
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient polymer solar cells cast from non-halogenated xylene/anisaldehyde solution
Christian
Sprau
a,
Felix
Buss
b,
Michael
Wagner
a,
Dominik
Landerer
a,
Manuel
Koppitz
a,
Alexander
Schulz
a,
Daniel
Bahro
a,
Wilhelm
Schabel
b,
Philip
Scharfer
b and
Alexander
Colsmann
*a
aLight Technology Institute, Karlsruhe Institute of Technology (KIT), Engesserstrasse 13, 76131 Karlsruhe, Germany. E-mail: christian.sprau@kit.edu; alexander.colsmann@kit.edu
bInstitute of Thermal Process Engineering, Thin Film Technology, Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
First published on 16th July 2015
Abstract
Several high performance polymer:fullerene bulk-heterojunction photo-active layers, deposited from the non-halogenated solvents o-xylene or anisole in combination with the eco-compatible additive p-anisaldehyde, are investigated. The respective solar cells yield excellent power conversion efficiencies up to 9.5%, outperforming reference devices deposited from the commonly used halogenated chlorobenzene/1,8-diiodooctane solvent/additive combination. The impact of the processing solvent on the bulk-heterojunction properties is exemplified on solar cells comprising benzodithiophene-thienothiophene co-polymers and functionalized fullerenes (PTB7:PC71BM). The additive p-anisaldehyde improves film formation, enhances polymer order, reduces fullerene agglomeration and shows high volatility, thereby positively affecting layer deposition, improving charge carrier extraction and reducing drying time, the latter being crucial for future large area roll-to-roll device fabrication.
Broader contextOrganic bulk-heterojunction solar cells are on the cusp of commercialization. Printing and coating techniques are widely considered enablers of low-cost solar module fabrication with excellent carbon footprints. For the transfer of lab-scale processes to an environmentally friendly and sustainable industrial large-scale fabrication of polymer solar cells by printing, non-halogenated solvents and processing additives are mandatory prerequisites. As the choice of solvents is pivotal to the complex formation of the bulk-heterojunction and hence to the device performance, both academia and the solar industry have fostered strong research on “green” device processing in order to advance the market-readiness of organic solar modules. |
1. Introduction
Recent efforts to push organic photovoltaics towards market-readiness mainly focused on the development of new photo-active polymers and intensive optimization of lab-scale devices, nowadays yielding power conversion efficiencies (PCEs) beyond 10%.1 Yet, the main challenge will be the transfer of the optimized lab-scale processes to large-area industrial solar cell fabrication.2,3 In contrast to lab-processing, the use of non-halogenated, eco-friendly and non-hazardous solvents for the processing of the photo-active layers is pivotal for any industrial fabrication. Common lab-scale research utilizes halogenated solvents, such as chlorobenzene (CB), o-dichlorobenzene (o-DCB) or chloroform, which typically show excellent solubility of most photo-active polymers. Unfortunately, the large-scale use of those solvents has a dramatic impact on the environment and is hazardous to the human health. Therefore, a strong need for alternative solvents has been recognized by the community, recently focusing on novel high-performance polymer:fullerene bulk-heterojunction (BHJ) solar cells, deposited from non-chlorinated mono-, bis- or tris-substituted benzene analogues.4–11 While most reports in the literature have focused on the eco-friendliness of the main solvents, little attention has been paid to solvent additives which are often used to control the film drying in order to yield a favorable nano-morphology. Indisputably, the morphology of organic solar cells depends on the processing conditions and critically determines the basic physical processes in the photo-active layer, including light absorption, exciton dissociation, charge carrier transport, charge carrier extraction and recombination. Typically, solvent additives have a higher boiling point than the main solvent and can show selective solubility of one of the two blend components,12 with the complex BHJ formation processes still being discussed.13–16 Most additive studies evolved around the use of alkanedithiols and halogenated alkanes.12,17 For the deposition of most novel polymer:fullerene BHJs, the iodated solvent additive 1,8-diiodooctane (DIO) is used. Alternative additives such as the chlorinated 1-chloronaphthalene (CN) or the non-halogenated 1-methylnaphthalene (MN), 1,2,3,4-tetrahydronaphthalene (THN), diphenyl ether (DPE) and 1-methyl-2-pyrrolidone (NMP) have been studied,5,7,8,10,18–20 however, all-together disregarding health and environmental hazards (Table 1).| Additive | Hazard statements |
|---|---|
| DIO | H413 chronic aquatic toxicity |
| CN | H302 acute toxicity, oral |
| H315 skin irritation | |
| H319 eye irritation | |
| H335 specific target organ toxicity – single exposure | |
| H400 acute aquatic toxicity | |
| MN | H302 acute toxicity, oral |
| H315 skin irritation | |
| H319 eye irritation | |
| H334 respiratory sensitisation | |
| H335 specific target organ toxicity – single exposure | |
| H411 chronic aquatic toxicity | |
| THN | H315 skin irritation |
| H319 eye irritation | |
| H351 carcinogenicity | |
| H304 aspiration hazard | |
| H411 chronic aquatic toxicity | |
| DPE | H319 eye irritation |
| H411 chronic aquatic toxicity | |
| NMP | H315 skin irritation |
| H319 eye irritation | |
| H360 reproductive toxicity | |
| H335 specific target organ toxicity – single exposure | |
| AA | None |
Besides environmental concerns, more volatile solvent additives than DIO may be beneficial for future roll-to-roll processing due to reduced drying times and temperatures.
In this work, we investigate the deposition of various high-performing polymer:fullerene BHJs from the non-halogenated and production relevant solvents o-xylene or anisole in combination with the eco-compatible additive p-anisaldehyde (AA). The respective solar cells yield PCEs outperforming devices fabricated from common halogenated solvent/additive combinations such as CB and DIO.
2. Experimental
Device fabrication
Organic solar cells were fabricated according to the device architecture depicted in Fig. 1a. Patterned indium tin oxide (ITO, R□ ≈ 15 Ω□) coated glass substrates were cleaned by sequential ultrasonication in acetone and isopropanol (10 min). Then the substrates were transferred into a nitrogen glovebox for the fabrication and characterization process. A zinc oxide (ZnO) electron extraction layer was spin cast from nanoparticle dispersion (Nanograde Ltd, 1 wt% in isopropanol, 4000 rpm, 30 s) and thermally annealed on a hotplate (80 °C, 10 min). Poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7, 1-Material Inc., Mw = 125 kg mol−1, ĐM = 2.5) and [6,6]-phenyl C71-butyric acid methyl ester (PC71BM, Solenne, 99%) were dissolved (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 w/w, polymer concentration 10 g L−1) in either chlorobenzene (CB, anhydrous, 99.8%,), o-xylene (anhydrous, 97%) or anisole (anhydrous, 99.7%) and stirred overnight at elevated temperatures (CB: 50 °C, o-xylene and anisole: 85 °C). All solvents and additives were used as purchased (Sigma-Aldrich). The solvent additives, either 1,8-diiodooctane (DIO, 98%) or p-anisaldehyde (AA, 98%), were then added to the stock solution. The non-filtered warm solution was spun onto the samples (1500 rpm, 60 s) to form the photo-active layer. After initial film drying at room temperature (10 min), the films were annealed on a hotplate (60 °C, 20 min) to remove the residual additive. Then the devices were transferred into a vacuum chamber (base pressure 10−6 mbar) to deposit the molybdenum oxide hole extraction layer (MoO3, 10 nm) and the silver top electrode (Ag, 100 nm) by thermal evaporation, using a shadow mask defining the active solar cell area (3 × 3.5 mm2). The same device architecture and fabrication procedure were employed to investigate all other polymer:PC71BM photo-active layers. Poly[[4,8-bis[5-(2-ethylhexyl)thiophen-2-yl]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7-Th, 1-Material Inc., Mw = 108 kg mol−1, ĐM = 2.5), poly[[2,7-(5,5-bis-(3,7-dimethyloctyl)-5H-dithieno[3,2-b:2′,3′-d]pyran)-alt-4,7-(5,6-difluoro-2,1,3-benzothiadiazole)]] (PDTP-DFBT, 1-Material Inc., Mw = 37 kg mol−1, ĐM = 2.4) and poly[(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di(2-octyldodecyl)-2,2′;5′,2′′;5′′,2′′′-quaterthiophen-5,5′′′-diyl)] (PffBT4T-2OD, 1-Material Inc., Mw = 100 kg mol−1, ĐM = 2.5) were dissolved and mixed with PC71BM according to the blend ratios and polymer concentrations listed in Table 4. All solids were sufficiently dissolved in all solvents and the solutions were deposited without filtering. PTB7-Th:PC71BM solutions and films were prepared following the PTB7:PC71BM deposition protocol. PDTP-DFBT:PC71BM solutions were stored overnight on a hotplate (85 °C) and spun (1200 rpm, 60 s; 800 rpm from o-DCB solution) from warm solution (60 °C). PffBT4T-2OD:PC71BM solutions were stirred overnight (110 °C) and spun (800 rpm, 120 s) onto warm substrates (solution and substrates were stored on a 95 °C hotplate prior to spin casting) followed by subsequent annealing (80 °C, 5 min).
:1.5 w/w, polymer concentration 10 g L−1) in either chlorobenzene (CB, anhydrous, 99.8%,), o-xylene (anhydrous, 97%) or anisole (anhydrous, 99.7%) and stirred overnight at elevated temperatures (CB: 50 °C, o-xylene and anisole: 85 °C). All solvents and additives were used as purchased (Sigma-Aldrich). The solvent additives, either 1,8-diiodooctane (DIO, 98%) or p-anisaldehyde (AA, 98%), were then added to the stock solution. The non-filtered warm solution was spun onto the samples (1500 rpm, 60 s) to form the photo-active layer. After initial film drying at room temperature (10 min), the films were annealed on a hotplate (60 °C, 20 min) to remove the residual additive. Then the devices were transferred into a vacuum chamber (base pressure 10−6 mbar) to deposit the molybdenum oxide hole extraction layer (MoO3, 10 nm) and the silver top electrode (Ag, 100 nm) by thermal evaporation, using a shadow mask defining the active solar cell area (3 × 3.5 mm2). The same device architecture and fabrication procedure were employed to investigate all other polymer:PC71BM photo-active layers. Poly[[4,8-bis[5-(2-ethylhexyl)thiophen-2-yl]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7-Th, 1-Material Inc., Mw = 108 kg mol−1, ĐM = 2.5), poly[[2,7-(5,5-bis-(3,7-dimethyloctyl)-5H-dithieno[3,2-b:2′,3′-d]pyran)-alt-4,7-(5,6-difluoro-2,1,3-benzothiadiazole)]] (PDTP-DFBT, 1-Material Inc., Mw = 37 kg mol−1, ĐM = 2.4) and poly[(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di(2-octyldodecyl)-2,2′;5′,2′′;5′′,2′′′-quaterthiophen-5,5′′′-diyl)] (PffBT4T-2OD, 1-Material Inc., Mw = 100 kg mol−1, ĐM = 2.5) were dissolved and mixed with PC71BM according to the blend ratios and polymer concentrations listed in Table 4. All solids were sufficiently dissolved in all solvents and the solutions were deposited without filtering. PTB7-Th:PC71BM solutions and films were prepared following the PTB7:PC71BM deposition protocol. PDTP-DFBT:PC71BM solutions were stored overnight on a hotplate (85 °C) and spun (1200 rpm, 60 s; 800 rpm from o-DCB solution) from warm solution (60 °C). PffBT4T-2OD:PC71BM solutions were stirred overnight (110 °C) and spun (800 rpm, 120 s) onto warm substrates (solution and substrates were stored on a 95 °C hotplate prior to spin casting) followed by subsequent annealing (80 °C, 5 min).
 | ||
| Fig. 1 (a) Solar cell device architecture. (b) Active layer components PTB7 and PC71BM. (c) Chemical structures of the solvents and additives investigated in this work. | ||
Characterization
Current density–voltage (J–V) curves were measured using a source-meter unit (Keithley 238) under illumination from a spectrally monitored solar simulator (Oriel 300 W, 1000 W m−2, ASTM AM 1.5G), calibrated by a KG5 filtered silicon reference cell (91150-KG5, Newport). Layer thicknesses were measured using a tactile stylus profiler (Dektak XT, Bruker). Absorbance spectra were recorded using a UV-Vis-NIR spectrophotometer (Cary 5000, Agilent Technologies) in two-beam transmission mode. The respective photo-active layers on glass substrates were prepared following the procedure described above. Atomic force microscopy (AFM, Dimension ICON, Bruker) images were recorded on solar cells next to the top electrodes in tapping mode (TESP tip).QCM measurements
Quartz Crystal Microbalance (QCM) measurements were carried out at 25 °C in a temperature-controlled chamber equipped with a Q-Sense ALD holder connected to a Q-Sense E1 system. Quartz crystals with a gold surface (QSense QSX301) were cleaned by sequential ultrasonication in toluene (9 min) and ethanol (9 min). Prior to coating, the resonant frequencies of the uncoated crystals were determined. For each drying experiment, the PTB7:PC71BM solution (50 μl) was spin coated onto a quartz crystal (3000 rpm, 60 s) under ambient conditions at room temperature. Immediately after coating or after a subsequent heating step on a hot plate (1 min, 60 °C), the crystals were transferred to the measurement chamber, where the drying process of the film was monitored by recording the frequency change Δfn/n of the first five harmonics (n = 1, 3, 5, 7, 9) over time. For the calculation of the areal mass, the Sauerbrey equation was used: | (1) |
3. Results and discussion
3.1 Device performance
In order to allow comparison with the literature, we initially studied organic solar cells comprising the well investigated polymer:fullerene combination PTB7:PC71BM as high performing model systems, enabling PCEs to exceed 7%.22 Therefore, we utilized the inverted device architecture glass/ITO/ZnO/PTB7:PC71BM/MoO3/Ag depicted in Fig. 1. The photo-active layer was spin cast from either o-xylene (boiling point bp = 144 °C), anisole (bp = 154 °C) or, for reference, chlorobenzene (CB, bp = 132 °C). Fig. 2a shows the corresponding current density–voltage (J–V) curves. The key performance data short-circuit current density (Jsc), fill factor (FF), open circuit voltage (Voc) and PCE are summarized in Table 2. The moderate photovoltaic device performances (PCE ≈ 3%), when using pure solvents, are in accordance with the common literature and reflect the solar cells' moderate Jsc and FF.15,22–24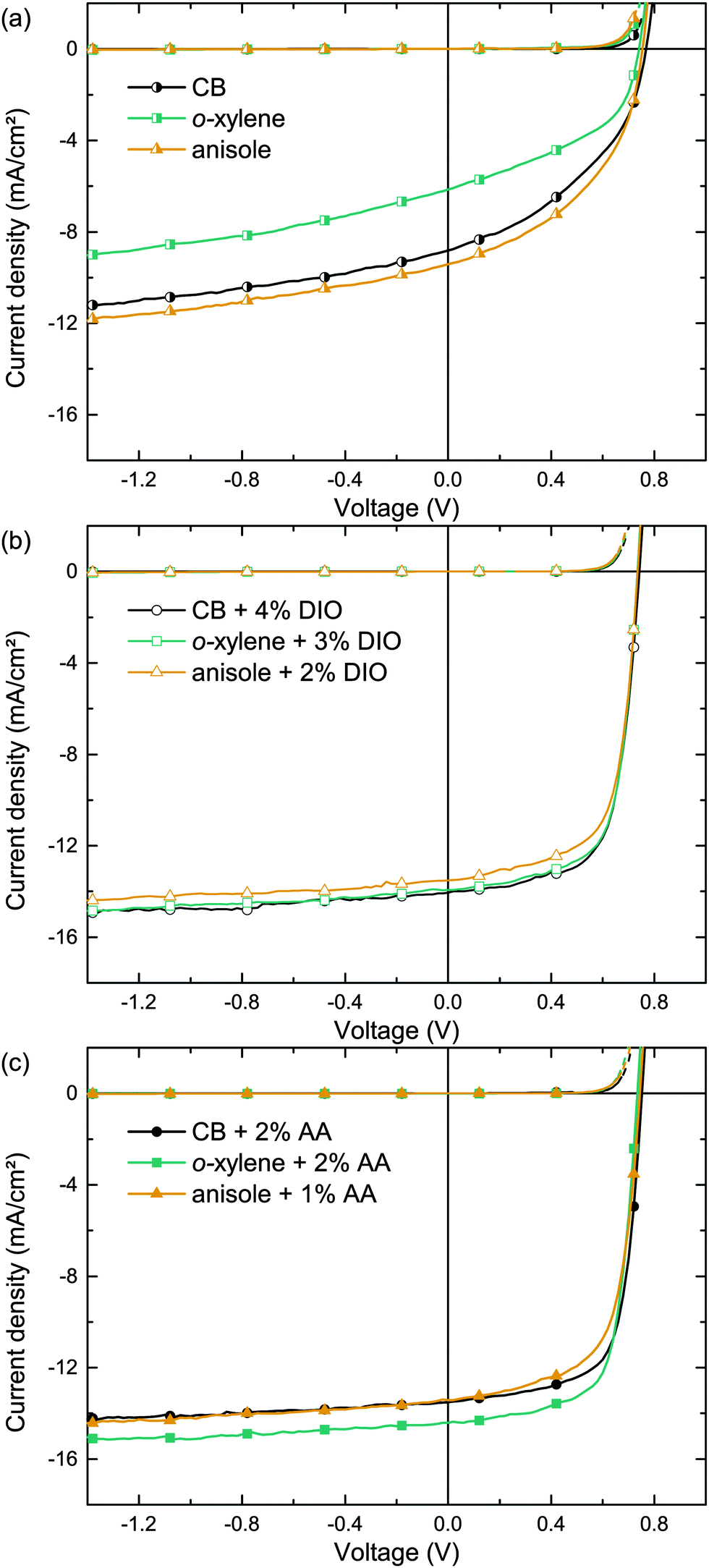 | ||
| Fig. 2 Typical J–V curves of the solar cells under AM1.5 illumination (solid lines) and in the dark (dashed lines) with PTB7:PC71BM photo-active layers spin cast from (a) pure solvents, (b) solvent/DIO mixtures and (c) solvent/AA mixtures. The amount of the additive has been optimized for each solvent/additive combination individually (data not provided here). 1 out of 15 data points is marked with a symbol to guide the eye. | ||
| Main solvent | Additive | t (nm) | J SC (mA cm−2) | V OC (mV) | FF (%) | PCEa (%) | R q (nm) | α 1/α2 |
|---|---|---|---|---|---|---|---|---|
| a PCE of the best performing devices in parentheses. | ||||||||
| CB | — | 90 | 8.8 ± 0.1 | 762 ± 7 | 42 ± 1 | 2.8 ± 0.1 (2.9) | 5.1 | 1.03 |
| o-Xylene | — | 90 | 6.2 ± 0.2 | 736 ± 5 | 45 ± 1 | 2.0 ± 0.6 (2.2) | 12.7 | 0.96 |
| Anisole | — | 80 | 9.3 ± 0.2 | 750 ± 1 | 46 ± 2 | 3.2 ± 0.2 (3.5) | 5.1 | 1.01 |
| CB | 4% DIO | 85 | 14.0 ± 0.1 | 739 ± 4 | 67 ± 2 | 6.9 ± 0.2 (7.3) | 2.5 | 1.07 |
| o-Xylene | 3% DIO | 85 | 14.2 ± 0.2 | 737 ± 5 | 67 ± 2 | 7.0 ± 0.2 (7.1) | 2.6 | 1.07 |
| Anisole | 2% DIO | 80 | 13.4 ± 0.2 | 738 ± 3 | 66 ± 1 | 6.5 ± 0.1 (6.7) | 1.9 | 1.08 |
| CB | 2% AA | 90 | 13.6 ± 0.6 | 748 ± 5 | 69 ± 2 | 7.0 ± 0.3 (7.5) | 2.7 | 1.07 |
| o-Xylene | 2% AA | 85 | 14.3 ± 0.1 | 738 ± 4 | 70 ± 1 | 7.4 ± 0.1 (7.6) | 2.4 | 1.07 |
| Anisole | 1% AA | 80 | 13.5 ± 0.2 | 747 ± 4 | 65 ± 1 | 6.6 ± 0.2 (6.9) | 4.1 | 1.05 |
Whereas BHJs from pure solvents did not yield efficient polymer solar cells, the addition of the processing additive DIO enhanced both Jsc and FF significantly. Fig. 2b depicts the J–V curves of solar cells deposited from CB, o-xylene and anisole with individually optimized DIO concentrations as noted in the graph. The corresponding key performance data are summarized in Table 2. The BHJ deposition from all three solvent/DIO combinations yields high PCEs of ≈7% due to an excellent FF >65% and a Jsc ≈ 14 mA cm−2. Whereas the solar cells that were deposited from pure solvents show different properties, the performance of all devices that were deposited utilizing the additive DIO yields very similar J–V curves. The voltage dependent current densities under reverse bias when using pure solvents indicate poor charge carrier extraction and field dependent recombination. In contrast, upon using the additive DIO, the current density under reverse bias saturates, indicating excellent extraction of free charges and low recombination losses even at low internal fields, being in good agreement with previous studies.24
This drastic device performance improvement upon using the additive DIO emphasizes the importance of the solvent additive for efficient PTB7:PC71BM solar cells. Although DIO is used in small amounts only, it is a hazardous substance that is not suitable for industrial large-area device processing. Furthermore, DIO residues are suspected to be remaining in the active layer after film formation, due to the high DIO boiling point (bp = 332.5 °C).
Being able to dissolve fullerenes in high concentrations25 and having a higher boiling point (>200 °C) than most main solvents, non-halogenated (substituted) benzaldehydes appear as an interesting alternative material class.26 Being a representative of this material class, we have focused on the eco-compatible additive AA (bp = 248 °C) to improve the formation of the photo-active layer. The J–V curves of the corresponding solar cells comprising photo-active layers deposited from CB, o-xylene or anisole with an individually optimized additive concentration are shown in Fig. 2c. The key performance data are summarized in Table 2. The drastic PCE enhancement over devices that were deposited from pure main solvents is comparable to the effect of DIO on the device performance. Best Jsc and FF were yielded upon BHJ deposition from o-xylene/AA, resulting in an average PCE of 7.4%.
We note that, with respect to future process upscaling, we have performed preliminary doctor blading experiments on PTB7:PC71BM in o-xylene/AA solution with the devices yielding similar performance (data not provided here).
3.2 Film characterization
In order to investigate the impact of the solvent/additive combinations on the film formation, we first analyzed the absorption of PTB7:PC71BM photo-active layers. Fig. 3 shows the absorbance spectra, which were normalized to the PC71BM absorption peak at 377 nm, thereby improving the comparability of the spectra by eliminating minor thickness variations and haze effects. When cast from the pure main solvents CB, o-xylene, or anisole (Fig. 3a), a distinct difference in the polymer absorption profile can be observed: the two local absorption maxima at 680 nm (1) and 630 nm (2) show different relative amplitudes α1 and α2. The respective ratios of the 680 nm and 630 nm absorption maxima α1/α2 are listed in Table 2. Upon film deposition from CB or anisole, the 680 nm peak is more pronounced than the 630 nm peak, that is, the peak ratio α1/α2 is >1, whereas the absorbance of the o-xylene sample at 680 nm is lower than the absorbance at 630 nm and the long wavelength absorption shoulder is less pronounced. The long wavelength absorption shoulder of the polymer usually corresponds to π–π interaction, and a more pronounced absorption shoulder is attributed to an enhanced polymer order.27,28 We conclude that films cast from o-xylene may exhibit a lower degree of polymer order that hampers charge carrier transport and may therefore partly account for the lower FF and Jsc observed on these devices. Upon using either process additive, AA or, for reference, DIO (Fig. 3b), the absorbance spectra were equal within the measurement accuracy and the long wavelength shoulders of all solvent/additive combinations were equally pronounced. The increase in polymer order may foster an improved charge carrier transport and therefore yield a higher FF and Jsc which is reflected in the increased α1/α2 and which is most pronounced when deposited from o-xylene in combination with either additive.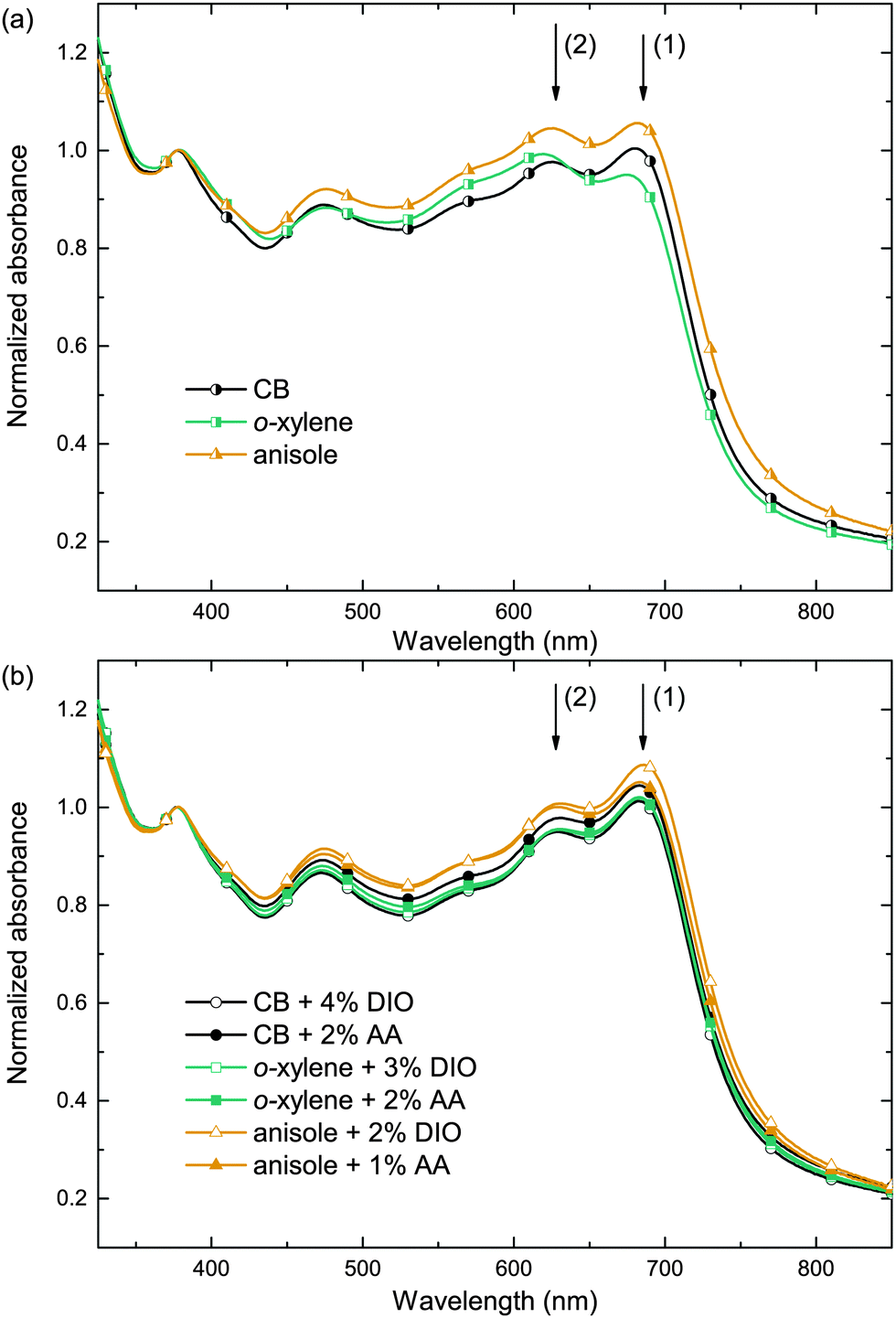 | ||
| Fig. 3 Absorbance spectra of the PTB7:PC71BM layer cast from (a) pure solvents and (b) solvent/additive mixtures. The spectra are normalized to the absorption peak of PC71BM at 377 nm. The arrows (1) and (2) indicate the main PTB7 absorption maxima. 1 out of 40 data points is marked with a symbol to guide the eye. | ||
To further analyze and compare the impact of the solvent additives DIO and AA on the film properties, we studied the film surface topography by atomic force microscopy (AFM) as depicted in Fig. 4. When depositing the PTB7:PC71BM film from the pure solvents CB, o-xylene or anisole, we observed protruding nano-spheres with a lateral size of about 150–200 nm and a height of approximately 10–20 nm above the average film surface. For the films cast from o-xylene, these features are even larger, measuring about 300 nm in diameter, with a typical height of about 30 nm. In contrast, when using the additives DIO or AA together with any of the three main solvents, these features are not visible in the AFM images and the surface is rather smooth.
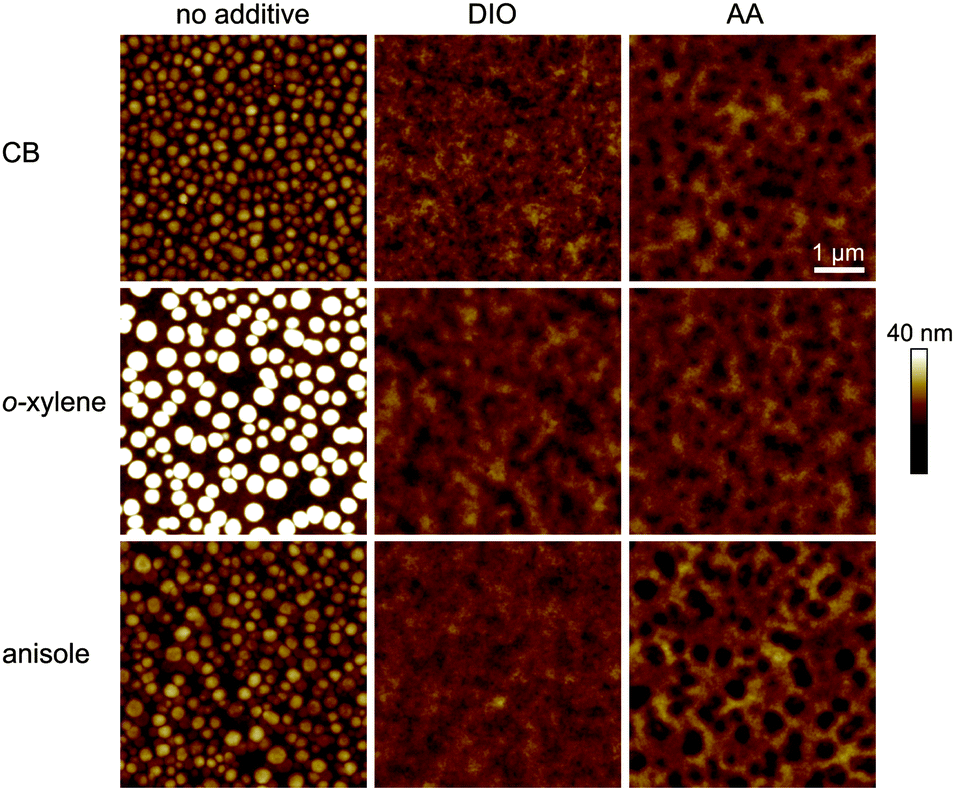 | ||
| Fig. 4 Topography (AFM, 5 × 5 μm2) of the PTB7:PC71BM photo-active layers cast from different solvent/additive combinations with individually optimized additive concentrations. The same color scale is used for all images. Whereas spherical (fullerene) agglomerates with a diameter of 100–300 nm are clearly visible in films cast from pure solvents, the surface is rather smooth upon addition of DIO or AA to the solution. | ||
These observations are in good agreement with earlier AFM and transmission electron microscopy (TEM) studies on PTB7:PC71BM films that were cast from CB or CB/DIO.15,22,23 By X-ray diffraction experiments, these spherical features in PTB7:PC71BM films were identified to be almost pure fullerene agglomerates embedded in a polymer:fullerene matrix, with the 200 nm agglomerates when cast from CB being reduced to 30 nm upon deposition from CB/DIO.23 In additive free bulk-heterojunctions, the fullerene agglomeration therefore leads to enhanced recombination, with the detailed processes still being discussed.23,24,29 These interpretations match our optoelectronic device study in Section 3.1, where we found reduced recombination as well as enhanced FF and Jsc for devices with photo-active layers that were deposited from solvent/DIO or solvent/AA.
We note that we found shallow depression features with a diameter of about 150–300 nm instead (depth 5–10 nm), upon film deposition from CB/AA, o-xylene/AA and, most pronounced, from anisole/AA. Taking into account the excellent performance of the corresponding solar cells, we attribute these features to surface inhomogeneities that form during the drying process rather than material separation.
3.3 Drying analysis
The low DIO vapor pressure at room temperature (pDIO* = 3.1 × 10−4 mbar at T = 25 °C)30 renders DIO practically useless for large-scale high-throughput fabrication of organic solar cells. Having about the same diffusion coefficient in air (DDIO = 5.4 × 10−6 m2 s−1, DAA = 7.1 × 10−6 m2 s−1 at T = 25 °C)31 but a higher vapor pressure (pAA* = 4.3 × 10−2 mbar at T = 25 °C) AA appears to be more suitable for fast and complete film drying.To analyze and compare the drying of PTB7:PC71BM/o-xylene solutions containing either additive, DIO or AA (2% v/v), we investigate the evaporation kinetics of additive residues from freshly prepared PTB7:PC71BM layers on a quartz crystal microbalance (QCM). On the QCM, we record the resonance frequencies over time. An increasing resonance frequency reflects a loss of mass due to solvent/additive evaporation. Fig. 5a and b shows the relative resonance frequency change of the first harmonics (n = 1, 3, 5, 7, 9) Δfn/n of both material systems over time. These resonance frequencies during additive evaporation are given relative to the resonance frequency of the uncoated crystal. We note that the time frame differs by a factor of 10, that is, 40 h for o-xylene/DIO and 4 h for o-xylene/AA. For the as-cast o-xylene/DIO film we find a linearly increasing Δf within the first 5 h of the experiment. During the early stages of drying, henceforth referred to as constant rate period, the thermodynamics at the vapor–liquid interface and the gas-phase mass transport are dominant. The observed linear increase is characteristic of single solvents that evaporate during the constant rate period, inferring that the high-boiling additive DIO is still present in the as-cast film whereas o-xylene has evaporated earlier, during spin casting. Simultaneous evaporation of two solvents would yield a drying curve that is either non-linear or features a kink at the transition from low- to high-boiling solvent dominated drying.32,33 At low additive content, that is, at a later stage of drying, which we henceforth refer to as the falling rate period, the diffusion coefficient in polymer solutions can decrease by orders of magnitudes, hampering the removal of trace solvent. The measurements were discontinued after 40 h when only little frequency variation was noted, that is, the film was effectively dry. The experiment was repeated with another film that was annealed on a hotplate (1 min at 60 °C) directly after spin coating, leading to a reduced constant rate period of ≈2 h. Here, after initial sample drying on the hotplate, the subsequent drying at 25 °C shows the same constant evaporation rate as the as-cast film, confirming the presence of only DIO in the film.
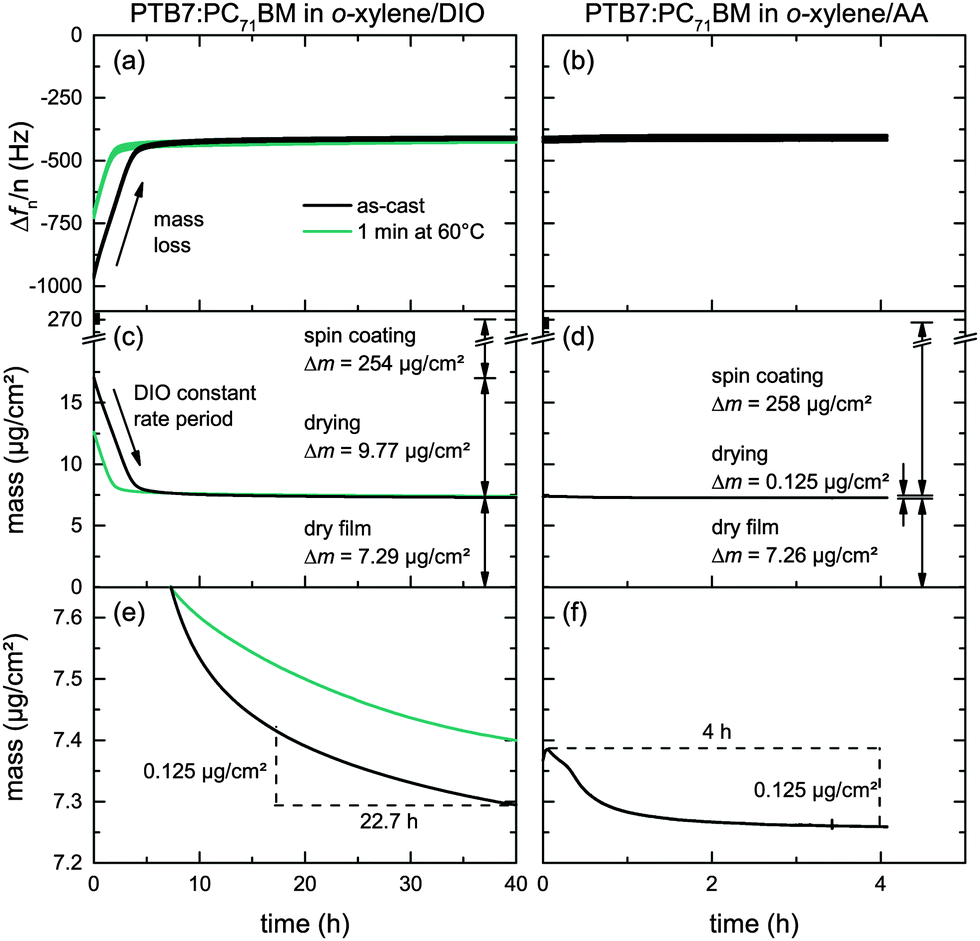 | ||
| Fig. 5 Drying characteristics of PTB7:PC71BM at 25 °C cast from o-xylene/DIO and o-xylene/AA (2% v/v). (a and b) QCM measurements after spin coating. The changes in the resonance frequencies reflect mass loss. (c and d) Drying curves calculated from the QCM frequency change using the Sauerbrey equation. Whereas only very little mass loss is observed for o-xylene/AA films, o-xylene/DIO films show a constant initial mass decrease, characteristic of evaporation of the additive at the vapor–liquid interface. (e and f) Zoom into the relevant mass regime of the falling rate period of the drying curve, where the mass loss is determined by the diffusion of the additive inside the PTB7:PC71BM matrix. AA diffuses faster than DIO. | ||
In contrast, QCM measurements of films deposited from o-xylene/AA showed almost no frequency change over time and no constant rate period. We conclude that AA had almost completely evaporated earlier, during spin coating.
To quantitatively determine and compare the evaporation rates and the corresponding mass loss from the resonance frequency change, we use the Sauerbrey equation. These mass losses and the corresponding thickness decrease due to additive evaporation are depicted in Fig. 5c and d and summarized in Table 3 for both solvent mixtures versus time. Fig. 5e and f shows a zoom in the relevant mass regime of the falling rate period. Within 4 hours of drying, about 125 ng cm−2 of the additive AA evaporated from the PTB7:PC71BM film at room temperature, yielding an average evaporation rate of 31 ng cm−2 h−1. In contrast, it takes 22.7 hours for the same amount of DIO to evaporate, yielding an average evaporation rate of 5.5 ng cm−2 h−1. Only at the end of the measurements, that is, after 4 h of AA evaporation and 40 h of DIO evaporation, the evaporation rates of both additives have equalized (2.5 ng cm−2 h−1). If we assume additive diffusion within the film to limit the additive evaporation and a dry film at the end of the experiment, we can conclude that the diffusion coefficient of DIO is lower than the diffusion coefficient of AA in the PTB7:PC71BM matrix.
| Additive | Drying time (h) | Solvent mass Δm–Δmdry (ng cm−2) | Thickness difference t–tdry (nm) |
|---|---|---|---|
| DIO | 0 | 9766 | 53 |
| 5 | 600 | 3.5 | |
| 10 | 244 | 1.5 | |
| 20 | 101 | 0.8 | |
| 30 | 41 | 0.4 | |
| 40 | 0 | 0 | |
| AA | 0 | 125 | 1.1 |
| 2 | 6 | 0.2 | |
| 4 | 0 | 0 | |
Moreover, the relationship of mass with frequency change allows for calculating the film thickness of the dry layers tdry. In all experiments the frequencies Δfn/n approached about −410 Hz at the end of the measurement procedures, which corresponds to a dry film thickness of 60 nm, assuming a density of 1.2 g cm−3 for typical organic semiconductors.34 This allowed us to determine the mass of the initial wet layer before solvent and additive evaporation, taking into account the initial composition of the PTB7:PC71BM/o-xylene/additive solutions and the respective densities. After spin coating, about 96 wt% of the o-xylene/DIO solution had evaporated from the PTB7:PC71BM layer, whereas 99.9 wt% of the o-xylene/AA solution had evaporated under equal conditions.
3.4 Other photo-active polymers
To demonstrate the general applicability of the o-xylene/AA solvent combination for the fabrication of highly efficient organic solar cells, we transferred the deposition process to various other commercially available polymer:fullerene blends that are known to perform very well after deposition from common hazardous halogenated solvents. We note that the commercially available polymers may have a different molecular weight than the respective polymers in earlier reports and may therefore yield a somewhat different device performance.As a structural advancement of PTB7, the polymer PTB7-Th was reported in the literature, enabling enhanced PCEs exceeding 9%, when deposited from CB/DIO on top of a modified ZnO interfacial layer, or yielding PCEs of more than 10%, when using an organic interlayer instead.35,36 Following the experimental protocol of Section 3.1, the J–V curves of these solar cells deposited from either CB/DIO, o-xylene or o-xylene/AA are depicted in Fig. 6. All key performance data are listed in Table 4. Whereas PTB7-Th:PC71BM reference solar cells cast from CB and 4% DIO (v/v) show PCEs of 7.5%, which is in good agreement with literature known results on plain ZnO,35 the PCEs of solar cells that were processed from o-xylene and 2% AA (v/v) improved to 8.3%.
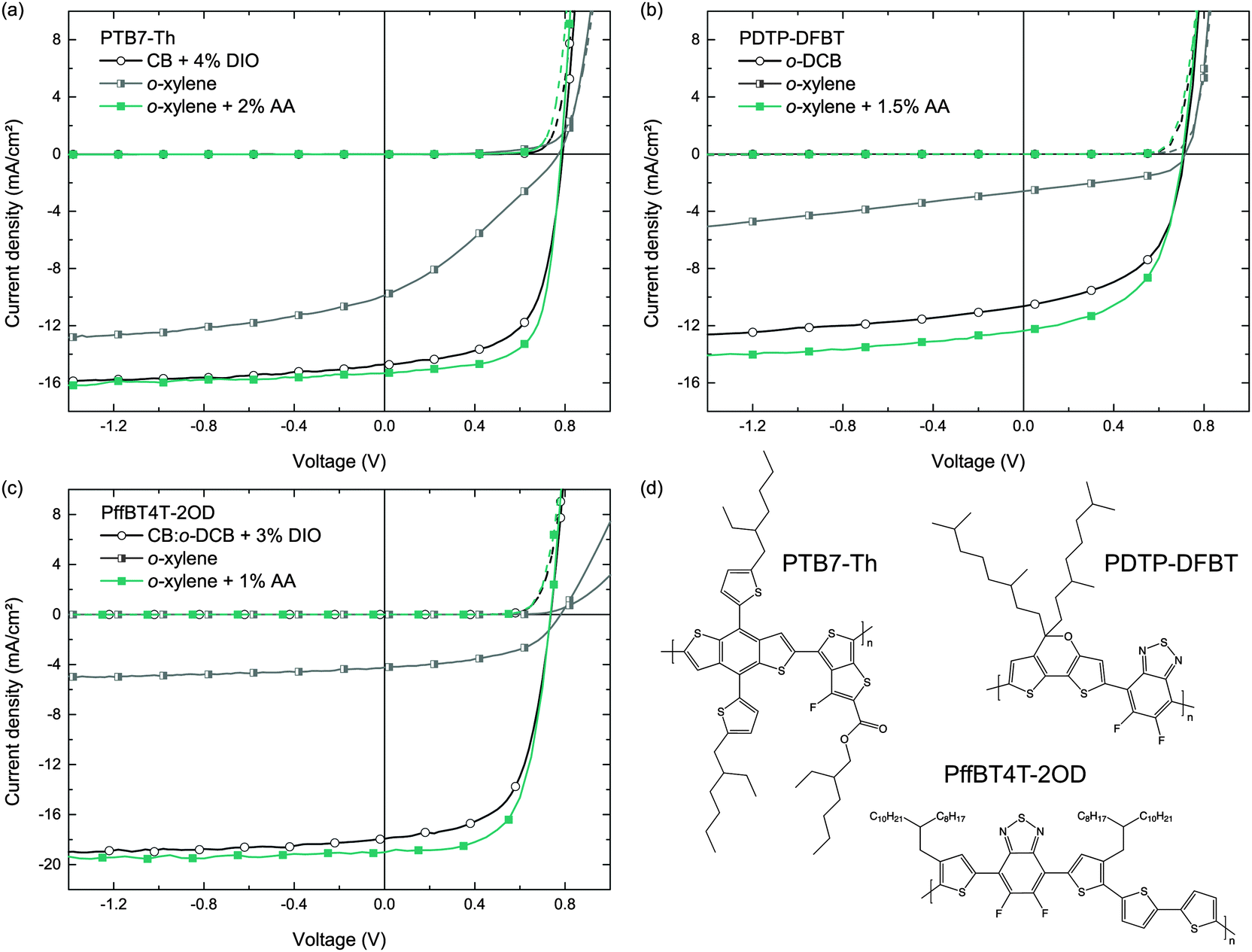 | ||
| Fig. 6 Typical J–V curves of solar cells with high-performance polymer:PC71BM BHJs comprising the polymers (a) PTB7-Th, (b) PDTP-DFBT and (c) PffBT4T-2OD, all deposited from o-xylene/AA. Dashed lines represent the J–V curves in the dark. 1 out of 10 data points is marked with a symbol to guide the eye. (d) Chemical structures of the photo-active polymers. | ||
| Polymer | Blend ratio | c (g L−1) | Main solvent | Additive | t (nm) | J SC (mA cm−2) | V OC (mV) | FF (%) | PCEa (%) |
|---|---|---|---|---|---|---|---|---|---|
| a PCE of the best performing device in parentheses. b Cells with up to 600 nm thickness and similar device performance. | |||||||||
| PTB7-Th | 1:1.5 |
10 | CB | 4% DIO | 125 | 15.1 ± 0.2 | 791 ± 3 | 62 ± 1 | 7.5 ± 0.1 (7.6) |
| 1:1.5 |
10 | o-Xylene | — | 120 | 10.0 ± 0.1 | 772 ± 1 | 31 ± 1 | 2.3 ± 0.1 (2.4) | |
| 1:1.5 |
10 | o-Xylene | 2% AA | 100 | 15.3 ± 0.2 | 783 ± 1 | 69 ± 1 | 8.3 ± 0.1 (8.5) | |
| PDTP-DFBT | 1:2 |
8 | o-DCB | — | 95 | 10.6 ± 0.2 | 712 ± 3 | 53 ± 1 | 4.0 ± 0.1 (4.2) |
| 1:1.5 |
9.6 | o-Xylene | — | 90 | 2.6 ± 0.1 | 720 ± 4 | 45 ± 1 | 0.9 ± 0.1 (0.9) | |
| 1:1.5 |
9.6 | o-Xylene | 1.5% AA | 105 | 12.4 ± 0.1 | 704 ± 5 | 54 ± 1 | 4.7 ± 0.2 (5.0) | |
| PffBT4T-2OD | 1:1.2 |
10 | CB:o-DCB 1:1 |
3% DIO | 440 | 17.5 ± 0.4 | 736 ± 3 | 60 ± 1 | 7.7 ± 0.3 (8.1) |
| 1:1.2 |
9 | o-Xylene | — | 450 | 4.0 ± 0.2 | 775 ± 15 | 48 ± 2 | 1.5 ± 0.1 (1.7) | |
| 1:1.2 |
9.5 | o-Xylene | 1% AA | 300b | 18.0 ± 1.0 | 738 ± 3 | 68 ± 3 | 9.0 ± 0.2 (9.5) | |
Similar observations were made for photo-active blends comprising the narrow bandgap polymer PDTP-DFBT and PC71BM, which can yield PCEs of up to 8% when cast from pure o-DCB,37 and which were also employed in highly efficient tandem solar cells.38–40 It was pointed out in the literature that the deposition of PDTP-DFBT:PC71BM BHJs from o-DCB, omitting any additives, is sufficient to yield high PCEs. However, we found that this concept does not apply to the deposition from the non-chlorinated solvent o-xylene. In contrast, adding AA to the o-xylene solution (1.5% v/v), we yielded PDTP-DFBT:PC71BM (1:1.5) solar cells with PCEs of up to 5.0%, clearly outperforming both reference devices deposited from pure o-DCB (PCE = 4%, blend ratio 1:2) or o-xylene (PCE = 1%). We note that we found significantly higher PDTP-DFBT solubility in o-xylene than in o-DCB, making o-xylene – besides the environmental aspect – much more favorable towards improved processability of this polymer.
As a third example, we investigated o-xylene/AA for the deposition of a blend comprising PffBT4T-2OD and PC71BM. Very recently, this polymer was reported to enable PCEs exceeding 10%. It exhibits high crystallinity and performs very well in thick active layers (≈300 nm).1 Following the previously published preparation protocol, we built reference solar cells incorporating a PffBT4T-2OD:PC71BM photo-active blend from a CB:o-DCB 1:1 solvent mixture with the addition of 3% DIO, resulting in an average device efficiency of 7.7%. Using the solvent o-xylene plus 1% of AA instead, we yielded PCEs of 9.0% (hero device: 9.5%), with the improvement originating mainly from a higher FF. Again, without employing AA, the efficiency of the solar cells is drastically lower.
4. Conclusions
The eco-compatible solvent/additive combination o-xylene/AA is an excellent alternative to the commonly used halogenated and environmentally harmful CB/DIO for the deposition of a broad selection of highly efficient polymer:PC71BM bulk-heterojunctions. The corresponding solar cells outperform the reference devices, yielding up to 9.5% power conversion efficiency (PffBT4T-2OD:PC71BM). As compared to DIO, the higher volatility of the additive AA reduces the drying time of the photo-active layer significantly, enabling smaller drying ovens or higher web-speeds for future roll-to-roll fabrication plants.Acknowledgements
This work was supported by the European Commission through the project MatHero, funded within the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 604603. We acknowledge support by the Center for Functional Nanostructures (CFN). The AFM was made available through funding by the Federal Ministry of Education and Research (Project TAURUS, grant no. 03EK3504). Part of this work was supported by the German Research Foundation (DFG) within the Priority Program 1355 “Elementary processes of organic photovoltaics”. We thank Nanograde Ltd for supplying zinc oxide nanoparticles and Joshua Fragoso for fruitful discussions.Notes and references
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- F. C. Krebs and M. Jørgensen, Sol. Energy Mater. Sol. Cells, 2013, 119, 73 CrossRef CAS PubMed.
- R. Po, A. Bernardi, A. Calabrese, C. Carbonera, G. Corso and A. Pellegrino, Energy Environ. Sci., 2014, 7, 925 CAS.
- O. Synooka, K.-R. Eberhardt and H. Hoppe, RSC Adv., 2014, 4, 16681 RSC.
- B. R. Aïch, S. Beaupré, M. Leclerc and Y. Tao, Org. Electron., 2014, 15, 543 CrossRef PubMed.
- P.-T. Tsai, C.-Y. Tsai, C.-M. Wang, Y.-F. Chang, H.-F. Meng, Z.-K. Chen, H.-W. Lin, H.-W. Zan, S.-F. Horng, Y.-C. Lai and P. Yu, Org. Electron., 2014, 15, 893 CrossRef CAS PubMed.
- C.-C. Chueh, K. Yao, H.-L. Yip, C.-Y. Chang, Y.-X. Xu, K.-S. Chen, C.-Z. Li, P. Liu, F. Huang, Y. Chen, W.-C. Chen and A. K.-Y. Jen, Energy Environ. Sci., 2013, 6, 3241 CAS.
- K.-S. Chen, H.-L. Yip, C. W. Schlenker, D. S. Ginger and A. K.-Y. Jen, Org. Electron., 2012, 13, 2870 CrossRef CAS PubMed.
- S. Venkatesan, Q. Chen, E. C. Ngo, N. Adhikari, K. Nelson, A. Dubey, J. Sun, V. Bommisetty, C. Zhang, D. Galipeau and Q. Qiao, Energy Technol., 2014, 2, 269 CrossRef CAS PubMed.
- A. Lange, W. Schindler, M. Wegener, K. Fostiropoulos and S. Janietz, Sol. Energy Mater. Sol. Cells, 2013, 109, 104 CrossRef CAS PubMed.
- G. Susanna, L. Salamandra, C. Ciceroni, F. Mura, T. M. Brown, A. Reale, M. Rossi, A. Di Carlo and F. Brunetti, Sol. Energy Mater. Sol. Cells, 2015, 134, 194 CrossRef CAS PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619 CrossRef CAS PubMed.
- N. Shin, L. J. Richter, A. a. Herzing, R. J. Kline and D. M. DeLongchamp, Adv. Energy Mater., 2013, 3, 938 CrossRef CAS PubMed.
- L. J. Richter, D. M. DeLongchamp, F. a. Bokel, S. Engmann, K. W. Chou, A. Amassian, E. Schaible and A. Hexemer, Adv. Energy Mater., 2015, 5, 1400975 Search PubMed.
- F. Liu, W. Zhao, J. R. Tumbleston, C. Wang, Y. Gu, D. Wang, A. L. Briseno, H. Ade and T. P. Russell, Adv. Energy Mater., 2014, 4, 1301377 Search PubMed.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef CAS PubMed.
- Y. Kim, H. R. Yeom, J. Y. Kim and C. Yang, Energy Environ. Sci., 2013, 6, 1909 CAS.
- X. Guo, M. Zhang, C. Cui, J. Hou and Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 8190 CAS.
- W. Zhao, L. Ye, S. Zhang, M. Sun and J. Hou, J. Mater. Chem. A, 2015, 3, 12723 CAS.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS PubMed.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65 CrossRef CAS PubMed.
- A. Foertig, J. Kniepert, M. Gluecker, T. Brenner, V. Dyakonov, D. Neher and C. Deibel, Adv. Funct. Mater., 2014, 24, 1306 CrossRef CAS PubMed.
- I. Burgués-Ceballos, F. Machui, J. Min, T. Ameri, M. M. Voigt, Y. N. Luponosov, S. A. Ponomarenko, P. D. Lacharmoise, M. Campoy-Quiles and C. J. Brabec, Adv. Funct. Mater., 2014, 24, 1449 CrossRef PubMed.
- R. Tuttle, M. L. Reitz and B. E. Woodworth, US Pat., 20100043876A1, 2010 Search PubMed.
- H.-Y. Chen, J. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371 CrossRef CAS PubMed.
- J. A. Bartelt, J. D. Douglas, W. R. Mateker, A. El Labban, C. J. Tassone, M. F. Toney, J. M. J. Fréchet, P. M. Beaujuge and M. D. McGehee, Adv. Energy Mater., 2014, 4, 1301733 Search PubMed.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. W. Samuel, Nat. Commun., 2013, 4, 2867 Search PubMed.
- C. L. Yaws, The Yaws Handbook of Vapor Pressure: Antoine Coefficients, 1st edn, 2007 Search PubMed.
- VDI Heat Atlas, VDI-Gesellschaft Verfahrenstechnik und Chemieingenieurwesen, Springer-Verlag, Berlin, Heidelberg, 2010 Search PubMed.
- B. Schmidt-Hansberg, M. Baunach, J. Krenn, S. Walheim, U. Lemmer, P. Scharfer and W. Schabel, Chem. Eng. Process., 2011, 50, 509 CrossRef CAS PubMed.
- B. Schmidt-Hansberg, M. Sanyal, N. Grossiord, Y. Galagan, M. Baunach, M. F. G. Klein, A. Colsmann, P. Scharfer, U. Lemmer, H. Dosch, J. Michels, E. Barrena and W. Schabel, Sol. Energy Mater. Sol. Cells, 2012, 96, 195 CrossRef CAS PubMed.
- J. W. Kiel, B. J. Kirby, C. F. Majkrzak, B. B. Maranville and M. E. Mackay, Soft Matter, 2010, 6, 641 RSC.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174 CrossRef CAS PubMed.
- L. Dou, C. Chen, K. Yoshimura, K. Ohya, W. Chang, J. Gao, Y. Liu, E. Richard and Y. Yang, Macromolecules, 2013, 46, 3384 CrossRef CAS.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- J. You, C.-C. Chen, Z. Hong, K. Yoshimura, K. Ohya, R. Xu, S. Ye, J. Gao, G. Li and Y. Yang, Adv. Mater., 2013, 25, 3973 CrossRef CAS PubMed.
- C.-C. Chen, W.-H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |