity of silver to microbes and algae. The greatest source of AgNPs to the environment is via wastewater discharges, meaning there is a potential risk to the microbial biofilms in secondary wastewater treatment. This study explores the behaviour of AgNPs in primary wastewater treatment, primarily using small-angle neutron scattering. The results show that AgNPs coated in a non-ionic stabiliser (PVP) undergo rapid settling in wastewater and will be removed to sewage sludge, rather than continuing to the secondary treatment stage. This settling behaviour seems to be controlled by hitherto unexplored synergistic interactions between hydrophilic parts of the non-ionic stabiliser and anionic components in the wastewater and is therefore generic. |
Introduction
Background
Humankind's appetite for engineered nanomaterials, ENMs, (i.e., excluding the commodity nanomaterials carbon black and pyrogenic/fumed silica which account for >96% of overall production) has recently been estimated at around 0.4 Mt per year representing, by the time this paper is published, a market worth some €3 bn ($4 bn).1–4 Roughly two-thirds of all ENMs produced are metal oxide nanoparticles (e.g., aluminium oxide, barium titanate, cerium oxide, iron oxide, titanium dioxide, zinc oxide).4–7 Pure metal nanoparticles (e.g., copper, gold, palladium, platinum, ruthenium, silver) currently account for <1% of ENM production but this is predicted to rise, particularly with advances in catalysis and an increasing number of applications in healthcare and the electronics industry. Indeed, in the last decade, worldwide demand for silver nanoparticles (AgNPs), the subject of this study, is estimated to have increased from <10 t per year to over 300 t per year (~1% of all silver produced).5,8–11AgNPs have found a multitude of uses; in recent history in the photographic industry, nowdays as anticorrosive pigments for primers and coatings, in conductive inks and flexible touch screens, but most notably in portable water purification, food packaging, high-performance clothing, wound dressings, and some medical devices, along with other less mainstream consumer health products.8,11,12 This is because silver has long been known to be a potent, broad-spectrum, bactericide. It is also an effective viricide, algaecide and fungicide. The presence of AgNPs in these products, rather than bulk silver, aids fabrication, lowers costs, and conveys greater efficacy. The problem, of course, is that at any point in the lifecycle of these products the AgNPs may be released to the environment, whether by accidental release during manufacture, through abrasion or cleaning during use, or after degradation of the matrix following disposal.13,14 The challenges for environmental nanoscience and (nano-)ecotoxicology are then: where do the AgNPs go, in what concentrations, what happens to them, and what are the consequences?15–18
For a large proportion of manufactured NPs, not just AgNPs, their major route of release into the natural environment is via sewage and industrial wastewater discharges, and from urban drainage.19 This means our wastewater treatment plants (WWTP) unwittingly act as ‘gateways’, controlling release of NPs and their transformation products from domestic and industrial sources to the aquatic or terrestrial environments: either via the treated effluent which is discharged into surface waters or, via sewage sludge disposal to land.20–22 AgNPs (and CuNPs too) are of particular concern to the wastewater treatment industry because these biocidal NPs pose a threat to the viability of the mixed microbial communities in the biofilms present in secondary wastewater treatment.23–28 Those biofilms break down organic pollutants (e.g., organic chemicals, drug metabolites, etc.) in the effluent received from primary treatment and are critical to the final quality of the treated discharge. Without the biofilms the aquatic environment would be further impacted. Thus the efficacy of primary wastewater treatment at removing harmful NPs, and the chemical transformation of NPs throughout wastewater treatment more generally, become key considerations.
Ref. 23–28 show that whilst ionic (dissolved) silver is most toxic to the microbes, nanoparticulate silver also has some toxicity. What is less clear, however, is whether that toxicity is due to the AgNPs themselves (or their surface coatings), and/or because they promote the formation of reactive oxygen species (ROS). The dissolution of coated AgNPs has been shown to be inversely related to size,29,30 water hardness, pH, and NOM (natural organic matter) concentration,31 but also directly related to DOC (dissolved organic carbon) concentration.32 However, it has also been shown that humic acids (a form of NOM) can reduce silver ions under environmentally relevant conditions to form non-anthropogenic AgNPs.33 Another key process is sulphidation of the AgNP to form Ag2S.34–36 In the case of AgNPs this oxidative dissolution process retards the release of silver ions (in contrast, it promotes the release of copper ions from CuNPs37) but is, again, also strongly size-dependent (smaller particles being transformed more).
Previous work
There are now several published studies on the fate and behaviour (as distinct from the application) of NPs in wastewater treatment, split between those that report on NP interactions with wastewater, and those that are concerned with NPs in the sludge (biosolids). Here we are primarily concerned with the former.Chang et al.38 looked at the coagulation of silica NPs in the effluent from a semiconductor chemical–mechanical planarization plant by polyaluminium chloride. These NPs would almost certainly have carried adsorbed surfactant coatings but there is no information on what the surfactants were. NP removal to the sewage sludge was found to be minimal. Jarvie et al. have also studied the behaviour of silica NPs, both uncoated (uncapped) and coated (capped; with a nonionic surfactant), in real municipal wastewater but in a laboratory model settlement tank.39 In contrast to Chang et al., the uncoated NPs were found to be quite stable, remaining in dispersion, but the coated NPs rapidly agglomerated and settled. From this they inferred that there must have been a specific interaction between the surfactant coating and components of the wastewater. Limbach et al.40 studied the agglomeration of both uncoated and coated (with an anionic surfactant or anionic polymer) ceria NPs in a laboratory model WWTP. Though the majority of NPs were eventually removed to the sewage sludge these authors also noted the significant influence that the particle coatings could have on the process, highlighting “complex interactions between dissolved species and the nanoparticles”. Kiser et al.,41 Westerhoff et al.42 and Wang et al.43 have investigated the fluxes of titanium entering and leaving some commercial WWTPs in the Southwest US, and conducted laboratory studies with uncoated titania NPs in a model WWTP. Again, the majority (~70–90%), but not all, of the NPs were removed to the sewage sludge. More recently, Lombi et al.44 have looked at the chemical stability of zinc oxide NPs (two uncoated, one coated with nonionic caprylic/capric triglycerides) in a laboratory model anaerobic digester (i.e., they had already assumed these NPs would be removed to the sewage sludge at the primary treatment stage). All three types of NP were converted to the sulphide, but the transformation of the coated particles was found to be substantially retarded relative to the uncoated NPs. Ma et al.36 also examined the fate of zinc oxide NPs, this time uncoated, added to the primary sludge and the activated sludge basin in a pilot WWTP. The study concluded that the NPs were significantly chemically transformed during treatment, seemingly corroborating the findings of Lombi et al.
Fewer studies have concentrated on metallic NPs in wastewater. Kaegi et al.45 have investigated the behaviour of AgNP coated with a non-ionic surfactant in a pilot WWTP fed from a municipal source. More than 85% of the AgNP were determined to have been removed to the sewage sludge. However, the authors also highlighted a rapid (~2 hours) and substantial (~60–90%) transformation of the AgNPs into Ag2S under increasingly anaerobic conditions. They also noted a residual ~10% of AgNPs that either transformed much more slowly or not at all. This was ascribed to the protective effects of the surface coating and/or passivated particle surfaces. In their work, Ma et al.36 also studied AgNPs but coated with non-ionic polymer (poly(vinyl pyrolidone), PVP). However, the fate of these AgNPs was the same as that of their zinc oxide NPs, suggesting that, in contrast, the surface coating offered little or no protection against sulphidation. However, this work was performed on samples that had been allowed to ‘age’ for a much longer length of time. In a more recent study, Kaegi et al.35 have extended their work to encompass the whole wastewater system. AgNPs coated with non-ionic surfactant were actually dosed into a trunk sewer, whilst AgNPs coated with PVP, or capped with citrate, were added to wastewater samples extracted from the sewer. The results demonstrated that AgNPs discharged to the sewer network are very likely to be efficiently delivered to the WWTP with only partial transformation. This was ascribed to the greater surface area for interaction presented by suspended sediment particles than by the sewer biofilms (which control the extent of sulphidation enroute), rather than to a difference in binding affinity. AgNP removal efficiency in the wastewater experiments was very high (~99%) irrespective of the particle size or surface coating. Impellitteri et al.46 and Doolette et al.47 have also investigated the fate of PVP-coated AgNPs at the primary treatment stage. The former study reports over 97% of the AgNPs as being removed to the sludge. Both studies identify sulphidation as the dominant transformation once the AgNPs are in the sludge. In their study, Wang et al.43 also looked at carboxy-terminated (i.e., anionic) polymer coated AgNP and found that whilst removal efficiency was less than for titania NPs, 88% of AgNPs were still removed to the sewage sludge. In contrast, Hou et al.48 studied citrate-capped (i.e., anionic) AgNPs in a laboratory model WWTP and found that over 90% would pass through primary treatment. Though Tejamaya et al.49 have reported instability in dispersions of citrate-capped AgNPs, this was in a standard OECD toxicology test media, not wastewater. The organic makeup, ion composition, ionic strength and pH of the two media are rather different (compare Tables S1 and S2 in ref. 49 with the present Table S5†). Very recently Johnson et al.50 have measured the total fluxes of silver entering and leaving nine commercial WWTPs, of three different process types, in the UK. For silver particles between 2–450 nm in size (i.e., the filter fraction including AgNP's) around 50% were found to be removed to the sewage sludge.
Thus, with one or two exceptions, the literature would appear to indicate that oxide/metallic NPs will usually be removed to the sewage sludge at the primary clarification stage of wastewater treatment. However, the efficiency of removal, and the extent of any chemical transformation, of the NPs would appear to be strongly influenced by the nature (e.g., how attached, thickness, charge) of any surface coating, how that interacts with other species present in wastewater, the specific environmental conditions (e.g., aerobic vs. anaerobic), and the duration of exposure to the environment.
Present work
Against this background, in this work we have systematically explored the behaviour of one type of AgNP, coated with the non-ionic polymeric stabiliser PVP, in five different aqueous matrices: pure water, untreated wastewater (sewage), wastewater dosed with ferric chloride, and pure water dosed with two very different concentrations of the anionic polymer poly(sodium 4-styrene sulphonate), PSS. Ferric chloride and PSS are sometimes used as flocculants in industrial wastewater treatment to enhance clarification (solids removal), a process known as advanced primary treatment. Ferric chloride may also be used in a tertiary treatment stage designed to strip phosphorus from the effluent. Thus our study covers the gamut of chemical conditions that the AgNPs are likely to encounter before entering, and once inside, a wastewater treatment plant. The choice of AgNP was largely a pragmatic one; to minimise sulphide formation and dissolution effects, and thus attempt to decouple the redox chemistry from the colloid chemistry, we chose passivated AgNPs, but then a stabilising moiety was necessary to successfully disperse the nanoparticles in water (particularly given the ten-fold difference in density). Of the three most widely available stabilisers – citrate, PVP, and poly(ethylene oxide) (PEO) – the non-ionic stabilisers, and PVP in particular, have been shown to be more successful at promoting dispersion than charged stabilisers like citrate.49 Also, whilst a charge-stabilized nanoparticle would be expected to aggregate in the presence of background electrolyte or a counter-charged flocculant, the expectation from a sterically-stabilized nanoparticle is much less obvious. Our selection of PVP-stabilised AgNPs for this work also allowed us to investigate the uniqueness of the central result from our earlier work with silica NPs stabilized by a short-chain PEO-based surfactant.39In order to mimic the processes occurring in primary wastewater treatment, our measurements have been performed following established methodology, employing settlement microcosms in the form of tall cuvettes containing fresh wastewater. Small-angle neutron scattering (SANS) data have then been collected near the top (i.e., nearest to the point of AgNP dosing) and the bottom of the cuvettes, allowing us to monitor the change in the AgNP size and concentration as a function of both time and position in the water column. The details of this are described in the experimental section at the end of this paper, and in the ESI.† Additional laboratory bench-top batch-sampling studies have also been conducted in order to cross-validate the SANS experiments. Our results enable us to propose a possible mechanism, hitherto unexplored in environmental nanoscience, behind the heteroaggregation responsible for the removal of non-ionic coated nanoparticles.
Results
SANS measures the angular variation in the intensity, I(Q), of neutrons scattered from a sample, the diffraction angle being ‘encoded’ in a vector, Q. The intensity is proportional to the size and number concentration of the NPs, the Q-dependence of the scattering is determined by the shape of the nanoparticles.The behaviour of the AgNPs in the different matrices can be qualitatively observed from the treated SANS data (Fig. 1, top and middle panes). These data are shown after subtraction of an essentially Q-independent background arising from the respective matrices (see point i below). From these data we can make the following observations:
 | ||
| Fig. 1 Example SANS data at different times from: (Top left pane) 18 mg mL−1 AgNP in nanopure water (filled symbols + continuous lines) or screened wastewater (open symbols + dotted lines) recorded at the top measuring position. (Top right pane) 18 mg mL−1 AgNP in screened wastewater dosed to 30 mg L−1 FeCl3 recorded at the top measuring position (open symbols + dotted lines). The signal from just the wastewater, displaced by 3 cm−1, is shown as a continuous line. (Middle left pane) 37 mg mL−1 AgNP in nanopure water (open symbols + dotted line) and when dosed with 0.59 mg L−1 PSS (filled symbols + continuous lines) recorded at the top measuring position. (Middle right pane) 37 mg mL−1 AgNP in nanopure water (open symbols + dotted line) and when dosed with 1091 mg L−1 PSS (filled symbols + continuous lines) recorded at the top measuring position. The inset shows the same data in log–log representation to highlight the similar Q-dependency of the scattering. (Bottom left pane) 18 mg mL−1 AgNP in nanopure water (filled symbols) or screened wastewater (open symbols) recorded at the bottom measuring position. The dotted lines on this graph are fits to the composite polydisperse spheres model (see text). (Bottom right pane) 18 mg mL−1 AgNP in nanopure water (filled symbols) or screened wastewater (open symbols) recorded at the bottom measuring position. The dotted lines on this graph are fits to the fractal cluster model (see text). | ||
(i) There is no appreciable signal from the wastewater alone (top-right panel, continuous line), nor from pure water (not shown), the former demonstrating that suspended solids in the wastewater do not produce an interference signal (they are either too dilute, too low contrast, or outside the measurement range of the instrument);
(ii) the dispersion stability of the AgNP in pure water on timescales exceeding 1.5 hours (top-left panel, filled symbols) is very high, the small loss in intensity with time indicating few NPs have settled out;
(iii) by contrast, the AgNP are very unstable in wastewater (top-left panel, unfilled symbols) with a two-thirds reduction in intensity in as little as 12 minutes, and approaching a 90% reduction in intensity after 2 hours;
(iv) adding ferric chloride to the wastewater appears to promote some additional instability of the AgNP (top-right pane) – though these data were measured after longer times – but it has no effect on the wastewater itself on the timescale of the experiments (Fig. S8†);
(v) adding PSS to the AgNP in pure water also promotes instability that progresses with time and which is more pronounced at higher PSS concentrations (middle-panes);
(vi) as there is no appreciable change in the Q-dependence of the scattering there does not appear to be any change in the shape of the AgNP in pure water, without and with, added PSS (top-left pane and middle-panes). The same may also be true of the nanoparticles in wastewater but the poorer signal-to-noise in these data makes it difficult to be certain without a quantitative analysis.
The data presented in the top and middle panes of Fig. 1 were all measured at the top of the sample cuvettes. The SANS data from near the bottom of the cuvettes are analogous, but for any given time have slightly higher intensity (bottom panes). This implies that the AgNP have settled.
To put these observations on a quantitative footing we have least-squares fitted the SANS data to analytical functions describing the shape and organisation of the nanoparticles in the different matrices (see Experimental). Example fits are shown in the bottom-panes of Fig. 1 to a polydisperse spheres function (eqn (2) and (3)) and a fractal cluster function (eqn (2)–(4)). For each matrix-time-position measurement this procedure yielded an apparent particle size and particle volume fraction (and cluster fractal dimension, where appropriate). The apparent volume fraction was then corrected by rescaling it against the known volume fraction of particles in a reference sample measured under similar conditions (see ESI†). Across all the measurements performed there is an excellent correlation between the particle sizes and volume fractions derived from the two different functions, though the fractal cluster model tended to return slightly larger primary particle sizes and slightly smaller apparent volume fractions (see Fig. S7†).
With knowledge of the final particle volume fractions, eqn (5) could then be used to determine the mass concentration of AgNPs as a function of time in each of the different matrices. These are shown in the top-panes of Fig. 2. Filled symbols correspond to the top of the cuvette, and open symbols the bottom. As inferred earlier, at a given time in a given matrix, the AgNP concentration is slightly higher nearer the bottom of the cuvette. In all instances the AgNP concentration decreases non-linearly with time, with the greatest change occurring in the first 30 minutes. Some of this change will be due to the differential settling velocities of different-sized NPs (see the PSD in Fig. S1†). These data can be adequately described by power law decays,‡ the characteristic parameters for which are given in Table 1, and clearly illustrate a wide variation in dispersion stability between the different matrices. The principal source of uncertainty in these data is the uncertainty associated with the volume fraction, ϕparticles. This is very difficult to estimate. However as a guide, using the model-fitting parameters returned for the AgNP in water system (the most stable), the value of σ/<ϕparticles> ~7%, where σ is one standard deviation. It is reasonable to assume that this figure will be slightly higher in the systems exhibiting less stability (where the signal-to-noise was poorer), but even if doubled to 15% the differences between the concentration-time datasets for different media are still statistically significant.
 | ||
| Fig. 2 Time dependence of: (Top left pane) AgNP concentration in nanopure water, screened wastewater, and screened wastewater dosed to 30 mg L−1 FeCl3 recorded at the top (filled symbols) and bottom (open symbols) measuring positions. (Top right pane) AgNP concentration in nanopure water dosed to 0.59 mg L−1 and 1091 mg L−1 PSS recorded at the top (filled symbols) and bottom (open symbols) measuring positions. (Bottom left pane) AgNP settling velocity in nanopure water, screened wastewater, and screened wastewater dosed to 30 mg L−1 FeCl3. Also shown are the settling velocities derived from the bench-top settling studies performed at much lower AgNP concentrations (see text). (Bottom right pane) AgNP settling velocity in nanopure water dosed to 0.59 mg L−1 and 1091 mg L−1 PSS. The dotted lines in all four graphs are power law trendlines to guide the eye. Error bars shown represent one standard deviation (see text). | ||
| Sample | Measuring | Average | Average | Loss | [AgNP] trendline | ν s trendline | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Matrix | Position | D | R (nm) | (%) | A | n | R 2 | B | m | R 2 |
| Water | Top | 2.55 ± 0.03 | 10.4 ± 6.2 | 27 | 12.586 | −0.066 | 0.816 | 101.620 | −1.016 | 0.999 |
| Bottom | 2.54 ± 0.02 | 10.3 ± 6.2 | 20 | 14.330 | −0.046 | 0.896 | 22.757 | −1.044 | 0.980 | |
| Screened sewage | Top | 3.38 ± 0.8 | 13.6 ± 8.2 | 74 | 4.480 | −0.280 | 0.888 | 203.630 | −0.941 | 0.992 |
| Bottom | 3.38 ± 0.8 | 12.9 ± 7.7 | 78 | 4.338 | −0.318 | 0.684 | 66.346 | −0.528 | 0.775 | |
| Screened sewage + FeCl3 | Top | 2.91 ± 0.4 | 7.6 ± 4.6 | 87 | 1.779 | −0.423 | 0.980 | 311.470 | −0.955 | 1.000 |
| Bottom | 3.26 ± 0.7 | 10.9 ± 6.5 | 84 | 2.534 | −0.384 | 0.924 | 82.012 | −0.815 | 0.998 | |
| Water + low [PSS] | Top | 2.58 ± 0.01 | 9.5 ± 5.7 | 25 | 27.043 | −0.061 | 0.972 | 91.442 | −0.853 | 0.996 |
| Bottom | 2.58 ± 0.01 | 9.5 ± 7.7 | 16 | 30.555 | −0.037 | 0.851 | 17.869 | −0.895 | 0.853 | |
| Water + high [PSS] | Top | 2.58 ± 0.01 | 9.7 ± 5.8 | 54 | 18.267 | −0.163 | 0.964 | 149.660 | −0.719 | 0.995 |
| Bottom | 2.59 ± 0.01 | 9.8 ± 5.9 | 35 | 23.835 | −0.091 | 0.807 | 37.304 | −0.851 | 0.970 | |
From a wastewater treatment industry perspective a more insightful parameter is the settling velocity of the particles, νs. Efficient primary clarification typically requires ~100 < νs < ~1000 mm h−1 depending on the range of particle sizes in the influent,51,52 with the residence (hydraulic retention) time of the wastewater in the settling tanks typically engineered to be ~1 hour to a few hours, depending on influent load. We are able to calculate experimental settling velocities for the AgNPs from our data using the change in AgNP concentration relative to the known dosing concentration using eqn (6). The results are shown in the bottom-panes of Fig. 2.
For comparison, the much larger Tween™-capped SiO2NPs in our previous work were determined to have a settling velocity νs ~ 80 mm h−1,39 but these were of course much less dense. By contrast, the Stokes settling velocity of the AgNP in pure water calculated from eqn (7) (for radius, R = 10 nm) is just νs,stokes ~ 6.7 × 10−3 mm h−1 (equivalent to 1.8 nm s−1, or about 20 μm in a 3 h SANS experiment).§
In all matrices, but particularly in wastewater, the derived νs exceeds 100 mm h−1 during the first hour at the top measuring position (equivalent to where influent wastewater would enter the settling tank). The corresponding efficiency of AgNP removal is shown in Table 1, as calculated from the trendlines shown in the bottom panes of Fig. 2 at t = 1 h. It can be seen from Table 1 that the wastewater is almost three times more effective at removing the AgNP as pure water alone, and almost twice as effective as pure water with the high dose of PSS. The low dose of PSS essentially had no effect. Adding ferric chloride to the wastewater enhances AgNP removal by around 10% compared to the wastewater alone.
To cross-validate the results from the SANS experiments we have also performed a bench-top settling experiment using real wastewater. In order that the AgNP concentrations could be determined by ICPMS we used a much lower, and more environmentally-relevant, particle concentration (~hundreds of μg L−1). Nevertheless, the results (see Table S6†) directly correspond with those from the SANS experiments: after 2 hours just over 60% of AgNPs were removed from the top of the water column. The derived settling velocities – which actually represent a vertical positional average over some 60 mm as compared to just 8 mm in the SANS experiments – are plotted in the bottom-left pane of Fig. 2 (star symbols) and lie between the trendlines from the SANS experiments in wastewater.
In the SANS experiments we deliberately did not measure the scattering at the very bottom of the cuvette, but the design of our bench-top experiment made it relatively straightforward to extract and analyse this fraction of the water column. As one would anticipate, we find particles removed from the top of the water column accumulate at the bottom.
Whilst our settling velocities may not be directly transferrable to a WWTP where there will be other mitigating factors, they should certainly be indicative, and are most definitely internally self-consistent.
Discussion
At this point we remind the reader that the only parameter that this study has in common with our earlier work is that the adsorbed stabiliser is non-ionic; the particle interfaces are chemically distinct with different degrees of surface ionisation (where exposed) and curvature, the chemical structure and molecular weight (i.e., length) of the stabilisers are different, and the wastewater was sourced from different WWTPs at different times. Yet it is clear that something in the wastewater is as effective at colloidally destabilising the PVP-capped AgNPs in this study as it was the Tween™-capped SiO2NPs in our earlier work. We have previously ruled out the background ionic strength of wastewater (<0.01 M NaCl equivalent) as an explanation for the rapid settlement of the NPs39 and discuss this further later on. Another important result from our earlier work was that uncapped SiO2NPs did not settle (in wastewater or pure water).Mechanism of NP removal
The most logical explanation for our findings, therefore, is that there are specific interactions between the PVP or Tween™ coatings and some component(s) of the wastewater. In this respect it is interesting to note that the fractal dimension of the AgNP ‘flocs’ in Table 1 appear to jump from ~2.5 in pure water to just over 3 in wastewater (though the poor signal-to-noise of the latter data gives rise to large uncertainties) with only a small variation in primary particle size. This change in fractal dimension could be consistent with a change from AgNPs with ‘normal’ PVP coatings to AgNPs with ‘entangled’ PVP coatings, or perhaps even the formation of a weak network entrapping the nanoparticles; but there is clearly no significant particle clustering taking place, else the SANS analysis would detect it. There are three pieces of evidence that shed some light on a possible mechanism behind our findings: first, the fact that at the pH of wastewater the uncapped SiO2NPs from our previous work would have had negatively-charged interfaces; second, the stability of those NPs in wastewater; and third, the stability of Hou et al.'s48 citrate-capped AgNPs (i.e., NPs that also had an interface with a dense population of close-bound negative charges) in wastewater. For these negatively-charged NPs to be electrostatically stabilized (citrate is too short to be an effective steric barrier) implies that they are not interacting with positively-charged components in wastewater and that, in turn, to explain the instability of the capped NPs our attention should be directed to those uncharged or negatively-charged components present. Here, the ability of a sufficiently high concentration of anionic PSS to destabilise the PVP-capped AgNPs (Fig. 1, middle panes) suggests that it is in fact negatively-charged components that may be the key.Limbach et al.40 have implicated peptones – mixtures of polypeptides and amino acids derived from partially-hydrolysed proteins – in the stabilisation of uncapped ceria NPs. We also note that the isoelectric points of all but two amino acids are less than the pH of our wastewater, and so the net charge on amino acids and peptides would be negative in our experiments. Similarly there are many anionic polysaccharides, derived from the degradation of plant biomass that will find their way into wastewater. But why should anionic components of wastewater interact with the non-ionic coatings on our NPs?
A possible insight comes from the seemingly unrelated areas of detergency and fabric conditioning. These applications deliberately exploit synergistic interactions between mixtures of short-chain ionic and non-ionic surfactants, but this can also lead to co-adsorption of the different species at an interface. An excellent demonstration of this effect has been provided by Penfold et al.,53 who showed that the anionic surfactant sodium dodecyl sulphate (SDS) could be made to adsorb on a negatively-charged silica surface in water in the presence of the non-ionic surfactant hexaethylene glycol monododecyl ether (C12E6), provided there was a molar excess of C12E6. In the absence of C12E6 there was no adsorption of the SDS, as one would expect. (The same authors also demonstrated similar behaviour with a cationic surfactant in place of the SDS at a different pH54). The amount of SDS adsorbed was small, <1 mole%, even when the amount in solution was tens of mole%, and the adsorbed SDS was only located at the outer region of the mixed adsorbed layer, but it nonetheless indicates that anionic and non-ionic species can interact under specific conditions. Penfold et al. ascribed the co-adsorption to favourable packing of the (hydrophobic) alkyl ‘tails’ but also to headgroup interactions, noting that in ionic/non-ionic surfactant mixtures the mixing process dilutes what would otherwise be unfavourable electrostatic interactions between the charged SDS headgroups. However, this also provides an environment in which those charged headgroups can then interact through van der Waals forces with weak dipoles in the headgroups of the uncharged surfactant molecules. We believe this is the mechanism that is ultimately responsible for the instability of our non-ionic surfactant-coated NPs in wastewater, particularly if the co-adsorbing species also interacts with other oppositely-charged components (cationic polysaccharides, mineral particles, etc.).
Though Penfold et al. studied two surfactants adsorbing from bulk solution, it is not unreasonable to consider that a pre-adsorbed layer of non-ionic surfactant would constitute a local excess and so promote analogous behaviour. If so, one might also speculate that the same would apply to non-ionic surfactants interacting with a pre-adsorbed layer of ionic surfactant, such as in the systems studied by Limbach et al.40 and Wang et al.43
Challenging our interpretation
But how robust is this interpretation? We shall now consider potential alternative explanations.Whilst the mass concentration of AgNPs on dosing was high (18 mg ml−1), because of the high density of silver the volume fraction of AgNPs was a mere 0.19% (i.e., ϕ ~ 0.002). This is important, because the thermodynamics of colloidal systems are expressed in terms of volume fraction55 in much the same way that the colligative properties of a chemical system are normally expressed in terms of molality. A volume fraction of 0.19% is a very low value. Some measure of this may be gained from the impact of ϕ on the interparticle structure factor S(Q), because if interparticle interactions between the AgNPs were important they would manifest themselves in the SANS through eqn (2). In Fig. S9† we provide calculations of S(Q) and I(Q) for dispersions of spherical particles having the same physical characteristics as the AgNPs used in our study. The cases of both uncharged and charged particles are considered, but it is clear that at the particle volume fraction we have used interparticle interactions are negligible in both cases. Furthermore, in our earlier work39 with (larger) SiO2NPs we used a lower volume fraction of 0.10% (an equivalent mass concentration of 2.5 mg ml−1 for that system) yet observed completely analogous behaviour to that described in the present study. But the final, perhaps most compelling, piece of supporting evidence, are the results from our bench-top settling experiment. Performed at a very much lower particle concentration, they demonstrate the same behaviour of the AgNPs, to broadly similar extents, over the same timescales, as found from the SANS experiments (Fig. 2, bottom-left pane).
The absence of homo-aggregation between our AgNPs is also supported by the results of the SANS data model-fitting. First, there is no systematic increase in the derived particle size with time (Fig. S10†), as would be expected if the AgNPs were aggregating into clusters. Second, the fractal dimensions for the floc structures we report in Table 1 are not what would be expected for aggregate structures formed through either diffusion-limited (D ~ 1.8) or reaction-limited (D ~ 2.1) particle cluster mechanisms. And the absence of homo-aggregation is further strengthened by the similarity of the results from the bench-top settling experiments performed at much lower AgNP concentration.
In situations where the NPs have patchy steric coatings it is sometimes possible to get interparticle bridging by the stabiliser. However this requires the NPs to be able to approach within each other's sphere of influence, often enough, and, in the case of NPs with charged surfaces, compression of the surrounding electrical double layers (else the NPs will repel one another). In our earlier work39 we demonstrated that uncoated SiO2NPs did not settle in wastewater, indicating that the ionic composition and ionic strength of wastewater was not sufficient to coagulate those (negatively) charge-stabilised NPs through compression of their double layers. We then performed a series of coagulation studies on the non-ionic surfactant-coated SiO2NPs using La(NO3)3; i.e., a cation an order of magnitude more effective at coagulating charged particles than Ca2+ (Schultze–Hardy rule). The results showed that only when the ionic strength of the medium was much higher than that of the wastewater could we even begin to coagulate the coated SiO2NPs on anything like the same timescale as the wastewater alone was achieving. This observation is indirectly reinforced by our present data where we added FeCl3 to the wastewater – there is only a small enhancement in AgNP removal over what wastewater alone achieved. Also reinforcing our observations in this regard are the stability of Hou et al.'s citrate-capped AgNPs in wastewater,48 and the very recent work of Zhou et al. with uncapped ZnO and TiO2 NPs.60 The ionic strength of wastewater is approximately equivalent to ~0.01 M NaCl, for which the double layer thickness would be of the order of 2 nm. This happens to be about the same as the radius-of-gyration of the PVP-coating on our AgNPs; i.e., the AgNPs would need to approach very close to one another indeed.
A related mechanism would be interparticle bridging by other components in wastewater. But again, the same physical constraints in respect of stabiliser coverage, double layer thickness and NP encounter frequency discussed above, would still apply. The most likely agents for such bridging would be humic/fulvic acids and similar charged ‘polymer-like’ species, but in the case of our AgNPs only anionic species could adsorb to the Ag2O surface (and an equivalent mechanism, but with cationic components in wastewater, would need to have occurred in our SiO2NP work). Though it is tempting to identify the effect of added PSS on our AgNPs (Fig. 1, middle-panes) as proof of this mechanism there are in fact significant issues. First, our PSS data were measured in water, not wastewater, and so one would not expect (any) significant loss of the PVP stabiliser (or other transformation of the NPs) to facilitate the bridging. Second, the rate of settling in the AgNP-PSS-water systems is a lot slower, and particle removal is less efficient, than in the AgNP-wastewater systems, even though the Rg of the PSS flocculant is approximately six times that of the PVP. Taking all these factors together, it seems unlikely that destabilisation of our AgNPs by interparticle bridging as a result of incomplete PVP coatings could be a significant mechanism, at least on the timescales of primary clarification.
In the last couple of decades mainstream colloid science has recognised the importance of what has become known as the hydrophobic interaction, an entropy-driven effect where the association of hydrophobic moieties disrupts the usual cage-like structure formed by hydrogen-bonding water molecules. The effect is now implicated in processes such as the folding of proteins, for example. The strongest hydrophobic interactions occur at elevated temperatures, in systems of long linear aliphatic organic molecules, but Song et al.61 have provided evidence that the hydrophobic interaction is likely responsible for the attachment of non-ionic (including PVP) coated AgNPs to C18 chemically-hydrophobised glass beads. However, attachment efficiencies did not exceed 16% even under the most favourable coverage conditions with a PVP stabiliser 67% longer (Mw ~ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) than that on our AgNPs. Thus, whilst we cannot rule out hydrophobic interactions in the systems we have studied, it seems unlikely that they could be the dominant mechanism responsible for NP removal. More problematic in any case is the fact that, in wastewater, any truly hydrophobic substances (mimicking the hydrophobised glass beads), such as fat particles and oil droplets, will be solubilised. Substances like fatty acids, of course, meet our description of ‘anionic components’. There is some literature on the adsorption of alkanes into non-ionic surfactant layers at the macroscopic oil–water interface,62 but this showed adsorption to increase with activity; i.e., the adsorption of short alkanes was favoured. This process would not therefore produce a hydrophobic interaction strong enough to drive aggregation.
000 g mol−1) than that on our AgNPs. Thus, whilst we cannot rule out hydrophobic interactions in the systems we have studied, it seems unlikely that they could be the dominant mechanism responsible for NP removal. More problematic in any case is the fact that, in wastewater, any truly hydrophobic substances (mimicking the hydrophobised glass beads), such as fat particles and oil droplets, will be solubilised. Substances like fatty acids, of course, meet our description of ‘anionic components’. There is some literature on the adsorption of alkanes into non-ionic surfactant layers at the macroscopic oil–water interface,62 but this showed adsorption to increase with activity; i.e., the adsorption of short alkanes was favoured. This process would not therefore produce a hydrophobic interaction strong enough to drive aggregation.
Environmental perspective
One final observation that we may make is that the total naturally-occurring NP background in our wastewater sample must be considerably less than the minimum AgNP concentration we derive, 1.2 mg ml−1 (Fig. 2, top-left pane), otherwise the SANS from the wastewater alone (Fig. 1, top-right pane) would not be a flat line. This upper bound on the NP concentration is consistent with the recent analytical work of Johnson et al.50 that measured the average concentration of ‘colloidal’ silver (defined as being in the size range 2–450 nm) in a range of UK wastewaters at a very low ~10−8 mg ml−1. This concentration of AgNPs is well below levels that have been shown to be toxic to microbes present in biofilms in secondary wastewater treatment. For example, Amaout26 has shown that nitrite production by N. europaea can be inhibited by as much as 15% at AgNP concentrations of 0.002–0.02 mg ml−1, depending on the surface coating (citrate and gum Arabic both inhibiting more effectively than PVP). At even higher AgNP concentrations cell lysis was observed. Within mixed microbial communities some individual communities were initially affected by the addition of AgNPs more than others, but overall there was greater resilience, perhaps evidence of adaptation to the change in chemical environment. These findings appear to be borne out by Giska.27 Sheng et al.24 observed that whilst isolated cultures could be wiped out by 0.001 mg ml−1 of AgNP (Thiotrichales was particularly susceptible), mixed biofilms were still resilient at 0.2 mg ml−1. And Doolette et al.47 have noted that whilst transformed (into Ag–S species) AgNPs “did not affect the dominant populations of nitrifiers and methogenic organisms in aerobic and anaerobic generated sludges… a subtle shift in niche populations was observed” (the latter being more evident in anaerobically-digested sludges).Together, these studies, and the findings we report here, would seem to indicate that AgNPs entering WWTPs in raw sewage pose no significant risk to secondary treatment biofilms at the present time. Where there may be greater cause for concern, however, is in the post-treatment disposal of the sewage sludge. This is because the sewage sludge is effectively a sink for the AgNPs and the practice of adding it to land as an agricultural fertiliser means that there is the potential for AgNPs, or their oxidation products (e.g., Ag2S), to accumulate in the soil. As Johnson et al.50 have noted, this aspect of the lifecycle of AgNPs is not yet well researched, though Whitley et al.56 have recently pointed to the profound impact that the presence or absence of the surface coating can have on the partitioning of NPs to soil pore water.
Experimental
Materials
000 g mol−1) was sourced from Sigma-Aldrich (Gillingham, Dorset, UK). Iron(III) chloride hexahydrate flocculant was sourced from Merck Millipore (Watford, Hertfordshire, UK). Nanopure water (18 MΩ resistivity) was produced in-house (Barnstead International Diamond system).
Small-angle neutron scattering (SANS)
| Q = (4π sin(θ))/λ | (1) |
550 mg L−1 PSS solution (‘high [PSS]’, to give 1091 mg L−1 in the cuvette) were added to nanoparticle dispersions in wastewater or nanopure water. These flocculant concentrations were chosen to be representative of those used in the wastewater treatment industry.65–69
| I(Q) = Kϕ1ϕ2(ρ1 − ρ2)2P(Q)S(Q) + B | (2) |
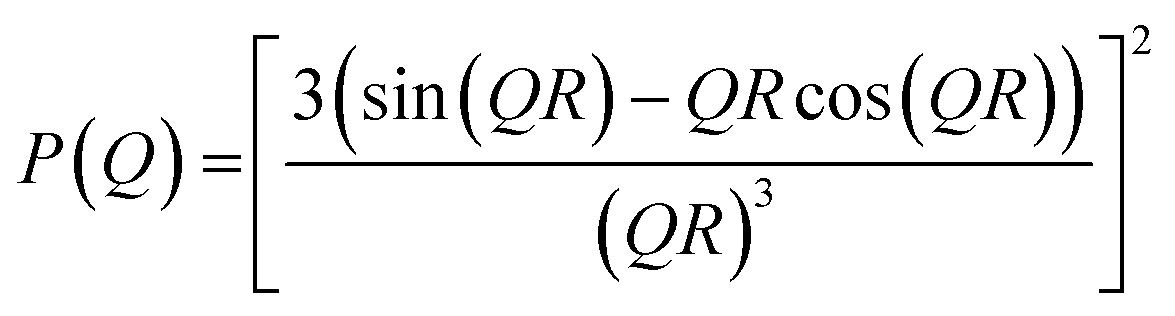 | (3) |
At the particle concentrations used in this work we found it unnecessary to invoke specific interparticle interaction potentials during the data fitting; i.e., it was assumed that S(Q) = 1. In earlier work64,73 we have also shown that these sorts of system can sometimes be described in terms of fractal cluster models where eqn (3) is used to describe the spherical ‘building blocks’ of the cluster and74
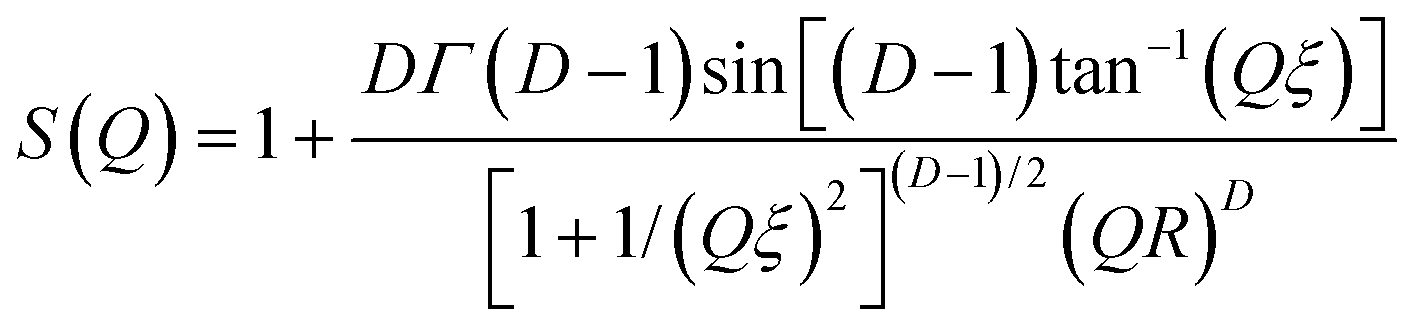 | (4) |
Here D is the fractal dimension and ξ is related to the overall size of the cluster. Unfortunately these two quantities are correlated and so to facilitate the fitting we chose to fix the latter at ξ = 73 nm (i.e., >5R), the maximum length scale accessible on the LOQ diffractometer. Density correlations at this distance are expected to be negligible.75
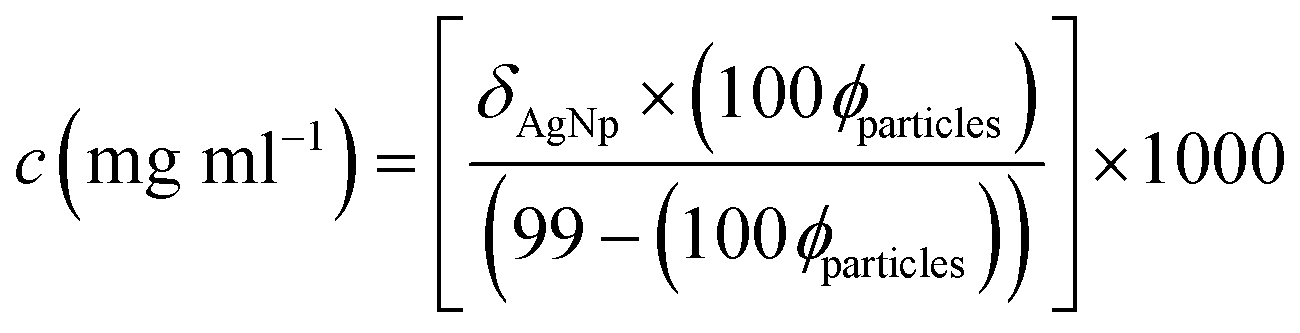 | (5) |
| νs,expt (mm h−1) = Δc × (100/d) × (1/t) | (6) |
By contrast, the Stokes settling velocity νs,stokes of a nanoparticle of radius R nm is76
 | (7) |
Bench-top settling studies
After some mixing, achieved by simply inverting the burette, a 10 mL (t = 0 min) sample was immediately extracted from the top of the burette by syringe. After a given time, a further 10 ml sample was extracted from the top of the burette, and then a 10 ml sample was drawn off from the bottom of the burette through the tap. Samples were recovered in this way after 15, 45 and 120 minutes using a separate burette for each time point. Each sample was immediately stabilised by the addition of ultrapure nitric acid (Ultrex analytical grade, JT Baker, UK) before subsequent digestion in aqua regia on a hotplate. This method is based on US EPA Method 200.2 and achieves good recoveries. Samples after digestion were optically transparent confirming the suitability of the method.
Additional experimental details may be found in the ESI.†
Conclusions
This study builds on our earlier work, and demonstrates that the rapid settling of NPs coated with non-ionic stabilisers in wastewater is a generic heteroaggregation effect. On entering WWTPs such NPs will be efficiently removed to the sewage sludge at the primary treatment stage.The origin of the effect is, we believe, a synergistic interaction between the hydrophilic part of the stabilising moiety and anionic components present in wastewater (e.g., peptones, polysaccharides), possibly even leading to entrapment of the NPs in a diffuse ‘polymer-like’ network which then interacts more widely with other (oppositely charged) components to enhance the settling velocities. This interaction has previously been ignored in discussions of nanoparticle behaviour in environmental matrices such as wastewater.
However, as others have noted,16,18 the natural environment is a complex chemical reactor where NPs are potentially subject to many different biotic and abiotic transformations; NPs entering the environment with surface coatings may lose them (and vice versa), or indeed gain different or mixed coatings. Thus determining a mechanism that controls the fate of NPs in the environment does not in itself necessarily determine which NPs are subject to that control. This of course has profound implications for modelling NP transport, whether in wastewater treatment or throughout the wider environment.
Our work also demonstrates that, in contrast to the predictions of the Stokes equation, NP settling velocities in complex environmental matrices are in fact time-dependent (though the change in settling velocity with time eventually reaches a steady-state, see Fig. S11†). This is a known phenomenon, due in part to the size polydispersity of the NPs but also, and particularly for wastewater treatment, the presence of density stratifications in the settling tank.77 The mechanism we have outlined, and thus the settling velocities derived, will also be dependent on the concentration of both the NPs and the wastewater components with which they interact.
Though real wastewater treatment is a much more dynamic environment than that we have modelled, WWTPs are normally operated in such a way as to give primary clarification the greatest ability to succeed. Our results could potentially help plant operators adapt their procedures to enhance the removal of ENMs alongside the normal colloidal and mesoscale detritus.
Acknowledgements
We thank H. Al-Obaidi for taking the TEMs of the NPs; D. Myatt and L. Clifton for making the DLS measurements; S. Waller and B. Holsman for the design of the Biological Containment Vessels; the ISIS Facility for funding construction of the vessels and providing neutron beam time (Experiment RB1010250); and Thames Water Utilities Limited for granting access to the treatment works. The work described was funded by the UK Science & Technology Facilities Council, the UK Natural Environment Research Council, and by the EU Framework Programme 7 NanoFATE project. The data analysis benefitted from use of the SasView software originally developed by the DANSE project under US National Science Foundation award DMR-0520547. Finally we would like to thank the anonymous Referee's and the Editor. Their careful critique led us to re-examine several aspects of our presentation and as a result the revised manuscript has benefitted significantly.Notes and references
- Nanoscale Chemicals and Materials, SRI Consulting. December 2010; http://www.ihs.com/products/chemical/planning/scup/nanoscale-chemicals.aspx (accessed 29/07/2014) Search PubMed.
- Global Nanomaterials Opportunity and Emerging Trends, Lucintel. March 2011; http://www.lucintel.com/LucintelBrief/GlobalNanomaterialsopportunity-Final.pdf (accessed 29/07/2014) Search PubMed.
- World Nanomaterials to 2016, Study # 2871, The Freedonia Group, Inc. May 2012; http://www.freedoniagroup.com/World-Nanomaterials.html (accessed 29/07/2014) Search PubMed.
- Types and uses of nanomaterials, European Commission Document 52012SC0288. October 2012; http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52012SC0288 (accessed 29/07/2014) Search PubMed.
- C. O. Hendren, X. Mesnard, J. Dröge and M. R. Wiesner, Environ. Sci. Technol., 2011, 45, 2562 CrossRef CAS PubMed.
- F. Piccinno, F. Gottschalk, S. Seeger and B. Nowack, J. Nanopart. Res., 2012, 14, 1109 CrossRef.
- The Global Market for Metal Oxide Nanoparticles to 2020, Future Markets, Inc. March 2013; http://www.reportsnreports.com/reports/236441-the-global-market-for-metal-oxide-nanoparticles-to-2020.html (accessed 29/07/2014) Search PubMed.
- Roadmap Report on Nanoparticles, NanoRoadMap. November 2005; http://nanoparticles.org/pdf/PerezBaxEscolano.pdf (accessed 29/07/2014) Search PubMed.
- European Commission, Memo/12/732. October 2012; http://europa.eu/rapid/press-release_MEMO-12-732_en.htm (accessed 29/07/2014).
- Nanosilver: Safety, Health and the Environmental Effects and Role of Antimicrobial Resistance, Silver Nanotechnology Working Group, The Silver Research Consortium, LLC. May 2012; https://www.silverinstitute.org/site/wp-content/uploads/2013/05/SNWGSubmissiontoSCENIHR.pdf (accessed 29/07/2014) Search PubMed.
- N. Seltenrich, Environ. Health Perspect., 2013, 121, A220 CrossRef PubMed.
- Global Market for Nano Silver, Research and Markets. May 2012; http://www.researchandmarkets.com/reports/2124534/global_market_for_nano_silver.pdf (accessed 29/07/2014) Search PubMed.
- T. Benn, B. Cavanagh, K. Hristovski, J. D. Posner and P. Westerhoff, J. Environ. Qual., 2010, 39, 1875 CrossRef CAS PubMed.
- F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145 RSC . Review.
- B. Nowack and T. D. Bucheli, Environ. Pollut., 2007, 150, 5 CrossRef CAS PubMed . Review.
- G. V. Lowry, E. M. Hotze, E. S. Bernhardt, D. D. Dionysiou, J. A. Pedersen, M. S. Wiesner and B. Xing, J. Environ. Qual., 2010, 39, 1867 CrossRef CAS PubMed.
- D. Lin, X. Tian, F. Wu and B. Xing, J. Environ. Qual., 2010, 39, 1896 CrossRef PubMed.
- E. S. Bernhardt, B. P. Colman, M. F. Hochella, B. J. Cardinale, R. M. Nisbet, C. J. Richardson and L. Yin, J. Environ. Qual., 2010, 39, 1954 CrossRef CAS PubMed.
- A. B. A. Boxall, K. Tiede and Q. Chaudry, Nanomedicine, 2007, 2, 919 CrossRef CAS PubMed.
- V. L. Colvin, Nat. Biotechnol., 2003, 21, 1166 CrossRef CAS PubMed.
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Environ. Sci. Technol., 2006, 40, 4336 CrossRef CAS PubMed.
- J. Evans, Chem. World, 2006, 3, 58 CAS.
- N. Musee, M. Thwala and N. Nota, J. Environ. Monit., 2011, 13, 1164 RSC . Review.
- Z. Sheng and Y. Liu, Water Res., 2011, 45, 6039 CrossRef CAS PubMed.
- A. García, L. Delgado, J. A. Torà, E. Casals, E. González, V. Puntes, X. Font, J. Carrera and A. Sánchez, J. Hazard. Mater., 2012, 199–200, 64 CrossRef PubMed.
- C. L. Arnaout, PhD Thesis, Duke University, 2012; http://dukespace.lib.duke.edu/dspace/bitstream/handle/10161/5852/Arnaout_duke_0066D_11593.pdf (accessed 29/07/2014) Search PubMed.
- J. R. Giska, MSc Thesis, Oregon State University, 2013, http://ir.library.oregonstate.edu/xmlui/bitstream/handle/1957/39835/GiskaJonathanR2013.pdf (accessed 29/07/2014) Search PubMed.
- A. K. Suresh, D. A. Pelletier and M. J. Doktycz, Nanoscale, 2013, 5, 463 RSC.
- R. Ma, C. Levard, S. M. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752 CrossRef CAS PubMed.
- J. Liu, Z. Wang, F. D. Liu, A. B. Kane and R. H. Hurt, ACS Nano, 2012, 6, 9887 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169 CrossRef CAS PubMed.
- L. R. Pokhrel, B. Dubey and P. R. Scheuerman, Environ. Sci.: Nano, 2014, 1, 45 RSC.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Environ. Sci. Technol., 2011, 45, 3895 CrossRef CAS PubMed.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866 CrossRef CAS PubMed.
- R. Ma, C. Levard, J. D. Judy, J. M. Unrine, M. Durenkamp, B. Martin, B. Jefferson and G. V. Lowry, Environ. Sci. Technol., 2014, 48, 104 CrossRef CAS PubMed.
- R. Ma, J. Stegemeier, C. Levard, J. G. Dale, C. W. Noack, T. Yang, G. E. Brown and G. V. Lowry, Environ. Sci.: Nano, 2014, 1, 347 RSC.
- M. R. Chang, D. J. Lee and J. Y. Lai, J. Environ. Manage., 2007, 85, 1009 CrossRef CAS PubMed.
- H. P. Jarvie, H. Al-Obaidi, S. M. King, M. J. Bowes, M. J. Lawrence, A. F. Drake, M. A. Green and P. J. Dobson, Environ. Sci. Technol., 2009, 43, 8622 CrossRef CAS PubMed ; and accompanying ESI†.
- L. K. Limbach, R. Bereiter, E. Mueller, R. Krebs, R. Gaelli and W. J. Stark, Environ. Sci. Technol., 2008, 42, 5828 CrossRef CAS PubMed.
- M. A. Kiser, P. Westerhoff, T. Benn, Y. Wang, J. Pérez-Rivera and K. Hristovski, Environ. Sci. Technol., 2009, 43, 6757 CrossRef CAS PubMed.
- P. Westerhoff, G. Song, K. Hristovski and M. A. Kiser, J. Environ. Monit., 2011, 13, 1195 RSC.
- Y. Wang, P. Westerhoff and K. D. Hristovski, J. Hazard. Mater., 2012, 201–202, 16 CrossRef CAS PubMed.
- E. Lombi, E. Donner, E. Tavakkoli, T. W. Turney, R. Naidu, B. W. Miller and K. G. Scheckel, Environ. Sci. Technol., 2012, 46, 9089 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, B. Sinnet, S. Zuleeg, H. Hagendorfer, M. Burkhardt and H. Siegrist, Environ. Sci. Technol., 2011, 45, 3902 CrossRef CAS PubMed.
- C. A. Impellitteri, S. Harmon, R. G. Silva, B. W. Miller, K. G. Scheckel, T. P. Luxton, D. Schupp and S. Panguluri, Water Res., 2013, 47, 3878 CrossRef CAS PubMed.
- C. L. Doolette, M. J. McLaughlin, J. K. Kirby, D. J. Batstone, H. H. Harris, H. Ge and G. Cornelis, Chem. Cent. J., 2013, 7, 46 CrossRef PubMed.
- L. Hou, K. Li, Y. Ding, Y. Li, J. Chen, X. Wu and X. Li, Chemosphere, 2012, 87, 248 CrossRef CAS PubMed.
- M. Tejamaya, I. Römer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011 CrossRef CAS PubMed.
- A. C. Johnson, M. D. Jurgens, A. J. Lawlor, I. Cisowska and R. J. Williams, Chemosphere, 2014, 112, 49 CrossRef CAS PubMed.
- T. Maruejouls, P. Lessard, B. Wipliez, G. Pelletier and P. A. Vanrolleghem, Water Sci. Technol., 2011, 64(9), 1898 CrossRef CAS PubMed.
- R. Y. G. Andoh and R. P. M. Smisson, Water Sci. Technol., 1996, 33(9), 127 CrossRef.
- J. Penfold, E. Staples, I. Tucker and R. K. Thomas, Langmuir, 2002, 18, 5755 CrossRef CAS.
- J. Penfold, E. J. Staples, I. Tucker and L. J. Thompson, Langmuir, 1997, 13, 6638 CrossRef CAS.
- Fundamentals of Interface and Colloid Science, Volume IV: Particulate Colloids, ed. J. Lyklema, Academic Press, 2005 Search PubMed.
- A. R. Whitley, C. Levard, E. Oostveen, P. M. Bertsch, C. J. Matocha, F. V. D. Kammer and J. M. Unrine, Environ. Pollut., 2013, 182, 141 CrossRef CAS PubMed.
- H. Dong and I. M. C. Lo, Water Res., 2013, 47, 2489 CrossRef CAS PubMed.
- http://soft-matter.seas.harvard.edu/index.php/Origins_of_surface_charge .
- A. Carrè and V. Lacarrière, Contact Angle, Wettability and Adhesion, ed. K. L. Mittal, 2006, vol. 4, pp. 1–14, VSP/Brill Search PubMed.
- X.-H. Zhou, B.-C. Huang, T. Zhou, Y.-C. Liu and H.-C. Shi, Chemosphere, 2015, 119, 568 CrossRef CAS PubMed.
- J. E. Song, T. Phenrat, S. Marinakos, Y. Xiao, J. Liu, M. R. Wiesner, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2011, 45, 5988 CrossRef CAS PubMed.
- R. Aveyard, B. P. Binks, P. D. I. Fletcher and J. R. McNab, Langmuir, 1995, 11, 2515 CrossRef CAS.
- http://www.isis.stfc.ac.uk/instruments/loq/ .
- H. P. Jarvie and S. M. King, Environ. Sci. Technol., 2007, 41, 2868 CrossRef CAS PubMed ; and accompanying ESI†.
- P. Carter, J. Worrell, G. Daigger, E. Allen and G. Land, Proc. Water Environ. Fed., WEFTEC 2003 Sessions 21 – 30, 2003, vol. 21, p. 312 Search PubMed.
- T. Pavón-Silva, V. Pacheco-Salazar, J. C. Sánchez-Meza, G. Roa-Morales and A. Colín-Cruz, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 108 CrossRef PubMed.
- I. Bekri-Abbes, S. Bayoudh and M. Baklouti, Desalination, 2007, 204, 198 CrossRef CAS.
- A. S. Landim, G. R. Filho and R. M. N. de Assunção, Polym. Bull., 2007, 58, 457 CrossRef CAS.
- E. Michiels, F. Michiels, D. A. Topchiev and G. G. Kardash, U.S. Pat. Number 5423990, 1995 Search PubMed.
- R. K. Heenan, S. M. King, R. Osborn and H. B. Stanley, Rutherford Appleton Laboratory Report, 1989, RAL-89-128 Search PubMed; S. M. King and R. K. Heenan, Rutherford Appleton Laboratory Report, 1995, RAL-95-005 Search PubMed.
- G. D. Wignall and F. S. Bates, J. Appl. Crystallogr., 1987, 20, 28 CrossRef CAS.
- http://www.sasview.org/ .
- S. M. King and H. P. Jarvie, Environ. Sci. Technol., 2012, 46, 6959 CrossRef CAS PubMed ; and accompanying ESI†.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781 CrossRef.
- J. Cai, N. Lu and C. M. Sorensen, J. Colloid Interface Sci., 1995, 171, 470 CrossRef CAS.
- H. Lamb, Hydrodynamics, Cambridge University Press, 6th edn, 1932, Reprinted 2006 Search PubMed.
- A. Doostmohammadi, S. Dabiri and A. M. Ardekani, J. Fluid Mech., 2014, 750, 5 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation data for the silver nanopowder; TEMs of the silver nanopowder; estimation of the radius-of-gyration of the PVP stabiliser; schematic representation of a silver nanoparticle; neutron scattering length densities and contrasts; SANS characterisation data for the silver nanopowder dispersions; SANS model comparisons; characterisation data for the wastewater; results of the bench-top settling experiments; effect of FeCl3 on the SANS from wastewater; impact of particle volume fraction on colloidal interactions; resilience to dissolution of the AgNPs; rate of change of the NP settling velocities; temperature history during the SANS experiments. See DOI: 10.1039/c4en00151f |
| ‡ Though logarithmic trendlines actually return slightly better statistical R2-factors than the power law trendlines shown when describing the concentration-time data, the long time behaviour of these functions is unphysical for some datasets (resulting in negative concentrations). Either type of trendline is however arbitrary. |
| § It would take AgNPs settling at this velocity 50 days to transit the 8 mm high window illuminated by the neutron beam. |
| This journal is © The Royal Society of Chemistry 2015 |