
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directed synthesis of {CuII2ZnII2} and {CuII8ZnII8} heterometallic complexes†
María José
Heras Ojea
a,
Claire
Wilson
a,
Simon J.
Coles
 b,
Floriana
Tuna
c and
Mark
Murrie
*a
b,
Floriana
Tuna
c and
Mark
Murrie
*a
aWestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow G12 8QQ, UK. E-mail: mark.murrie@glasgow.ac.uk
bDepartment of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
cSchool of Chemistry and Photon Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 14th October 2015
Abstract
Two new heterometallic complexes [Cu2Zn2(H4L)2(CH3COO)2Cl2]·2.5CH3OH·0.5H2O (1) and [Cu8Zn8(OH)8(H4L)8](Cl)2(ClO4)6·16H2O (2) have been synthesised following two different preparative routes, by using the polydentate ligand Bis–tris propane (H6L = 2,2′-(propane-1,3-diyldiimino)bis[2-(hydroxymethyl)propane-1,3-diol]). Herein, we describe the synthesis, structure, and magnetic properties of 1, a tetramer which forms in the absence of base and 2, a hexadecanuclear complex with a remarkable double-concentric ring structure that forms in the presence of base. Antiferromagnetic coupling between Cu(II) ions is observed in both compounds despite the long distances between paramagnetic metal centres, due to the involvement of diamagnetic Zn(II) ions in the magnetic exchange pathway.
Introduction
From aesthetic reasons to numerous potential future applications, supramolecular chemistry has focused its efforts on developing alternative synthetic methodologies which control the formation of molecular architectures with large and complex shapes.1–4 The importance in controlling the assembly process of large molecular complexes lies in the synthesis of nanomaterials and molecular machines with possible uses in assorted fields such as nanoelectronics or nanobiochemistry.5–7 One of the latest efforts that the synthetic community focuses on is the preparation of multifunctional heterometallic systems by the combination of different transition metal and/or lanthanides ions.8–10 Several polydentate organic ligands which dispose multiple donor atoms and/or well-defined coordination pockets have been successfully used to control the nuclearity and the assembly of well-defined heterometallic polynuclear complexes.11–15 Previous studies carried out in our group highlighted the capability of the flexible polydentate ligand 2,2′-(propane-1,3-diyldiimino)bis[2-(hydroxylmethyl) propane-1,3-diol] (bis–tris propane, H6L) to control and direct the assembly of the metal atoms in the synthesis of 3d/3d′ homo-and heterometallic polynuclear compounds.16–18 The skeleton of H6L is mainly composed of alkyl groups, which provides for high flexibility, and includes a {N2O6} unit that defines multiple binding positions (see Scheme 1). However, despite its potential as a ligand in coordination compounds, it has been used mainly as a biological buffer in biochemistry or molecular biology. The heterometallic {Mn18Cu6} complexes [Mn18Cu6O14(H2L)6Cl2(H2O)6]Cl6 and [Mn18Cu6O14(H2L)6Cl6]Cl2 are the only reported examples of 3d/3d′ heterometallic complexes obtained with this ligand so far.18 Unfortunately, the magnetic studies of these compounds revealed dominant intramolecular antiferromagnetic exchange, leading to small spin ground states. To try to reduce the number of antiferromagnetic interactions, and therefore increase the magnitude of the spin ground state, we proposed a strategy based on the replacement of the paramagnetic Cu(II) centers by diamagnetic Zn(II) ions. During the development of this strategy, we investigated the chemistry of the bis–tris propane ligand in the formation of Cu/Zn heterometallic complexes yielding: [Cu2Zn2(H4L)2(CH3COO)2Cl2]·2.5CH3OH·0.5H2O (1) and [Cu8Zn8(OH)8(H4L)8](Cl)2(ClO4)6·16H2O (2). The unusual arrangements of both complexes led us to study their magneto-structural properties. Notably, the {Cu8Zn8} complex is the largest reported Cu/Zn-containing heterometallic complex and contains an unprecedented double-concentric ring structure.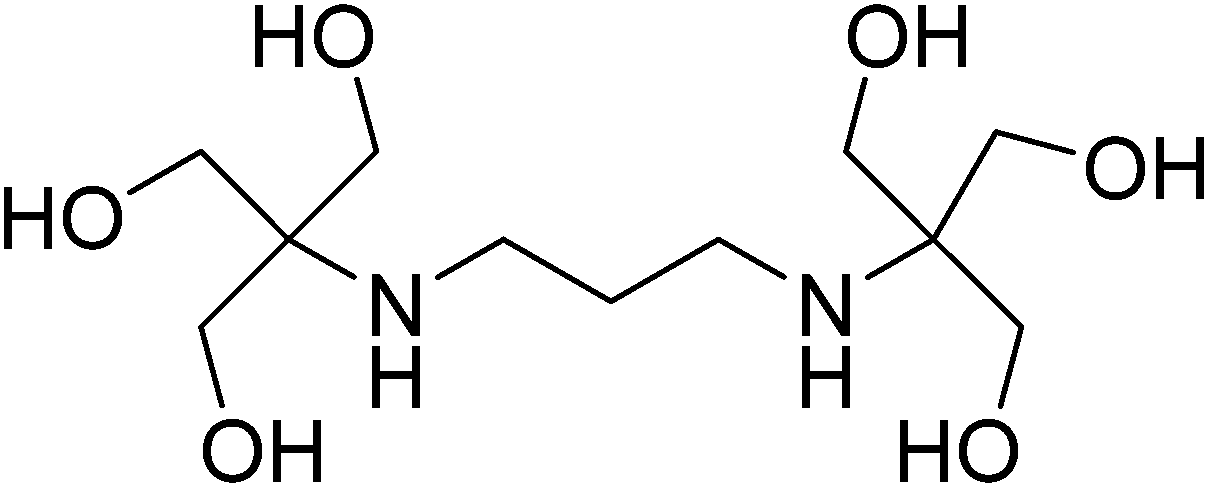 | ||
| Scheme 1 Bis–tris propane, H6L. | ||
Experimental section
All reagents and solvents were obtained from commercial suppliers and used without further purification. The polydentate ligand H6L used in the synthetic routes is the commercial reagent 2,2′-(propane-1,3-diyldiimino)bis[2-(hydroxymethyl)propane-1,3-diol] (H6L). Perchlorate salts are potentially explosive, and so the compounds should be prepared in small quantities and handled with care.Crystallographic data were collected for both 1 and 2 at 100 K using Mo-Kα radiation (λ = 0.71073 Å). For 1 a Bruker APEX II CCD diffractometer with an Oxford Cryosystems n-Helix device mounted on a sealed tube generator was used and for 2 a Rigaku AFC12 goniometer equipped with an (HG) Saturn724+ detector mounted on an FR-E+ SuperBright rotating anode generator with HF Varimax optics (100 μm focus).19 Both structures were solved using SUPERFLIP20 and refined using full-matrix least squares refinement on F2 using SHELX2014![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21,22 within OLEX2.23 The crystals of 1 grew as stacks of very thin plates which could not be separated. Therefore, data were collected from a multiply twinned crystal and the structure was refined as a 2-component twin; corresponding to a 180° rotation about [001] identified using ROTAX,24 twin component factor refined to 0.140(8). SQUEEZE25,26 was used to identify solvent accessible voids in the structure, calculated to contain 23 electrons per cell, however, this resulted in no significant improvement and was not used finally. There remain some large peaks of residual electron density, possibly due to further twin components which have not been accounted for. Due to the overall poor quality of the data only the Cu, Zn and Cl atom sites were refined with anisotropic adps, all other atoms were refined with isotropic adps. Hydrogen atoms were placed in geometrically calculated positions and refined as part of a riding model except OH hydrogens which were refined as part of a rigid rotating group. Hydrogen atoms for the solvent lattice molecules were not included explicitly in the model but were included in the cell contents and all values derived from them. The structure of 2 also contains a large void space without identifiable electron density. SQUEEZE in PLATON25,26 was used to calculate the solvent accessible volume, locating two large voids per unit cell each of 437 Å3, calculated to contain 81 electrons each. Disorder present in the structure was modelled with partially occupied atom sites and suitable geometric restraints; O72B was modelled over two sites with occupancy 0.8/0.2, O71 over three sites with occupancy 0.6/0.2/0.2 and O11b over two sites with occupancy 0.75/0.25. Occupancies were determined by competitive refinement and later fixed at the closest simpler values. Five lattice H2O sites are included in the model, two of them 0.5 occupied and hydrogen atoms for these sites were not included explicitly but are included in the unit cell contents and values derived from it. Hydroxide hydrogen atoms were located in difference Fourier maps and refined with a distance restraint. Through consideration of charge balance, the electron density and resulting adps, as well as the close proximity of the two partially Cl− sites, the ClO4− and Cl− anions were modelled as follows: both ClO4− anions were refined as 0.75 occupied, with isotropic adps for the oxygen atoms, a single common Uiso for O5c to O8c. The three Cl− ions lie on the 4-fold axis, Cl3 is chemically fully occupied (0.25 per asymmetric unit) and the second site is split over two partially occupied sites the occupancy of which refines to 0.75/0.25. Suitable distance restraints were applied to the disordered fragments and the ClO4− anions and isotropic adps retained for partially occupied atoms except Cl. The IR spectra were measured using a FTIR-8400S SHIMADZU IR spectrophotometer. The microanalyses and mass spectrometry analyses were performed by the analytical services of the School of Chemistry at the University of Glasgow. Magnetic measurements of complexes 1 and 2 were performed on polycrystalline samples using a Quantum Design MPMS-XL SQUID magnetometer. Data were corrected for the diamagnetic contribution of the sample holder by measurements and for the diamagnetism of the compounds by using Pascal's constants. EPR measurements were performed at the University of Glasgow. X-band EPR spectra were recorded on a Bruker ELEXSYS E500 spectrometer. Simulations were performed using Bruker's Xsophe software package.27
21,22 within OLEX2.23 The crystals of 1 grew as stacks of very thin plates which could not be separated. Therefore, data were collected from a multiply twinned crystal and the structure was refined as a 2-component twin; corresponding to a 180° rotation about [001] identified using ROTAX,24 twin component factor refined to 0.140(8). SQUEEZE25,26 was used to identify solvent accessible voids in the structure, calculated to contain 23 electrons per cell, however, this resulted in no significant improvement and was not used finally. There remain some large peaks of residual electron density, possibly due to further twin components which have not been accounted for. Due to the overall poor quality of the data only the Cu, Zn and Cl atom sites were refined with anisotropic adps, all other atoms were refined with isotropic adps. Hydrogen atoms were placed in geometrically calculated positions and refined as part of a riding model except OH hydrogens which were refined as part of a rigid rotating group. Hydrogen atoms for the solvent lattice molecules were not included explicitly in the model but were included in the cell contents and all values derived from them. The structure of 2 also contains a large void space without identifiable electron density. SQUEEZE in PLATON25,26 was used to calculate the solvent accessible volume, locating two large voids per unit cell each of 437 Å3, calculated to contain 81 electrons each. Disorder present in the structure was modelled with partially occupied atom sites and suitable geometric restraints; O72B was modelled over two sites with occupancy 0.8/0.2, O71 over three sites with occupancy 0.6/0.2/0.2 and O11b over two sites with occupancy 0.75/0.25. Occupancies were determined by competitive refinement and later fixed at the closest simpler values. Five lattice H2O sites are included in the model, two of them 0.5 occupied and hydrogen atoms for these sites were not included explicitly but are included in the unit cell contents and values derived from it. Hydroxide hydrogen atoms were located in difference Fourier maps and refined with a distance restraint. Through consideration of charge balance, the electron density and resulting adps, as well as the close proximity of the two partially Cl− sites, the ClO4− and Cl− anions were modelled as follows: both ClO4− anions were refined as 0.75 occupied, with isotropic adps for the oxygen atoms, a single common Uiso for O5c to O8c. The three Cl− ions lie on the 4-fold axis, Cl3 is chemically fully occupied (0.25 per asymmetric unit) and the second site is split over two partially occupied sites the occupancy of which refines to 0.75/0.25. Suitable distance restraints were applied to the disordered fragments and the ClO4− anions and isotropic adps retained for partially occupied atoms except Cl. The IR spectra were measured using a FTIR-8400S SHIMADZU IR spectrophotometer. The microanalyses and mass spectrometry analyses were performed by the analytical services of the School of Chemistry at the University of Glasgow. Magnetic measurements of complexes 1 and 2 were performed on polycrystalline samples using a Quantum Design MPMS-XL SQUID magnetometer. Data were corrected for the diamagnetic contribution of the sample holder by measurements and for the diamagnetism of the compounds by using Pascal's constants. EPR measurements were performed at the University of Glasgow. X-band EPR spectra were recorded on a Bruker ELEXSYS E500 spectrometer. Simulations were performed using Bruker's Xsophe software package.27
Synthetic methods
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) (cm−1) = 3362, 1581, 1427, 1265, 1047, 1022, 889, 682, 613. Elemental analysis (C26H54Cl2Cu2N4O16Zn2·2.25H2O) [%], found: C 29.67, H 5.25, N 5.06; calcd: C 29.80, H 5.63, N 5.35. MS (ESI+, m/z): 344.9 [Cu(H5L)]+, 366.07 [Cu(H4L)Na]+, 688.7 [Cu2(H4L)(H5L)]+, 751.07 [Cu2(H3L)(H4L)Zn]+, 787.05 [ClCu2(H4L)2Zn]+, 850.96 [ClCu2(H3L)2Zn2]+, 888.4 [Cl2Cu2(H3L)(H4L)Zn2]+ (see Fig. S1 of ESI†).
:H2O, 1:1) over several days. Yield (crystals) 2% (10 mg). IR: (cm−1) = 3225, 2888, 1640, 1462, 1269, 1044, 1013, 685, 621. Elemental analysis of the crystalline product (C88H200Cl8Cu8N16O80Zn8·5H2O) [%], found: C 24.97, H 5.03, N 5.14; calcd: C 25.36, H 5.08, N 5.38. MS (ESI+, m/z): 344.1 [Cu(H5L)]+, 376.06 [ClCu(H6L)]+, 444.0 [Cu8(H3L)5(H4L)3Na(OH)8Zn8]4+, 687.2 [Cu2(H4L)(H5L)]+, 751.11 [Cu2(H3L)(H4L)Zn]+, 787.09 [ClCu2(H4L)2Zn]+, 851.0 [Cu16(H3L)8(H4L)8(OH)16Zn16]8+ (see Fig. S1 of ESI†).
(cm−1) = 3362, 1581, 1427, 1265, 1047, 1022, 889, 682, 613. Elemental analysis (C26H54Cl2Cu2N4O16Zn2·2.25H2O) [%], found: C 29.67, H 5.25, N 5.06; calcd: C 29.80, H 5.63, N 5.35. MS (ESI+, m/z): 344.9 [Cu(H5L)]+, 366.07 [Cu(H4L)Na]+, 688.7 [Cu2(H4L)(H5L)]+, 751.07 [Cu2(H3L)(H4L)Zn]+, 787.05 [ClCu2(H4L)2Zn]+, 850.96 [ClCu2(H3L)2Zn2]+, 888.4 [Cl2Cu2(H3L)(H4L)Zn2]+ (see Fig. S1 of ESI†).
:H2O, 1:1) over several days. Yield (crystals) 2% (10 mg). IR: (cm−1) = 3225, 2888, 1640, 1462, 1269, 1044, 1013, 685, 621. Elemental analysis of the crystalline product (C88H200Cl8Cu8N16O80Zn8·5H2O) [%], found: C 24.97, H 5.03, N 5.14; calcd: C 25.36, H 5.08, N 5.38. MS (ESI+, m/z): 344.1 [Cu(H5L)]+, 376.06 [ClCu(H6L)]+, 444.0 [Cu8(H3L)5(H4L)3Na(OH)8Zn8]4+, 687.2 [Cu2(H4L)(H5L)]+, 751.11 [Cu2(H3L)(H4L)Zn]+, 787.09 [ClCu2(H4L)2Zn]+, 851.0 [Cu16(H3L)8(H4L)8(OH)16Zn16]8+ (see Fig. S1 of ESI†).
Results and discussion
The reaction between H6L, Cu(CH3COO)2·H2O and ZnCl2 gives the tetranuclear compound [Cu2Zn2(H4L)2(CH3COO)2Cl2]·2.5CH3OH·0.5H2O (1). The chloride and acetate anions present in the starting materials complete the first coordination sphere of the complex. Thin blue plate-like crystals of 1 were obtained by layering the reaction solution with Et2O. However, when a mild base was added to a combination of H6L, ZnCl2, and Cu(ClO4)2·6H2O under analogous conditions to the synthesis of 1, the high nuclearity complex [Cu8Zn8(OH)8(H4L)8](Cl)2(ClO4)6·16H2O (2) was obtained. In this case, the uncoordinated chloride and perchlorate anions present in the starting materials balance the charge of the {Cu8Zn8}8+ cation. Unlike complex 1, compound 2 is initially isolated in good yield as a precipitate (checked by elemental analysis and IR), and subsequently as a crystalline product by vapour diffusion with Et2O into a MeOH:H2O solution of the precipitate.
Several different synthetic routes alternative to those described above, which involved alterations in starting salts, stoichiometric ratios between H6L:salt:base, or the type of base used show the stability and the preference of the complexes for certain reaction conditions. Although 1 has been only isolated following the synthetic process mentioned above, we have been able to obtain 2 through several reactions (e.g. using Cu(CH3COO)2·H2O and ZnCl2 as starting materials), in which the presence of base (e.g. Et3N or dipropylamine) was the only common element. This reveals the key role of base in the formation of the larger heterometallic system 2. Moreover, the coordination of the acetate ions to Zn(II) in 1 suggests that the starting salt also influences the final complex.
X-ray crystallographic analysis
Selected crystallographic experimental details for complexes 1 and 2 are shown in Table 1. Complex 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit of 1 contains two half molecules of [Cu2Zn2(H4L)2(CH3COO)2Cl2], two and a half molecules of methanol, and a half molecule of water. Distances and angles between the symmetry inequivalent molecules do not vary significantly (see Fig. S2 of the ESI†). The maximum difference between bond distances is dZn–O = 0.039 Å, with σcomb = 0.017 (2.3σ). The largest variation in the Zn(II) coordination geometry is the angle defined by {(CH3CO)O–Zn–O(H4L)} (3°). The structure of this tetranuclear complex comprises two {Cu(H4L)} units and two Zn(II) ions that are linked by the two doubly deprotonated H4L2− ligands (see Fig. 1). Each H4L2− is coordinated to one Cu(II) centre through O,N-donor atoms in a [4 + 1] quasi-square-based pyramidal environment (average τCu = 0.06).28 One of the residual hydroxyl groups of H4L2− fills the axial position of the pentacoordinated Cu(II) centres; the three remaining ligand arms are uncoordinated. One chloride and one monodentate acetate anion complete the coordination environment of each Zn(II) centre leading to a distorted tetrahedral geometry. All the metal centres are in the same plane linked together by four oxygen atoms of two H4L2− units to form a “diamond-like” structure, in which Cu(II)/Zn(II) ions occupy alternately its four vertices. The average distances between the different metal ions within this macrocycle are d(Cu⋯Zn) = 3.434(4) Å, and d(Cu⋯Cua) = 5.724(3) Å. The dihedral planes defined for Cu1–O104–Zn1 (in blue) and Cu1a–O110a–Zn1 (in green) form an angle of α(Cu⋯Zn⋯Cua) = 115° (Fig. 2).
. The asymmetric unit of 1 contains two half molecules of [Cu2Zn2(H4L)2(CH3COO)2Cl2], two and a half molecules of methanol, and a half molecule of water. Distances and angles between the symmetry inequivalent molecules do not vary significantly (see Fig. S2 of the ESI†). The maximum difference between bond distances is dZn–O = 0.039 Å, with σcomb = 0.017 (2.3σ). The largest variation in the Zn(II) coordination geometry is the angle defined by {(CH3CO)O–Zn–O(H4L)} (3°). The structure of this tetranuclear complex comprises two {Cu(H4L)} units and two Zn(II) ions that are linked by the two doubly deprotonated H4L2− ligands (see Fig. 1). Each H4L2− is coordinated to one Cu(II) centre through O,N-donor atoms in a [4 + 1] quasi-square-based pyramidal environment (average τCu = 0.06).28 One of the residual hydroxyl groups of H4L2− fills the axial position of the pentacoordinated Cu(II) centres; the three remaining ligand arms are uncoordinated. One chloride and one monodentate acetate anion complete the coordination environment of each Zn(II) centre leading to a distorted tetrahedral geometry. All the metal centres are in the same plane linked together by four oxygen atoms of two H4L2− units to form a “diamond-like” structure, in which Cu(II)/Zn(II) ions occupy alternately its four vertices. The average distances between the different metal ions within this macrocycle are d(Cu⋯Zn) = 3.434(4) Å, and d(Cu⋯Cua) = 5.724(3) Å. The dihedral planes defined for Cu1–O104–Zn1 (in blue) and Cu1a–O110a–Zn1 (in green) form an angle of α(Cu⋯Zn⋯Cua) = 115° (Fig. 2).
 | ||
| Fig. 1 Structure of 1. C, grey; Cl, green; Cu, turquoise; N, blue; O, red; Zn, lavender; H omitted for clarity. | ||
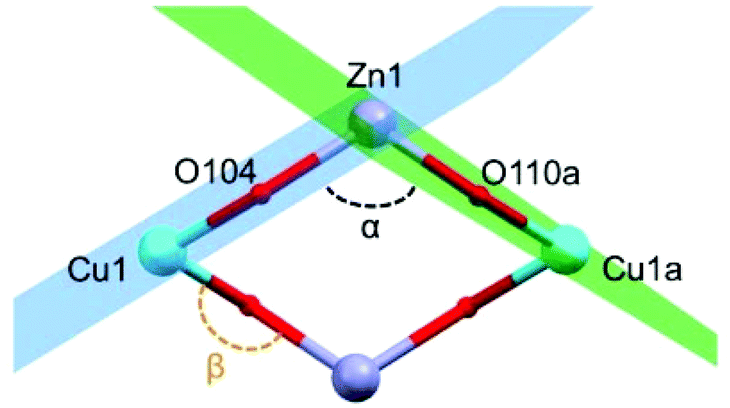 | ||
| Fig. 2 Detail of the metal alkoxide core of 1. Cu, turquoise; O, red; Zn, lavender. | ||
| Complex | 1 | 2 |
|---|---|---|
| Empirical formula | C28.5H65Cl2Cu2N4O19Zn2 | C88H232Cl8Cu8N16O96Zn8 |
| Formula weight | 1094.55 | 4365.76 |
| Temperature/K | 100(2) | 100(2) |
| Crystal system | Triclinic | Tetragonal |
| Space group |
P |
P4/n |
| a/Å, b/Å, c/Å | 10.9593(12), 13.8454(15), 17.4039(19) | 26.5574(19), 26.5574(19), 13.1186(10) |
| α/°, β/°, γ/° | 109.186(7), 90.177(7), 108.009(7) | 90, 90, 90 |
| Volume/Å3 | 2355.8(5) | 9252.5(15) |
| Z | 2 | 2 |
| ρ calc/mg m−3 | 1.543 | 1.567 |
| μ/mm−1 | 2.080 | 2.126 |
| F(000) | 1132.0 | 4496.0 |
| 2θ range for data collection | 4.224 to 50.054° | 4.364 to 50.048° |
| Index ranges | −13 ≤ h ≤ 13, −16 ≤ k ≤ 16, −20 ≤ l ≤ 20 | −29 ≤ h ≤ 23, −13 ≤ k ≤ 31, −15 ≤ l ≤ 15 |
| Reflections collected | 44970 |
23159 |
| Data/restraints/parameters | 44970/9/276 |
8104/38/491 |
| GOF on F2 | 1.020 | 1.042 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.1284, wR2 = 0.3062 | R 1 = 0.0900, wR2 = 0.2302 |
| Final R indexes [all data] | R 1 = 0.2216, wR2 = 0.3782 | R 1 = 0.1522, wR2 = 0.2549 |
| Largest diff. peak/hole/e Å−3 | 3.53/−1.43 | 0.99/−1.15 |
Compound 2 crystallises in the tetragonal space group P4/n. The asymmetric unit of 2 contains a quarter molecule of [Cu8Zn8(OH)8(H4L)8]8+, two perchlorate anions each of 3/4 occupancy, two chloride anions each of 1/4 occupancy, and four water molecules. The structure of this {Cu8Zn8} system consists of eight Cu(II) ions and eight Zn(II) ions linked by hydroxide and H4L2− bridging ligands, adopting an unusual “double-concentric ring” structure (see Fig. 3). The external ring is formed by {Cu(H4L)} units, whereas the internal one is composed of Zn(II) centres and hydroxide ligands. Each Cu(II) centre is coordinated to two H4L2− anions through O- and/or N-donor atoms: one of the H4L2− ligands acts as a chelating ligand (see Fig. 3 right) and the second one, which is coordinated to the neighbouring Cu(II) ion, completes the coordination sphere, leading to a [4 + 2] distorted octahedral environment. The alkoxide arms of the chelating H4L2− units also act as bridges between the Cu(II) ion and two Zn(II) ions; the remaining hydroxyl arms are unbound. In addition to the coordination of the alkoxide groups, each Zn(II) ion is bound to two hydroxide ligands leading to a distorted tetrahedral geometry. In comparison to the {Cu2Zn2} complex 1, the different Cu(II) centres are linked to each other through one CH2OH arm, separated by 5.685(2) Å (see Fig. 3). A possible weak Cu1⋯Cu2 exchange mechanism related to this additional CH2OH arm could be important to understand the differences in the magnetic behaviour exhibited by both compounds 1 and 2 (vide infra). The Zn⋯Cu average distance is d(Cu⋯Zn) = 3.389(2) Å. The heterometallic complex 2 is further stabilized by intramolecular H-bonds between O(OH)⋯H(H4L2−), and between the central Cl−⋯H(OH) (see Fig. S3 and S4 of the ESI†). In both structures, Cu(II) centres are encapsulated by a H4L2− ligand in a very similar arrangement to the heterometallic {Mn18Cu6} complexes [Mn18Cu6O14(H2L)6Cl2(H2O)6]Cl6 and [Mn18Cu6O14(H2L)6Cl6]Cl2, and also [LnCu3(H2edte)3(NO3)][NO3]2 (H4edte = 2,2′,2′′,2′′′-(ethane-1,2-diyldinitrilo)tetraethanol).29,30 The metallo-ligand assembly in these Cu/Zn complexes reveals a clear tendency of the H4L2− ligand to encapsulate the Cu(II) ions in a typical [4 + 1] coordination environment. The predisposition of Cu(II) ions to occupy the central pocket of the ligand defines potential additional binding sites for different metal ions present in the reaction media, thus providing some directing influence in the synthesis of heterometallic complexes.
 | ||
| Fig. 3 Structure of the cation (left) and detail (right) of 2. C, grey; Cu, turquoise; N, blue; O, red; Zn, lavender; H omitted for clarity. | ||
A detailed search of the Cambridge Structural Database (CSD) reveals the rarity of the structures of both 1 and 2: there is only one complex with a similar structure to 1 and there are no reported complexes comparable to 2. The complex [Cu2Zn2(NH3)2Br2(DEA)4]Br2·CH3OH (H2DEA = diethanolamine) displays an analogous {Cu2Zn2} metal core to 1, with relatively similar structural parameters.29 The unprecedented large antiferromagnetic coupling between Cu(II) centres through the unusual Cu–O–Zn–O–Cu exchange pathway exhibited by [Cu2Zn2(NH3)2Br2(DEA)4]Br2·CH3OH (J = −17.5 cm−1, corresponding to the Hamiltonian Ĥ = −2J·Ŝ1·Ŝ2), along with the uncommon structures of 1 and 2 makes their characterization and magnetic studies attractive.
Magnetic properties
In both compounds 1 and 2, the intramolecular distances between the different Cu(II) ions are quite large (5.724(3) Å for 1, and 5.685(2) Å for 2), so magnetic independence of the metal ions could be expected. However considering the surprising strong antiferromagnetic coupling displayed by [Cu2Zn2(NH3)2Br2(DEA)4]Br2·CH3OH, the magnetic properties of both complexes were studied.29 The magnetic susceptibility of both complexes was investigated in applied fields of 10000 Oe (1) and 5000 Oe (2). The plots of χMT versus T of 1 and 2 are shown in Fig. 4. The experimental value of χMT at 260 K of 0.85 cm3 mol−1 K for complex 1 is consistent with that expected for two uncoupled Cu(II) ions (0.83 cm3 mol−1 K, S = 1/2, g = 2.11). The g = 2.11 value is reasonable for Cu(II) ions, and is consistent with that obtained from the EPR experiments (see Fig. S6 of ESI†).31 The χMT value decreases with temperature reaching a minimum of 0.05 cm3 mol−1 K at 1.8 K. The displayed behaviour is characteristic of antiferromagnetically coupled compounds. Using the program PHI (see eqn (S1) of ESI†) the χMT versus T data were fitted (shown as a black solid line in Fig. 4) to give J = −11.5 cm−1, and a monomeric impurity term of 2.76% (R = 99.51%) (fixed parameters in the fit: g = 2.11; temperature-independent paramagnetism TIP = 1.2 × 10−4 cm3 mol−1).32
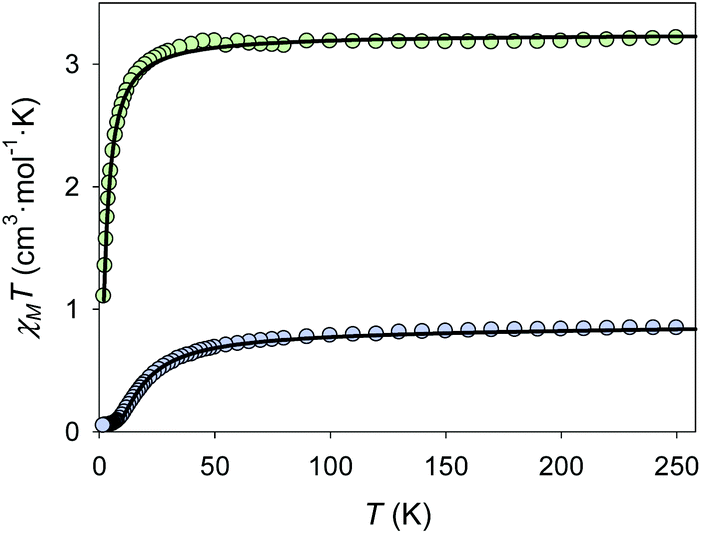 | ||
| Fig. 4 Temperature dependence of χMT for complex 1 (blue) and 2 (green) in an applied field of 10000 (1) and 5000 (2) Oe. The black lines correspond to the fit (see text for details). | ||
The molecular arrangement of compound 1 (see Fig. 1 and 2) defines the triatomic units O–Zn–O, which connect the two Cu(II) ions, as the unique pathway of the displayed antiferromagnetic behaviour. The possibility of antiferromagnetic exchange between the different molecules in the crystal lattice was discarded due to the large values of Cu1⋯Cu1a intermolecular distances (shortest intermolecular distance is d(Cu⋯Cua) = 7.377(4) Å, see Fig. 5). The similar experimental J values shown by 1 (−11.5 cm−1) and {Cu2Zn2(DEA)4} (−17.5 cm−1) suggest an analogous exchange coupling between the Cu(II) ions to that reported for {Cu2Zn2(DEA)4}. The slight decrease in the J value could be due to some structural differences displayed by the atoms involved in the magnetic exchange coupling, such as the different coordination environment of the Cu(II) ions. The pentacoordinated Cu(II) centres of 1 are arranged in a square-based pyramidal environment (C4v), whereas in {Cu2Zn2(DEA)4} they are hexacoordinated in an elongated octahedral geometry (D4h). Therefore, a change in the orbital overlap between the Zn(d), O(p) and Cu(d) centers could occur, leading to a change of the J value. Besides this distinct change in geometry around the Cu(II) centers, a structural comparative study between 1 and {Cu2Zn2(DEA)4} shows that the distances between Cu⋯Cu are slightly higher in 1 and that the average angle between Cu–O–Zn (β(CuOZn)) and the one defined for the dihedral planes (α(Cu⋯Zn⋯Cua)) are also larger in 1 (see Fig. 1 above and Table S1 of ESI†). Despite the slight decrease in the experimental J value compared to {Cu2Zn2(DEA)4}, the coupling between the Cu(II) ions in 1 is still quite unusual. For compound 2, the experimental value of χMT at 260 K of 3.23 cm3 mol−1 K is consistent with that expected for eight Cu(II) ions (3.24 cm3 mol−1 K, S = 1/2, g = 2.08). The g value is reasonable for Cu(II) ions.31 The χMT value decreases with temperature reaching a minimum of 1.10 cm3 mol−1 K at 2 K. The displayed behaviour is characteristic of weak antiferromagnetic exchange, resulting in an S = 0 ground state. The best data fit (see eqn (S2) ESI†) is consistent with an octanuclear complex of Cu(II) (Si = 1/2) with g and J fitting values equal to g = 2.08 and J = −1.23 cm−1 (R = 91.32%). There is a remarkable difference in the experimental J value between 2 (−1.23 cm−1) and 1 (−11.5 cm−1), but also compared to [Cu2Zn2(NH3)2Br2(DEA)4]Br2·CH3OH. It should be highlighted that due to the new coordination arrangement, the average angle formed by the dihedral planes defined as α(Cu⋯Zn⋯Cua) is close to 90° (88.65°), and the β(CuOZn) average angle is also smaller (see Table S1 and Fig. S7 of the ESI†). Numerous studies performed on different families of polynuclear hydroxo and alkoxo Cu(II) complexes reveal the importance of magneto-structural correlations to understand their magnetic behaviour, such as the predisposition to display ferromagnetic coupling when the Cu–O–Cu′ angles tend to 90°.33,34 In addition, the fall of the J value may be a consequence of the existence of an additional CH2OH arm that connects the Cu(II) atoms (vide supra): this coordination allows the appearance of several magnetic exchange pathways in which might participate up to five different C, N, and/or O atoms. Weak antiferromagnetic coupling between metal centers that form a ring structure is not unusual, as similar behaviour is displayed by other related rings of different transition metal ions.35
 | ||
| Fig. 5 Intermolecular hydrogen bonding interactions in complex 1. The hydrogen bonds between the central amino and hydroxyl units are shown as dashed black lines. Cu1⋯Cu1a intermolecular distances are highlighted as dashed orange lines. C grey; Cl, green; Cu, turquoise; N, blue; O, red; Zn, lavender; H omitted. | ||
Conclusions
In conclusion, we have confirmed the ability of the bis–tris propane ligand to direct the synthesis of two new 3d/3d′ heterometallic complexes. Slight modifications of the synthetic route have led to the synthesis of [Cu2Zn2(H4L)2(CH3COO)2Cl2]·2.5CH3OH·0.5H2O (1) and [Cu8Zn8(OH)8(H4L)8](Cl)2(ClO4)6·16H2O (2). It has been shown that the presence of base in the reaction media is essential for the formation of large polynuclear systems such as 2. Although a range of ring-like systems have been reported, we note that this is the first example of heterometallic ring formed by the assembly of two concentric homometallic rings. Complex 2 could also be considered a new member of the antiferromagnetic even-membered rings family which are interesting models for the study of one-dimensional magnetic systems, magnetic anisotropy or different quantum effects in molecular magnetism.36–39 Moreover, despite the long intramolecular Cu⋯Cu distances, both complexes display an interesting and quite unusual exchange pathway: the antiferromagnetic coupling between Cu(II) ions observed suggests a possible contribution of Zn(d) orbitals in the magnetic exchange. The ability of the diamagnetic Zn(II) ions to facilitate exchange interactions between different paramagnetic centres opens the possibility of using closed shell metal ions as additional linkers to the organic ligands typically used in molecular magnetism.40Competing interest
The authors declare no competing financial interest.Acknowledgements
The UK Engineering and Physical Sciences Research Council are thanked for financial support (grant ref. EP/I027203/1). The data which underpin this work are available at http://dx.doi.org/10.5525/gla.researchdata.201. We thank the EPSRC UK National Crystallographic Service for single crystal data collection. We thank Stephen Sproules for performing the EPR measurements and EPR data analysis for 1.Notes and references
- J. T. Brockman, J. C. Huffman and G. Christou, Angew. Chem., Int. Ed., 2002, 41, 2506 CrossRef CAS.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978 CrossRef CAS PubMed.
- M. Murugesu, M. Habrych, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2004, 126, 4766 CrossRef CAS PubMed.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- R. S. Forgan, D. C. Friedman, C. L. Stern, C. J. Bruns and J. F. Stoddart, Chem. Commun., 2010, 46, 5861 RSC.
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed.
- R. S. Forgan, R. A. Smaldone, J. J. Gassensmith, H. Furukawa, D. B. Cordes, Q. Li, C. E. Wilmer, Y. Y. Botros, R. Q. Snurr, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 406 CrossRef CAS PubMed.
- P. Bag, A. Chakraborty, G. Rogez and V. Chandrasekhar, Inorg. Chem., 2014, 53, 6524 CrossRef CAS PubMed.
- V. Baskar, K. Gopal, M. Helliwell, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Dalton Trans., 2010, 39, 4747 RSC.
- R. W. Saalfrank, R. Prakash, H. Maid, F. Hampel, F. W. Heinemann, A. X. Trautwein and L. H. Böttger, Chem. – Eur. J., 2006, 12, 2428 CrossRef CAS PubMed.
- E. Colacio, J. Ruiz-Sanchez, F. J. White and E. K. Brechin, Inorg. Chem., 2011, 50, 7268 CrossRef CAS PubMed.
- L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Inorg. Chem., 2009, 48, 3323 CrossRef CAS PubMed.
- T. Glaser, Chem. Commun., 2011, 47, 116 RSC.
- I. Oyarzabal, J. Ruiz, J. M. Seco, M. Evangelisti, A. Camón, E. Ruiz, D. Aravena and E. Colacio, Chem. – Eur. J., 2014, 20, 14262 CrossRef CAS PubMed.
- L. Zhang, R. Clérac, P. Heijboer and W. Schmitt, Angew. Chem., Int. Ed., 2012, 51, 3007 CrossRef CAS PubMed.
- A. Ferguson, A. Darwish, K. Graham, M. Schmidtmann, A. Parkin and M. Murrie, Inorg. Chem., 2008, 47, 9742 CrossRef CAS PubMed.
- A. Ferguson, M. Schmidtmann, E. K. Brechin and M. Murrie, Dalton Trans., 2011, 40, 334 RSC.
- V. A. Milway, F. Tuna, A. R. Farrell, L. E. Sharp, S. Parsons and M. Murrie, Angew. Chem., Int. Ed., 2013, 52, 1949 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- R. I. Cooper, R. O. Gould, S. Parsons and D. J. Watkin, J. Appl. Crystallogr., 2002, 35, 168 CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194 CrossRef.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- G. R. Hanson, K. E. Gates, C. J. Noble, M. Griffin, A. Mitchell and S. Benson, J. Inorg. Biochem., 2004, 98, 903 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- E. A. Buvaylo, V. N. Kokozay, O. Y. Vassilyeva, B. W. Skelton, J. Jezierska, L. C. Brunel and A. Ozarowski, Chem. Commun., 2005, 4976 RSC.
- F. J. Kettles, V. A. Milway, F. Tuna, R. Valiente, L. H. Thomas, W. Wernsdorfer, S. T. Ochsenbein and M. Murrie, Inorg. Chem., 2014, 53, 8970 CrossRef CAS PubMed.
- P. de Hoog, P. Gamez, O. Roubeau, M. Lutz, W. L. Driessen, A. L. Spek and J. Reedijk, New J. Chem., 2003, 27, 18 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107 CrossRef CAS.
- M. Handa, N. Koga and S. Kida, Bull. Chem. Soc. Jpn., 1988, 61, 3853 CrossRef CAS.
- J. L. Atwood, E. K. Brechin, S. J. Dalgarno, R. Inglis, L. F. Jones, A. Mossine, M. J. Paterson, N. P. Power and S. J. Teat, Chem. Commun., 2010, 46, 3484 RSC.
- A. Cornia, M. Affronte, A. G. M. Jansen, G. L. Abbati and D. Gatteschi, Angew. Chem., Int. Ed., 1999, 38, 2264 CrossRef CAS.
- F. Meier and D. Loss, Phys. Rev. B: Condens. Matter, 2001, 64, 224411 CrossRef.
- F. Meier and D. Loss, Phys. Rev. Lett., 2001, 86, 5373 CrossRef CAS.
- P. King, T. C. Stamatatos, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2006, 45, 7379 CrossRef CAS PubMed.
- A. M. Ako, B. Burger, Y. Lan, V. Mereacre, R. Clérac, G. Buth, S. Gómez-Coca, E. Ruiz, C. E. Anson and A. K. Powell, Inorg. Chem., 2013, 52, 5764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, ESI+-MS spectra, intramolecular hydrogen-bonding interactions, further magnetic data, and a table of selected structural details of 1 and 2. CCDC 1415818–1415819. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03344f |
| This journal is © The Royal Society of Chemistry 2015 |