
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrokinetic effects in perhydro-N-ethylcarbazole dehydrogenation and H2 productivity optimization by using egg-shell catalysts†
Willi
Peters
a,
Alexander
Seidel
a,
Stefan
Herzog
a,
Andreas
Bösmann
a,
Wilhelm
Schwieger
a and
Peter
Wasserscheid
*ab
aLehrstuhl für Chemische Reaktionstechnik, University Erlangen-Nürnberg (FAU), Egerlandstrasse 3, 91054 Erlangen, Germany. E-mail: peter.wasserscheid@fau.de
bForschungszentrum Jülich, “Helmholtz-Institut Erlangen-Nürnberg” (IEK-11), Nägelsbachstrasse 49b, 91052 Erlangen, Germany
First published on 14th August 2015
Abstract
This contribution deals with pore diffusion influences on the dehydrogenation kinetics of perhydro-N-ethylcarbazole (H12-NEC). The reaction is of high interest in the context of hydrogen storage in the N-ethylcarbazole (NEC)/perhydro-N-ethylcarbazole (H12-NEC) Liquid Organic Hydrogen Carrier (LOHC) system. The hydrogen content of H12-NEC is 5.8 wt% and total dehydrogenation releases for each mL of H12-NEC more than 600 mL of H2. Further optimization of H12-NEC dehydrogenation catalysis requires a better understanding of the role of mass transfer effects. Pore diffusion effects have been studied by preparing egg-shell catalysts (Pt/γ-alumina layer on α-alumina core) of different active layer thicknesses (24 –88 μm). It has been found that even at very thin catalyst layers (24 μm) the kinetic regime is limited to 235 °C, thus pore diffusion effects the dehydrogenation in almost all commercial catalysts strongly.
Broader contextOne of the main challenges we face in the transition to renewable energy sources is the lack of long term electrical energy storage technologies. A very promising storage technique is hydrogen storage in Liquid Organic Hydrogen Carrier (LOHC). Thus, hydrogen generated through electrolysis can easily be stored in the liquid state at ambient conditions through reversible hydrogenation/dehydrogenation of organic compounds. The LOHC system N-ethylcarbazole/perhydro-N-ethylcarbazole is among the most attractive candidates for this purpose. Our contribution deals with the dehydrogenation of H12-NEC and highlights in detail for the first time mass transfer effects that influence and limit the observed catalyst productivity. By preparing catalyst materials with defined active layer thicknesses we could boost the precious metal productivity to a value of 10.9 gH2 gPt−1 min−1 (TOF = 17.5 molH2 molPt−1 s−1 at a hydrogen yield of 20.9%) which is – to the best of our knowledge – the highest hydrogen productivity from a LOHC system reported so far. From these findings we anticipate that egg-shell catalyst materials will play a key role in the on-going industrial realization of the LOHC technology as they allow a very efficient utilization of the precious metal component in the hydrogen release process. |
Introduction
A major obstacle for the replacement of a larger share of fossil fuels by renewable and unsteady energy sources (wind and solar power) is the lack of efficient electrical energy storage technologies. Short time energy storage of small energy amounts has been made available by supercapacitors. Midterm energy storage in the MW h range has been achieved efficiently through battery technologies. Even so these technologies still need further improvement, the real challenge remains in long term energy storage processes of large amounts of energy (e.g. energy storage in the GW h to TW h range).1,2Hydrogen has been considered a suitable long-term energy carrier since the beginning of the last century. However, due to its physical properties, high pressures (e.g. 700 bar) or low temperatures (e.g. −253 K) are necessary to reach significant material based energy densities (1.18 and 2.33 kW h L−1).3,4 These concepts are the most mature hydrogen storage technologies available. Small scale (e.g. automotive) and large scale (underground) storage has proven its applicability.4–7 Because of these storage conditions and the missing infrastructure for storage and transportation of compressed and liquefied hydrogen a broad variety of research efforts has focused on chemical hydrogen storage. In the recent decades, the chemical conversion of hydrogen and CO2 to classical fuels by the Fischer–Tropsch process (fuels, diesel)8 or the Sabatier reaction (SNG)4,9 has gained interest. These synthetic fuels produced from renewable hydrogen do offer long-term energy storage at ambient or moderate conditions and high potential to replace fossil fuels in transportation due to the existing infrastructure for storage and transportation.10,11 However, applying these synthetic fuels would still lead to CO2 emissions at the location of energy use.
Liquid Organic Hydrogen Carrier (LOHC) systems
A more recent approach that avoids this drawback is the application of Liquid Organic Hydrogen Carrier (LOHC) systems.12–20 The latter offer the possibility of hydrogen storage by reversible catalytic hydrogenation and dehydrogenation. Thus, the storage cycle neither needs CO2 nor releases CO2. Hydrogen storage at ambient conditions is possible at a high material based energy density (up to 2.25 kW h L−1 depending on the LOHC system) for long periods of time as hydrogen is firmly bond in the form of homocyclic or heterocyclic compounds. The above mentioned storage density does not take into account the total system volume (e.g. dehydrogenation reactor, tank volume). The search for suitable LOHC compounds and mixtures thereof is continued, since some of the components (e.g. N-ethylcarbazole) are solid at ambient conditions. In order to release H2, high temperatures (typically 150 to 320 °C depending on the LOHC system), continuous heat supply and the presence of a suitable catalyst are required. The liquid hydrocarbon nature of LOHCs makes its handling, transportation and storage in the same infrastructure possible that is used today for liquid fuels. Therefore, no new infrastructure is needed.An early studied LOHC system is the toluene (hydrogen lean)/methylcyclohexane (hydrogen rich) couple. Extensive research on the hydrogenation and dehydrogenation kinetics of this system has been published.21–23 More recently, polycyclic LOHCs with a nitrogen heteroatom have been proposed with the N-functionality causing much milder dehydrogenation conditions.18 The most prominent storage pair among these systems is N-ethylcarbazole/perhydro-N-ethylcarbazole (NEC/H12-NEC) with a material based energy density of 1.82 kW h L−1.24–27 Due to its higher boiling point (above 275 °C)28–30 and lower dehydrogenation temperature, H12-NEC dehydrogenation is carried out as a liquid phase reaction.
This contribution deals with a detailed kinetic analysis of the dehydrogenation of H12-NEC with a special focus on pore and film diffusion influences on the observed reaction rate in the three-phase reaction system (heterogeneous catalyst/LOHC/hydrogen).
Perhydro-N-ethylcarbazole dehydrogenation kinetics
The full dehydrogenation of 1 mL liquid H12-NEC (physico-chemical data refer to values at ambient conditions) leads to the formation of more than 600 mL hydrogen after cooling to ambient conditions and ca. 0.85 mL NEC. Such enormous gas formation and gas flow rates will reasonably effect mass transfer processes in porous catalyst systems. However, the so far published data on the kinetics of H12-NEC dehydrogenation do not discuss this important fact for the interpretation of the observed kinetics. So far, all reported kinetic data for H12-NEC dehydrogenation have been collected from batch mode experiments. Sotoodeh et al. did not report any mass transport effects and determined the activation energy for the dehydrogenation of H12-NEC to be 127.6 kJ molH2−1, that of H8-NEC to be 67.1 kJ molH2−1 and that of H4-NEC to be 144.3 kJ molH2−1 on powder Pd/SiO2 catalysts.26,30–32 Yang et al. studied H12-NEC dehydrogenation using Rh, Ru, Pt and Pd on alumina as powder catalysts. In this study the reaction temperature was fixed at 180 °C and no determination of activation energy was carried out.33For a precise determination of reaction kinetics the use of continuous reactors is always favorable. Continuous catalytic experiments reach (if the catalyst is stable) after a certain time of constant reaction conditions steady-state conditions (typically after three times the hydrodynamic residence time τ = VR/![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) ). Additionally, temperature programmed experiments are possible under continuous operation conditions, resulting in highly resolved kinetic data.
). Additionally, temperature programmed experiments are possible under continuous operation conditions, resulting in highly resolved kinetic data.
Therefore, we applied in this study a continuous dehydrogenation set-up (see later for details) to study H12-NEC dehydrogenation. Moreover, to identify effects of pore diffusion, we decided to compare catalyst beds consisting of catalyst pellets of identical size but coated with active catalytic layers of different thicknesses. The application of catalyst pellets instead of catalyst powder reflects a practical requirement, namely that the very large gas volume formed in the reactor would cause bubble formation, pulsation and large fluctuations in residence times when using a powder catalyst in larger continuous reactors.
The catalyst pellets with defined thicknesses of active layer were obtained by coating a layer of porous γ-alumina on dense α-alumina spheres. At constant density of precious metal in the catalyst layer (gPt mm3), a dependency of observed rate on the layer thickness gives a clear indication on pore diffusion effects if the nature of the precious metal, e.g. particle size, remains constant for the different layer thicknesses. If the layer thickness is larger than the mean diffusional length of H12-NEC prior to its complete conversion, a certain part of the catalyst is not fully applied and the observed productivity per metal decreases.
Thin coatings with porous catalyst materials are well known to the broad public since the introduction of the three-way catalyst for automotive off-gas treatment.34,35 Moreover, porous coatings are often applied to add catalytic functionalities to foams, micro reactor walls, and membranes.36–41 Among the preparation techniques, sol–gel coating, slurry coating and a combination thereof (hybrid coating) are frequently used. The advantages of these methods are inexpensive coating precursors and inexpensive coating equipment. The coating can be tailored by adjusting the composition of the coating suspension (washcoat) or by variation of the coating technique.38,42–45
The washcoat composition of choice to achieve coating thicknesses above 50 μm is a hybrid washcoat based on the sol–gel recipe. The latter uses a nm-sized precursor (Boehmite–AlOOH) in combination with a diluted acid (e.g. HNO3, pH: 2–4) to form a stable dispersion. To create a hybrid washcoat, μm-sized particles (γ-alumina, 2–5 μm) are added to the dispersion (sol) prior to the coating or while coating. In this way, layer thicknesses of up to 400 μm have been achieved on flat surfaces.46,47 Agrafiotis et al. prepared in this way thick, homogeneous coatings on cordierite monoliths.40 Meille et al. coated flat stainless steel with a hybrid alumina washcoat in a one-step procedure achieving stable coatings up to 200 μm.37,45,48
Prior to coating, the support is usually mechanically (roughness), chemically (etching), or thermally (oxidation) pretreated, to create a rough surface for good coating adhesion.46
The coating itself proceeds in one or multiple steps using different coating methods. Dip coating, drop coating (for microreactors), spin coating and brush coating (for flat substrates), as well as spray and electrostatic sol spray coating are applied depending on the different structures to be coated. The application of simple dip coating is widespread, too.46,49 The coating process is followed by a drying period (formation of the gel) and a high temperature treatment (calcination, sintering).39,45,50,51
Coating stability after calcination is tested by impact, bulk crush and drum attrition tests. The latter shows the highest impact on coated spheres due to the applied hear forces.52 For flat surfaces the stability was tested via bending or traction of the coating.37
Despite this preliminary work, synthesis of stable and homogeneous porous coatings on dense α-alumina spheres is not a trivial task. Earlier studies by Ould Chikh et al. resulted in brittle coatings which suffered severe damages when shear forces were applied.48,52 Therefore, prior to the kinetic investigation into H12-NEC dehydrogenation, we had to optimize the synthesis of stable γ-alumina coatings on dense α-aluminum for the purpose of our study.
For our kinetic experiments with the obtained coated catalyst spheres we placed the latter into a fixed-bed reactor (see details later) and performed continuous, temperature programmed H12-NEC dehydrogenation experiments. To obtain highly resolved kinetic data as function of temperature (Arrhenius plots), 40 different temperatures were investigated between 220 and 266 °C.
Experimental
α-Alumina spheres, Denstone 99® – 3 mm (1/8′′), were kindly provided by St. Gobain. Those spheres show a minimum crush strength of 491 N.53 The washcoat was prepared using the powder precursor Disperal® and the γ-alumina powder Puralox SCFa 140, both kindly provided by Sasol-Germany.54,55 The weight ratio of binder to filler was set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The total amount of solid species in water was set to 20 wt%. Diluted HNO3 was added until a pH of 2 was reached. Prior to the addition of the filler, the binder sol has been stirred for at least one hour.
:2. The total amount of solid species in water was set to 20 wt%. Diluted HNO3 was added until a pH of 2 was reached. Prior to the addition of the filler, the binder sol has been stirred for at least one hour.
A simple in-house made pan coater was used to provide the washcoat via a small spray pistol. In contrast to Ould-Chikh et al., the filler was added to the dispersion (sol) prior to the spray coating, creating a hybrid suspension. The slope of the pan was adjusted to be about 15. At the lower point of the pan the spray was applied onto the spheres in a pulsed mode, whereas at the upper part a heat gun continuously dried the spheres at 80–100 °C. The pan rotated at 30–50 rpm. After a coating period of 15 min the spheres were dried in a ventilated oven for 10 min at 220 °C. If necessary, multiple coating steps have been applied. After the coating procedure the calcination took place at 550 °C for 6 h.
Platinum deposition on the washcoat was realized via ion exchange impregnation, using hexachloroplatinic acid from Alfa Aesar. Thereafter, the catalysts were calcined at 330 °C for 6 h and were reduced prior to the reaction at 300 °C for 2 h with hydrogen (10 vol% diluted in nitrogen).
Dehydrogenation setup
All experiments were performed in continuous mode in a vertical tubular reactor with a length of 7 cm and an inner diameter of 0.8 cm (Fig. 1). 30 catalyst spheres were mixed with 47 uncoated nonporous α-alumina spheres to form a diluted catalyst bed with a height of 6 cm. On top and below the catalyst bed two layers of uncoated spheres (each 0.5 cm in height) were placed. The reactor was heated via a ventilated oven. The H12-NEC flow was provided via a HPLC-pump and the resulting hydrogen flow rate was measured via a H2-mass flow meter (MFM). Four different H12-NEC – flow rates were applied (0.2/0.4/0.6/0.8 mL min−1) during each experiment. At a flow rate of 0.6 mL min−1 a temperature programmed dehydrogenation experiment was performed. The reaction pressure was kept at 1 barabs. A downward and an upward temperature ramp with the rate of 0.5 K min−1 was started at 260 °C, leading down to 220 °C and back up to 260 °C. Steady state conditions were reached before and after the temperature programmed dehydrogenation at 260 °C and at 220 °C (operation for 30 min under constant conditions). The excellent agreement of the data from the downward and the upward ramp show reproducibility and the quasi steady-state conditions of the experiment (see later for details). Dehydrogenation of H12-NEC starts at temperatures as low as 150 °C. This specific temperature range (220–260 °C) has been chosen to maximise catalyst productivity (H2 flow rate per platinum mass). | ||
| Fig. 1 Continuous dehydrogenation setup schematic. | ||
Results and discussion
Sphere coating and catalyst synthesis
The coating and impregnation processes of the applied catalyst spheres are illustrated in Fig. 2. | ||
| Fig. 2 Sphere coating: top down: (A) – uncoated; (B) – coated; (C) – impregnated; (D) – coating layer; (E) – impregnated coating, partially removed. | ||
The uncoated spheres offer a surface roughness suitable for the adhesion of coatings (Fig. 2A). The applied coatings show a good homogeneous texture (Fig. 2B). After the impregnation with hexachloroplatinic acid a fine crack structure became visible (Fig. 2C). A closer look using Scanning Electron Microscopy is shown in Fig. 2D. Fig. 2E shows the impregnated coating partially removed from the spheres.
The coating layer shows cracks and some micrometer cavities. Those results seem to be comparable to the work of Schwarz et al., who described a homogeneous coating layer with some larger cavities due to low contents of water during the coating procedure.37 Even so the water content of our dispersion was set to 80 wt%, the surface remained in a dry state, where it did not become soaked with the dispersion. This state was achieved by adding the hybrid suspension in a spray pulse mode on continuously dried alumina spheres.
A variation of the filler particle size (not shown here) confirmed the findings of Ould Chick et al. and Agrafotis et al. These authors stated a good coating stability for a mean filler particle size of 2–5 μm. Smaller particle sizes, however, exhibited a very brittle coating, even during the coating procedure itself.39,52
By mechanical removing of the coating layer after the impregnation (Fig. 2E), a white core sphere was exposed, indicating no impregnation of the core sphere with platinum. Thus, it can be reasonably assumed that measured catalytic activity stems only from the porous, metal-containing egg shell coating layer. The relevant layer density was measured from the layer thickness and the respective mass increase in the coating process. In this way, a coating density of 1.04 to 1.32 g cm−3 (coating layer mass divided by coating layer volume) was determined. The determined specific surface area of the coating layer was 145 m2 g−1.
To confirm the mechanical stability of our coating layers a single sphere crush test was applied after calcination using a Mesmesin AFG-1000 N apparatus. For comparison, the crush strength of commercial γ-alumina spheres (2–3 mm in diameter), kindly provided by Sasol-Germany, was determined with the same apparatus. The commercial γ-alumina spheres were crushed at a strength of 100 N with a large deviation (±25 N). Coated α-alumina spheres exceeded those values significantly. At a force of 100 N up to 90% of the tested spheres showed no visual damage. Even with a force of 400 N the damage on the coating was limited to the contact area of the crush piston. Thus, coated γ-alumina layers are significantly reinforced by the dense α-alumina support and present a suitably mechanical stable catalyst for the here presented kinetic investigations.
For our systematic coating study, coating thicknesses were varied in the range of 24 to 88 μm. Layer thicknesses were measured using an optical microscope (Nikon Eclipse 50i). A layer thickness of 200 μm was achieved but was found to be brittle after multiple uses in the reaction. Therefore experiments with this thick layer were excluded from our kinetic analysis (Table 1).
| Catalyst nomenclature/μm | Coating thickness/μm | Coating thickness deviation/μm | Platinum-content/mg |
|---|---|---|---|
| 24 | 24 | 5.0 | 0.36 |
| 33 | 33 | 4.4 | 0.82 |
| 44 | 44 | n/a | 1.00 |
| 88 | 88 | n/a | 1.87 |
H12-NEC dehydrogenation results
Fig. 3 shows as an example, the dehydrogenation of H12-NEC for a catalyst with a 33 μm active layer thickness. The flow rate of released hydrogen is plotted against temperature for the H12-NEC flow rates 0.4 mL min−1 and 0.6 mL min−1. Each experiment included a downward and an upward temperature ramp embedded by measurements at steady-state reaction conditions at 220 °C and 260 °C. At 250 to 260 °C, some additional measurements were performed at 0.2 and 0.8 mLH12-NEC min−1. Combined steady-state and temperature ramp measurements are presented in Fig. 3. | ||
| Fig. 3 Dehydrogenation of H12-NEC as a function of temperature: reaction temperature between 220 °C and 266 °C; H12-NEC flow rates between 0.2 mL min−1 and 0.8 mL min−1; catalyst spheres with 33 μm coating thickness; 30 spheres applied ≅0.82 mg of total Pt. | ||
Since the temperature ramp was set to 0.5 K min−1, we assumed quasi steady-state reaction conditions. This assumption is clearly supported by the comparison of the upward and the downward temperature ramp: the hydrogen release for the downward and the upward temperature ramp displayed only minor deviations (see ESI† for details). Additionally, we performed multiple steady-state measurements in between 220 and 260 °C which reassured our assumption.
The quasi steady-state conditions have been made possible by the homogeneous heating of the ventilated oven and the fast response time of the reactor due to a small reactor diameter (10 mm). Owing to the high dynamics in the reactor due to the three phase reaction system, some fluctuations in pressure and hydrogen flow rate are observed. Up to temperatures of 245 °C the standard deviation in hydrogen flow rate was ±2–4 mLH2 min−1, above 245 °C deviations of ±4–8 mLH2 min−1 were observed.
Fig. 4 shows the effect of H12-NEC flow variation on produced hydrogen for catalysts with different coating thicknesses. The reaction temperature was set over the whole experiment to 260 °C.
 | ||
| Fig. 4 Dehydrogenation of H12-NEC – effect of H12-NEC flow variation on hydrogen yields: reaction temperature: 260 °C; catalyst: 24 μm, 33 μm, 44 μm, 88 μm; 30 catalyst spheres; platinum mass between 0.36–1.87 mg. | ||
For hydrogen yields in excess of 33% of the totally releasable hydrogen the conversion of octahydro-N-ethylcarbazole (H8-NEC) to tetrahydro-N-ethylcarbazole (H4-NEC) has to be taken into account next to the conversion of H12-NEC to H8-NEC. As the consecutive hydrogenation steps are significantly slower than the initial one, we here restrict the discussion of reaction rates to small hydrogen yields, i.e. to the dehydrogenation from H12-NEC to H8-NEC.
With increasing feed flow rate a decreasing residence time and therefore a lower degree of H12-NEC conversion was observed. The hydrogen flow rate rises as expected with an increasing thickness of the active coating reflecting the higher amount of precious metal catalyst present in thicker layers. For better comparison of catalysts with different total Pt-contents, productivity was defined as the mass of released hydrogen per mass of platinum and minute (eqn (1)).
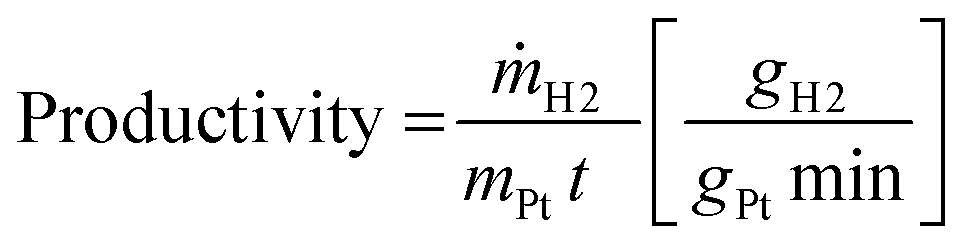 | (1) |
 | ||
| Fig. 5 Catalyst productivity as function of H12-NEC flow rate: reaction temperature: 260 °C ± 1 °C; catalysts: 24 μm, 33 μm, 44 μm, 88 μm; 30 catalyst spheres; Pt mass in reactor: 0.36–1.87 mg; productivity defined as gram H2 released per gram Pt. | ||
As expected, maximum productivity is reached at the highest flow rate (0.8 mLH12-NEC min−1) as a consequence of low H12-NEC conversion (see Fig. 4). The comparison of catalysts with different layer thicknesses reveals that productivities follow a clear trend with significantly lower productivity for the catalysts with layer thicknesses above a layer thickness of 33 μm. With the 33 μm active layer, the productivity has been found to reach a remarkably high value of up to 10.9 gH2 gPt−1 min−1 (T = 262 °C, H12-NEC flow rate = 0.8 mL min−1, H2-yield = 20.9%). This corresponds to a Turn-Over-Frequency (TOF) of 17.5 molH2 molPt−1 s−1. To the best of our knowledge, these values represent hitherto unreached productivities for LOHC dehydrogenation catalysis. Based on the heating value (LHV) of the released hydrogen, this level of productivity corresponds to a power output of 21.7 kW gPt−1. The highest value to date has been reported by Kariya et al. for the dehydrogenation of cyclohexane in a spray-pulse reactor (7.7 gH2 gPt−1 min−1 at 350 °C and a H2-yield of 15.7%).56
The observed dependency of hydrogen productivity on catalyst layer thickness is an unambiguous sign for pore diffusion influenced dehydrogenation kinetics with thicker films. Obviously, the relatively slow diffusion of the H12-NEC makes the Pt further away from the outer surface of the catalyst pellet less effective in the dehydrogenation catalysis. Thus, productivity which is a quantity referenced against the total amount of Pt in the reactor decreases with a larger degree of pore diffusion influence.
Fig. 6 evaluates the productivity taking into account the corresponding hydrogen yield. As illustrated by eqn (2) hydrogen yield represents in this context the share of released hydrogen divided by the total quantity of releasable hydrogen from H12-NEC.
 | (2) |
 | ||
| Fig. 6 Productivity and corresponding yield: reaction temperature: 260 °C ± 1 °C; catalysts: 33 μm, 88 μm; 30 catalyst spheres; Pt mass: 0.82 mg and 1.87 mg. | ||
As mentioned above productivity and H2 yield show an opposite dependency on H12-NEC flow rate and residence time. Fig. 6 highlights in addition the effect of the two different layer thicknesses, 33 μm and 88 μm, on productivity and H2 yield.
Two observations are obvious from Fig. 6: (a) at the same H12-NEC flow rate (0.8 mL min−1) the catalyst productivity of the 33 μm catalyst (10.9 gH2 gPt−1 min−1, YH2 = 20.9%) outperforms the 88 μm catalyst (5.0 gH2 gPt−1 min−1, YH2 = 21.9%) significantly; (b) at the same flow rate of 0.4 mLH12-NEC min−1 the 88 μm catalyst exceeds the 33 μm catalyst in H2-yield significantly (35% vs. 26%), while the difference in productivity is less pronounced (4 vs. 7.3 gH2 gPt−1 min−1).
Thus, depending on the applications scenario, different coating thicknesses might be optimal: for applications where high hydrogen yields and high power densities are in the focus (e.g. for all potential mobile applications), thicker catalyst layers are suitable. By this selection, slightly more Pt is needed as this choice compromises on Pt productivity. For stationary applications where investment cost is the key parameter for optimization, small active layer thicknesses are preferred as this maximizes Pt productivity and thus minimizes the investment in platinum. On the other hand this choice leads to a larger dehydrogenation reactor for a given hydrogen output. Thus, the coating thickness should be adjusted to the specific application scenario.
To conclude this section, Fig. 7 displays catalyst productivities as a function of the catalyst layer thickness at different dehydrogenation temperatures.
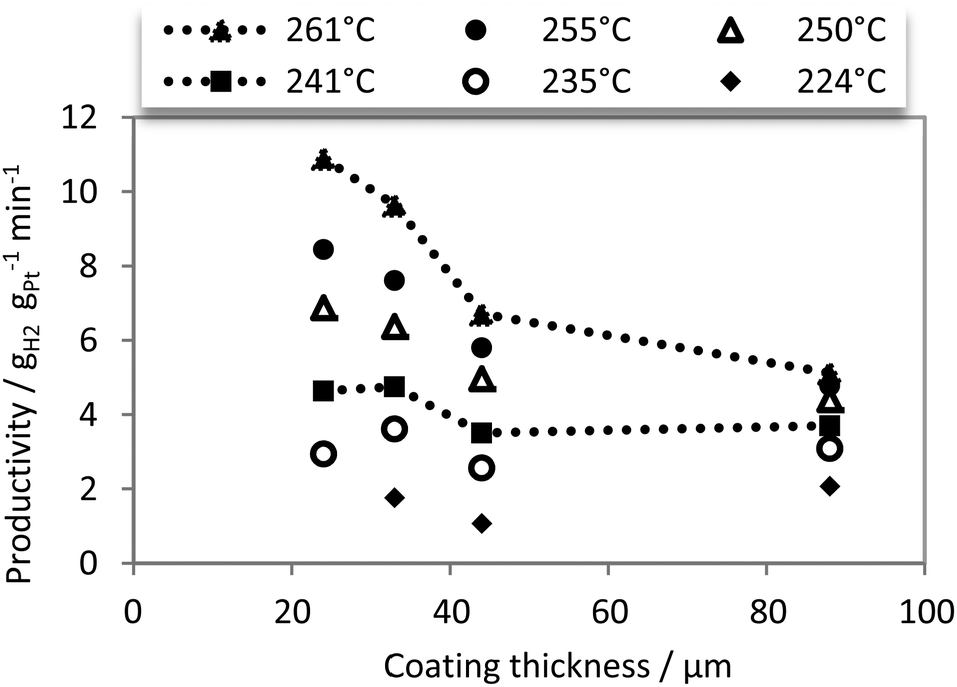 | ||
| Fig. 7 Productivity as a function of diffusion length: reaction temperature: 224–261 °C; H12-NEC flow rate 0.6 mL min−1; catalysts: 24 μm, 33 μm, 44 μm, 88 μm; 30 catalyst spheres; Pt mass: 0.36–1.87 mg. | ||
In good agreement with an increasing pore diffusion influence with increasing reaction temperature, a significant improvement of productivity from thick to thin coatings is observed for temperatures above 250 °C. This indicates a mass transport limitation for high temperatures.
Observable activation energy EA
For a more detailed discussion of the obtained kinetic data, Fig. 8 shows the obtained Arrhenius plot derived from all experimental data of this study.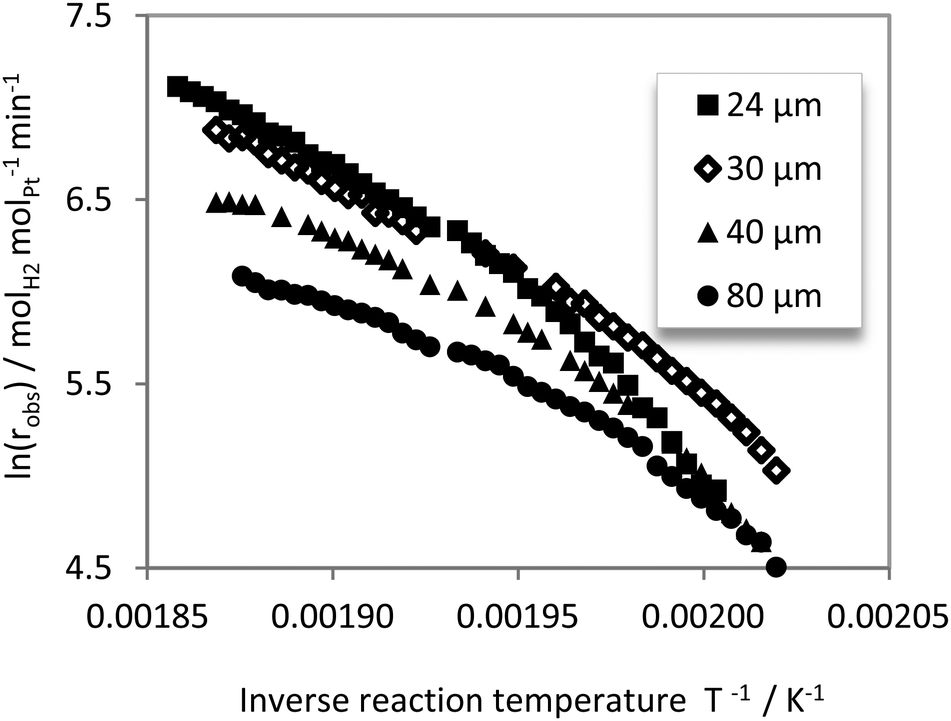 | ||
| Fig. 8 Arrhenius plot derived from all experimental data obtained in this study: reaction temperatures: 224–261 °C; H12-NEC flow rate 0.6 mL min−1. | ||
All catalysts under investigation display a clear dependency of the slope in the Arrhenius plot (activation energy) on the reaction temperature. For low temperatures, a much steeper slope has been determined than for higher temperatures, indicating mass transport limitation at higher reaction temperatures. Notably, the catalyst with the thickest coating (88 μm) shows a flatter slope across the considered temperature range compared to catalysts with thinner coatings. With thinner coatings the transition temperatures from kinetic to mass transfer influenced regime moves to higher temperatures, as expected. All these results clearly indicate a strong influence of pore diffusion on the observable reaction rates. This is discussed in detail for the 24 μm catalyst and the 88 μm catalyst in the following. The kinetic data have been evaluated using a graphical fit procedure to determine the different slopes in the Arrhenius plot. All slopes have been determined from at least eight data points with a fit accuracy of at least 98%.
In case of the 24 μm catalyst three different regimes with three different slopes were determined. A kinetic regime (EA,obs = 204 kJ molH2−1) is followed at temperatures higher than 235 °C by a narrow transition range. From 245 °C on, the system is in a pore diffusion regime as indicated by an EA,obs of 81 kJ molH2−1 (see Fig. 9).
 | ||
| Fig. 9 Arrhenius plot for the 24 μm catalyst: reaction temperature: 224–261 °C; H12-NEC flow rate 0.6 mL min−1; active layer of catalyst: 24 μm; 30 catalyst spheres; Pt mass 0.36 mg; H2-yield <33%; constant slopes (kinetic vs. pore diffusion regime) are displayed as dashed lines. | ||
The Arrhenius plot for the 88 μm catalyst displays lower activation energies (see Fig. 10). A regime limited only by the intrinsic kinetics was not observed in this case down to a reaction temperature of 224 °C. The observed slope in the lower temperature range (EA,obs = 144 kJ molH2−1) is similar to the one found for the transition from kinetic to pore diffusion limitation in case of the 24 μm catalyst (EA,obs = 130 kJ molH2−1, Fig. 9). For the catalyst with the 88 μm active layer, the pore diffusion regime (EA,obs = 87 kJ molH2−1) was observed above T = 233 °C. Above 247 °C a stronger flattening of the curve indicates a growing influence of external mass transport limitation on the observable kinetics (EA,obs = 50 kJ molH2−1).
 | ||
| Fig. 10 Arrhenius plot for the 88 μm catalyst: reaction temperature: 224–261 °C; H12-NEC flow rate 0.6 mL min−1; active layer of catalyst: 88 μm; 30 catalyst spheres; Pt mass = 1.87 mg; H2-yield <33%; constant slopes (pore diffusion regime vs. transition regimes) are displayed as dashed lines. | ||
All these results highlight the relevance of mass transfer influences on the observable kinetics in H12-NEC dehydrogenation. Even for the catalyst with a coating thickness of as low as 24 μm, intrinsic kinetics are only observable below a temperature of 235 °C. To avoid pore diffusion influences two different options are straight forward: (a) to reduce further on the thickness of the catalyst layer; however, this would lead to lower volumetric productivity in the reactor as less and less active catalyst would be provided per reactor volume. (b) To lower the temperature below 224 °C for the catalyst materials with thicker coatings, however, this would lead to lower hydrogen evolution due to the strong temperature dependency of dehydrogenation kinetics (as evidenced by the high EA in the kinetic regime).
All activation energies given in Table 2 were determined with at least three different catalyst materials with only little deviations (below 5%).
| Regime | Kinetic | Transition | Pore diffusion | Transition |
|---|---|---|---|---|
| Detected in catalyst | 24/33/44 μm | 24/33/44/88 μm | 24/33/44/88 μm | 88 μm |
| E A/kJ molH2−1 | 203 | 136 | 85 | 50 |
| E A deviation/% | 2.7 | 5.6 | 4.1 | — |
| k 0/L molPt−1 min−1 | 5.25 × 1022 | 8.60 × 1015 | 2.55 × 1010 | 4.32 × 1006 |
Our data on the different mass transfer influences as a function of temperature and catalyst coating thicknesses are summarized in Fig. 11. Exponential fits have been applied to describe the transition temperatures between the different regimes, which is reasonable due to the exponential dependency of reaction kinetics on temperature. The obtained fits were very accurate indicating (a) that the quality of synthesised egg-shell catalysts was obviously very high (homogeneity in thickness and texture); (b) that the collected kinetic data is of high quality.
 | ||
| Fig. 11 Reaction regime map: reaction temperature: 224–261 °C; H12-NEC flow rate 0.6 mL min−1; catalysts: 24 μm, 33 μm, 44 μm, 88 μm; 30 catalyst spheres; platinum mass: 0.36–1.87 mg. | ||
Another conclusion from the data in Fig. 11 is that for each catalyst layer thickness the temperature regimes for different kinetic and diffusional regimes are different. Consequently, proper combinations of layer thickness and reaction temperature have to be adjusted for the operation mode of choice.
Finally, it should be noted that even so we could describe the kinetics of H12-NEC dehydrogenation with heterogeneous Pt-catalysts quite satisfactorily using standard evaluation techniques of reaction engineering other phenomena might influence the transport of substrates and heat within the catalyst. For example, it is well possible that the observed effective kinetics is influenced by oscillations in the porous system caused by the significant amount of hydrogen gas formed in the catalyst. Datsevich, et al. have reported similar phenomena in the past for heterogeneously catalyzed liquid phase reactions involving hydrogen gas.57
Conclusion
Perhydro-N-ethylcarbazole (H12-NEC)/N-ethylcarbazole (NEC) is considered as an attractive Liquid Organic Hydrogen Carrier (LOHC) system. The storage of renewable electrical energy in the form of chemically bound hydrogen using this system has been evaluated by several papers and found an interesting concept for energy transportation18 and decentralized combined heat and power applications.19,58 This justifies detailed kinetic studies of the catalytic hydrogenation and dehydrogenation of the respective hydrogen-lean and hydrogen-rich storage compounds. This contribution deals with the dehydrogenation of H12-NEC and highlights in detail for the first time mass transfer effects that influence and limit the observed dehydrogenation kinetics. For this purpose, different dehydrogenation catalyst materials with defined active layer thicknesses have been prepared. The materials under investigation consisted of an inert non-porous α-alumina core and an active, porous γ-alumina shell of defined thickness, impregnated with platinum. Catalyst pellets with active coatings thicknesses between 24 and 88 μm were realized by a hybrid sol–gel suspension process with a filler particle size of 2–5 μm. The coatings have been prepared on the α-alumina core using a spray pulse technique.The catalysts were successfully tested in H12-NEC dehydrogenation in a temperature programmed reaction mode resulting in highly resolved kinetic data. By recording hydrogen release between 220 and 260 °C at different H12-NEC feed flow rates, four different reaction regimes have been identified. Remarkably, for all catalyst coating thicknesses limitations by mass transfer processes were identified in the temperature range under investigation. Depending on the thickness, limitations were detected at different transition temperatures with thinner layers leading to higher temperatures for the same type of transition. Even for the thinnest active layer (24 μm) influence of pore diffusion has been found for temperatures above 235 °C.
The intrinsic activation energy (203 kJ molH2−1) of the Pt-catalysed H12-NEC dehydrogenation and the activation energy for the pore diffusion regime (85 kJ molH2−1) have been determined. By minimizing pore diffusion limitation by thin catalyst layers, a maximum productivity of 10.9 gH2 gPt−1 min−1 (TOF = 17.5 molH2 molPt−1 s−1) was achieved at hydrogen yields of 20.9% for a catalyst with a 33 μm coating. This value is much higher than earlier reported productivities in the literature (7.7 gH2 gPt−1 min−1, 350 °C, H2-yield = 15.7%). This difference highlights the role of pore diffusion influences on the reachable precious metal related productivity in LOHC dehydrogenation processes. From our experiments it can be anticipated that future industrial applications of the LOHC technology will strongly build on egg-shell catalyst designs for maximizing efficiency and reducing cost of the related hydrogen storage systems.
Acknowledgements
The authors acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) within the Excellence Cluster “Engineering of Advanced Materials” in the framework of the excellence initiative. The present work was also supported by BMW Forschung und Technik GmbH. We thank Christian Schmidt, Torsten Frank, Marco Di-Pierro and Martin Eypasch for helpful discussions and technical input. P.W. acknowledges support by the European Research Council (ERC) through his Advanced Investigator Grant (No. 267376).References
- J. P. Barton and D. G. Infield, IEEE Trans. Energy Conver., 2004, 19(2), 441 CrossRef.
- C. Abbey and G. Joos, IEEE Trans. Ind. Appl., 2007, 43(3), 769 CrossRef.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608 CrossRef CAS PubMed.
- U. Eberle, B. Müller and R. von Helmolt, Energy Environ. Sci., 2012, 5(10), 8780 Search PubMed.
- F. Crotogino, in Large-Scale Hydrogen Underground Storage for Securing Future Energy Supplies, ed. F. Crotogino, S. Donadei, U. Bünger and H. Landinger, Forschungszentrum Jülich GmbH, Zentralbibliothek, Verlag, Jülich, 2010 Search PubMed.
- A. Yamashita, M. Kondo, S. Goto and N. Ogami, SAE 2015 World Congress & Exhibition, SAE International, 400 Commonwealth Drive, Warrendale, PA, United States, 2015 Search PubMed.
- D. A. J. Rand and R. M. Dell, Hydrogen Distribution and Storage, Hydrogen Energy, RSC Publishing, Cambridge, UK, 2008, ch. 5 Search PubMed.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Chem. Ing. Tech., 2015, 87(4), 489 Search PubMed.
- G. Gahleitner, Int. J. Hydrogen Energy, 2013, 38, 2039 CrossRef CAS PubMed.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Chem. Ing. Tech., 2013, 85(4), 489 CrossRef CAS PubMed.
- M. Bornschlegl, M. Drechsel, S. Kreitlein and J. Franke, in Sustainable Automotive Technologies 2013, ed. J. Wellnitz, A. Subic and R. Trufin, Springer International Publishing, Cham, 2014, p. 227 Search PubMed.
- A. A. Shukla, S. Karmarkar and R. B. Biniwale, Int. J. Hydrogen Energy, 2012, 37(4), 3719 CrossRef CAS PubMed.
- R. B. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33(1), 360 CrossRef CAS PubMed.
- M. Ichikawa and G. Walker, Organic liquid carriers for hydrogen storage, Solid State Hydrogen Storage, Woodhead Publishing Limited, Cambridge, England, 2008, ch. 18 Search PubMed.
- A. C. Cooper and G. P. Pez, DOE Hydrogen Programm, 2006.
- J. Graetz, D. Wolstenholme, G. P. Pez, L. Klebanoff, S. McGrady and A. C. Cooper, Development of Off-Board Reversible Hydrogen Storage Materials, Hydrogen Storage Technologies/Materials and Applications, CRC Press, 2013, ch. 8 Search PubMed.
- A. C. Cooper, A. R. Scott, F. Donald, J. Cunningham, M. Ford, F. C. Wilhelm, V. Monk, H. Cheng and G. P. Pez, DOE Hydrogen Programm, 2008, vol. 6, p. 602 Search PubMed.
- D. Teichmann, W. Arlt and P. Wasserscheid, Int. J. Hydrogen Energy, 2012, 37(23), 18118 CrossRef CAS PubMed.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4(8), 2767 CAS.
- G. W. H. Scherer, Systems and Economic Analysis of the Seasonal Storage of Electricity with Liquid Organic Hydrides, Dissertation, Zurich, 1997 Search PubMed.
- M. R. Usman, Energy Sources, 2011, 33, 2231 CrossRef CAS PubMed.
- J. K. Ali and A. Baiker, Appl. Catal., A, 1997, 155(1), 41 CrossRef CAS.
- F. Alhumaidan, D. L. Cresswell and A. Garforth, Ind. Eng. Chem. Res., 2011, 50, 2509 CrossRef CAS.
- N. Brückner, K. Obesser, A. Bösmann, D. Teichmann, W. Arlt, J. Dungs and P. Wasserscheid, ChemSusChem, 2014, 7(1), 229 CrossRef PubMed.
- M. Sobota, I. Nikiforidis, M. Amende, B. S. Zanón, T. Staudt, O. Höfert, Y. Lykhach, C. Papp, W. Hieringer, M. Laurin, D. Assenbaum, P. Wasserscheid, H.-P. Steinrück, A. Görling and J. Libuda, Chem. – Eur. J., 2011, 17(41), 11542 CrossRef CAS PubMed.
- F. Sotoodeh and K. J. Smith, J. Catal., 2011, 279(1), 36 CrossRef CAS PubMed.
- W. Peters, M. Eypasch, T. Frank, J. Schwerdtfeger, C. Körner, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2015, 8(2), 641 CAS.
- S. P. Verevkin, V. N. Emel’yanenko, A. A. Pimerzin and E. E. Vishnevskaya, J. Phys. Chem. A, 2011, 115(10), 1992 CrossRef CAS PubMed.
- S. P. Verevkin, V. N. Emel’yanenko, A. Heintz, K. Stark and W. Arlt, Ind. Eng. Chem. Res., 2012, 51(37), 12150 CrossRef CAS.
- F. Sotoodeh, L. Zhao and K. J. Smith, Appl. Catal., A, 2009, 362(1–2), 155 CrossRef CAS PubMed.
- F. Sotoodeh and K. J. Smith, J. Phys. Chem. C, 2013, 117(1), 194 CAS.
- F. Sotoodeh, B. J. M. Huber and K. J. Smith, Int. J. Hydrogen Energy, 2012, 37(3), 2715 CrossRef CAS PubMed.
- M. Yang, Y. Dong, S. Fei, H. Ke and H. Cheng, Int. J. Hydrogen Energy, 2014, 39, 18976 CrossRef CAS PubMed.
- P. Avila, M. Montes and E. E. Miró, Chem. Eng. J., 2005, 109(1–3), 11 CrossRef CAS PubMed.
- V. Tomašić, Catal. Today, 2007, 119(1-4), 106 CrossRef PubMed.
- P. Jiang, G. Lu, Y. Guo, Y. Guo, S. Zhang and X. Wang, Surf. Coat. Technol., 2005, 190, 314 CrossRef CAS PubMed.
- T. Schwarz, S. Schirrmeister, H. Döring and E. Klemm, Chem. Ing. Tech., 2010, 82(6), 921 CrossRef CAS PubMed.
- C. Agrafiotis, A. Tsetsekou, C. J. Stournaras, A. Julbe, L. Dalmazio, C. Guizard, G. Boretto, M. Debenedetti and F. Parussa, Appl. Catal., B, 2001, 34(2), 149 CrossRef CAS.
- C. Agrafiotis and A. Tsetsekou, J. Eur. Ceram. Soc., 2002, 22(4), 423 CrossRef CAS.
- C. Agrafiotis, A. Tsetsekou, C. J. Stournaras, A. Julbe, L. Dalmazio and C. Guizard, J. Eur. Ceram. Soc., 2002, 22(1), 15 CrossRef CAS.
- P. Kerleau, Y. Swesi, V. Meille, I. Pitault and F. Heurtaux, Catal. Today, 2010, 157(1–4), 321 CrossRef CAS PubMed.
- C. J. Brinker, G. C. Frye, A. J. Hurd and C. S. Ashley, Thin Solid Films, 1991, 201(1), 97 CrossRef CAS.
- C. J. Brinker and A. J. Hurd, J. Phys. III, 1994, 4, 1231 CrossRef.
- V. Meille, S. Pallier, B. Santa Cruz, V. Gabriela, M. Roumanie and J.-P. Reymon, Appl. Catal., A, 2005, 286, 232 CrossRef CAS PubMed.
- V. Meille, S. Pallier and P. Rodriguez, Colloids Surf., A, 2009, 336, 104 CrossRef CAS PubMed.
- V. Meille, Appl. Catal., A, 2006, 315, 1 CrossRef CAS PubMed.
- C. Agrafiotis and A. Tsetsekou, J. Eur. Ceram. Soc., 2000, 20, 815 CrossRef CAS.
- S. Ould-Chikh, N. Brodusch, N. Crozet, M. Hemati and L. Rouleau, Powder Technol., 2013, 237, 255 CrossRef CAS PubMed.
- C. Agrafiotis, Solid State Ionics, 2000, 136–137(1–2), 1301 CrossRef CAS.
- G. W. H. Scherer, J. Am. Ceram. Soc., 1990, 73, 3 CrossRef CAS PubMed.
- M. Valentini, G. Groppi, C. Cristiani, M. Levi, E. Tronconi and P. Forzatti, Catal. Today, 2001, 69(1–4), 307 CrossRef CAS.
- S. Ould-Chikh, B. Celse, M. Hemati and L. Rouleau, Powder Technol., 2009, 190(1–2), 19 CrossRef CAS PubMed.
- Saint-Gobain NorPro, Denstone Support Media, 2005.
- Sasol Germany GmbH, DISPERAL: Product Information Data Sheet, Hamburg, Germany, 2007 Search PubMed.
- Sasol Germany GmbH, PURALOX-CATALOX.GB - High purity activated aluminas, Hamburg, Germany, 2007 Search PubMed.
- N. Kariya, A. Fukuoka, T. Utagawa, M. Sakuramoto, Y. Goto and M. Ichikawa, Appl. Catal., A, 2003, 247(2), 247 CrossRef CAS.
- L. B. Datsevich, Appl. Catal., A, 2004, 262(2), 149 CrossRef CAS PubMed.
- D. Teichmann, K. Stark, K. Müller, G. Zöttl, P. Wasserscheid and W. Arlt, Energy Environ. Sci., 2012, 5(10), 9044 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02024g |
| This journal is © The Royal Society of Chemistry 2015 |