
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLa5Ti2Cu1−xAgxS5O7 photocathodes operating at positive potentials during photoelectrochemical hydrogen evolution under irradiation of up to 710 nm†
Takashi
Hisatomi
ab,
Shintaro
Okamura
a,
Jingyuan
Liu
a,
Yuki
Shinohara
a,
Koichiro
Ueda
ab,
Tomohiro
Higashi
ab,
Masao
Katayama
ab,
Tsutomu
Minegishi
ab and
Kazunari
Domen
*ab
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, 113-8656 Tokyo, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
bJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan
First published on 22nd September 2015
Abstract
A photoelectrochemical (PEC) cell based on a series-connected photocathode and photoanode made of particulate semiconductors is a potentially scalable and inexpensive device for renewable solar hydrogen production via PEC water splitting without any external power supply. The realisation of such PEC devices hinges on the development of photoelectrodes that operate at a small applied voltage. In this study, solid solutions of La5Ti2CuS5O7 (LTC) and La5Ti2AgS5O7 (LTA) were synthesised, and their physical, optical, and PEC properties in the water splitting reaction were discussed. LTC and LTA formed a La5Ti2Cu1−xAgxS5O7 solid solution (LTC1−xAx) over the whole compositional range. The indirect bandgap energy of LTC1−xAx changed nonlinearly with respect to composition, attaining its minimum value (ca. 1.8 eV) at a composition of x ≈ 0.16. Photoelectrodes of Al-doped LTC1−xAx solid solution powder fabricated using the particle transfer method exhibited a photocathodic response regardless of the Ag content. 1%Al-LTC0.9A0.1 photocathodes exhibited the best PEC properties in the hydrogen evolution reaction and yielded a hypothetical half-cell solar-to-hydrogen energy conversion efficiency of 0.25% at +0.6 V vs. RHE, three times higher than the previously reported 1%Sc-LTC. In addition, 1%Al-LTC0.9A0.1 photocathodes were fairly stable at +0.7 V vs. RHE without any protective modifications. Owing to the positive operational electrode potential of 1%Al-LTC0.9A0.1, unassisted PEC water splitting was accomplished using series-connected photoelectrodes made of 1%Al-LTC0.9A0.1 and BaTaO2N, particulate semiconductors with absorption edge wavelengths of 710 and 660 nm, respectively, at a Faradaic efficiency of unity and a solar-to-hydrogen energy conversion efficiency of approximately 0.1%.
Broader contextA scalable and inexpensive system for solar fuel production is essential for the sustainability of our society. Photoelectrochemical water splitting is one of the most cost-effective and industrially valuable processes for renewable solar hydrogen production. The hydrogen thus produced does not depend on fossil fuels and can be used to fix carbon dioxide to produce various chemicals by catalytic processes in the chemical industry. The development of particulate semiconductor photoelectrodes capable of decomposing water without an external power supply will have a significant impact on the realisation of practical photoelectrochemical water splitting devices, because photoelectrodes made of particulate semiconductors can be fabricated by inexpensive and scalable solution processes. In this article, Al-doped La5Ti2Cu1−xAxS5O7 powder is presented as a photocathode material that allows photoelectrochemical hydrogen production at +0.7 V vs. RHE, a markedly positive potential for single-semiconductor photocathode materials. Unassisted PEC water splitting is demonstrated using a series-connected Al-doped La5Ti2Cu1−xAxS5O7 photocathode and a BaTaO2N photoanode made of particulate semiconductors with visible light absorption of up to 710 and 660 nm, respectively, at a Faradaic efficiency of unity. The present work establishes an important milestone in the development of scalable PEC systems and broadens the prospects for PEC solar water splitting technology. |
Introduction
Photoelectrochemical (PEC) water splitting has been studied extensively as a means of renewable hydrogen production from water and solar energy on a large scale.1–6 The hydrogen thus produced does not depend on fossil fuels and leaves only water as a by-product when used as a fuel to drive fuel cells and in a combustion process. Hydrogen is also an important feedstock for various chemicals in the chemical industry. In particular, the reduction of carbon dioxide using solar-derived hydrogen is considered to have significant impact on the energy efficiency of the process.6 The storage and transportation of hydrogen in the form of liquid-phase organic hydrides have also been studied.7–9A technoeconomical analysis published in 2013 suggests that the cost of hydrogen produced by PEC water splitting can be low enough to be competitive with the steam methane reforming process.2 However, a solar-to-hydrogen energy conversion efficiency (STH) of at least 10% is needed to realise this ideal scenario. Achieving an STH of 10% at a feasible quantum efficiency clearly requires harvesting of long-wavelength photons.10 In addition, the cost and scalability of PEC systems are important for practical applications. It was found that 80% of the cost of hydrogen would originate from the capital cost when panel array PEC cells based on integrated thin films are used.2 The development of particulate photoelectrode devices capable of decomposing water into hydrogen and oxygen without external bias can solve this problem, because photoelectrodes made of particulate semiconductors can in principle be fabricated by inexpensive and scalable printing processes.
In 2004, Meignen et al. reported La5Ti2CuS5O7 (LTC) and La5Ti2AgS5O7 (LTA) to be semiconducting oxysulphides with bandgap energies of 2.0 and 2.2 eV, respectively.11 LTC has a longer absorption edge wavelength than LTA because Cu 3d orbitals form the top of the valence band and shift the valence band maximum negatively.12 The present authors' group reported that LTC and LTA powders modified with catalysts were active in hydrogen and oxygen evolution reactions from aqueous solutions containing electron donors (sulphide and sulphite ions) and acceptors (silver ions) as sacrificial reagents, respectively.12,13 LTC powder can also be processed into photoelectrodes for PEC water splitting.14–16 It was revealed that p-type doping of LTC, such as doping the Ti4+ sites with Sc3+ and Ga3+, boosted the photocathodic current by a factor of eight. Moreover, the onset potential of the photocathodic current in Sc-doped LTC photocathodes modified with Pt as a hydrogen evolution catalyst was +0.9 V vs. RHE in the PEC hydrogen evolution reaction, one of the most positive potentials ever reported for any single p-type semiconductor photocathode. A photocathode with such a positive onset potential is indeed ideal for application to p/n PEC cells for unassisted water splitting, because the operating current and potential are determined by the intersection of the current–potential curves of the photocathode and the photoanode used.1,5 Our subsequent studies revealed the unique semiconducting properties of LTC, allowing for one-dimensional charge transfer15 and the utilization of both photoexcited electrons and holes depending on the band structure at the junction of the LTC particles and the back contact material.16 However, the photocurrent produced by Sc-doped LTC was at most 0.2–0.3 mA cm−2 at the reversible hydrogen evolution potential (0 V vs. RHE) under simulated sunlight (AM1.5G),15 which negated the unique and promising properties of LTC photocathodes. Some Cu(I)-based photocathode materials such as Cu2O,17,18 CuFeO2,19 CuNb3O8,20 Cu5Ta11O30,21 Cu3Ta7O19,21 CuRhO2,22 Cu(In,Ga)S2,23 CuInS2,24 Cu2ZnSnS4,25 and Cu(In,Ga)Se2,26 often modified with n-type overlayers, can exhibit much higher photocathodic current or a photocurrent onset potential as positive as LTC. However, the electrode potential used to produce hydrogen was still close to 0 V vs. RHE. This fact points out the difficulty in developing photocathode materials suitable for application in p/n PEC cells.
In our previous study, LTA exhibited twice the hydrogen evolution rate of LTC despite the shorter absorption edge wavelength.12 Thus, the photocurrent produced by LTC in PEC hydrogen evolution would be expected to improve upon forming solid solutions of LTC and LTA (La5Ti2Cu1−xAgxS5O7, hereafter LTC1−xAx). In the present study, a series of LTC1−xAx solid solution powders was synthesised by solid state reactions in sealed evacuated tubes to examine the powders' physical and optical properties. Subsequently, LTC1−xAx doped with Al as a p-type dopant was processed into photoelectrodes using the particle transfer method to determine its PEC properties in the water splitting reaction. LTC0.9A0.1 was found to have a narrower indirect bandgap energy than LTC and LTA. 1%Al-doped LTC0.9A0.1 exhibited a photocathodic current of approximately 1 mA cm−2 at 0 V vs. RHE and 0.4 mA cm−2 at +0.6 V vs. RHE, more than double that for the previously reported 1%Sc-doped LTC.15 Moreover, p/n PEC cells comprising a photocathode of 1%Al-doped LTC0.9A0.1 powder developed in this study and a photoanode of BaTaO2N powder,27 which had absorption edge wavelengths at 710 and 660 nm, respectively, enabled unassisted PEC water splitting into hydrogen and oxygen at a Faradaic efficiency of unity under visible light irradiation.
Experimental
LTC1−xAx solid solution powders were synthesised by solid state reactions in sealed evacuated tubes.14 La2O3 (99.99%, Kanto Chemical Co., Inc.), freshly calcined at 1273 K for 10 h, La2S3 (99.9%, Kojundo Chemical Laboratory Co., Ltd), TiO2 (99.99%, Rare Metallic Co., Ltd), freshly calcined at 1073 K for 1 h, Cu2S (99%, Kojundo Chemical Laboratory Co., Ltd), Ag2S (99%, Kojundo Chemical Laboratory Co., Ltd), Al2O3 (99.99%, 0.3 μm, Soekawa Chemicals), freshly calcined at 1273 K for 6 h, and S (99.99%, Kojundo Chemical Laboratory Co., Ltd) were stored and used as raw materials in a glovebox under a nitrogen atmosphere. La2O3, La2S3, TiO2, Cu2S, Ag2S, and S were mixed in the glovebox at a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4:1 − x:x:0.25. Here, sulphur was added excessively to suppress the generation of oxide impurities. It was found in our previous study that the PEC properties of the LTC powder decreased due to the presence of oxide impurities.14 The precursor mixture was sealed in evacuated quartz tubes and heated at 1273 K for 48 h. After heating, the samples were ground into powder. When LTC1−xAx was doped with Al, the amount of TiO2 was reduced and corresponding amounts of Al2O3 were added. The Al-doped La5Ti2Cu1−xAgxS5O7 samples will hereafter be referred to as Al-LTC1−xAx.
:3:4:1 − x:x:0.25. Here, sulphur was added excessively to suppress the generation of oxide impurities. It was found in our previous study that the PEC properties of the LTC powder decreased due to the presence of oxide impurities.14 The precursor mixture was sealed in evacuated quartz tubes and heated at 1273 K for 48 h. After heating, the samples were ground into powder. When LTC1−xAx was doped with Al, the amount of TiO2 was reduced and corresponding amounts of Al2O3 were added. The Al-doped La5Ti2Cu1−xAgxS5O7 samples will hereafter be referred to as Al-LTC1−xAx.
X-ray diffraction (XRD) patterns were acquired using an X-ray diffractometer (RINT-Ultima-III, RIGAKU). The crystal structure was depicted using the VESTA software package,28 and the crystallographic information files for LTC and LTA.11 Diffuse reflectance spectra (DRS) were recorded using an ultraviolet-visible-infrared diffuse reflectance spectrometer (V-670, JASCO). The indirect bandgap energy was estimated from the following equation:
| (αhν)n = A(hν − Eg) |
Electrodes of LTC1−xAx were prepared using the particle transfer method.14,29 A Au layer thicker than 2 μm was deposited by vacuum evaporation on LTC1−xAx powder piled on a glass plate. Then, carbon tape supported by a glass plate was used to peel off the Au layer and LTC1−xAx powder composite from the original glass plate. Excess particles were removed by sonication. LTC1−xAx photoelectrodes consisted of only a few layers of particles as shown in Fig. S1 in the ESI.† Accordingly, the thickness of the LTC1−xAx particle layer was a few micrometres at most. This is in fact key to minimize the number of grain boundaries and ensure adequate mechanical and electric contacts between LTC1−xAx particles and the underlying Au conductive layer. An electric wire was soldered to the electrode and covered with epoxy resin. Pt was deposited on the LTC1−xAx photoelectrodes by potentiostatic photodeposition under simulated sunlight (AM1.5G, SAN-EI Electric Co., Ltd, XES-40S2-CE) in an aqueous solution (100 mL) of Na2SO4 (0.1 M), K2C2O4 (0.1 M), and H2PtCl6 (3.5 × 10−6 M).15 A Ag/AgCl electrode and Pt wire were used as reference and counter electrodes, respectively. The pH of the electrolyte solution was adjusted to 10 by an aqueous NaOH solution. During the photodeposition reaction, the electrolyte solution was stirred while bubbling Ar.
The current–potential curves were measured using a three-electrode system with Ag/AgCl and Pt electrodes as reference and counter electrodes, respectively. A 0.1–0.5 M Na2SO4 solution (100 mL) was used as electrolyte solution. The pH of this solution was adjusted to 10 by an aqueous NaOH solution (0.1 M). A solar simulator was used as a light source. The potential of the electrode was controlled by an automatic polarization system (Hokutodenko, HSV-110). The incident photon-to-current efficiency (IPCE) was measured at 0 and +0.65 V vs. RHE under a 300 W Xe lamp (MAX-302, Asahi Spectra) equipped with a series of band-pass filters.
A p/n PEC cell was prepared by connecting Pt-loaded Al-LTC0.9A0.1 (Pt/Al-LTC0.9A0.1) and cobalt-species-loaded BaTaO2N (Co/BaTaO2N) photoelectrodes27 fabricated separately. The photoelectrodes were mounted side by side on a glass plate. The pH of the aqueous Na2SO4 solution (0.1 M) used as the electrolyte solution was adjusted to 11 with an aqueous NaOH solution (5 M). A p/n PEC cell of Pt/Al-LTC0.9A0.1 and Co/BaTaO2N was immersed in the electrolyte solution in an airtight reactor with a flat window and a cooling jacket. The reactor was purged with Ar and maintained at atmospheric pressure. The p/n PEC cell was irradiated with visible light from a 300 W Xe lamp through a cutoff filter (λ > 420 nm) or the simulated sunlight. During the measurement, the photocurrent and the electrode potential were monitored using two automatic polarization systems: one was used for chronoamperometry in the two-electrode configuration with no applied voltage to record the photocurrent passing between the Pt/LTC0.9A0.1 and Co/BaTaO2N photoelectrodes and the other was used for measuring the rest potential to record the electrode potential of the p/n PEC cell vs. a Ag/AgCl reference electrode. The electrode potential of the p/n PEC cell is equal to those of the Pt/LTC0.9A0.1 and Co/BaTaO2N photoelectrodes. The amounts of H2 and O2 evolved were quantified using an on-line micro-gas chromatograph (3000A, Agilent).
Results and discussion
Fig. 1 and 2 show the XRD patterns and the lattice spacing for the (714) diffraction of LTC1−xAx prepared by a solid state reaction. Fig. 1 also shows the simulated XRD patterns for LTC and LTA, available in the Inorganic Crystal Structure Database (ICSD), as a reference.11 LTC and LTA were obtained as the major phases at compositions of x = 0 and 1.0, respectively, similarly to our previous work.12 The XRD patterns for LTC1−xAx with varying compositions were similar to those for LTC and LTA, but shifted to lower angles with increasing Ag content. This reflects the fact that the Ag+ ion has a larger ionic radius than the Cu+ ion. The dependence of the lattice spacing of the (714) planes on the composition could be approximated linearly by following Vegard's law. These observations indicated that LTC1−xAx was not a physical mixture of LTC and LTA, but rather formed a solid solution over the whole composition range. The lattice constants of LTC and LTA differ by less than 1%.11 Therefore, they can form a solid solution. Likewise, Al-doped LTC1−xAx formed a solid solution. As shown in Fig. S2 in the ESI,† the diffraction patterns were attributed to the LTC and LTA structures, and the lattice spacing of the (714) planes was fitted with a straight line over the whole composition range. | ||
| Fig. 1 XRD patterns for LTC1−xAx. (a) LTC, (b) LTC0.9A0.1, (c) LTC0.7A0.3, (d) LTC0.5A0.5, (e) LTC0.25A0.75, and (f) LTA. The reference XRD patterns for LTC and LTA refer to ICSD #99612 and #99613, respectively.11 | ||
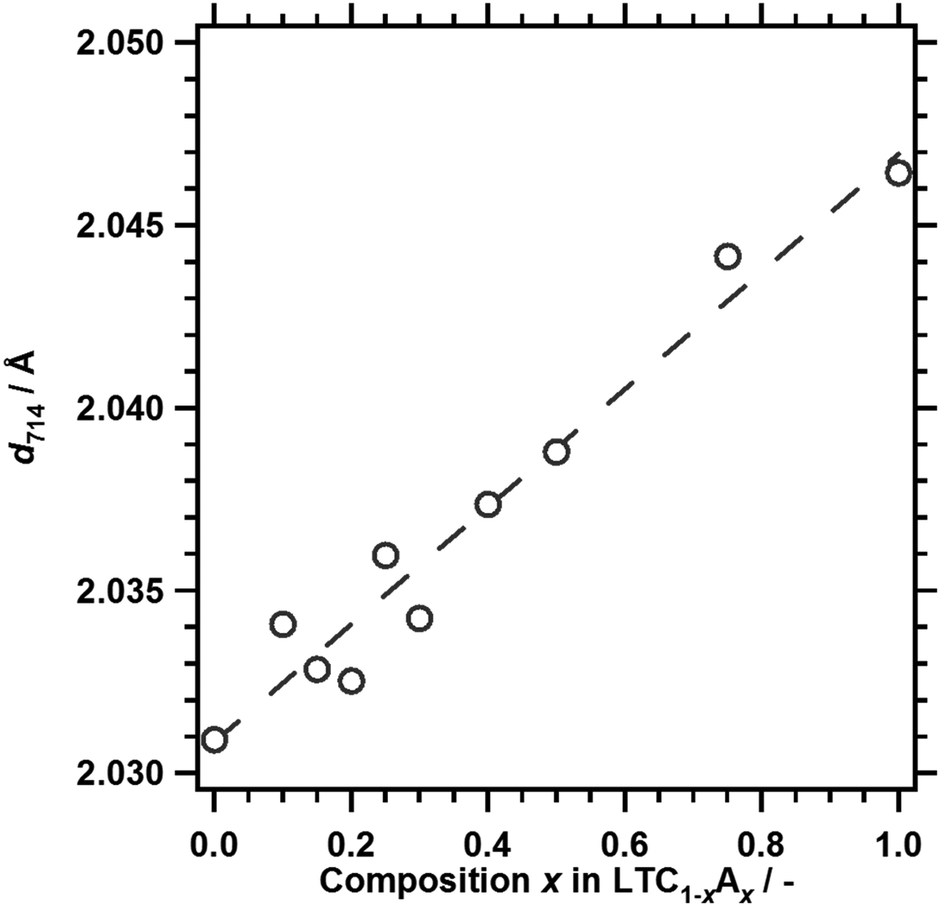 | ||
| Fig. 2 The lattice spacing of the (714) planes, d714, of LTC1−xAx, estimated from the XRD patterns. | ||
Fig. 3 shows the DRS of LTC1−xAx solid solutions. The onset values for light absorption by LTC and LTA from the background absorption were comparable to those obtained in our previous study.12 Substituting Ag for 10% of the Cu shifted the absorption edge toward longer wavelengths. However, further substitution shortened the absorption edge wavelength. As a consequence, LTC and LTC0.7A0.3 exhibited virtually the same absorption edge wavelength. The dependence of the optical properties of LTC1−xAx solid solutions on the Ag content can be plainly observed by comparing the indirect bandgap energies, as shown in Fig. 4. Clearly, the bandgap energy exhibited a nonlinear dependence on the composition of the LTC1−xAx solid solution. Some solid solution semiconductors, such as (Ga1−xZnx)(N1−xOx),30–32 Al1−xInxN,33 and AgxGu1−xGaSe2,34 are known to behave similarly. The bandgap energies of solid solutions of semiconductors A and B (A1−xBx) can be empirically expressed in the following manner:
| Eg,AB = xEg,A + (1 − x)Eg,B − bx(1 − x) = bx2 + (Eg,A + Eg,B − b)x + Eg,A + Eg,B, |
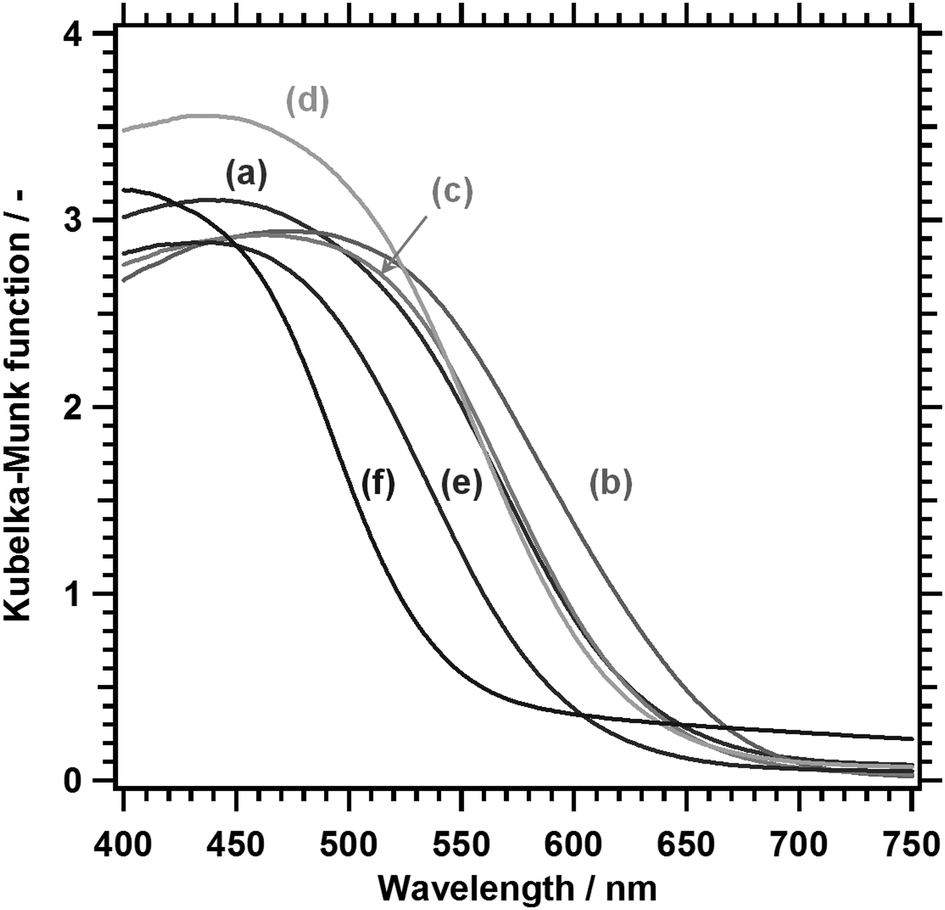 | ||
| Fig. 3 DRS of LTC1−xAx. (a) LTC, (b) LTC0.9A0.1, (c) LTC0.7A0.3, (d) LTC0.5A0.5, (e) LTC0.25A0.75, and (f) LTA. | ||
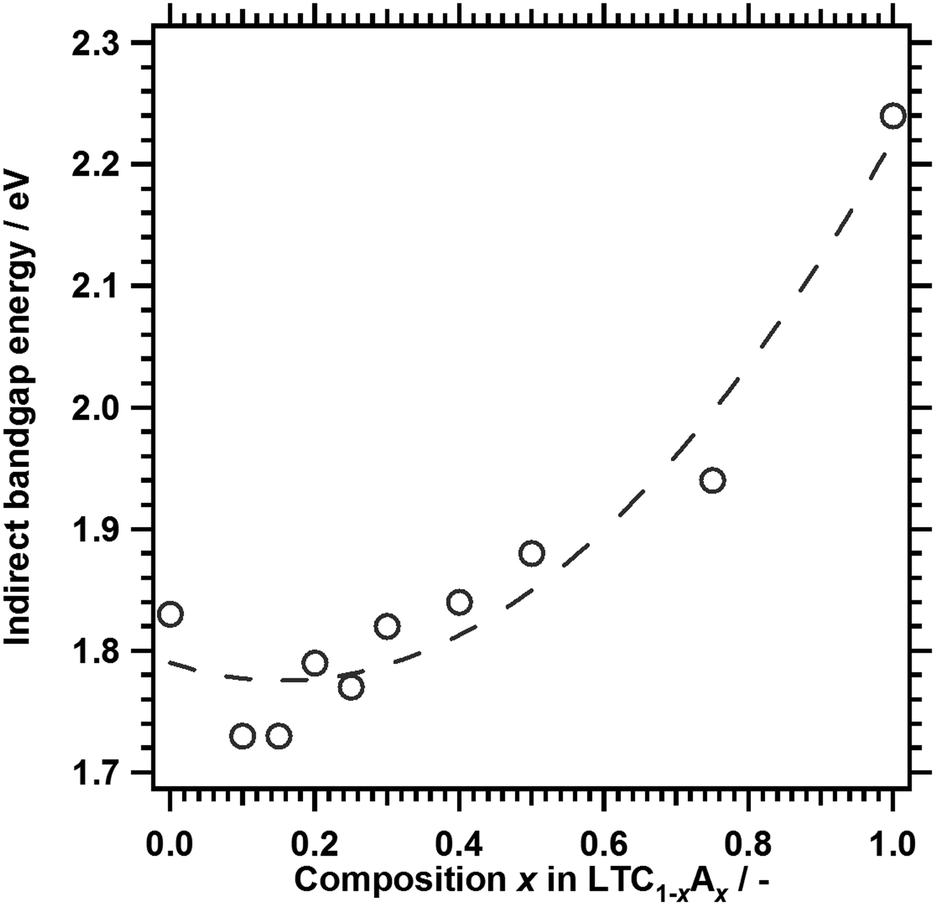 | ||
| Fig. 4 Indirect bandgap energies of LTC1−xAx. The dashed curve, which represents a quadratic fit, is included as a guide to the eye. | ||
On the basis of the outcome of density functional theory (DFT) calculations in our previous study,12 it is thought that the valence and conduction bands of LTC1−xAx are spatially localised around one-dimensional chains of (Cu1−xAgx)S4 tetrahedra and Ti(O,S)6 octahedra, respectively. LTC has a narrower bandgap energy than LTA because Cu 3d orbitals with a more negative potential than S 3p and O 2p make a major contribution to the valence band edge of LTC. Therefore, LTC1−xAx with x ≈ 0.16 would not be expected to have a narrower bandgap energy than LTC. The bowing of the bandgap energies of LTC1−xAx is not due to the local inhomogeneity of the solid solution, because LTC1−xAx can have a narrower bandgap energy than LTC and LTA. Rather, this can be explained by the density of the Cu 3d orbitals and the lattice constants of the LTC1−xAx solid solution system. The density of states and the width of a band consisting of Cu 3d orbitals become fewer and thinner with decreasing Cu content. Because the valence band edge of LTC1−xAx is composed mainly of Cu 3d orbitals, the decrease in Cu content will shift the valence band edge of LTC1−xAx positively and increase the bandgap energy. On the other hand, it is empirically known that for most solid solutions of metal pnictogenide or chalcogenide semiconductors, the bandgap energy decreases as the lattice constant increases. The lattice constants of LTC1−xAx increased linearly with increasing Ag content (Fig. 2), reflecting the larger lattice constants of LTA as compared to those of LTC.11 Therefore, in terms of the lattice constant, narrowing of the bandgap can be expected with increasing Ag content of LTC1−xAx. These two factors affect the bandgap energy of LTC1−xAx in opposite ways. As a result, the bandgap energy of LTC1−xAx may show a nonlinear dependence on composition. Structural optimisation and band structure calculations are in progress for a more conclusive discussion.
PEC properties of LTC0.9A0.1 photoelectrodes with different Al doping amounts were studied by taking into account their longer absorption edge wavelengths as compared to those of LTC and LTA. Fig. 5 shows the current–potential curves measured under simulated sunlight in an aqueous Na2SO4 solution at pH 10. The LTC0.9A0.1 photoelectrodes functioned as photocathodes and generated a photocathodic current regardless of whether they were Al doped or not. The photocathodic current increased significantly upon Al doping, particularly in the positive potential regions near the onset of the photocathodic current. This is consistent with our previous study on LTC in that doping lower-valence cations in the Ti4+ sites enhanced the photocathodic response most likely because of an increase in the majority carrier concentration.14 However, excessive doping (2%) weakened the enhancement presumably because of the formation of defect species. Transient photocurrent spikes owing to charging and discharging of surface states35 were observed near the onset potential of the photocathodic current. Similarly to previously reported LTC photocathodes, 1%Al-LTC0.9A0.1 generated a photocathodic current at 0.8–0.9 V vs. RHE, which is markedly positive compared to many other single semiconductor photocathode materials.14 The onset potential is positive enough to construct p/n PEC cells in combination with existing sunlight-driven photoanodes such as BiVO4,36 Ta3N5,37 and BaTaO2N,27 without photovoltaic assistance. The photocathodic current increased as the electrode potential became more negative. The hypothetical half-cell solar-to-hydrogen energy conversion efficiency (HC-STH) peaked at +0.6 V vs. RHE and was estimated to be 0.25%, which represents a threefold improvement over that for Sc-LTC.15 HC-STH is lower than existing state-of-the-art photocathodes, such as planar and nanorod-type Si38 and Cu(In,Ga)Se2-based thin films.24 However, it should be noted that the optimum operational positive potential is unique to 1%Al-LTC0.9A0.1 and suitable for the construction of p/n PEC cells.
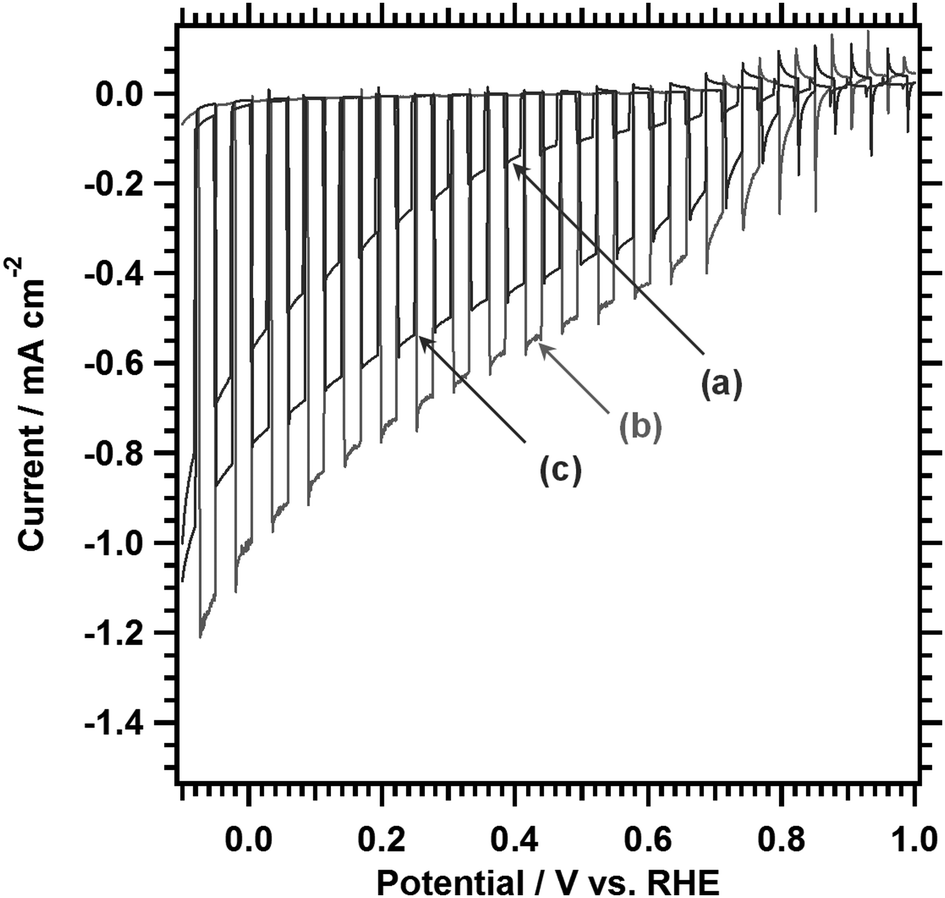 | ||
| Fig. 5 Current–potential curves for photocathodes made of LTC0.9A0.1 doped with (a) 0%, (b) 1%, and (c) 2% Al measured under chopped simulated sunlight in an aqueous solution of Na2SO4 (0.5 M, pH 10). The photocathodes were modified with Pt by photodeposition. The electrode potential was swept from a negative to positive potential at 10 mV s−1. | ||
The durability of 1%Al-LTC0.9A0.1 photocathodes in the positive potential region was examined. Fig. 6 shows a typical current–time profile for a 1%Al-LTC0.9A0.1 photocathode measured at +0.7 V vs. RHE. In the initial hour, the photocurrent was decreased by approximately 40%. The photocurrent became relatively stable in the following measurement, maintaining a photocathodic current of 0.12 mA cm−2 after 16 h of illumination. Fig. S5 (ESI†) shows SEM images of 1%Al-LTC0.9A0.1 photoelectrodes before and after a 1.5 hour potentiostatic PEC hydrogen evolution reaction at +0.7 V vs. RHE. Rod-like particles of 1%Al-LTC0.9A0.1 were observed before and after the PEC measurement. Neither a significant loss of 1%Al-LTC0.9A0.1 particles nor a change in morphology because of segregation of reduced Cu or Ag typical of unprotected Cu2O photocathodes was detected.17 Thus, the photoelectrode was mechanically and chemically stable at this timescale and under these experimental conditions. 1%Al-LTC0.9A0.1 is a rare photocathode material that can maintain a photocathodic response at a potential as positive as +0.7 V vs. RHE without any protecting modifications. In our preliminary experiments, the stability of the 1%Al-LTC0.9A0.1 photocathode was sensitive to the kinds of ionic species present in the reaction solution. This suggests that the decrease in photocurrent might be attributable in part to the adsorption of specific ionic species. It is believed that the use of appropriate surface modifications and supporting electrolytes improves the stability of 1%Al-LTC0.9A0.1 photocathodes.
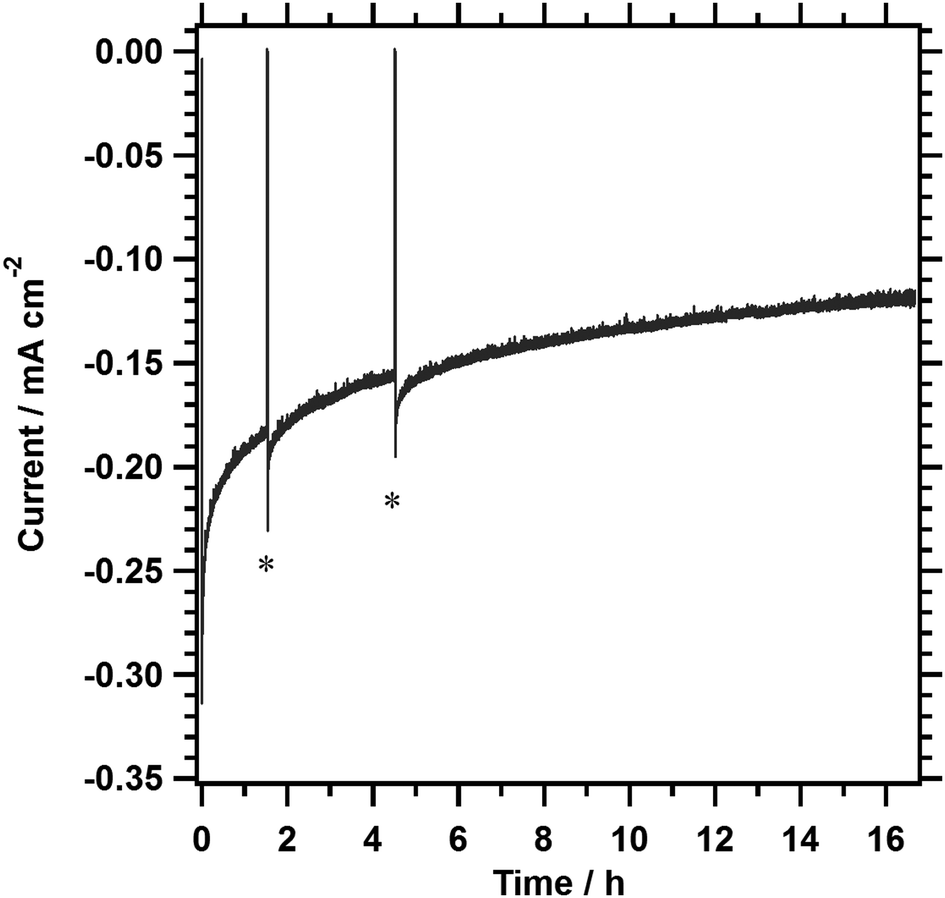 | ||
| Fig. 6 A current–time profile for 1%Al-LTC0.9A0.1 measured at +0.7 V vs. RHE under simulated sunlight illumination in an aqueous solution of Na2SO4 (0.5 M, pH 10). The photocathodes were modified with Pt by photodeposition. Illumination was stopped temporarily at the time points marked by asterisks. | ||
Fig. 7 shows the dependence of the photocathodic current for 1%Al-LTC1−xAx photoelectrodes at 0 and +0.6 V vs. RHE under simulated sunlight irradiation in an aqueous Na2SO4 solution at pH 10. 1%Al-doped LTC1−xAx photoelectrodes functioned as photocathodes and generated a photocathodic current regardless of the composition. The photocathodic current for 1%Al-LTC was significantly increased by substituting Ag for 10% of the Cu. Further substitution decreased the photocathodic current. The photocathodic response of 1%Al-LTA was rather weak despite the ability to absorb sunlight up to ca. 550 nm and the high hydrogen evolution activity of the LTA powder suspension. It is known that for some Cu-based chalcogenide semiconductors, the replacement of Cu with Ag tends to weaken the p-type semiconducting character or even convert these p-type semiconductors into n-type semiconductors.34 Similarly, Al-LTA may almost lose its functionality as a photocathode.
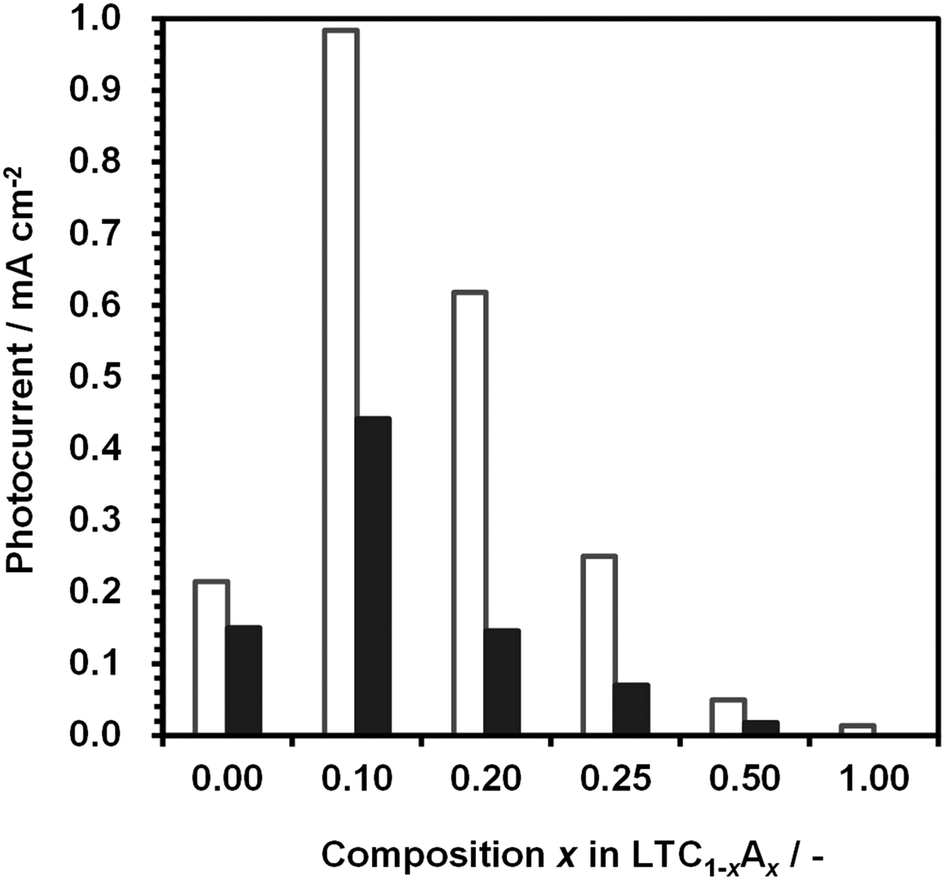 | ||
| Fig. 7 Dependence of the photocurrent of 1%Al-LTC1−xAx on the Ag content, measured under simulated sunlight in an aqueous solution of Na2SO4 adjusted to pH 10. Open and filled bars represent the photocurrents at 0 and +0.6 V vs. RHE, respectively. The photocathodes were modified with Pt by photodeposition. | ||
To discuss the cause of the increase in the photocathodic current upon the replacement of 10% of Cu with Ag in LTC, IPCE spectra of 1%Al-LTC0.9A0.1 and 1%Al-LTC were recorded (Fig. 8). As a general trend, the IPCE increased as the illumination wavelength decreased and the electrode potential became more negative because of the shorter light penetration depth and the wider depletion layer. Both of these trends enabled more efficient electron collection at surface active sites. 1%Al-LTC0.9A0.1 showed an appreciable photoresponse under illumination of up to 710 nm at 0 and +0.65 V vs. RHE while 1%Al-LTC did not. This result agreed well with the light absorption properties of these two materials (Fig. S2, ESI†): 1%Al-LTC0.9A0.1 has a narrower bandgap energy than 1%Al-LTC. In addition, the IPCE of 1%Al-LTC0.9A0.1 was higher than that of 1%Al-LTC over the whole wavelength region measured. This suggests that charge separation and migration in 1%Al-LTC0.9A0.1 particles were more efficient than in 1%Al-LTC. The band distribution and effective masses of charge carriers will be evaluated by DFT calculation to reveal the behaviour of photoexcited carriers in LTC1−xAx solid solutions.
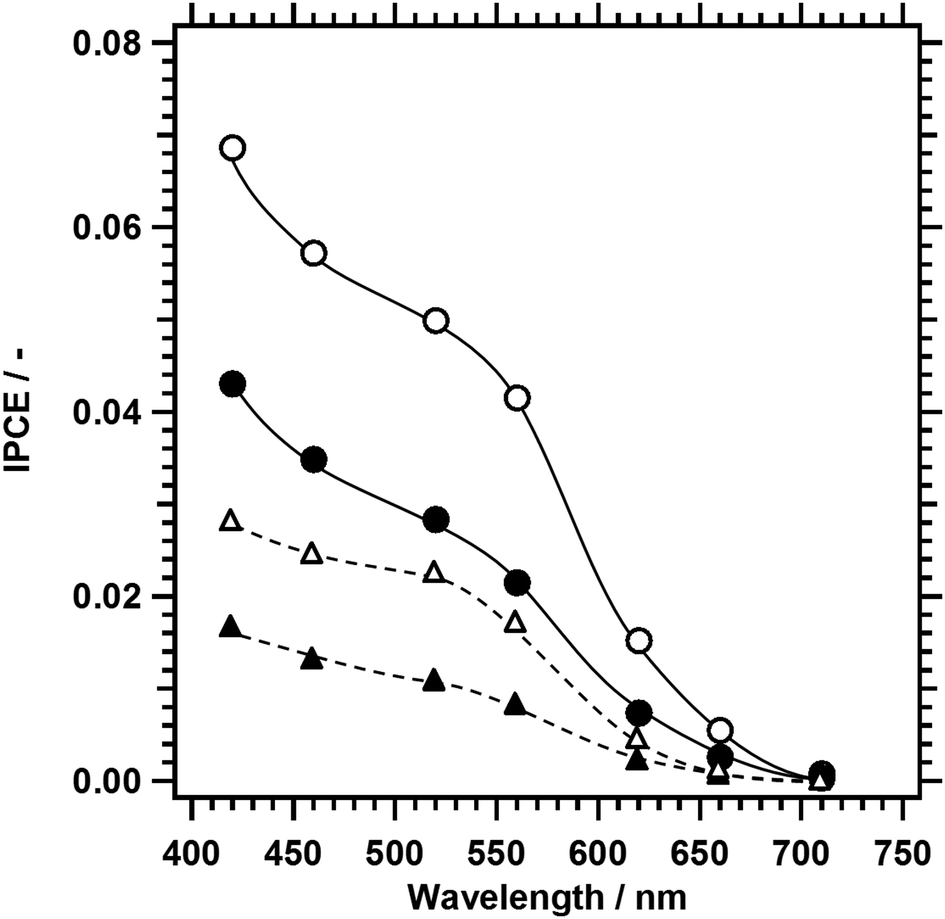 | ||
| Fig. 8 IPCE spectra of 1%Al-LTC and 1%Al-LTC0.9A0.1 recorded at 0 and +0.65 V vs. RHE in an aqueous solution of Na2SO4 adjusted to pH 10. The triangles and circles represent 1%Al-LTC and 1%Al-LTC0.9A0.1, while the open and closed symbols represent 0 and +0.65 V vs. RHE, respectively. The photocathodes were modified with Pt by photodeposition. | ||
Given that 1%Al-LTC0.9A0.1 is capable of generating an appreciable photocathodic current at +0.7 V vs. RHE, unassisted PEC water splitting is feasible using a 1%Al-LTC0.9A0.1 photocathode and a photoanode with a photoanodic current onset potential more negative than +0.7 V vs. RHE. In this study, 1%Al-LTC0.9A0.1 and BaTaO2N photoelectrodes were used as the photocathode and photoanode of a p/n PEC cell for unassisted water splitting because of their ability to absorb visible light up to 710 and 660 nm, respectively. The details of the preparation and the PEC properties of BaTaO2N photoanodes were reported elsewhere.27Fig. 9 shows the time course of the gas evolution on a p/n PEC cell of Pt/Al-LTC0.9A0.1 (0.43 cm2) and Co/BaTaO2N (2.32 cm2) placed side by side under visible light illumination from a Xe lamp (λ > 420 nm). Here, the Co/BaTaO2N photoanode used was larger than the Pt/Al-LTC0.9A0.1 photocathode and the pH value of the electrolyte solution was increased to pH 11 because the photoanode could generate a lower photoanodic current in an aqueous solution without a buffer capacity at lower pH. Hydrogen and oxygen were generated at a stoichiometric ratio of 2:1 over 10 h. The operational electrode potential was stabilised at 0.7 V vs. RHE during irradiation. The amounts of the gases detected by gas chromatography matched well with those calculated from the charge recorded as photocurrent, indicating that the faradaic efficiencies of hydrogen and oxygen evolution were unity on the Pt/Al-LTC0.9A0.1 photocathode and the Co/BaTaO2N photoanode, respectively. The STH was estimated under simulated sunlight irradiation (AM1.5G) to be approximately 0.1% in the initial stage of the reaction. This STH was lower than those of state-of-the-art photovoltaic-powered (photo)electrochemical water-splitting devices (STH of up to 12.4%),39–42 but comparable to those of standalone PEC water-splitting devices, such as p-GaN/p-In0.2Ga0.8N nanowires prepared on a Si substrate (0.2%, and higher at stronger illumination)43 and SrTiO3:La,Rh/Au/BiVO4 photocatalyst sheets (0.2%) mounted on a glass plate,44 and photocatalyst powder suspension Z-scheme systems, such as ZrO2/TaON + WO3 + IO3−/I− (estimated to be 0.2%),45 SrTiO3:Rh + BiVO4 + Fe3+/Fe2+ (0.1%),46 and SrTiO3:La,Rh + Ta3N5 (0.037%).47 Our results represent an important milestone in the development of inexpensive and efficient PEC solar water-splitting devices in that particulate semiconductor materials that function under an irradiation of up to 710 and 660 nm are shown to be used for the PEC hydrogen and oxygen evolution reactions, respectively. In principle, particulate semiconductors with reasonable semiconducting properties can be prepared in bulk and processed into photoelectrodes with large areas by scalable and inexpensive solution processes. In addition, there seems to be much room for improvement in the PEC properties of particulate semiconductors because the quality of particulate semiconductors has been generally believed to be worse than that of single-crystalline semiconductors. Compositional and surface modifications of LTC1−xAx are expected to further increase the photocathodic current near the onset potential and thereby produce higher-STH p/n PEC cells.
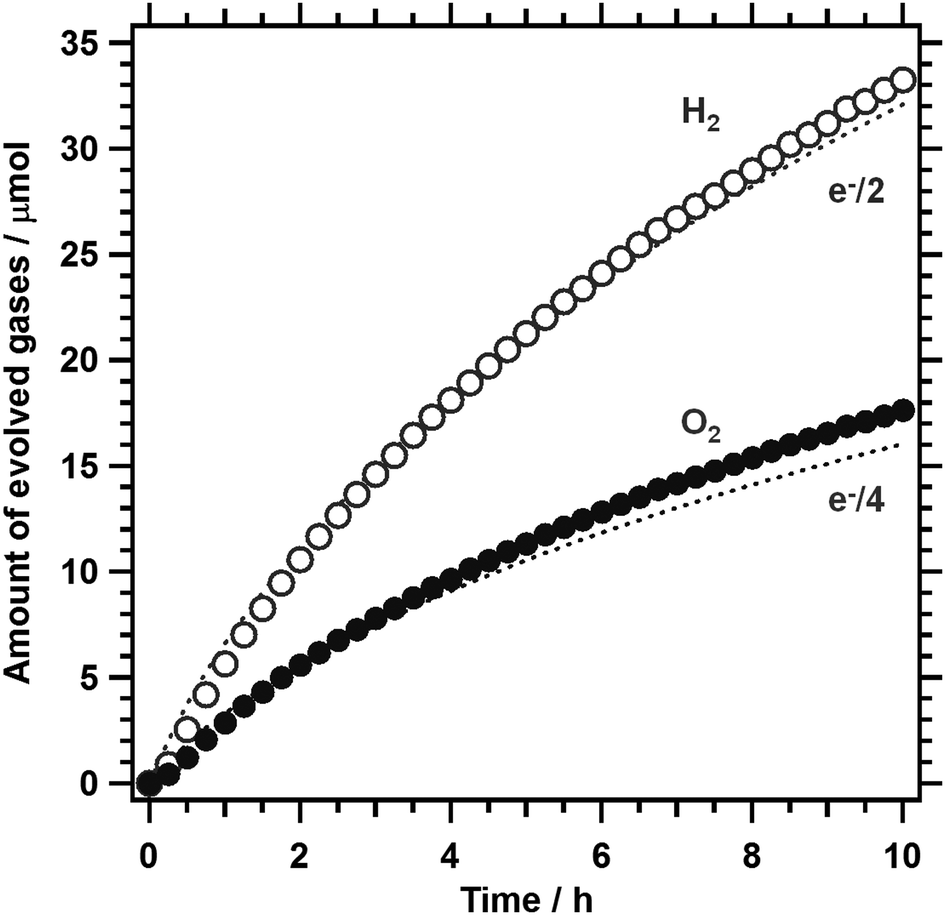 | ||
| Fig. 9 Unassisted PEC water splitting under visible light irradiation from a 300 W Xe lamp (λ > 420 nm) by a p/n PEC cell consisting of Pt/Al-LTC0.9A0.1 (0.43 cm2) and Co/BaTaO2N (2.32 cm2) placed side by side. The electrolyte used was a 0.1 M aqueous Na2SO4 solution whose pH was adjusted to 11. | ||
Conclusions
Solid solutions of LTC and LTA doped with Al were prepared and their structural, optical, and PEC properties in the water splitting reaction were studied. XRD analysis revealed that LTC and LTA formed LTC1−xAx solid solutions over the whole compositional range (0 ≤ x ≤ 1). The lattice spacing of LTC1−xAx solid solutions changed linearly with respect to the Ag content. It was found that the indirect bandgap energy depended on the Ag content with a bowing parameter of approximately 0.6 eV and was minimised at x ≈ 0.16. The LTC1−xAx photoelectrodes functioned as photocathodes in the PEC water splitting reaction. The PEC properties in the hydrogen evolution reaction were significantly enhanced by Al doping, peaking at a composition of 1%Al-LTC0.9A0.1. Incorporation of Ag into Al-LTC not only extended the wavelength region in which the photocathode functioned to ca. 710 nm, but it also increased the IPCE at the respective wavelengths, which points to an improvement in the carrier separation and transport properties of the semiconducting powder. 1%Al-LTC0.9A0.1 exhibited a HC-STH of 0.25% at +0.6 V vs. RHE and generated a photocathodic current over 16 h at +0.7 V vs. RHE. This characteristic functionality as a photocathode enabled a p/n PEC cell of 1%Al-LTC0.9A0.1 and BaTaO2N to accomplish unassisted PEC water splitting using only particulate semiconductors with PEC activity under an irradiation of up to 710 and 660 nm, respectively. The emergence of 1%Al-LTC0.9A0.1, characterised by an outstanding positive onset potential, a Faradaic efficiency of unity during hydrogen evolution, reasonable stability under operating conditions, and an absorption edge wavelength of 710 nm, establishes an important milestone in the development of potentially-scalable PEC systems and broadens the prospects for PEC solar water splitting technology.Acknowledgements
This work was financially supported by Grants-in-Aid for Specially Promoted Research (No. 23000009) and for Young Scientists (A) (No. 15H05494), and the A3 Foresight Program of Japan Society for the Promotion of Science (JSPS), and the Artificial Photosynthesis Project of the Ministry of Economy, Trade and Industry (METI) of Japan.Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983 CAS.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347 CAS.
- L. C. Seitz, Z. Chen, A. J. Forman, B. A. Pinaud, J. D. Benck and T. F. Jaramillo, ChemSusChem, 2014, 7, 1372–1385 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126 CAS.
- R. B. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33, 360 CrossRef CAS PubMed.
- P. Wang, T. Minegishi, G. Ma, K. Takanabe, Y. Satou, S. Maekawa, Y. Kobori, J. Kubota and K. Domen, J. Am. Chem. Soc., 2012, 134, 2469 CrossRef CAS PubMed.
- V. Kalousek, P. Wang, T. Minegishi, T. Hisatomi, K. Nakagawa, S. Oshima, Y. Kobori, J. Kubota and K. Domen, ChemSusChem, 2014, 7, 2690 CrossRef CAS PubMed.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95 CrossRef CAS.
- V. Meignen, L. Cario, A. Lafond, Y. Moëlo, C. Guillot-Deudon and A. Meerschaut, J. Solid State Chem., 2004, 177, 2810 CrossRef CAS PubMed.
- T. Suzuki, T. Hisatomi, K. Teramura, Y. Shimodaira, H. Kobayashi and K. Domen, Phys. Chem. Chem. Phys., 2012, 14, 15475 RSC.
- M. Katayama, D. Yokoyama, Y. Maeda, Y. Ozaki, M. Tabata, Y. Matsumoto, A. Ishikawa, J. Kubota and K. Domen, Mater. Sci. Eng., B, 2010, 173, 275 CrossRef CAS PubMed.
- J. Liu, T. Hisatomi, G. Ma, A. Iwanaga, T. Minegishi, Y. Moriya, M. Katayama, J. Kubota and K. Domen, Energy Environ. Sci., 2014, 7, 2239 CAS.
- G. Ma, J. Liu, T. Hisatomi, T. Minegishi, Y. Moriya, M. Iwase, H. Nishiyama, M. Katayama, T. Yamada and K. Domen, Chem. Commun., 2015, 51, 4302 RSC.
- G. Ma, Y. Suzuki, R. B. Singh, A. Iwanaga, Y. Moriya, T. Minegishi, J. Liu, T. Hisatomi, H. Nishiyama, M. Katayama, K. Seki, A. Furube, T. Yamada and K. Domen, Chem. Sci., 2015, 6, 4513 RSC.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- C. Li, T. Hisatomi, O. Watanabe, M. Nakabayashi, N. Shibata, K. Domen and J.-J. Delaunay, Energy Environ. Sci., 2015, 8, 1493 CAS.
- M. S. Prévot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359 CrossRef PubMed.
- U. A. Joshi and P. A. Maggard, J. Phys. Chem. Lett., 2012, 3, 1577 CrossRef CAS PubMed.
- O. Palasyuk, A. Palasyuk and P. A. Maggard, J. Solid State Chem., 2010, 183, 814 CrossRef CAS PubMed.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830 CrossRef CAS PubMed.
- W. Septina, Gunawan, S. Ikeda, T. Harada, M. Higashi, R. Abe and M. Matsumura, J. Phys. Chem. C, 2015, 119, 8576 CAS.
- J. Zhao, T. Minegishi, L. Zhang, M. Zhong, Gunawan, M. Nakabayashi, G. Ma, T. Hisatomi, M. Katayama, S. Ikeda, N. Shibata, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2014, 53, 11808 CrossRef CAS PubMed.
- L. Rovelli, S. D. Tilley and K. Sivula, ACS Appl. Mater. Interfaces, 2013, 5, 8018 CAS.
- H. Kumagai, T. Minegishi, N. Sato, T. Yamada, J. Kubota and K. Domen, J. Mater. Chem. A, 2015, 3, 8300 CAS.
- K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 2227 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120 RSC.
- K. Maeda, K. Teramura, T. Takata, M. Hara, N. Saito, K. Toda, Y. Inoue, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 20504 CrossRef CAS PubMed.
- M. Yoshida, T. Hirai, K. Maeda, N. Saito, J. Kubota, H. Kobayashi, Y. Inoue and K. Domen, J. Phys. Chem. C, 2010, 114, 15510 CAS.
- H. Chen, L. Wang, J. Bai, J. C. Hanson, J. B. Warren, J. T. Muckerman, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2010, 114, 1809 CAS.
- B.-T. Liou, S.-H. Yen and Y.-K. Kuo, Appl. Phys. A: Mater. Sci. Process., 2005, 81, 651 CrossRef CAS.
- L. Zhang, T. Minegishi, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2014, 16, 6167 RSC.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707 CAS.
- M. Zhong, T. Hisatomi, Y. Kuang, J. Zhao, M. Liu, A. Iwase, Q. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053 CrossRef CAS PubMed.
- Y. Li, L. Zhang, A. Torres-Pardo, J. M. González-Calbet, Y. Ma, P. Oleynikov, O. Terasaki, S. Asahina, M. Shima, D. Cha, L. Zhao, K. Takanabe, J. Kubota and K. Domen, Nat. Commun., 2013, 4, 2566 Search PubMed.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, J. Am. Chem. Soc., 2011, 133, 1216 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- J. Brillet, J.-H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593 CrossRef CAS PubMed.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- Q. Wang, Y. Li, T. Hisatomi, M. Nakabayashi, N. Shibata, J. Kubota and K. Domen, J. Catal., 2015, 328, 308 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327 CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen Wang, Chem. Mater., 2014, 26, 4144 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Composition dependence of lattice spacing and of indirect bandgap energy, and SEM images of photoelectrodes. See DOI: 10.1039/c5ee02431e |
| This journal is © The Royal Society of Chemistry 2015 |