
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic nickel complexes for selective CO2 carbon capture and sequestration†
F.
Möller
a,
K.
Merz
a,
C.
Herrmann
b and
U.-P.
Apfel
*a
aInorganic Chemistry I/Bioinorganic Chemistry, Ruhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de
bPhysical Chemistry I, Ruhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany
First published on 9th December 2015
Abstract
Herein we report a dinickel azacryptand complex that enables fast, selective, and tight CO2 binding from air. Exploiting the affinity of the cavitand towards azides, CO2 release was observed. Despite the stability of the azido complex, UV irradiation under atmospheric conditions proved to be a suitable pathway for N3− replacement by CO2.
The continuous emission of carbon dioxide from combustion processes is a major reason for the increase of the global greenhouse effect. Thus CO2 capture and sequestration technologies (CCS) are required to lower its concentration in our atmosphere and allow for storage and transportation.1 In addition, reversible systems are needed that allow for its successive release and application as a C1 building block in industrial processes. In industry, aqueous ethanolic amine solutions are commonly applied for CCS but show high energy consumption and severe corrosion problems.2 Furthermore, amino acids,3 metal oxides,4 and organic polymers5 were evaluated for this purpose. Additionally, metal–organic frameworks show good CO2 uptake capabilities,6,7 and are also reported to allow for CO2 reduction.8–10 However, although promising, they do not allow for a selective, non-competitive guest absorption/uptake of CO2versus e.g. H2, N2, or CH4 and increasing the CO2 affinity results in difficult desorption.11 Furthermore, water, a significant component of flue gases, was found to predominantly decrease CO2 capacities.12
In contrast, dinuclear metalloenzymes are known to selectively bind CO2 and catalyze hydrolysis as well as redox reactions at ambient conditions. Among those, the nickel containing ureases13 as well as the iron and nickel containing CO dehydrogenases (CODHNi)14 can bind CO2 during the conversion of urea to CO2/NH3 or the reduction of CO2 to CO, respectively. Commonly, such CO2 fixation is facilitated by a Ni–OH moiety. Whereas complexes comprising a Ni–OH center are widely known to facilitate CO2 fixation and Holm et al. showed their outstanding potential for fast CO2 fixation,15–17 only few systems exist that selectively bind CO2 between two nickel centers.18–22 General problems associated with such systems are either loose binding of the generated carbonate to nickel or formation of oligomers with bridging carbonates rendering the systems not applicable for reversible CCS and transportation. Contrary to those complexes, cryptands comprise well-defined metal binding cavities with tuneable metal–metal distances that allow for a strong binding of specific anions. Nelson and coworkers showed the uptake of carbonates into the cavity of dinickel azacryptands affording a stable dinickel complex with an anti–anti η1,η1-bound carbonate (Scheme 1).23 Furthermore, Sullivan and coworkers showed that in presence of H2, CO2 can be reduced to afford CO and CH4.24 However, neither CO2 uptake kinetics nor selectivity studies were reported in order to apply azacryptands for CCS. Additionally, Lu as well as Malthouse and coworkers showed evidence for a direct atmospheric CO2 uptake by dinuclear copper25 and zinc cryptands.26 The uptake capability of these bimetallic cryptands and of dinuclear metalloenzymes as well as the high stability of the obtained carbonate complexes within those frameworks pointed our interest towards the application of dinickel azacryptands as highly selective CO2 binding and transportation platform under ambient conditions.
 | ||
| Scheme 1 Carbonate uptake by bimetallic complexes. | ||
Reaction of the azacryptands 1 or 2 with Ni(ClO4)2 comprising the non-coordinative perchlorate anion afforded complexes 3 and 4 under inert atmosphere as crystalline material in 56% and 52% yield, respectively (Scheme 2). The composition of both complexes was unequivocally confirmed by structure analysis (Fig. 1 and S1†). Both complexes 3 and 4 are isostructural and reveal, although possessing two identical metal coordination sites, two nickel centers with different coordination modes. One nickel atom is coordinated by four amines and an additional chloride ligand. The chlorido ligand herein most likely originates from the reduction of a perchlorate catalyzed by a Ni–O species,27 and multiple synthetic attempts with independently generated starting materials gave similar results. In contrast, the second nickel center is coordinated by four amines as well as an additional acetonitrile and a water molecule. The Ni–O distances of 2.044 Å (in 3) and 2.035 Å (in 4) are in good agreement with literature data for nickel-bound water and are significantly longer than Ni–O distances observed for Ni–OH moieties.28 Charge balance is provided by three perchlorate anions. To exclude nickel oxidation due to reductive perchlorate decomposition within the complexes we performed SQUID measurements on 4, which indicate two nickel centers with S = 1, consistent with two isolated Ni(II) centers (Fig. S2†).
 | ||
| Scheme 2 Synthesis and reactivity of dinickel azacryptands 3 and 4. | ||
 | ||
| Fig. 1 Molecular structures of the apo-cryptand 3 (left) and its bicarbonate-binding equivalent 5 (right) (Counter ions and hydrogen atoms were omitted for clarity). | ||
Notably, whereas solutions of 4 were stable under atmospheric conditions, solutions of 3, kept in acetonitrile in air rapidly changed color (Fig. 2). The resulting species was thus isolated and revealed a significantly altered IR spectrum (Fig. S3†). Crystals obtained by concentrating an acetonitrile solution of 3 under air revealed the molecular structure of the resulting compound 5 as depicted in Fig. 1. It is noteworthy, the cavity of the azacryptand is occupied by a bicarbonate bridging the two nickel-centers with a Ni–OCO3 distance of 1.991 Å and 2.019 Å respectively. The Ni–Cl distance is significantly increased from 2.326 in 3 to 2.783 Å in 5 and the chloride is held in place by a hydrogen bond to the coordinated bicarbonate. As highlighted in Fig. 1, an internal hydrogen bond to an attached water molecule further stabilizes the bicarbonate ion between the two nickel(II) centers. Although a magnetic communication between both nickel centers can be expected, SQUID measurements unequivocally revealed two isolated nickel(II) centers in complex 5 with S = 1.
 | ||
| Fig. 2 Absorption spectra measured in acetonitrile at RT: green trace – complex 3 (LNi2); blue trace – after bubbling CO2 through the solution of 3 for 1 min (LNi2(HCO3)); black trace – after reaction of 5 with sodium azide for 6 h (LNi2(N3)); red trace – after UV-irradiation of 6 (LNi2(N3) + 2 h hν). | ||
In order to prove that the formation of 5 is the result of a selective uptake of CO2 from air, we performed binding experiments under 12CO2 and 13CO2 atmosphere. Treatment with either gas resulted in the formation of a bicarbonate-binding complex 5. A comparison of both IR spectra did not allow for a clear assignment of the carbonate vibrations since the bicarbonate bands coincide with the azacryptand ligand vibrations. Mass spectrometry, however, confirmed the successful uptake of the respective isotopically-enriched substrates (Fig. S4†). Notably, no reaction of complex 3 with SO2, NO, N2O, NO2, nor CO was observed by UV-vis spectroscopy.
The kinetics of the CO2 fixation process was furthermore followed by stopped-flow spectrophotometry at various temperatures and applying different concentrated CO2 solutions in acetonitrile obtained from saturated stock solutions (Fig. S5†).29 The reaction was monitored by the occurring absorbance changes at 470 nm. The reaction of 3 and CO2 is of first-order in CO2 concentration, yielding kobs, and the apparent association rate constant k2, 298 K = 0.067 ± 0.005 M−1 s−1 is obtained from the slope of the plot of kobsvs. CO2 concentration (Fig. S5c†). Only a small increase of k2 with increasing temperature is observed and an apparent enthalpy of the activation of ΔH‡ = 7.55 ± 1.07 kJ mol−1 is obtained from the Eyring plot (Fig. S6†). This small value reflects that the association of CO2 proceeds in more than one step. Therefore pre-equilibrium and activation enthalpies compose the apparent ΔH‡ value.
Contrary to 3, an uptake of CO2 into complex 4 was not possible. This fact demonstrates the different uptake capacities of both species. As no significant structural differences are observed at the metal centers in 3vs.4, we believe that the hydrophilic bicarbonate is repelled by the interaction with the hydrophobic tert-butyl group. Thus the CO2 uptake can easily be adjusted by different bridging elements.
Attempts to release the bicarbonate by purging solutions of 5 with nitrogen gas or heating up the same solutions did not result in the re-generation of 3 (Fig. S7†). Azacryptands are well known to support the binding of different anions with varying binding strength.30,31 To reverse the strong CO2 binding of 5, we therefore added different anions to complex 5 to test whether we would be able to release the tightly-bound bicarbonate. Whilst addition of Cl− or Br−, as well as SO2, NO, N2O, NO2, CO, or CS2 did not reveal any dissociation of the bicarbonate, attempts of reactivation through treatment with NaOH, HOAc, or HBF4 resulted in precipitation of Ni(OH)2, formation of [Ni(OAc)2(H2O)4] (Fig. S8†),32 or other, unidentifiable, decomposition products. However, when N3− or SCN− was allowed to react with 5, the formation of a single well-defined product was observed via UV-vis spectroscopy (Fig. 2 and S9†). The identity of the N3− reaction product was elucidated as compound 6 by single-crystal X-ray diffractometry and confirmed the exchange of the bicarbonate by an azide ligand (Scheme 3 and Fig. S10†). Furthermore, ESI-MS experiments as well as infrared spectra of this solution are in good agreement with a solution of 6, independently generated by reaction of 1 with NaN3 and Ni(ClO4)2. The release of CO2 from complex 6 is slow (kobs, 303.15 K = 0.00223 min−1) (Fig. S11†) and reveals the high stability of compound 5. Likewise, reaction of 5 and SCN− lead to exchange of HCO3− by SCN− visible in ESI-MS. Notably, although 4 is not capable to incorporate the bicarbonate anion, [Ni2(2)(N3)](ClO4)37 can be easily obtained by reaction of 4 with NaN3 (Fig. S10†) and shows that the cavity is not intrinsically blocked by the tert-butyl groups.
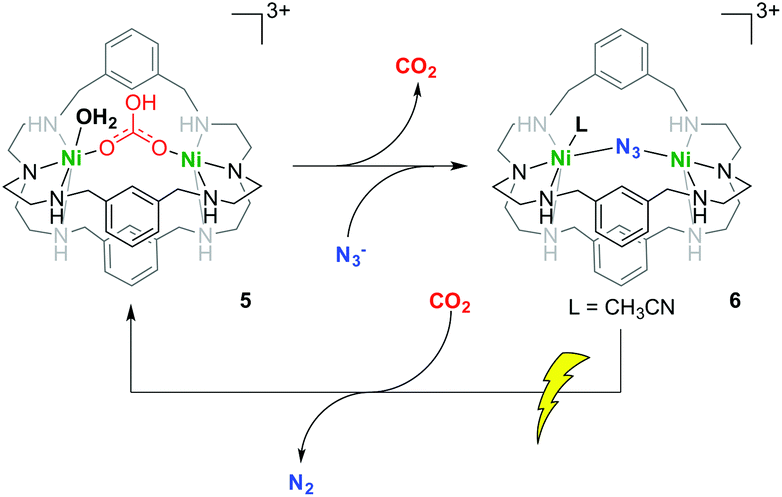 | ||
| Scheme 3 CO2 binding and release utilizing dinickel azacryptands 5 and 6. | ||
We wondered about the general nature of the released “CO2-species”. We therefore treated 13CO2-5 with the different substrates and investigated changes by 13C NMR spectroscopy, measured in an inert tube in deuterated acetonitrile, and using gas chromatography. This treatment unequivocally showed the release of CO2 as the only C1-species in all cases (Fig. S12†).
Similar reactivity was observed upon treatment of complex 5 with NaHCO3 and provides evidence for a reversible CO2 binding within the cryptand. Unfortunately, complex 6 did not allow for further ligand substitution reactions. It is, however, well known that Ni–N3 complexes decompose under UV irradiation.33 We therefore irradiated an acetonitrile solution of 6 with a 150 W Xe-lamp under air. This treatment revealed a change of the initial UV-vis bands and complete conversion was achieved within 2 hours (Fig. 2). Inspection of the UV-vis spectrum of the obtained compound showed close resemblance to the spectrum of complex 5 (Fig. 2).
Furthermore, the obtained IR and MS spectra are in good agreement with the formation of compound 5 through UV-irradiation of 6 (Fig. S13†). Although we are at this point not able to provide an exact mechanism for the uptake of CO2, its expelling by N3− and the subsequent cleavage of the resulting free azide, an overall pathway is presented in Scheme 3.
In conclusion, we have shown for the first time that nickel-azacryptands are superior compounds for highly selective CO2 capture under ambient conditions. Due to their defined CO2-hosting cavity, typical problems associated with gas selectivity and low binding strength in the presence of moisture can be avoided as compound 5 is thermodynamically stable in the presence of excess SO2, NO, N2O, NO2, CO, or CS2. As shown with complex 4, the CO2 binding affinity can be easily adjusted by manipulation of the bridging elements connecting both coordination cavities. Complex 5 showed high stability even at higher temperatures. We were able to show a selective release of CO2 upon treatment with pseudo-halogenides. Especially interesting is the reaction of 5 with NaN3 to give 6. This reaction proved to be very useful since irradiation with a standard UV Xe-lamp emptied the cryptands’ cavity and allowed for subsequent binding of CO2.
The well-defined cavity of azacryptands provides a highly sophisticated system for carbon capture and sequestration in a reversible manner. In addition, it shows that azacryptands can serve as molecular reactors allowing specific reactions to proceed in an elaborate, protected environment. Further investigations concerning the mechanism of N3− uptake, photocleavage, as well as manipulation of the substitution kinetics and reduction experiments are currently under way.
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (Liebig grant) and through the Deutsche Forschungsgemeinschaft (Emmy Noether grant to UPA, AP242/2-1) as well as the Cluster of Excellence “Ruhr Explores Solvation” (RESOLV, EXC 1069)). FM gratefully acknowledges financial support by the Deutsche Bundesstiftung Umwelt. We thank Eckhard Bill, Andreas Göbels and Maurice van Gastel (MPI-CEC) for their support with SQUID data and valuable discussions. We thank Eric Victor (Brown Univ.) and Manuela Winter (RUB) for valuable support and discussions.References
- R. Sabouni, H. Kazemian and S. Rohani, Environ. Sci. Pollut. Res., 2014, 21, 5427–5449 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- X. Wang, N. G. Akhmedov, Y. Duan and B. Li, Energy Fuels, 2015, 29, 3780–3784 CrossRef CAS.
- R. A. Rahman, P. Mehrani, D. Y. Lu, E. J. Anthony and A. Macchi, Energy Fuels, 2015, 29, 3808–3819 CrossRef CAS.
- R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC.
- H.-H. Wang, L.-N. Jia, L. Hou, W. Shi, Z. Zhu and Y.-Y. Wang, Inorg. Chem., 2015, 54, 1841–1846 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed.
- Y. Liu, Z. U. Wang and H.-C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239–259 CrossRef CAS.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- P. A. Karplus, M. A. Pearson and R. P. Hausinger, Acc. Chem. Res., 1997, 30, 330–337 CrossRef CAS.
- P. A. Lindahl, Biochemistry, 2002, 41, 2097–2105 CrossRef CAS PubMed.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Inorg. Chem., 2011, 50, 10070–10081 CrossRef CAS PubMed.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222–1227 CrossRef CAS PubMed.
- O. Troeppner, D. Huang, R. H. Holm and I. Ivanovic-Burmazovic, Dalton Trans., 2014, 43, 5274–5279 RSC.
- E. Carmona, J. M. Marin, P. Palma, M. Paneque and M. L. Poveda, Inorg. Chem., 1989, 28, 1895–1900 CrossRef CAS.
- N. Kitajima, S. Hikichi, M. Tanaka and Y. Morooka, J. Am. Chem. Soc., 1993, 115, 5496–5508 CrossRef CAS.
- M. Ito and Y. Takita, Chem. Lett., 1996, 25, 929–930 CrossRef.
- A. A. Lozano, M. Saez, J. Perez, L. Garcia, L. Lezama, T. Rojo, G. Lopez, G. Garcia and M. A. D. Santana, Dalton Trans., 2006, 3906–3911 RSC.
- J. P. Wikstrom, A. S. Filatov, E. A. Mikhalyova, M. Shatruk, B. M. Foxman and E. V. Rybak-Akimova, Dalton Trans., 2010, 39, 2504–2514 RSC.
- Y. Dussart, C. Harding, P. Dalgaard, C. McKenzie, R. Kadirvelraj, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 2002, 1704–1713 RSC.
- G. G. Morgan, K. Fennell, M. J. L. Kishore and J. A. Sullivan, ChemCatChem, 2013, 5, 951–958 CrossRef CAS.
- J.-M. Chen, W. Wei, X.-L. Feng and T.-B. Lu, Chem. – Asian J., 2007, 2, 710–719 CrossRef PubMed.
- A. A. C. Wild, K. Fennell, G. G. Morgan, C. M. Hewage and J. P. G. Malthouse, Dalton Trans., 2014, 43, 13557 RSC.
- L. W. Collins, Inorg. Chim. Acta, 1980, 39, 53–56 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- A. Gennaro, A. A. Isse and E. Vianello, J. Electroanal. Chem., 1990, 203–215 CrossRef CAS.
- B. Dietrich, J. Guilhem, J.-M. Lehn, C. Pascard and E. Sonveaux, Helv. Chim. Acta, 1984, 67, 91–104 CrossRef CAS.
- J. Caballero-Jimenez, F. Habib, D. Ramirez-Rosales, R. Grande-Aztatzi, G. Merino, I. Korobkov, M. K. Singh, G. Rajaraman, Y. Reyes-Ortega and M. Murugesu, Dalton Trans., 2015, 44, 8649–8659 RSC.
- R. E. Cramer, W. V. Doorne and R. Dubois, Inorg. Chem., 1975, 14, 2462–2466 CrossRef CAS.
- H. Henning, K. Hofbauer, K. Handke and R. Stich, Angew. Chem., Int. Ed., 1997, 36, 408–410 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, X-ray crystallographic analysis, UV-Vis spectra, SQUID and kinetic data, detailed experimental conditions. CCDC 1418422–1418427. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04267d |
| This journal is © The Royal Society of Chemistry 2016 |