
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenyl-triazine oligomers for light-driven hydrogen evolution†
K.
Schwinghammer‡
abc,
S.
Hug‡
abc,
M. B.
Mesch
d,
J.
Senker
d and
B. V.
Lotsch
*abc
aMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany. E-mail: b.lotsch@fkf.mpg.de
bDepartment of Chemistry, University of Munich, LMU, Butenandtstr. 5-13, 81377 Munich, Germany
cNanosystems Initiative Munich and Center of Nanoscience, Schellingstr. 4, 80799 Munich, Germany
dInorganic Chemistry III, University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
First published on 15th September 2015
Abstract
The design of stable, yet highly tunable organic photocatalysts which orchestrate multi-step electron transfer reactions is at the heart of the newly emerging field of polymer photocatalysis. Covalent triazine frameworks such as the archetypal CTF-1 have been theorized to constitute a new class of photocatalytically active polymers for light-driven water splitting. Here, we revisit the ionothermal synthesis of CTF-1 by trimerization of 1,4-dicyanobenzene catalyzed by the Lewis acid zinc chloride and demonstrate that the microporous black polymer CTF-1 is essentially inactive for hydrogen evolution. Instead, highly photoactive phenyl-triazine oligomers (PTOs) with higher crystallinity as compared to CTF-1 are obtained by lowering the reaction temperature to 300 °C and prolonging the reaction time to >150 hours. The low reaction temperature of the PTOs largely prevents incipient carbonization and thus results in a carbon-to-nitrogen weight ratio close to the theoretical value of 3.43. The oligomers were characterized by MALDI-TOF and quantitative solid-state NMR spectroscopy, revealing variations in size, connectivity and thus nitrile-to-triazine ratios depending on the initial precursor dilution. The most active PTO samples efficiently and stably reduce water to hydrogen with an average rate of 1076 (±278) μmol h−1 g−1 under simulated sunlight illumination, which is competitive with the best carbon nitride-based and purely organic photocatalysts. The photocatalytic activity of the PTOs is found to sensitively depend on the polymerization degree, thus suggesting a prominent role of the unreacted nitrile moieties in the photocatalytic process. Notably, PTOs even show moderate hydrogen production without the addition of any co-catalyst.
Broader contextThe increasing demand of renewable energy resources, such as hydrogen generated by photocatalytic water reduction, necessitates the exploration of efficient, stable, non-toxic and abundant photocatalysts. While organic photocatalysts fulfil these requirements, rational catalyst design remains challenging both experimentally and computationally due to the scarcity of structure–property–activity insights, particularly in disordered polymeric systems. This case in point is illustrated by the archetype covalent triazine framework CTF-1, which was computationally predicted to satisfy the (thermodynamic) criteria for exhibiting activity for photocatalytic hydrogen evolution, but shown to be nearly inactive here. We demonstrate further that triazine-based oligomers populated with terminal nitrile groups, rather than the extended framework, are highly active photocatalysts for the hydrogen evolution reaction. The nitrile groups likely act as solubilizing agents and conduits for charge transfer between the oligomer, co-catalyst and reactants, which puts the role of functional groups embedded in the carbon nitride backbone in the spotlight. This work thus illustrates the potential of well-defined oligomeric systems as a new generation of metal-free photocatalysts and provides a guideline for the rational design of novel organic photocatalysts by molecular engineering. |
Introduction
The direct conversion of solar energy to chemical energy, exemplified by light-induced water splitting into hydrogen and oxygen, enables the sustainable generation of storable chemical fuels and is hence a green energy cycle.1 However, the development of a cost-effective process for the generation of hydrogen from water has yet to be developed.1 Since the discovery of water splitting on an n-type semiconducting TiO2 electrode by Fujishima and Honda in 1972,2 research largely focused on inorganic semiconductors that often entail costly and environmentally critical fabrication steps.3 The development of carbon-based photocatalysts has become an active research field, which has produced the first promising examples of polymers,4 graphite oxide,5 carbon nitrides6–8 and COFs9 as predominantly carbon-based photocatalysts. In particular, the incorporation of triazine units into polymeric systems has been shown to correlate with efficient hydrogen evolution as the triazine nitrogen, with its free electron pair and electron poor character, may act as the active site.6,7,9–12 An example of such a triazine system is the covalent triazine framework CTF-1 (see Scheme 1) which was first synthesized in 2008 by Kuhn et al.13 CTF-1 is built from alternating triazine and phenyl building blocks forming a 12-membered hexagonal ring system, resulting in layers which are stacked in a close-to-eclipsed AA-type fashion.13 The black colored and highly porous product is obtained from 1,4-dicyanobenzene by a trimerization reaction under inert ionothermal conditions at 400 °C for 40 hours.13 However, incipient carbonization occurs due to the elevated reaction temperature, as evidenced by the larger carbon content and the dark color of the product.13 In 2012, Ren et al.14 achieved the synthesis of yellow colored and thus largely carbon-free, but non-porous CTF-1 through a super acid-catalyzed (microwave-assisted) synthesis. | ||
| Scheme 1 Reaction mechanism of CTF-1 starting from 1,4-dicyanobenzene, which leads to the formation of oligomers via trimerization and, ultimately, to an extended 2D CTF (right). The idealized crystal structure of fully condensed, crystalline CTF-1 is shown in an AA-type eclipsed configuration (viewed along the c-direction). | ||
Such colored compounds may be capable of light-induced hydrogen evolution drawing on recent experimental and theoretical results delineating the structural and photophysical properties of CTF-1-type materials.14,15 Moreover, based on first principles calculations, Zhao and co-workers16 predicted that certain CTFs including CTF-1 should be promising visible-light photocatalysts according to the suitable band gap of CTF-1 (2.42 eV) and the position of its band potentials which are dependent on the nitrogen content of the CTFs, the number of ring systems, and the extent of π–π interactions.
Here, we revisit the synthesis of CTF-1 and demonstrate that conditions circumventing the formation of amorphous carbon invariably yield low molecular weight triazine-based compounds, henceforth referred to as phenyl-triazine oligomers (PTO), with intriguing optical and electronic properties. These oligomers are studied with respect to their photocatalytic activity for hydrogen evolution and compared to the archetypal CTF-1 and the related molecular model system 2,4,6-tris(p-cyanophenyl)-1,3,5-triazine, henceforth referred to as “the trimer”.
In contrast to black CTF-1 which shows negligible catalytic activity, the most active PTO samples exhibit an average, but non-optimized hydrogen evolution rate of 1076 (±278) μmol h−1 g−1. Thus, PTOs are competitive with the commonly used Melon (g-C3N4) and other purely organic photocatalysts6–11,17–19 with regards to hydrogen evolution under simulated sunlight, and long-term stability. Notably, PTOs are even active without the addition of any co-catalyst.19 Their high activity and potential to act in a Pt-free environment (121 μmol h−1 g−1) renders triazine-based photocatalysts and PTOs in particular promising candidates for truly abundant photocatalysts.
Results and discussion
CTF-1 synthesized according to literature protocols was tested for hydrogen evolution under simulated sunlight (for experimental details see below and Table S8, ESI†) but was found to show very low activity. We rationalize the low activity of CTF-1 with the large carbon content in the sample, as the synthesis of CTF-1 seems to be a delicate balance between incomplete polymerization resulting mostly in oligomers, and the competitive formation and degradation of the framework yielding amorphous carbon at elevated temperatures. At the originally applied synthesis temperature of 400 °C, a large amount of carbon is formed, which may well hinder light penetration to the photocatalyst.13In addition, the thermodynamic driving force for electron transfer in the extended system may be too low to support efficient hydrogen evolution.
Synthesis
We therefore attempted to synthesize non-carbonized CTF-1. While the synthesis of yellow colored CTF-1 according to Ren et al.14 requires water-free conditions, we modified the original ionothermal approach,13 thereby obtaining PTOs rather than the extended CTF-1 polymer. The syntheses of the PTOs were carried out in a fashion analogous to the original ionothermal synthesis using zinc chloride melts, as established by Antonietti and co-workers.13 As already stated in that work, syntheses at temperatures below 350 °C yielded mainly soluble products after two days of reaction time. Due to the slow reaction kinetics at 350 °C we prolonged the reaction times to increase the conversion, in accordance with van't Hoff's law.20 Indeed, reaction times of 96 h for syntheses at 350 °C and of 168 h at 300 °C led to oligomeric products which were insoluble in organic solvents (THF, acetone, ethanol, chloroform and dichloromethane), water and hot 1 M HCl. During the organic washing procedure (and later on by Soxhlet extraction) with THF, smaller oligomers and unreacted starting material were washed off (organic phase of PTO, see Fig. S3 and S4, ESI†). The detailed synthesis conditions are shown in Table 1. The products are identified by adding to the prefix PTO first the synthesis temperature and then the equivalents of zinc chloride with respect to 1,4-dicyanobenzene used in the synthesis.| Sample | Eq. ZnCl2 | Synthesis temperature/°C | Synthesis timea/h |
|---|---|---|---|
| a Heating at a rate of 60 °C h−1 and cooling at 10 °C h−1. b Synthesized according to Kuhn et al.13 | |||
| PTO-300-1 | 1 | 300 | 168 |
| PTO-300-2.5 | 2.5 | 300 | 168 |
| PTO-300-5 | 5 | 300 | 168 |
| PTO-300-10 | 10 | 300 | 168 |
| PTO-300-15 | 15 | 300 | 168 |
| PTO-350-1 | 1 | 350 | 96 |
| PTO-350-10 | 10 | 350 | 96 |
| CTF-1b | 1 | 400 | 48 |
Characterization
First, we analyzed the crystallinity of the as-obtained materials by powder XRD measurements and compared the results with CTF-1 (Fig. 1a and c). The diffraction patterns of PTO-350-1 and the as-prepared CTF-1 sample are largely reminiscent of CTF-1,13 albeit with less intense reflections. At the lower reaction temperature of 300 °C and prolonged reaction time, materials with higher crystallinity are obtained which differ from the reference and cannot simply be assigned to the precursors. While PTO-300-1 still shows similarities to the framework due to the presence of the prominent 100 reflection, the samples synthesized with a larger amount of ZnCl2 (e.g. PTO-300-15) display diffraction patterns completely different from CTF-1, yet faintly resembling the trimer. We believe that the powder diffraction patterns of the PTO samples differ from the one of CTF-1 due to the formation of oligomers. These oligomers exhibit a decreased layer distance compared to CTF-1 (Table S1, ESI†). Furthermore, it seems that the oligomers formed at a one-to-one precursor to ZnCl2 ratio (PTO-300-1) differ from the ones synthesized in an excess of zinc chloride (>1 equivalent).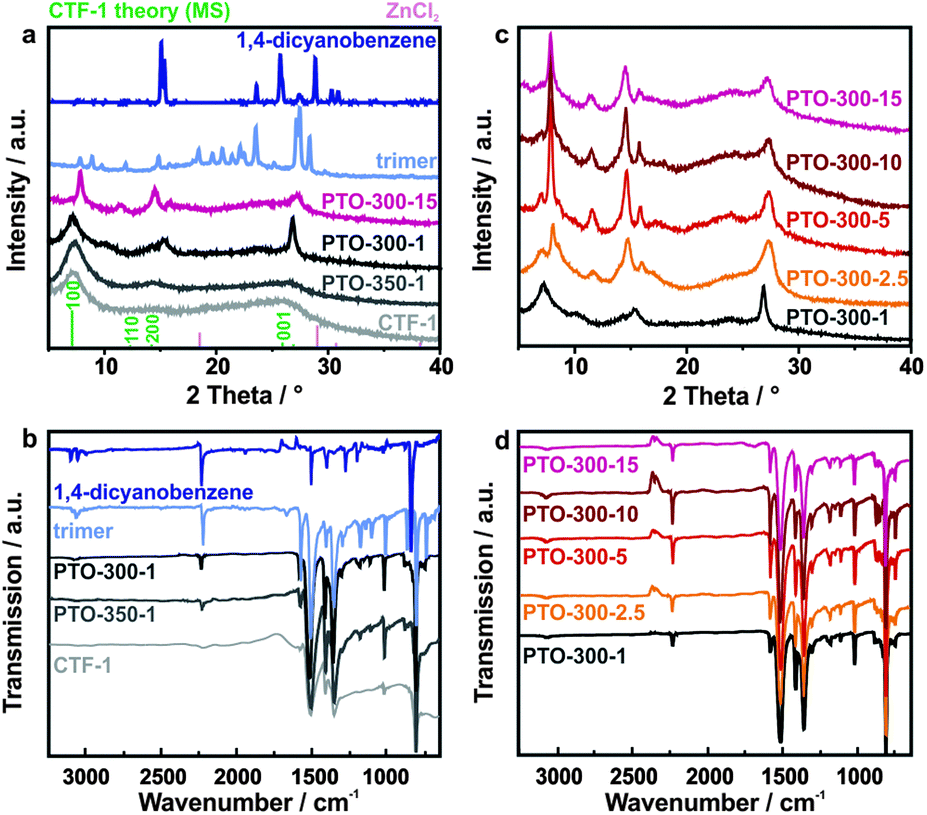 | ||
| Fig. 1 (a) PXRD patterns and (b) IR spectra of the PTOs: PTO-350-1, PTO-300-1 and PTO-300-15 in comparison to the as-synthesized CTF-1, the trimer and 1,4-dicyanobenzene. The diffraction patterns of ZnCl2 and the simulated pattern of CTF-113 are added for clarity. (c) PXRD patterns and (d) IR spectra of the PTOs synthesized at 300 °C with different amounts of ZnCl2. | ||
We used IR spectroscopy as a tool to survey the progress of the trimerization reaction based on the characteristic IR signals of the nitrile groups of the starting material, which are expected to form triazine rings. The IR spectra are depicted in Fig. 1b and d. The materials synthesized between 300–350 °C show the same prominent bands as CTF-1 and the trimer. The characteristic bands for the triazine unit are found at 1506 cm−1 (C–N stretching mode)21,22 and 1355 cm−1 (in-plane ring stretching vibrations),23,24 confirming the successful formation of triazine rings under these synthesis conditions. The lower temperature clearly leads to incomplete polymerization as indicated by the additional signal at 2227 cm−1 representing unreacted nitrile groups in the sample.
To further confirm the formation of CTF-1-like materials, we conducted elemental analysis (Table S2, ESI†). The elemental analysis (EA) data of the as-synthesized PTOs synthesized at 300 °C reveal values very close to the theoretical ones for CTF-1 and the ones reported by Ren et al.14 for yellow colored CTF-1. The results are in sharp contrast to the materials synthesized above 300 °C and the ones found by Antonietti and co-workers13 where a higher carbon content (and C/N ratio) is observed due to partial carbonization of the products.13 An incipient carbonization can also be detected for the materials synthesized in an excess of zinc chloride (PTO-300-15). No statement on the extent of trimerization of the PTOs and thus the amount of unreacted nitrile groups can be made based on EA because the precursor of the synthesis (1,4-dicyanobenzene) has the same elemental composition and, hence, C/N ratio as one formula unit of CTF-1 or any intermediate oligomer.
To gain more insights into the local chemical environment in the different materials, XPS measurements were conducted (Fig. S5 and S6, ESI†). The nitrogen environments of the low temperature products (PTO-300-1 and 15) are in agreement with the as-synthesized CTF-1 sample showing a dominant N 1s peak at around 398.9 eV. According to the literature,25,26 this as well as the weak signal at 400.3 eV observed for CTF-1 can be assigned to nitrogen atoms within the triazine units (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) as well as terminal nitrile groups. More likely, however, the signal at 400.3 eV arises from pyrrolic nitrogen containing decomposition products since this signal is more prominent in the CTF-1 sample. The C 1s XPS measurements of the three samples reveal two peaks. The first signal at 284.6 eV is attributed to the carbon atoms of the phenyl rings as well as adventitious carbon (C–C) which is used for calibration.27 The second one appearing at around 286.8 eV for CTF-1 and PTO-300-1 and at 286.3 eV for PTO-300-15 is assigned to the carbon atoms in the triazine (NC–N) and nitrile moieties (–C
C) as well as terminal nitrile groups. More likely, however, the signal at 400.3 eV arises from pyrrolic nitrogen containing decomposition products since this signal is more prominent in the CTF-1 sample. The C 1s XPS measurements of the three samples reveal two peaks. The first signal at 284.6 eV is attributed to the carbon atoms of the phenyl rings as well as adventitious carbon (C–C) which is used for calibration.27 The second one appearing at around 286.8 eV for CTF-1 and PTO-300-1 and at 286.3 eV for PTO-300-15 is assigned to the carbon atoms in the triazine (NC–N) and nitrile moieties (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).28 The C–C carbon signal of CTF-1 is more dominant in comparison to the triazine/nitrile peak due to a higher amorphous carbon content which was already verified by elemental analysis. For the different samples, no obvious shifts of the binding energy of C 1s and N 1s core electrons are observable, suggesting that the chemical states of both carbon and nitrogen atoms in the PTOs are the same as in the more highly condensed CTF-1 sample.
N).28 The C–C carbon signal of CTF-1 is more dominant in comparison to the triazine/nitrile peak due to a higher amorphous carbon content which was already verified by elemental analysis. For the different samples, no obvious shifts of the binding energy of C 1s and N 1s core electrons are observable, suggesting that the chemical states of both carbon and nitrogen atoms in the PTOs are the same as in the more highly condensed CTF-1 sample.
The results obtained by IR and XPS were further supported by 13C and 15N solid-state (ss) NMR spectroscopy. The 13C CP MAS ssNMR spectra of PTO-300-1, PTO-300-10 as well as CTF-1 (Fig. 2, left) show strong signals at 169, 138, 130 and 115 ppm. The signal at 169 ppm is assigned to triazine ring carbon atoms,29 while the signals at 138 and 130 ppm correspond to the phenyl ring. The signal at 115 ppm is characteristic for nitrile groups as well as the carbon of the phenyl ring to which the nitrile groups are bound. Although it is significantly weaker in CTF-1 it indicates residual nitrile groups in all materials. The 15N CP MAS ssNMR spectra of all samples (Fig. 2, right) show only one signal at around −125 ppm. From the chemical shift alone it is not possible to distinguish between the triazine unit and terminal nitrile groups, as their signals appear in the same region.30,31 Note that in PTO-300-10 the carbon peaks at 130 and 115 ppm as well as the nitrogen peak show a fine structure, indicating better crystallinity. This is also supported by the XRD measurements shown above. To summarize, the results from IR and ssNMR spectroscopy strongly suggest the successful formation of triazine rings already at a reaction temperature of 300 °C, while the phenyl rings are maintained. In addition, unreacted nitrile groups are found in all products, with higher relative quantities of free nitriles for the PTO samples. Whereas at 400 °C the polymerization process is almost complete, incomplete polymerization and the formation of oligomers are detected at lower temperatures.
 | ||
| Fig. 2 13C CP MAS ssNMR spectra of (a) CTF-1, (b) PTO-300-1 and (c) PTO-300-10. 15N CP MAS ssNMR spectra of (d) CTF-1, (e) PTO-300-1 and (f) PTO-300-10. Asterisks mark rotational spinning side bands. A contact time of 5 ms was used for all measurements and the spinning speed was set to 10 kHz (all PTO measurements), 20 kHz (13C of CTF-1) and 5 kHz (15N of CTF-1), respectively. | ||
Porosity data were calculated from argon sorption measurements and are summarized in Table S2 (ESI†). According to Kuhn et al.13 CTF-1 displays a relatively high surface area (SA) of 791 m2 g−1 due to microporosity, which is confirmed by sorption measurements of our CTF-1 sample (610 m2 g−1). In contrast, the PTOs synthesized below 400 °C show significantly lower values in the range of 6–116 m2 g−1, suggesting that either no distinct continuous 1D pore system is formed or pore blocking occurs. Similarly low surface areas (2–4 m2 g−1) were reported by Ren et al.14 for their yellow colored CTF-1s synthesized by a super acid-catalyzed (microwave-assisted) procedure. The surface areas of PTO-300 samples are slightly increased compared to the ones synthesized at 350 °C, whereas a higher salt concentration (>1 eq.) leads to lower surface areas irrespective of how much excess of salt melt was used.
Estimation of the degree of polymerization
While the chemical environments of the PTOs are similar to CTF-1 based on IR, ssNMR and XPS, appreciable differences between the samples are discernible, even in PXRD and EA. We assumed that these differences result from different degrees of polymerization and, hence, nitrile content of the PTOs synthesized at 300 °C compared to CTF-1 and PTO-350. Therefore, MALDI-TOF measurements were performed with PTO-300-1 and PTO-300-15 to further analyze the molecular sizes of the synthesized PTOs (Fig. S7 and S8, ESI†). No mass signal was detected for the starting material 1,4-dicyanobenzene (m/z = 128) and the polymer CTF-1 itself. For the PTOs, multiples of masses of 1,4-dicyanobenzene (n × 128 for n = 0, 1, 2, 3,…) are expected since the polymerization up to CTF-1 proceeds without the formation of side products. Taking into account the rules of the trimerization reaction—three nitrile groups forming one triazine unit—fragments of 1,4-dicyanobenzene triplets (trimers) with a mass of 384 m/z should be observable. Furthermore, the formation of chains or dendrimers (of the trimer units) leads to (3 + 2n) multiples of the mass of 1,4-dicyanobenzene (Fig. 3b, Tables S4 and S5, ESI†). A ring closure would lead to (12 + 7n) multiples of the 1,4-dicyanobenzene mass (Fig. 3b and Table S6, ESI†). In contrast to CTF-1, we obtained signals in the mass spectra for the PTO samples (Table S3 and Fig. S6 and S7, ESI†) which, in principle, already suggests that the molecular sizes of the PTOs are smaller than CTF-1 and more susceptible to ablation by ionization. The recorded mass spectra of PTO-300-1 and PTO-300-15 differ from each other in that they show small fragments (m/z ≤ 12 × 128) for the sample synthesized with a high zinc chloride content (PTO-300-15) and additionally larger ones (m/z > 12 × 128) for the PTO synthesized with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 precursor ratio (PTO-300-1). For both samples, multiples of odd numbers up to 11 of the masses of 1,4-dicyanobenzene were found which can be assigned to chain fragments or small dendrimers (Tables S3–S5, ESI†). Additionally, a signal at 12 times the mass of 1,4-dicyanobenzene was recorded which likely represents a 12-membered ring (Fig. 3b, Tables S3 and S6, ESI†). In the case of PTO-300-1, the mass spectra also show larger fragments of 16–23 multiples of 1,4-dicyanobenzene due to longer chains, larger dendrimers and double ring systems (Fig. 3a, Tables S3–S6, ESI†). It seems that the amount of zinc chloride is crucial for the obtained molecular size of the oligomers as well as the type of oligomeric fragments formed. A high dilution of the starting material leads to smaller, chain-like oligomers (e.g. PTO-300-15) with very low porosities and more favorable stacking interactions (see below and Table S2, ESI†). In contrast, higher concentration of 1,4-dicyanobenzene (PTO-300-1) favors the formation of larger oligomers featuring higher porosities due to the formation of ring-systems (see Table S2, ESI†). The polymerization degree of the PTO samples was also roughly estimated by means of IR spectroscopy. To this end, we relate the IR signal intensities and integrals of the nitrile (2227 cm−1) and triazine bands (1355 cm−1) and compare them with those of the trimer and CTF-1 (Fig. 1c and d and Tables S4–S7, ESI†). In the case of CTF-1, almost no nitrile signal is visible in the IR spectrum, which indicates a near-zero nitrile-to-triazine ratio due to the largely complete conversion to the trimerized product. On the contrary, the highest nitrile-to-triazine ratio is obtained for the trimer itself (see Tables S4 and S5, ESI†). The PTO samples invariably yielded lower nitrile-to-triazine ratios compared to the trimer. This is consistent with the assumption that the polymerization proceeded further than the trimer in the case of the PTO samples. More precisely, a relatively high nitrile-to-triazine ratio was calculated for PTOs-300 synthesized with large zinc chloride contents (e.g. PTO-300-15), which can only be explained by the formation of short chains or small dendrimers (Tables S4 and S5, ESI†). PTO-300-1 samples reveal lower ratios caused by ring closure or larger dendrimers (Tables S4 and S6, ESI†). To get a more quantitative picture of the degree of polymerization, 13C one pulse ssNMR spectra of PTO-300-1 and PTO-300-10 were recorded (Fig. S11, ESI†). As already described in a previous section the signals for the triazine units and the nitrile groups are well resolved (169 ppm for triazine as well as 120–110 ppm for nitrile groups and the benzene carbons to which the nitrile groups are bound). Using the integrals of the signals, the nitrile-to-triazine ratio can be calculated. Due to the significantly longer spin–lattice relaxation of the triazine carbons compared to the other carbon species the chosen measurement conditions lead to a slight underestimation of around 5% for the intensity of the triazine signal (for further details see the ESI†). The calculated ratio for the PTO-300-10 sample is 0.46, suggesting a mixture of trimers (0.50) and short chains or small dendrimers (<0.50). For PTO-300-1 it is much lower (0.21) pointing towards a higher degree of polymerization. Therefore, the formation of dendrimers or ring structures is favored under these conditions. This is well in line with the IR and MALDI-TOF data.
:1 precursor ratio (PTO-300-1). For both samples, multiples of odd numbers up to 11 of the masses of 1,4-dicyanobenzene were found which can be assigned to chain fragments or small dendrimers (Tables S3–S5, ESI†). Additionally, a signal at 12 times the mass of 1,4-dicyanobenzene was recorded which likely represents a 12-membered ring (Fig. 3b, Tables S3 and S6, ESI†). In the case of PTO-300-1, the mass spectra also show larger fragments of 16–23 multiples of 1,4-dicyanobenzene due to longer chains, larger dendrimers and double ring systems (Fig. 3a, Tables S3–S6, ESI†). It seems that the amount of zinc chloride is crucial for the obtained molecular size of the oligomers as well as the type of oligomeric fragments formed. A high dilution of the starting material leads to smaller, chain-like oligomers (e.g. PTO-300-15) with very low porosities and more favorable stacking interactions (see below and Table S2, ESI†). In contrast, higher concentration of 1,4-dicyanobenzene (PTO-300-1) favors the formation of larger oligomers featuring higher porosities due to the formation of ring-systems (see Table S2, ESI†). The polymerization degree of the PTO samples was also roughly estimated by means of IR spectroscopy. To this end, we relate the IR signal intensities and integrals of the nitrile (2227 cm−1) and triazine bands (1355 cm−1) and compare them with those of the trimer and CTF-1 (Fig. 1c and d and Tables S4–S7, ESI†). In the case of CTF-1, almost no nitrile signal is visible in the IR spectrum, which indicates a near-zero nitrile-to-triazine ratio due to the largely complete conversion to the trimerized product. On the contrary, the highest nitrile-to-triazine ratio is obtained for the trimer itself (see Tables S4 and S5, ESI†). The PTO samples invariably yielded lower nitrile-to-triazine ratios compared to the trimer. This is consistent with the assumption that the polymerization proceeded further than the trimer in the case of the PTO samples. More precisely, a relatively high nitrile-to-triazine ratio was calculated for PTOs-300 synthesized with large zinc chloride contents (e.g. PTO-300-15), which can only be explained by the formation of short chains or small dendrimers (Tables S4 and S5, ESI†). PTO-300-1 samples reveal lower ratios caused by ring closure or larger dendrimers (Tables S4 and S6, ESI†). To get a more quantitative picture of the degree of polymerization, 13C one pulse ssNMR spectra of PTO-300-1 and PTO-300-10 were recorded (Fig. S11, ESI†). As already described in a previous section the signals for the triazine units and the nitrile groups are well resolved (169 ppm for triazine as well as 120–110 ppm for nitrile groups and the benzene carbons to which the nitrile groups are bound). Using the integrals of the signals, the nitrile-to-triazine ratio can be calculated. Due to the significantly longer spin–lattice relaxation of the triazine carbons compared to the other carbon species the chosen measurement conditions lead to a slight underestimation of around 5% for the intensity of the triazine signal (for further details see the ESI†). The calculated ratio for the PTO-300-10 sample is 0.46, suggesting a mixture of trimers (0.50) and short chains or small dendrimers (<0.50). For PTO-300-1 it is much lower (0.21) pointing towards a higher degree of polymerization. Therefore, the formation of dendrimers or ring structures is favored under these conditions. This is well in line with the IR and MALDI-TOF data.
 | ||
| Fig. 3 (a) MALDI-TOF spectra of PTO-300-1 compared to PTO-300-15, measured in positive mode and in the mass range of 2000–3000. For both PTO materials signals have been detected in the mass spectra below the mass range of 2000. DB represents the mass of the precursor 1,4-dicyanobenzene (m/z = 128). (b) Examples of possible ring and chain-like oligomers with their calculated masses. (c) UV-Vis spectra and product color (inset) of the PTO samples synthesized at different temperatures compared to CTF-1. (d) UV-Vis spectra of the PTO samples synthesized at 300 °C with different precursor ratios from low (bottom) to high ZnCl2 content (top) and their corresponding Tauc plots (inset). | ||
Optical properties
Interestingly, the materials showed different colors compared to the samples synthesized at 400 °C (black), as depicted in Fig. 3c. While the samples synthesized at 350 °C are still rather dark and their color is independent of the amount of ZnCl2 used, the synthesis at 300 °C leads to products with yellow (PTO-300-1) or green-greyish complexion (PTO-300-15). The colors of the PTOs differ also from those of the starting materials and the trimer (colorless), which again suggests the formation of oligomeric products.UV-Vis spectra were recorded in order to evaluate the absorption characteristics of the materials as a function of their polymerization state (Fig. 3c and d). When comparing the samples synthesized at different temperatures, a strong absorption in the UV region (350 nm) is observed for the PTO samples synthesized at 300 °C, with a sharp absorption edge around 370 nm (3.2–3.3 eV, assuming a direct band gap), as well as a weak and broad absorption in the visible light range. According to Butchosa et al.15 these strong absorptions in the UV are caused by π → π* and n → π* excitations, resulting from absorptions within isolated molecular units and oligomer stacking, respectively. The long sloping wavelength tail extending beyond 700 nm in the samples synthesized at higher salt contents (Fig. 3d) likely occurs due to enhanced absorption caused by incipient carbonization. Likewise, the materials synthesized at elevated temperatures (>300 °C) show broad absorption in the whole visible region with no distinct absorption edge, which is expected judging from their dark sample color (Fig. 3c). Note, however, that theoretical calculations suggested a band gap of 2.42 eV for CTF-1.16 When comparing the UV-Vis spectra of the PTO samples synthesized at 300 °C but with different zinc chloride concentrations (Fig. 3d), a second absorption around 410 nm arises, which increases relative to the UV absorption and slightly red-shifts for the materials when synthesized in an excess of salt melt (>1 eq.). This feature may be associated with a reduced interlayer distance for the PTO-300 samples obtained with increasing salt content (Table S1, ESI†), or with more pronounced torsional angles in the otherwise planar systems as suggested by Butchosa et al.15 However, due to the sloping background caused by incipient carbonization,7 it is not possible to reliably extract the intensity of this absorption or the position of the absorption edge.
Photocatalytic hydrogen evolution
Since triazine-based organic semiconductors have been reported to reduce water to hydrogen under visible light irradiation, we reasoned that the incorporation of triazine units into organic frameworks may be a viable strategy to rationally design efficient organic photocatalysts.12 The high chemical and thermal stabilities of the CTFs, together with the suitable optical properties based on theoretical calculations and our experimental findings, should render CTFs suitable photocatalysts for the water-splitting reaction.16 Platinum-loaded (2.2 wt%) CTF-1 itself (0.02 μmol h−1) and the PTO-350 samples (0.45 μmol h−1) showed only negligible activity in a buffered (pH 7) aqueous triethanolamine (TEoA) solution when illuminated with simulated sunlight. These low activities may be explained by the large carbon content in the samples which results from incipient carbonization, or by a mismatch in energy levels. The excess of carbon absorbs a significant portion of the light which then cannot penetrate deeply into the photocatalyst. The low crystallinity of the materials may also hinder the photocatalytic process in that the light-induced charge carriers quickly recombine at traps rather than being able to diffuse to the surface of the material.In contrast, the samples synthesized below 350 °C (PTO-300) exhibit a stable and high hydrogen evolution rate for over 100 hours under the conditions described above (Fig. 4a and b and Fig. S17, ESI,† right). The most active PTO-300 materials were found to produce 10.8 (±2.8) μmol h−1 hydrogen on average when illuminated with simulated sunlight. The action spectrum of the PTO-300 samples suggest that the highest hydrogen contribution arises from wavelengths with an energy of 400 (±20) nm, revealing average estimated apparent quantum efficiencies of 5.5 (±1.1)% (Fig. 4c). Activities above 450 nm are negligible due to the weaker absorption in this wavelength range. In several cases, not only a constant hydrogen evolution rate was observed but even an increase in activity over time (e.g. Fig. S13, ESI†), likely due to the gradually improved wettability of the oligomers and, hence, dispersion. We have analyzed the recovered photocatalyst after operating for over tens of hours and can exclude hydrolysis or any photodegradation of the catalyst (Fig. S22–S24, ESI†). An additional vibration band at 1771 cm−1 is observed in the IR spectrum for the photocatalysts treated in a phosphate buffer (Fig. S22, ESI,† right) which can be explained by a possible phosphate coordination. No hydrolysis products are observed in the 13C CP MAS ssNMR spectra (Fig. S24, ESI†).
 | ||
| Fig. 4 (a) Stability test of 10 mg Pt-modified PTO-300-15 and PTO-300-1 in 10 vol% buffered (pH 7, 0.5 M, phosphate buffer) TEoA solution under irradiation with simulated sunlight (for PTO-300-15, red) and UV-Vis light (≥250 nm for PTO-300-1). Stable hydrogen evolution for over 100 h (PTO-300-1) shown as two slopes: the black slope represents a fresh PTO-300-1 suspension within the first 20 h and the grey represents the same suspension after 78 h of photocatalysis, demonstrating stable H2 evolution for at least 100 h. (b) Average H2 evolution rate of Pt-modified PTO-300 ≥ 2.5 with standard deviation, compared to PTO-300-1. (c) Action spectrum of Pt-PTO-300-5 (measured with a 40 nm FWHM band pass filter), overlaid on the UV-Vis spectrum of the photocatalyst. (d) Reproducible hydrogen evolution rate of Pt-PTO-300 ≥ 2.5 and of the best performing sample (Pt-PTO-300-2.5) in comparison with literature photocatalysts used as internal standards under identical reactor and illumination conditions (230 mL reactor volume, top-irradiation, equal irradiation surface and intensity 380 mW cm−2), as well as dispersion volume (10 mL of 10 vol% TEoA solution), 10 mg (2 mg for PTI nanosheets) Pt-modified photocatalyst (2.2 wt% Pt) under reported conditions. | ||
A factor significantly influencing the variation of activity in the series of PTOs is the ratio of precursor to ZnCl2 in the syntheses at 300 °C. Higher activities are observed for PTO-300 samples for which an excess of zinc chloride was used in the synthesis (>1 eq.) (Fig. 4b). In line with our previous observations described above, the large zinc chloride content leads to the formation of smaller oligomers, whereas a low zinc chloride environment favors the formation of larger PTO fragments which are photocatalytically less active (Fig. 4b). Similar observations have recently been reported for carbon nitrides,32 where oligomers of Melon showed an increased photocatalytic activity compared to the polymer. Furthermore, the activity of these oligomers depended on their size such that smaller oligomers of Melon showed a higher activity compared to the larger ones.32 We believe that the increased activity of the smaller oligomers compared to their larger relatives or the polymer might be associated with the amount of terminal groups. The terminal groups in the PTO samples are nitrile moieties which appear in larger quantities for the samples synthesized in excess of zinc chloride. A large nitrile content favors hydrogen bonding and thus, leads to a better dispersion of the photocatalyst during the photocatalytic experiment. Furthermore, it is expected that platinum, which is used as a co-catalyst, preferably coordinates to the terminal nitrile groups and thus promotes improved charge transfer.33 Another possibility is that the nitrile groups can be considered as active sites themselves, as the photocatalytic activity scales roughly with the relative amount of nitrile groups in the sample. Note, however, that we do not observe a linear correlation between the observed activity and the excess of zinc chloride used. Nevertheless, the interplay between triazines and nitriles or, in other words, a reasonably large conjugated backbone, is decisive since a certain amount of triazines is needed to exhibit a photocatalytic response. For instance, the trimer (0.08 μmol h−1) showed only minor activity even with UV-Vis (≥250 nm) illumination (Fig. S14, ESI†). Note that we observe quite significant batch-to-batch variations in the samples with ZnCl2-to-precursor ratios of >1 (Table S10 and Fig. S21, ESI†), which we attribute to the dilution and low diffusion of the precursor during the ionothermal reaction at 300 °C, and the resulting variations in the oligomer size, indicating that the synthesis is not yet fully optimized. For example, next to average hydrogen evolution rates around 10 μmol h−1, lower (3.4 μmol h−1) and higher hydrogen evolution rates of 30.3 μmol h−1 were observed, the latter with an estimated apparent quantum efficiency of up to 9.9 (±2.0)%. A fine-tuning of the synthesis parameters and the deliberate introduction of additional nitrile groups may therefore further improve the photocatalytic activity of these phenyl-triazine oligomers.
Another important aspect for gauging the intrinsic photocatalytic activities of the samples is to relate them to their specific surface areas, which may correlate with the number of exposed active sites. In this case, however, the PTOs with smaller surface areas showed significantly improved activity and we can therefore exclude surface area effects as dominant factors for the photocatalytic performance (Table S8, ESI†).
Further, the question arises as to whether residual zinc chloride is present as an impurity or quantitatively incorporated into the system and as such may influence the photocatalytic activity of the PTOs. Thomas and co-workers34 reported on possible Zn2+ incorporation in CTFs during the synthesis of CTF-0 (derived from 1,3,5-tricyanobenzene) when synthesized in the presence of large zinc chloride quantities. Minor amounts of Zn2+ ions within the structure might act as “dopants” as reported previously for carbon nitrides.35 However, this zinc doping procedure (stirring Melon in an acidic ethanol zinc chloride solution) most likely leads to an increased amount of nitrile groups from depolymerization and opening of the triazine rings as seen from the IR spectra. It is therefore doubtful whether the Zn-doping or the formation of nitriles was the reason for the improved activity of the carbon nitrides.35 Here, the presence of zinc chloride in the PTO is ≤0.1 wt% as established by ICP-AES analysis (Table S2, ESI†). As we cannot completely exclude that minute amounts of impurities may be the reason for the different activities of PTOs-300, we performed two different control experiments. Firstly, zinc chloride (5 mg, 10 mg and 17 mg) was added to a standardized PTO-300 suspension for photocatalysis, which resulted in a long-term decrease in activity presumably caused by chlorine poisoning of the platinum nanoparticles (Fig. S15, ESI,† left). Secondly, a PTO-300 sample was excessively washed with 0.1 M ethylenediaminetetraacetic acid (EDTA), which acts as a complexing agent for zinc ions (Table S9, ESI†), and compared the activities before and after EDTA treatment. Here again, no change in activity was detected after washing (Fig. S15, ESI,† left), which implies that Zn2+ doping is probably not the reason for the activity differences.
We therefore hypothesize that besides the different nitrile content variations in activity within the oligomers primarily arise from the different optical properties (more well-defined absorption in the visible, see Fig. 3d) as well as more favorable carrier dynamics. The latter may be most facile for oligomers with shorter lengths and smaller interlayer distances, which leads to stronger π-orbital overlap and hence more efficient extraction of the charge carriers at the interfaces. This is supported by the significantly enhanced dispersibility of the smaller oligomers with a higher degree of nitrile moieties.
In addition, several control experiments were performed to ensure that the evolution of hydrogen is indeed photocatalytic and not due to chemical reactions with the catalyst. Most fundamentally, the absence of light resulted in no activity, proving the hydrogen evolution to be photo-induced. Secondly, the activity of the photocatalyst was tested without the addition of the proton reduction catalyst platinum. In fact, unmodified PTO-300 also showed a moderate, yet significant hydrogen evolution rate (PTO-300-15: 1.21 μmol h−1 or 121 μmol h−1 g−1; PTO-300-1: 0.53 μmol h−1 or 53 μmol h−1 g−1), which indicates that PTO-300 acts as an actual photocatalyst rather than just a photosensitizer (Fig. S16, ESI†). Surprisingly, PTO-300 (121 μmol h−1 g−1) significantly outperforms other co-catalyst-free light element photocatalysts (e.g. Melon: 1–40 μmol h−1 g−1,8 msp-g-C3N4: 2 μmol h−1 g−1,17 B4.3C: 14.5 μmol h−1 g−1,19 and PTI: 0 μmol h−1 g−1).6,7,10 Yet, this modest activity is significantly increased and stabilized by platinum deposition. The absence of an electron donor resulted in negligible activity (0.01 μmol h−1) which underlines the importance of a sacrificial donor in the system to avoid recombination of the generated charge carriers and to enhance the driving force for hole collection as compared to the oxidation of pure water. The highest activities were observed when Pt-modified PTO-300 was suspended in an aqueous TEoA solution. However, other electron donors such as oxalic acid and EDTA also led to sustained hydrogen evolution (Fig. S17, ESI†).
We also tested PTOs-300 doped with 3 wt% Co3O4 nanoparticles and silver nitrate as sacrificial agents for water oxidation. However, no oxygen was detected (within the detection limit), even though triazine-based materials have been predicted to be oxygen evolution catalysts due to their suitable HOMO potential.16
PTO-300 synthesized from diluted zinc chloride melt is competitive (average rate of 1076 μmol h−1 g−1, maximum rate of 3030 μmol h−1 g−1) with highly condensed heptazine- and triazine-based carbon nitride photocatalysts, such as Melon (e.g. msp-g-C3N4: 1490 μmol h−1 g−1 or g-C3N4 doped with barbituric acid: 294 μmol h−1 g−1),17,36 crystalline PTI nanosheets (1750 μmol h−1 g−1),10 4-AP doped amorphous PTI (4907 μmol h−1 g−1),7 TFPT-COF (1970 μmol h−1 g−1)9 and boron carbide (B4.3C: 31 μmol h−1 g−1).19 As differing measurement set-ups (light-sources, intensities, irradiation type e.g. inner vs. top-irradiation) may give different results for the same photocatalyst in different laboratories we compare PTO-300 with four of the above reported photocatalysts measured under identical instrumental conditions, namely Melon (g-C3N4) synthesized from dicyandiamide for 4 hours at 600 °C,8a crystalline PTI/LiCl nanosheet suspension,10 16% 4-AP doped amorphous PTI7 and TFPT-COF9 (Fig. 4d). While PTO-300-1 (1.5 μmol h−1) is less active than Melon (6.4 μmol h−1), PTOs-300 synthesized in an excess of zinc chloride (10.8 (±2.8) μmol h−1) are superior to Melon and rival TFPT-COF (15.5 μmol h−1). Whereas some photocatalysts tested here (TFPT-COF and (amorphous doped) PTI) exhibit a significant decrease in their hydrogen evolution rate over time within an aqueous TEoA solution, PTO-300 shows long term stability.
Conclusions
We have demonstrated that a lower reaction temperature during ionothermal CTF-1 synthesis gives rise to oligomers of CTF-1 which differ in their length and connectivity. These oligomers are highly efficient hydrogen evolution photocatalysts which show an average hydrogen evolution rate of 10.8 (±2.8) μmol h−1 (10 mg photocatalyst) with simulated sunlight, whereas hardly any activity was observed for the CTF-1 polymer. We hypothesize that the suspicious absence of activity in CTF-1 is due to a reduced thermodynamic driving force for hydrogen evolution or to the incipient carbonization at its synthesis temperature of 400 °C, where light absorption by carbon prevents photon absorption by the framework, thus rendering hydrogen evolution unfeasible. For the triazine-based oligomers obtained at different precursor/zinc chloride ratios, distinct crystallinity, surface area, nitrile content as well as variations in activity were observed. Specifically, PTOs synthesized with an excess of zinc chloride (>1 equivalent relative to the precursor) display higher crystallinity, a reduced layer distance, smaller surface areas, lower hydrophobicity, smaller fragment sizes and a larger amount of terminal nitrile groups. These features were found to contribute to substantially improved photocatalytic activity compared to PTOs obtained with 1:1 precursor/zinc chloride ratio. Since the observed activity enhancement likely correlates with an increased amount of nitrile moieties and improved π-stacking of the oligomers, we believe that by controlling the polymerization progress, we can significantly enhance the charge transfer kinetics, which still is the limiting factor in organic photocatalysts. Finally, the oligomers show the highest activity observed for metal-free organic photocatalysts in the absence of any co-catalyst, thus boding well for the development of earth-abundant organic photocatalysts by controlled polymerization reactions and molecular engineering.6–10,17–19
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (projects LO1801/1-1, SE1417/5-1), the cluster of excellence “Nanosystems Initiative Munich” (NIM), and the Center for NanoScience (CeNS) is gratefully acknowledged. We thank Dr M. Konuma, C. Minke, C. Sondermann, V. Duppel and M.-L. Schreiber for their assistance with the sample characterization and Dr V. Vyas for the synthesis of the trimer. Dr V. W. Lau, F. Haase and L. Stegbauer are greatfully acknowledged for helpful discussions.References
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 8, 474 RSC.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255 CrossRef CAS PubMed.
- Y. Ham, K. Maeda, D. Cha, K. Takanabe and K. Domen, Chem. – Asian J., 2013, 8, 218 CrossRef CAS PubMed.
- K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435 CrossRef CAS PubMed.
- (a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed; (b) L. Seyfarth, J. Seyfarth, B. V. Lotsch, W. Schnick and J. Senker, Phys. Chem. Chem. Phys., 2010, 12, 2227 RSC.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789 RSC.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11011 Search PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS PubMed.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357 CrossRef CAS PubMed.
- C. Butchosa, T. O. McDonald, A. I. Cooper, D. J. Adams and M. A. Zwijnenburg, J. Phys. Chem. C, 2014, 118, 4314 CAS.
- X. Jiang, P. Wang and J. Zhao, J. Mater. Chem. A, 2015, 3, 7750 CAS.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- J. Liu, X. Wen, Y. Hou, F. Zuo, G. J. O. Beran and P. Feng, Angew. Chem., Int. Ed., 2013, 52, 3241 CrossRef CAS PubMed.
- J. H. van't Hoff, Die Gesetze des chemischen Gleichgewichtes für den verdünnten, gasförmigen oder gelösten Zustand, Engelmann, Leipzig, 2nd edn, 1915, p. 105 Search PubMed.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202 CrossRef CAS PubMed.
- V. G. Manecke and D. Wöhrle, Makromol. Chem., 1968, 120, 176 CrossRef PubMed.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319 CrossRef CAS.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970 CrossRef CAS.
- W. J. Gammon, O. Kraft, A. C. Reilly and B. C. Holloway, Carbon, 2003, 41, 1917 CrossRef CAS.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers, the Scienta ESCA300 Database, 1992 Search PubMed.
- J. A. Taylor, G. M. Lancaster and J. W. Rabalais, Appl. Surf. Sci., 1978, 1, 503 CrossRef CAS.
- (a) S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed; (b) M. Barber, J. A. Connor, M. F. Guest, I. H. Hillier, M. Schwarz and M. Stacey, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 551 RSC; (c) B. A. Sexton and N. R. Avery, Surf. Sci., 1983, 129, 21 CrossRef CAS.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288 CrossRef PubMed.
- S. Hug, M. Tauchert, S. Li, U. E. Pachmayr and B. V. Lotsch, J. Mater. Chem., 2012, 22, 13956 RSC.
- E. T. Haupt and D. Leibfitz, Spectrochim. Acta, Part A, 1989, 45, 119 CrossRef.
- V. W. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064 CrossRef CAS PubMed.
- (a) B. Y. Kukushkin, Platinum Met. Rev., 1998, 42, 106 Search PubMed; (b) V. W. Lau, I. Moudrakovski, T. Botari, S. Weinburger, M. B. Mesch, V. Duppel, J. Senker, V. Blum, B. V. Lotsch, 2015, submitted.
- P. Katekomol, J. Roeser, M. J. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542 CrossRef CAS.
- B. Yue, Q. Li, H. Iwai, T. Kako and J. Ye, Sci. Technol. Adv. Mater., 2011, 12, 034401 CrossRef.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details (methods and syntheses), characterization data of the organic phase of PTO and further XRD, XPS, NMR, TEM, BET, MALDI-TOF, IR, ICP, EDX and photocatalytic measurements as well as elemental analyses. See DOI: 10.1039/c5ee02574e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |