DOI:
10.1039/C5DT03060A
(Paper)
Dalton Trans., 2015,
44, 19282-19293
Pentanuclear [2.2] spirocyclic lanthanide(III) complexes: slow magnetic relaxation of the DyIII analogue†
Received
8th August 2015
, Accepted 4th October 2015
First published on 7th October 2015
Abstract
The reaction of LnCl3·6H2O (Ln = Dy3+, Tb3+ and Ho3+) with the multisite coordinating ligand N′-(2-hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide (LH3) in the presence of pivalic acid (PivH) leads to the formation of three isostructural homometallic pentanuclear complexes, [Dy5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2η1Piv)2(H2O)]·Cl·9·5H2O·5MeOH (1), [Tb5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2η1Piv)2(H2O)]·Cl·10.5H2O·2MeOH·2CHCl3 (2) and [Ho5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2η1Piv)2(H2O)]·Cl·14.5H2O·2CHCl3 (3). 1–3 are monocationic and are comprised of four doubly deprotonated [LH]2− ligands along with six pivalate ions. These complexes possess a [2.2] spirocyclic topology formed by the fusion of two triangles of LnIII ions at a common vertex. The magneto chemical analysis reveals the presence of antiferromagnetic exchange interactions at low temperature, and the DyIII complex 1 gives an out-of-phase signal with a small curvature in alternating current (ac) magnetic susceptibility measurement. Application of a 3000 G static field during ac measurement intensifies the signals, revealing a second slow relaxation process in the DyIII analogue.
Introduction
The synthesis of polynuclear clusters of 4f metal ions is of interest because of their potential utility in single-molecule magnets (SMMs), catalysis,1 luminescence,2 medical imaging,3 magnetic refrigeration,4 high-density data storage,5 spintronics6 and quantum computing.7 SMMs are characterized by a slow relaxation of magnetization below the blocking temperature, TB. Magnetic phenomena have been thoroughly explored in polynuclear 3d metal complexes8,9 and heterometallic 3d/4f complexes,10 and the recent years have seen the emergence of lanthanide complexes, particularly those involving Dy3+ or Tb3+. Here, the zero-field splitting of the mJ sub-states belonging to the J ground state produces the thermal energy barriers. Interest in lanthanide complex SMMs has been fueled by the seminal discovery by Winpenny and co-workers of complexes [Dy4K2O(OtBu)12] and [Dy5O(OiPr)13], possessing exceptionally high energy barriers for magnetization reversal.11 Homometallic 4f complexes of varying nuclearities (Ln,12 Ln2,13 Ln3,14 Ln4,15 Ln5,16 Ln6,17 Ln7,18 Ln8,19 Ln9,20 Ln10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and Ln1122) are now documented in the literature, and we have reported new SMMs involving 3d/4f23 and 4f24 metal ions. The five examples of pentanuclear lanthanide complexes known display three distinct structural topologies: pyramidal,16a–c butterfly16e and trigonal bipyramidal.16d These pentanuclear complexes evince SMM behavior including Winpenny's [Dy5O(OiPr)13] complex, which showed a slow relaxation of magnetization with a 528 ± 11 K thermal energy barrier.16a To pursue new polynuclear lanthanide complexes, we have designed a new linear alkyl hydrazone-based multidentate Schiff base ligand, N′-(2-hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide (LH3); upon reaction with LnCl3·6H2O (Ln = Dy3+, Tb3+ and Ho3+), this afforded homometallic pentanuclear [2.2] spirocyclic complexes 1–3, [Ln5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·xH2O·yMeOH·zCHCl3 (Dy3+, x = 9.5, y = 5, z = 0; Tb3+, x = 10.5, y = 2, z = 2 and Ho3+, x = 14.5, y = 0, z = 2). In these complexes two triangles of Ln3+ ions are fused through a common vertex. The synthesis, structure and magnetic properties of these complexes are discussed herein.
21 and Ln1122) are now documented in the literature, and we have reported new SMMs involving 3d/4f23 and 4f24 metal ions. The five examples of pentanuclear lanthanide complexes known display three distinct structural topologies: pyramidal,16a–c butterfly16e and trigonal bipyramidal.16d These pentanuclear complexes evince SMM behavior including Winpenny's [Dy5O(OiPr)13] complex, which showed a slow relaxation of magnetization with a 528 ± 11 K thermal energy barrier.16a To pursue new polynuclear lanthanide complexes, we have designed a new linear alkyl hydrazone-based multidentate Schiff base ligand, N′-(2-hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide (LH3); upon reaction with LnCl3·6H2O (Ln = Dy3+, Tb3+ and Ho3+), this afforded homometallic pentanuclear [2.2] spirocyclic complexes 1–3, [Ln5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·xH2O·yMeOH·zCHCl3 (Dy3+, x = 9.5, y = 5, z = 0; Tb3+, x = 10.5, y = 2, z = 2 and Ho3+, x = 14.5, y = 0, z = 2). In these complexes two triangles of Ln3+ ions are fused through a common vertex. The synthesis, structure and magnetic properties of these complexes are discussed herein.
Results and discussion
Synthesis
Ligand design is a crucial element in modulating the nuclearity and structural topology of lanthanide complexes. Recently, we have prepared rhombus-shaped Ln4 complexes, [Ln4(LH)2(μ2-O)4(H2O)8] (Ln = Dy3+ and Ho3+),25 incorporating an aroyl hydrazone-based Schiff base ligand (6-hydroxymethyl)-N′-((8-hydroxyquinolin-2-yl)-methylene)picolinohydrazide (Scheme 1a).
 |
| | Scheme 1 (a) Synthesis of rhombus-shaped tetranuclear lanthanide complexes25 (b) Synthesis of cubane-shaped tetranuclear lanthanide complexes.27 | |
Motivated by this work, as well as by the use of other hydrazone-based Schiff base ligands in the recent literature,14c,15d,17d,24,26 we have prepared a multisite coordinating semi-flexible alkyl hydrazone-based ligand, N′-(2-hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide (LH3). This was produced using a two-step synthetic protocol that involved the preparation of B1, which subsequently undergoes a condensation reaction with B2 to give LH3 (Scheme 2). The semi flexible ligand, LH3 provides five divergent coordinating sites: an alkyl hydrazone oxygen, an imine N, an amide N, a phenolic O and a flexible –CH2OH arm. The latter is a crucial element in the formation of the coordination geometry, as recently shown by us in the preparation of cubane-shaped tetranuclear lanthanide complexes (Scheme 1b).27 The pivalic acid co-ligands help to saturate the primary coordination spheres of the metals by bridging the metal centers, and confers lipophilicity to the complexes.
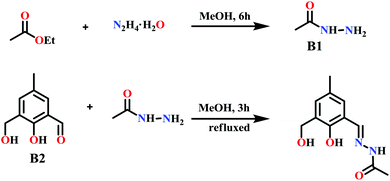 |
| | Scheme 2 Synthesis of the ligand LH3. | |
The reaction of LH3, LnCl3·6H2O, and pivalic acid in the stoichiometric ratio of 4:5:6 in the presence of 4 equivalents of triethylamine in methanol afforded pentanuclear complexes [Ln5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·xH2O·yMeOH·zCHCl3 [compound 1, Ln = Dy3+, x = 9.5, y = 5, z = 0; compound 2, Ln = Tb3+, x = 10.5, y = 2, z = 2 and compound 3, Ln = Ho3+, x = 14.5, y = 0, z = 2] (Scheme 3).
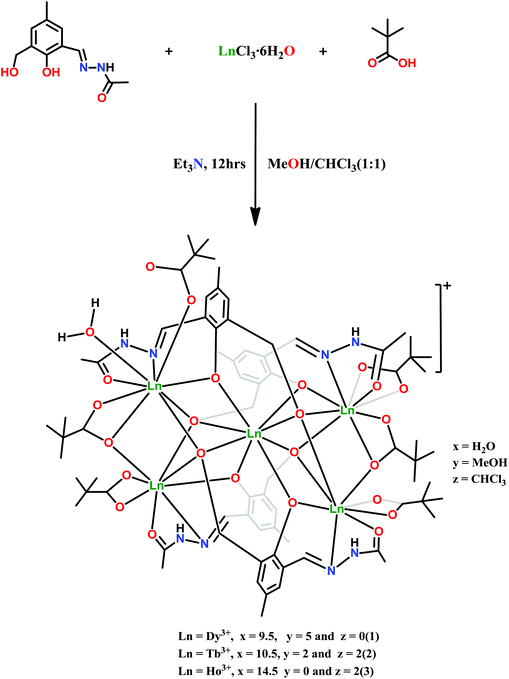 |
| | Scheme 3 Synthesis of [Ln5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·xH2O·yMeOH·zCHCl3 (Dy, x = 9.5, y = 5, z = 0; Tb, x = 10.5, y = 2, z = 2 and Ho, x = 14.5, y = 0, z = 2). | |
X-ray crystal structures
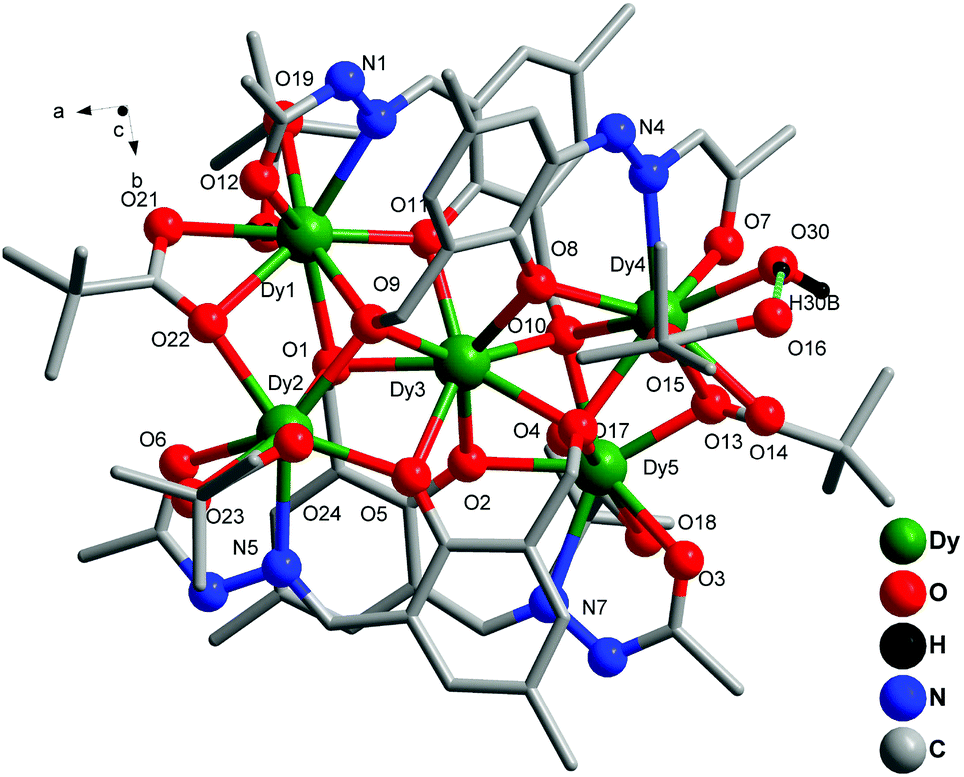
Single-crystal X-ray diffraction measurements reveal that compounds 1–3 are isostructural, and crystallize in the tetragonal space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) with Z = 4. The complexes are monocationic, with their charges each balanced by a chloride anion, and possess a [2.2] spirocycle of LnIII ions containing two fused triangular motifs. Complex 1 has been chosen as a representative example to illustrate the common structural features of these clusters. Selected bond parameters of 1 are summarized in Table 1. The molecular structure and selected bond parameters of the other complexes (2 and 3) are presented in the ESI (Fig. S1–S2† and Tables S1–S2†). A perspective view of the molecular structure of 1 is depicted in Fig. 1.
with Z = 4. The complexes are monocationic, with their charges each balanced by a chloride anion, and possess a [2.2] spirocycle of LnIII ions containing two fused triangular motifs. Complex 1 has been chosen as a representative example to illustrate the common structural features of these clusters. Selected bond parameters of 1 are summarized in Table 1. The molecular structure and selected bond parameters of the other complexes (2 and 3) are presented in the ESI (Fig. S1–S2† and Tables S1–S2†). A perspective view of the molecular structure of 1 is depicted in Fig. 1.
 |
| | Fig. 1 Molecular structure of 1 (selected hydrogen atoms, chloride and the solvent molecules have been omitted for clarity). | |
Table 1 Selected bond distances (Å) and bond angles (°) parameters for 1
|
Selected bond length around Dy1
|
Dy(3)–O(11) |
2.351(6) |
Dy(5)–O(10) |
2.409(6) |
| Dy(1)–O(11) |
2.310(6) |
Dy(3)–O(4) |
2.351(6) |
Dy(5)–O(18) |
2.456(9) |
| Dy(1)–O(12) |
2.396(7) |
Dy(3)–O(4) |
2.351(6) |
Dy(5)–N(7) |
2.508(9) |
| Dy(1)–O(20) |
2.409(7) |
Dy(3)–O(1) |
2.353(6) |
|
| Dy(1)–O(9) |
2.417(6) |
Dy(3)–O(2) |
2.381(7) |
Selected Bond angles around Dy
|
| Dy(1)–O(19) |
2.442(7) |
Dy(3)–O(5) |
2.414(6) |
Dy(2)–O(1)–Dy(1) |
97.9(2) |
| Dy(1)–O(21) |
2.447(6) |
|
Dy(3)–O(1)–Dy(1) |
94.3(2) |
| Dy(1)–O(1) |
2.457(6) |
Selected bond length around Dy4
|
Dy(5)–O(2)–Dy(3) |
97.2(2) |
| Dy(1)–N(1) |
2.519(8) |
Dy(4)–O(15) |
2.312(7) |
Dy(3)–O(4)–Dy(5) |
95.2(2) |
| Dy(1)–O(22) |
2.521(6) |
Dy(4)–O(8) |
2.373(6) |
Dy(3)–O(4)–Dy(4) |
96.9(2) |
|
|
Dy(4)–O(7) |
2.384(7) |
Dy(5)–O(4)–Dy(4) |
99.5(2) |
|
Selected bond length around Dy2
|
Dy(4)–O(4) |
2.396(6) |
Dy(2)–O(5)–Dy(3) |
95.3(2) |
| Dy(2)–O(24) |
2.320(7) |
Dy(4)–O(30) |
2.411(7) |
Dy(3)–O(8)–Dy(4) |
98.4(2) |
| Dy(2)–O(5) |
2.324(6) |
Dy(4)–N(4) |
2.494(8) |
Dy(3)–O(9)–Dy(1) |
95.9(2) |
| Dy(2)–O(22) |
2.325(6) |
Dy(4)–O(10) |
2.500(6) |
Dy(3)–O(9)–Dy(2) |
94.5(2) |
| Dy(2)–O(1) |
2.333(6) |
Dy(4)–O(14) |
2.505(8) |
Dy(1)–O(9)–Dy(2) |
96.18(9) |
| Dy(2)–O(6) |
2.402(7) |
Dy(4)–O(13) |
2.666(9) |
Dy(3)–O(10)–Dy(5) |
96.0(2) |
| Dy(2)–O(9) |
2.439(6) |
|
Dy(3)–O(10)–Dy(4) |
95.4(2) |
| Dy(2)–N(5) |
2.450(8) |
Selected bond length around Dy5
|
Dy(5)–O(10)–Dy(4) |
96.1(2) |
| Dy(2)–O(23) |
2.454(7) |
Dy(5)–O(13) |
2.258(7) |
Dy(1)–O(11)–Dy(3) |
98.3(2) |
|
Selected bond length around Dy3
|
Dy(5)–O(2) |
2.286(6) |
Dy(5)–O(13)–Dy(4) |
95.4(3) |
| Dy(3)–O(10) |
2.301(6) |
Dy(5)–O(17) |
2.356(11) |
Dy(2)–O(22)–Dy(1) |
96.4(2) |
| Dy(3)–O(8) |
2.321(6) |
Dy(5)–O(3) |
2.381(7) |
Dy(2)–O(1)–Dy(3) |
96.7(2) |
| Dy(3)–O(9) |
2.330(6) |
Dy(5)–O(4) |
2.387(6) |
|
|
The pentanuclear complex 1 is formed by the concerted coordination action of the four doubly deprotonated ligands [LH]2−. Four out of five potential coordinating sites of the ligand are coordinated to the metal centers: a phenolate oxygen which functions as a bridging ligand between two Dy3+ centers, a deprotonated hydroxymethyl arm which functions as a μ3-capping ligand among three DyIII centers, an imine nitrogen binding a Dy3+ center, and an acetohydrazide oxygen coordinating a Dy3+ center. The framework provided by the ligand [LH]2− is bolstered by the coordination of six pivalate ligands: one is η1-coordinated to Dy4, while three others chelate Dy1, Dy2 and Dy5 in an η2 fashion. The remaining two pivalate ligands exhibit both bridging and chelating modes (μ2–η2:η1) in their coordination that links Dy1 to Dy2 and Dy4 to Dy5. The pentanuclear [2.2] spirocyclic core is thus constructed by the coordination action of four [LH]2− (μ4–η3:η2:η1:η1) and six pivalate ligands. The coordination modes of the ligands are depicted in Fig. 2.
 |
| | Fig. 2 Binding modes of [LH]2− and the pivalate ligands with Dy3+ ions in 1. | |
The pentacationic pentanuclear core, [Dy5(μ3-O)4(μ2-O)4(μ2–η2η1Piv)2]5+ consists of two Dy3 triangles which are interconnected by sharing a common vertex, Dy3. The remaining four vertices of the two triangles are occupied by Dy1, Dy2, Dy4 and Dy5 (Fig. 3a). The edges of the triangles are formed by the pivalate ligands as well as phenolate oxygens of the ligand [LH]2− while the faces of the triangle are effectively capped by μ3-O, emanating from the flexible pendant hydroxymethyl arms (Fig. 3a). The capping μ3 oxygen atoms lie at an average of ∼1.219 Å away from the two triangular planes. The triangles are nearly equilateral, with Dy⋯Dy vertex lengths of 3.50–3.651 Å and Dy–Dy–Dy angles that range from 58.1°–62.2°. The two triangles are twisted at the Dy3 [2.2] spirocyclic node by a dihedral angle 55.85° (Fig. 3b).
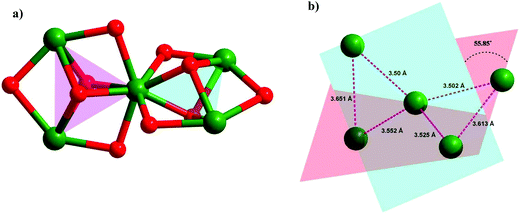 |
| | Fig. 3 (a) [2.2] spirocyclic core of the complex 1 (b). [2.2] spirocyclic core showing the dihedral angle between the two triangular motifs along with the distance between the Dy3+ centers. | |
The five crystallographically independent lanthanide centers present in 1 can be classified into three coordination geometry types (all distorted): triangular dodecahedron (Dy1, Dy5), square antiprism (Dy3), and mono-capped square-anti-prism (Dy2, Dy4) (Fig. 4). A minor variation exists in the coordination environment around Dy2 and Dy4: unlike the rest of the dysprosium centers, Dy4 is coordinated by a water molecule, which is engaged in strong hydrogen bonding with the oxygen atom of the η1-pivalate ligand (Fig. 1) (D–H⋯A distance 1.746 Å and angle 154.41°) accounting for the η1 binding mode rather than the anticipated η2 coordination mode of the pivalate ligand. The Dy–Ophenoxy bond lengths fall in the range of ∼2.286–2.414 Å which are slightly shorter than the Dy–Oalkoxy bond lengths (∼2.330–2.457 Å). These are in turn shorter than many Dy–Opiv bond distances that range from ∼2.258 to ∼2.660 Å. Bond lengths involving the coordinated imine nitrogens lie in the range of ∼2.449–2.519 Å. The Dy–Ophen–Dy (95.31°–98.36°) and Dy–Opiv–Dy angles (95.36–96.36°) are in a similar range.
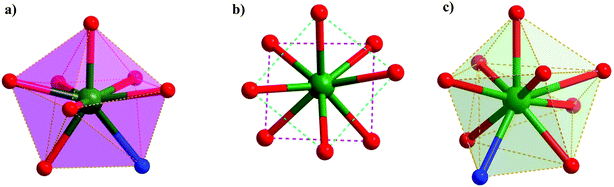 |
| | Fig. 4 Local geometry around the three types of dysprosium centres in 1: (a) Dy1 and Dy5 possessing a distorted triangular dodecahedral geometry, (b) Dy3 possessing a distorted square antiprism geometry and (c) Dy2 and Dy4 possessing a distorted monocapped square antiprism. | |
The [2.2] spirocyclic topology observed in complexes 1–3 is quite distinct from the pyramidal,16a–c butterfly16e or trigonal bipyramidal16d shaped homometallic Ln5 clusters reported previously (Fig. S9†). A comparison of the geometry around the metal centers, the structural topology around the metal ions and the SMM properties of the pentanuclear lanthanide families are summarized in Table 2.
Table 2 Structural and magnetic features of pentanuclear lanthanide assemblies
| Compound |
Core topology |
Coordination numbers (local geometries around Ln(III) centers) |
Magnetic properties |
Ref. |
| [Dy5(μ3-OH)6 (Acc)6 (H2O)10]·Cl9 |
Trigonal bipyramidal |
Eight coordinated (distorted square-antiprism) |
SMM |
16d
|
|
U
eff = 1.91 K |
| Acc = 1-amino cyclohexane-l-carboxylic acid. |
τ
0 = 1.01 × 10−6 s |
| [Dy5O(OiPr)13] |
Square pyramidal |
Six coordinated (octahedral) |
SMM |
16a
|
|
U
eff = 528 ± 11 K, 46.6 ± 0.7 K |
|
iPr = isopropyl |
|
τ
0 = 4.7 × 10−10 s, 3.8 × 10−6 s |
| [Dy5(OH)5(α-AA)4(Ph2acac)6] |
Square pyramidal |
Eight coordinated |
SMM |
16b
|
| α-AA = D-PhGly |
| [Dy5 (μ3-OH)3 (opch)6 (H2O)3] |
Butterfly |
Eight coordinated (distorted dodecahedral), nine coordinated (distorted mono-capped square-antiprismatic) and nine coordinated (distorted triplecapped prismatic) |
SMM |
16e
|
|
U
eff = 8.1 ; 37.9 K |
| H2opch = o-vanillin pyrazine acylhydrazone |
τ
0 = 1.7 × 10−5s; 9.7 × 10−8 s |
| [Dy5 (μ3-OH)3 (opch)6 (H2O)3](ClO4)2 |
Butterfly |
Eight coordinated (distorted dodecahedral), nine coordinated (distorted mono-capped square-antiprismatic) and nine coordinated (distorted triplecapped prismatic) |
SMM |
16e
|
|
U
eff = 197 K ; |
| H2opch = o-vanillin pyrazine acylhydrazone |
τ
0 = 3.2 × 10−9 s |
| [Dy5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl |
[2.2] spirocycle |
Eight coordinated (distorted triangulardodecahedral), eight coordinated (distorted square-antiprism) and nine coordinated (mono-capped-square-antiprism) |
Slow relaxation |
This work |
Thermogravimetric study of complex 1–3
Thermogravimetric analysis reveals that all the complexes 1–3 exhibit almost a similar decomposition pattern towards heat treatment involving a two-step weight loss process (Fig. S10–S12†). A small weight loss in the region of 60–120 °C was observed for all the complexes which is in part due to the loss of solvents of crystallization. In all the cases, some of the solvent molecules of crystallization are lost rapidly as the crystals are brought outside the mother liquor at room temperature. In the second step, above 300 °C, rapid weight loss of all the complexes was observed, confirming the decomposition of the complexes.
Magnetic studies
Direct current (dc) magnetic susceptibility data for 1–3 are presented as χmT vs. T curves at B = 0.1 Tesla, and molar magnetization Mmvs. applied field B diagrams at T = 2 K in Fig. 5. For the DyIII analogue 1, χmT reaches a value of 64.9 cm3 K mol−1 at 290 K which is slightly below the range 65.1–70.3 cm3 K mol−1 expected27a for five non-interacting Dy3+ (6H15/2, J = 15/2, gJ = 4/3) centers. The decrease of χmT with lowering temperature is gradual down to ∼100 K, and then more precipitous (approximate slope). This behavior and the low value of χmT at 290 K have their origin in the ligand field effect (i.e. depopulation of the mJ sublevels) and potential antiferromagnetic exchange interactions within the compound (note that the distinct deviation from linear behavior in χmT is already introduced at higher temperatures T ≈ 200 K). Analysis of the dc magnetic susceptibility data for 2 and 3 reveals similar behavior, and the χmT values at 290 K are also slightly lower than those expected for the respective number of non-interacting lanthanide centers. For 2, χmT reaches 58.1 cm3 K mol−1 at 290 K (expected27a 58.2–60.1 cm3 K mol−1 for five non-interacting TbIII (7F6, J = 6, gJ = 3/2) centers); for 3, χmT is 66.1 cm3 K mol−1 at 290 K (expected27a 66.3–69.0 cm3 K mol−1 for five non-interacting HoIII (5I8, J = 8, gJ = 5/4) centers).
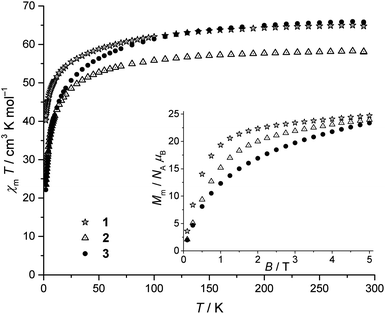 |
| | Fig. 5 Temperature dependence of χmT of 1–3 at 0.1 Tesla; inset: molar magnetization Mm as function of the applied field B at 2 K. | |
At 2 K, the molar magnetizations Mm of 1–3 (Fig. 5, inset) indicate saturation and thus hint roughly at the ground state of the LnIII centers in each compound expected due to ligand field effects on the spin–orbit ground term, since the weak exchange interactions of lanthanides at zero field are eliminated by the applied maximum fields: all of the extrapolated saturation values are roughly right in the center between the minimum magnetization of the corresponding five non-interacting LnIII centers (5gJmJ,minNAμB) and the maximum magnetization of such centers (5gJJNAμB). This indicates that neither all LnIII centers of a compound are characterized by a ground state of minimal mJ,min (±1/2 for 1, 0 for 2, 3) nor by the maximum mJ = J.
A comprehensive model of compounds 1–3 based exclusively on the magnetic susceptibility data is not feasible because the description of the compounds should include at least two different lanthanide sites (approximately D4d and D3h symmetric, eight- or nine-fold coordinated centers) and three different exchange pathways. Without a simpler model system such as {Gd5}, or complementary data such as from inelastic neutron scattering, degenerate preliminary solutions modeled by the computational framework CONDON 2.027b,c cannot be ruled out. One feature common among all of these preliminary solutions provides qualitative insight into χmT and Mm behavior presented in Fig. 5: all calculations reveal very weak ferromagnetic exchange interactions between the two outer lanthanides of each triangle (i.e. Ln1⋯Ln2 and Ln4⋯Ln5, Fig. 3b), and either antiferromagnetic (≈−0.3 cm−1) or almost nonexistent exchange interactions for the remaining pathways (Ln1⋯Ln3, Ln2⋯Ln3, Ln3⋯Ln4, and Ln3⋯Ln5). Note that although the bridging ligands are similar, and thus exchange interactions might be assumed to be similar as well, the various distances between the Ln pairs and the presence of three different site geometries, and thus ligand fields, explain the difference in the exchange interaction parameters.
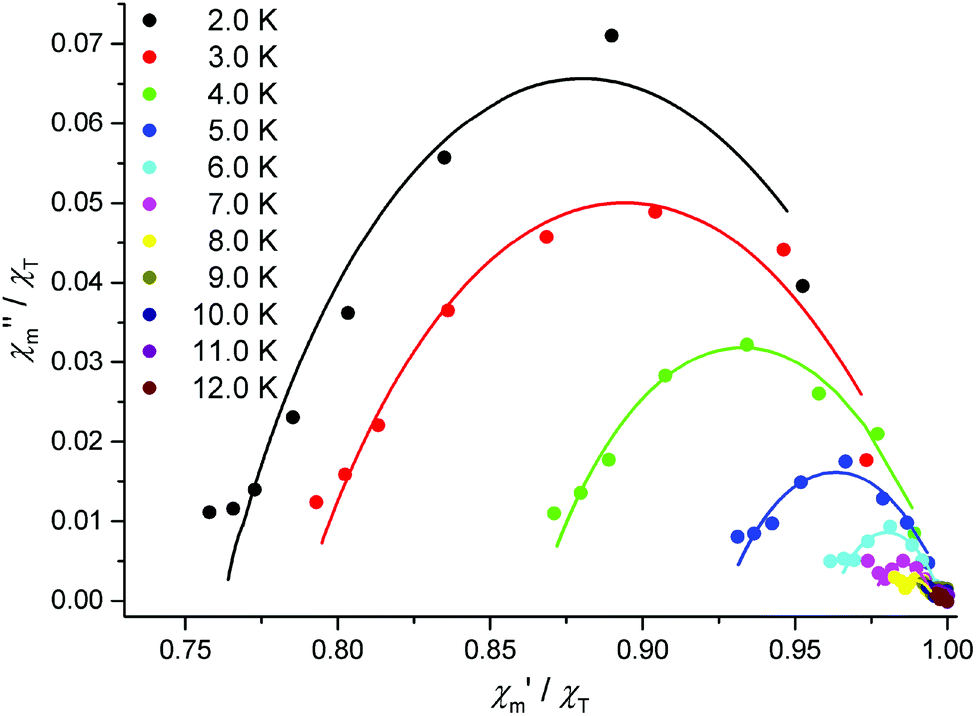
Alternating current (ac) molar magnetic susceptibility measurements on complexes 1–3 revealed out-of-phase signals for the DyIII analogue 1, but not for 2 and 3, at zero dc field. The very small curvatures within the Argand plane for complex 1 (Fig. S3†) reveal in-phase signals χ′m that are almost independent from the applied frequency (Fig. 6). The application of external static fields of up to 4000 G slightly shifted χ′m, but did not produce significantly greater curvatures for frequencies greater than 100 Hz. Similar behavior was observed by Thielemann et al.16b who ascribed it to a blocking temperature which lies significantly below the range explored by the magnetic measurements. Optimal slow relaxation magnetic measurements of complex 1 with respect to the experimental options at hand were obtained using a 3000 G static field, and a frequency range of 0.03 Hz–111 Hz (Fig. 7, insets Fig. 6). The in-phase (χ′m) and out-of-phase (χ′′m′) ac susceptibility components can be fitted to a Cole–Cole equation28 for each temperature (Fig. 7, solid lines; Fig. S3†). To determine the average relaxation times of the magnetization under the optimized conditions, the temperature-dependent fit parameters have been analyzed using an Arrhenius expression (τ = τ0exp(ΔU/kBT)). This results in an effective energy barrier ΔU = (5.2 ± 0.5) cm−1 and a time constant of τ0 = (2.6 ± 0.6) × 10−2 s (Fig. S5†). In the generalized Debye model, the distribution width of τ is parameterized by the scalar α. The nonzero mean value of α = 0.45 ± 0.08 reveals that several relaxation processes are active in this system. The time constant τ0 is anomalously large for SMM behavior, i.e. the entirely phenomenological parameter τ′0 does not fall within the typical SMM range, and indicates that rather a secondary relaxation process was observed in the presence of a bias field instead of the relaxation process indicated at zero field. This is supported by the occurrence of the minima in the Cole–Cole curves for higher frequencies (lower χ′m values) hinting at further subsequent semi-circles which we could not enhance with the experimental set-up at hand. Since some or all exchange interactions within lanthanide compounds are overridden by the application of an external field of 3000 G, the observed process may be connected to a forced alignment of the momenta albeit further evidence is needed to prove this hypothesis.
 |
| | Fig. 6 Left: in-phase magnetic susceptibility χm′ vs. T; right: out-of-phase magnetic susceptibility χm′′ vs. T of 1 at different frequencies f at zero dc bias field; insets: at 3000 G dc bias field (dashed lines are guides for the eye). | |
 |
| | Fig. 7 Normalized Cole–Cole plot of 1 at different temperatures and 3000 G dc field (solid circles), frequencies range from 0.03–111 Hz, χT is the isothermal susceptibility in the limit of lowest frequencies; fit to Cole–Cole equation (solid lines). | |
Conclusions
In summary, a series of isostructural homometallic pentanuclear Ln5 complexes that possess an unprecedented [2.2] spirocyclic topology were synthesized. The frameworks of these complexes are comprised of the multidentate Schiff base ligand, N′-(2-hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide, along with several bridging pivalate groups. The lanthanide centers present in the pentanuclear assembly can be grouped into three types based on their local coordination geometry: an eight-coordinate lanthanide in a distorted triangular dodecahedral geometry, an eight-coordinate lanthanide in a distorted square-antiprism geometry and a nine-coordinate lanthanide in a mono-capped square anti-prism geometry. Variable-temperature dc and ac magnetic susceptibility measurements reveal weak antiferromagnetic exchange interactions among the lanthanide centers of complexes 1–3. Compound 1 exhibited temperature-dependent out-of-phase ac molar magnetic susceptibility signals with small curvatures at zero dc field. The application of a static 3000 G field intensified the ac magnetic susceptibility component, and an Arrhenius analysis confirmed the slow magnetic relaxation of the DyIII analogue.
Experimental section
Solvents and other general reagents used in this work were purified according to the standard procedures.29 2,6-Bis(hydroxymethyl)-4-methylphenol, activated manganese(IV)dioxide (MnO2), DyCl3·6H2O, TbCl3·6H2O and HoCl3·6H2O were obtained from Sigma Aldrich Chemical Co. and were used as received. Acetyl hydrazide and 2-(hydroxymethyl)-6-carbaldehyde-4-methylphenol were prepared according to the literature procedure.23e Hydrazine hydrate (80%), pivalic acid and sodium sulphate (anhydrous) were obtained from S.D. Fine Chemicals, Mumbai, India and were used as such.
Instrumentation
Melting points were measured using a JSGW melting point apparatus and are uncorrected. IR spectra were recorded as KBr pellets on a Bruker Vector 22 FT IR spectrophotometer operating at 400–4000 cm−1. Elemental analyses of the compounds were obtained from Thermoquest CE instruments CHNS-O, EA/110 model. 1H NMR spectra were recorded in CD3OD solutions on a JEOL JNM LAMBDA 400 model spectrometer operating at 500.0 MHz, chemical shifts are reported in parts per million (ppm) and are referenced with respect to internal tetramethylsilane (1H). Powder X-ray diffraction (PXRD) patterns were recorded with a Bruker D8 advance diffractometer equipped with nickel-filtered Cu Kα radiation. Powder X-ray diffraction (PXRD) patterns of complexes 1–5 were in good agreement with the simulated patterns (ESI, Fig. S6–S8†). The difference in intensities could be due to the preferred orientation in the powder samples.
Magnetic measurements
Magnetic susceptibility data of 1–3 were recorded using a Quantum Design MPMS-5XL SQUID magnetometer for static field (DC) and dynamic field (AC) measurements. The polycrystalline samples were compacted and immobilized into PTFE capsules. DC susceptibility data were acquired as a function of the field (0.1–5.0 T) and temperature (2–290 K). AC susceptibility data were measured at zero field and in the presence of various static fields in the frequency range 0.03–1000 Hz (T = 1.8–50 K, Bac = 3 G, Bdc = 0–4000 G). All data were corrected for the contribution of the sample holder (PTFE capsule) and the diamagnetic contributions of compounds 1–3 calculated from tabulated values (−1.25 × 10−3 cm3 mol−1, −1.37 × 10−3 cm3 mol−1 and −1.32 × 10−3 cm3 mol−1, respectively).
X-ray crystallography
The crystal data for the compounds have been collected on a Bruker SMART CCD diffractometer (MoKα radiation, λ = 0.71073 Å). The program SMART30a was used for collecting frames of data, indexing reflections, and determining lattice parameters, SAINT30a for integration of the intensity of reflections and scaling, SADABS30b for absorption correction, and SHELXTL30c,d for space group and structure determination and least-squares refinements on F2. All the structures were solved by direct methods using the program SHELXS-9730e and refined by full-matrix least-squares methods against F2 with SHELXL-97.30e Hydrogen atoms were fixed at calculated positions and their positions were refined by a riding model. All the non-hydrogen atoms were refined with anisotropic displacement parameters. The lattice solvent molecules of the complexes 1–3 cannot be modeled satisfactorily due to the presence of very high disorder. PLATON/SQUEEZE30f,g routine was utilized to remove the severely disordered solvent molecules. The total electron count thus squeezed is 396, 646 and 780 respectively per unit cell which corresponds to 99, 161 and 195 electrons per molecule (Z = 4). These electron counts can be assigned to 5MeOH, H2O (expected 100) for 1, 2CHCl3, 2MeOH, H2O (expected 162) for 2 and 2CHCl3, 8H2O (expected 196) for 3. The crystallographic figures have been generated using Diamond 3.1e software.30h The crystal data and the cell parameters for compounds 1–3 are summarized in Table 3. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1031235–1031237.
Table 3 Crystal data and structure refinement parameters of 1–3
| |
1
|
2
|
3
|
| Formula |
C148H208Cl2Dy10N16O67 |
C148H206Cl2N16O69Tb10 |
C148H208Cl2Ho10N16O63 |
|
M/g |
4979.22 |
4967.45 |
4939.52 |
| Crystal system |
Tetragonal |
Tetragonal |
Tetragonal |
| Space group |
I |
I |
I |
|
a/Å |
40.368(2) |
40.479(5) |
40.446(5) |
|
b/Å |
40.368(2) |
40.479(5) |
40.446(5) |
|
c/Å |
11.922(7) |
11.943(5) |
11.940(5) |
|
α = β = γ (°) |
90 |
90 |
90 |
|
V/Å3 |
19429(2) |
19569(10) |
19532(9) |
|
Z
|
4 |
4 |
4 |
|
ρ
c/g cm−3 |
1.702 |
1.688 |
1.680 |
|
μ/mm−1 |
3.905 |
3.674 |
4.107 |
|
F(000) |
9752.0 |
9768.0 |
9664.0 |
| Cryst size (mm3) |
0.09 × 0.078 × 0.023 |
0.12 × 0.07 × 0.06 |
0.15 × 0.11 × 0.06 |
|
θ Range (°) |
2.26 to 26.80 |
2.25 to 28.04 |
2.25 to 20.39 |
| Limiting indices |
−43 ≤ h ≤ 51 |
−51 ≤ h ≤ 50 |
−49 ≤ h ≤ 29 |
| −44 ≤ k ≤ 51 |
−51 ≤ k ≤ 51 |
−48 ≤ k ≤ 49 |
| −15 ≤ l ≤ 14 |
−15 ≤ l ≤ 12 |
14 ≤ l ≤ 14 |
| Reflns collected |
76684 |
68807 |
53438 |
| Ind reflns |
21174 [R(int) = 0.0722] |
21266 [R(int) = 0.1230] |
18181[R(int) = 0.0923] |
| Completeness to θ (%) |
99.9 |
99.8 |
100.0 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/params |
21174/1/1068 |
21266/22/1101 |
18181/1/1081 |
| Goodness-of-fit on F2 |
1.029 |
1.028 |
1.027 |
| Final R indices [I > 2θ(I)] |
R
1 = 0.0501 |
R
1 = 0.0565 |
R
1 = 0.0676 |
| wR2 = 0.1065 |
wR2 = 0.1359 |
wR2 = 0.1536 |
|
R indices (all data) |
R
1 = 0.0671 |
R
1 = 0.0828 |
R1 = 0.1009 |
| wR2 = 0.1123 |
wR2 = 0.1532 |
wR2 = 0.1799 |
Synthesis
N′-(2-Hydroxy-3-(hydroxymethyl)-5-methylbenzylidene)acetohydrazide (LH3).
To a stirred solution of 2-(hydroxymethyl)-6-carbaldehyde-4-methylphenol (1.50 g, 9.02 mmol) (B2) in 40 mL ethanol, acetyl hydrazide (0.66 g, 9.02 mmol) (B1) was added dropwise over a period of 15 minutes and the resultant yellow colored solution was refluxed for 5 h. Then, the yellow solution was concentrated in vacuo to 15 mL and kept in a refrigerator at 0 °C overnight. A light yellow colored heavy precipitate was obtained which was filtered and washed with cold ethanol as well as diethyl ether before being dried. Yield: 1.6 g (79.8%). Mp: 180 °C. FT-IR (KBr) cm−1: 3398 ν(O–H); 3181 ν(N–H); 1661 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1624 ν(CN)imine; 1516 ν(CN)py. 1H NMR (CD3OD, δ, ppm, 500 MHz): 2.05 (s, 3H, –CH3acetyl), 2.27 (s, 3H, –CH3), 4.55 (s, 2H, –CH2OH), 5.46 (s, 1H, –OHphen), 7.02 (s, 1H, Ar–H), 7.22 (s, 1H, Ar–H), 8.06 (s, 1H, –NH), 8.17 (s, 1H, imine). Anal. Calcd for C11H14N2O3: C, 59.45; H, 6.35; N, 12.60. Found: C, 58.77; H, 6.01; N, 12.19. ESI-MS, m/z: (M + H)+. 223.09.
O); 1624 ν(CN)imine; 1516 ν(CN)py. 1H NMR (CD3OD, δ, ppm, 500 MHz): 2.05 (s, 3H, –CH3acetyl), 2.27 (s, 3H, –CH3), 4.55 (s, 2H, –CH2OH), 5.46 (s, 1H, –OHphen), 7.02 (s, 1H, Ar–H), 7.22 (s, 1H, Ar–H), 8.06 (s, 1H, –NH), 8.17 (s, 1H, imine). Anal. Calcd for C11H14N2O3: C, 59.45; H, 6.35; N, 12.60. Found: C, 58.77; H, 6.01; N, 12.19. ESI-MS, m/z: (M + H)+. 223.09.
General synthetic procedure for the preparation of complexes 1–3
All the pentanuclear complexes (1–3) have been synthesized according to the following procedure. LH3 (0.04 g, 0.18 mmol) was dissolved in 40 mL methanol. To this solution, under stirring, LnCl3·6H2O (0.23 mmol) was added and the reaction mixture was stirred for 20 minutes at room temperature. At this stage, triethylamine (0.096 mL, 0.73 mmol) was added dropwise and the stirring was continued for a further 10 minutes and pivalic acid (0.028 g, 0.27 mmol) was added dropwise to the mixture. The resulting yellow colored solution was continuously stirred for 12 hours at room temperature. Then, the solution was completely evaporated in vacuo to afford a light yellow colored solid mass which was washed 2–3 times with diethyl ether and dried. The solid mass was re-dissolved in MeOH/CHCl3 (1:1) and kept for crystallization. After about 12 days, needle-shaped yellow colored crystals suitable for X-ray crystallography were obtained by slow evaporation from the solvent mixture. Specific details of each reaction and the characterization data of the products obtained are given below.
[Dy5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·9.5H2O·5MeOH (1).
Quantities: LH3 (0.04 g, 0.18 mmol), DyCl3·6H2O (0.087 g, 0.23 mmol), Et3N (0.096 mL, 0.73 mmol), PivH (0.028 g, 0.27 mmol). Yield: 0.076 g, 63.3% (based on Dy3+). Mp: 200 °C (d). IR (KBr) (cm−1): 3433 (b), 2978 (s), 2950 (s), 2604 (s), 2497(s), 2343 (w), 1620 (s), 1574 (s), 1482 (s), 1429 (s), 1397 (s), 1309 (w), 1262 (w), 1228 (w), 1172 (w), 1072 (w), 1037 (s), 897 (w), 851 (w), 808 (s), 608 (w). Anal. Calcd for C79H122Cl Dy5 N8 O39.5 (2663.80): C, 35.62; H, 4.62 N, 4.21. Found: C, 34.96; H, 4.33 N, 4.09.
[Tb5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2 η1Piv)2(H2O)]·Cl·10.5H2O·2MeOH·2CHCl3 (2).
Quantities: LH3 (0.04 g, 0.18 mmol), TbCl3·6H2O (0.085 g, 0.23 mmol), Et3N (0.096 mL, 0.73 mmol), PivH (0.028 g, 0.27 mmol). Yield: 0.058 g, 46.03% (based on Tb3+). Mp: 200 °C (d). IR (KBr) (cm−1): 3441 (b), 2971 (s), 2930 (s), 2601 (s), 2499(s), 2339 (w), 1610 (s), 1570 (s), 1477(s), 1425 (s), 1393 (s), 1302 (w), 1252 (w), 1235 (w), 1178 (w), 1071 (w), 1032 (s), 895 (w), 854 (w), 803 (s), 610 (w). Anal. Calcd for C78H112Cl7N8O37.5Tb5 (2804.55): C, 33.40; H, 4.03; N, 4.00. Found: C, 32.93; H, 4.10 N, 4.04.
[Ho5(LH)4(η1-Piv)(η2-Piv)3(μ2–η2η1Piv)2(H2O)]·Cl·14.5H2O·2CHCl3 (3).
Quantities: LH3 (0.04 g, 0.18 mmol), HoCl3·6H2O (0.087 g, 0.23 mmol), Et3N (0.096 mL, 0.73 mmol), PivH (0.028 g, 0.27 mmol). Yield: 0.062 g, 48.4% (based on Ho3+). Mp: 200 °C (d). IR (KBr) (cm−1): 3447 (b), 2965 (s), 2933 (s), 2607 (s), 2497(s), 2331 (w), 1603 (s), 1579 (s), 1475(s), 1424 (s), 1390 (s), 1297 (w), 1251 (w), 1233 (w), 1172 (w), 1070 (w), 1029 (s), 899 (w), 844 (w), 798 (s), 611 (w). Anal. Calcd for C76H118Cl7Ho5N8O39.5 (2848.60): C, 32.04; H, 4.18; N, 3.93. Found: C, 31.73; H, 4.23; N, 3.61.
Acknowledgements
We thank the Department of Science and Technology (DST), India, for financial support, including support for a Single Crystal CCD X-ray Diffractometer facility at IIT-Kanpur. V. C. is grateful to the DST for a J. C. Bose fellowship. S. B. thanks the Council of Scientific and Industrial Research, India for the Senior Research Fellowship.
References
-
(a) P. W. Roesky, G. C. Melchor and A. Zulys, Chem. Commun., 2004, 738 RSC;
(b) P. W. Roesky and T. E. Muller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS PubMed; T. N. Parac-Vogt, K. Deleersnyder and K. Binnemans, Eur. J. Org. Chem., 2005, 1810 Search PubMed;
(c) F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104 RSC;
(d) X. Yu, S. Y. Seo and M. J. Tobin, J. Am. Chem. Soc., 2007, 129, 7244 CrossRef CAS PubMed.
-
(a) M. Romanelli, G. A. Kumar, T. J. Emge, R. E. Riman and J. G. Brennan, Angew. Chem., Int. Ed., 2008, 47, 6049 CrossRef CAS PubMed;
(b) C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS PubMed;
(c) S. L. Faulkner, S. Natrajan and D. Sykes, Dalton Trans., 2009, 3890 RSC;
(d) K. Binnemans, Coord. Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed;
(e) M. D. Ward, Coord. Chem. Rev., 2007, 251, 1663 CrossRef CAS PubMed;
(f) S. Sivakumar and M. L. P. Reddy, J. Mater. Chem., 2012, 22, 10852 RSC.
-
(a) A. Picot, A. D'Alé, P. L. Baldeck, A. Grichine, A. Duperray, C. Andraud and O. Maury, J. Am. Chem. Soc., 2008, 130, 1532 CrossRef CAS PubMed;
(b) M. Bottrill, L. Kwok and N. Long, J. Chem. Soc. Rev., 2006, 35, 557 RSC.
-
(a) S. Biswas, H. S. Jena, A. Adhikary and S. Konar, Inorg. Chem., 2014, 53, 3926 CrossRef CAS PubMed;
(b) S. Goswami, A. Adhikary, H. S. Jena and S. Konar, Dalton Trans., 2013, 42, 9813 RSC;
(c) J. A. Sheikh, A. Adhikary and S. Konar, New J. Chem., 2014, 38, 3006 RSC;
(d) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed;
(e) Y.-Z. Zheng, M. Evangelisti and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 3692 CrossRef CAS PubMed;
(f) Y.-Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057 CrossRef CAS PubMed;
(g) Y.-Z. Zheng, M. Evangelisti and R. E. P. Winpenny, Chem. Sci., 2011, 2, 99 RSC;
(h) J. M. Jia, S. J. Liu, Y. Cui, S. D. Han, T.-L. Hu and X. H. Bu, Cryst. Growth Des., 2013, 13, 4631 CrossRef CAS.
-
(a) M. Affronte, J. Mater. Chem., 2009, 19, 1731 RSC;
(b) A. R. Rocha, V. García-Suárez, S. W. Bailey, C. J. Lambert, J. Ferrerand and S. Sanvito, Nat. Mater., 2005, 4, 335 CrossRef CAS PubMed;
(c) M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef CAS PubMed.
-
(a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef CAS PubMed;
(b) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538 CrossRef CAS PubMed;
(c) M. Urdampilleta, S. Klyatskaya, J. P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef CAS PubMed;
(d) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
-
(a) A. Ardavan and S. J. Blundell, J. Mater. Chem., 2009, 19, 1754 RSC;
(b) F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119 RSC;
(c) P. C. E. Stamp and A. Gaita-Ariño, J. Mater. Chem., 2009, 19, 1718 RSC;
(d) J. Lehmann, A. Gaita-Ariño, E. Coronado and D. Loss, Nat. Nanotechnol., 2007, 2, 312 CrossRef CAS PubMed;
(e) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed.
-
(a) R. Sessoli, L. Hui, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi and G. Christou, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS;
(b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS PubMed;
(c) J.-P. Zhao, B. W. Hu, X. F. Zhang, Q. Yang, M. S. El Fallah, J. Ribas and X. H. Bu, Inorg. Chem., 2010, 49, 11325 CrossRef CAS PubMed.
-
(a) D. W. Boukhvalov, V. V. Dobrovitski, P. Kögerler, M. Al-Saqer, M. I. Katsnelson, A. I. Lichtenstein and B. N. Harmon, Inorg. Chem., 2010, 49, 10902 CrossRef CAS PubMed;
(b) T. Glaser, Chem. Commun., 2011, 47, 116 RSC;
(c) A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869 CrossRef CAS PubMed;
(d) A. K. Boudalis, M. Pissas, C. P. Raptopoulou, V. Psycharis, B. Abarca and R. Ballesteros, Inorg. Chem., 2008, 47, 10674 CrossRef CAS PubMed;
(e) G. Lazari, T. C. Stamatatos, C. P. Raptopoulou, V. Psycharis, M. Pissas, S. P. Perlepes and A. K. Boudalis, Dalton Trans., 2009, 3215 RSC;
(f) M. Shanmugam, S. Vaidya, A. Upadhyay, S. K. Singh, T. Gupta, S. Tewary, S. Langley, J. Walsh, K. S. Murray and G. Rajaraman, Chem. Commun., 2015, 51, 3739–3742 RSC.
-
(a) S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420 CrossRef CAS PubMed;
(b) C. M. Zaleski, E. C. Depperman, J. W. Kampf, M. L. Kirk and V. L. Pecoraro, Angew. Chem., Int. Ed., 2004, 43, 3912 CrossRef CAS PubMed;
(c) T. K. Prasad, M. V. Rajasekharan and J. P. Costes, Angew. Chem., Int. Ed., 2007, 46, 2851 CrossRef CAS PubMed;
(d) G. P. Guedes, S. Soriano, L. A. Mercante, N. L. Speziali, M. A. Novak, M. Andruh and M. G. F. Vaz, Inorg. Chem., 2013, 52, 8309 CrossRef CAS PubMed;
(e) J. Ruiz, G. Lorusso, M. Evangelisti, E. K. Brechin, S. J. A. Pope and E. Colacio, Inorg. Chem., 2014, 53, 3586 CrossRef CAS PubMed;
(f) M. A. Palacios, S. T.-Padilla, J. Ruiz, J. M. Herrera, S. J. A. Pope, E. K. Brechin and E. Colacio, Inorg. Chem., 2014, 53, 1465 CrossRef CAS PubMed;
(g) A. Upadhyay, S. K. Singh, C. Das, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2014, 50, 8838 RSC;
(h) Y. F. Zeng, G. C. Xu, X. Hu, Z. Chen, X. H. Bu, S. Gao and E. C. Sañudo, Inorg. Chem., 2010, 49, 9734 CrossRef CAS PubMed;
(i) S.-D. Han, S.-J. Liu, Q.-L. Wang, X.-H. Miao, T.-L. Hu and X.-H. Bu, Cryst. Growth Des., 2015, 15, 2253 CrossRef CAS;
(j) S.-J. Liu, Y.-F. Zeng, L. Xue, S.-D. Han, J.-M. Jia, T.-L. Hu and X. H. Bu, Inorg. Chem. Front., 2014, 1, 200 RSC.
-
(a) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673 CrossRef CAS PubMed;
(b) R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530 CrossRef CAS PubMed.
-
(a) E. Lucaccini, L. Sorace, M. Perfetti, J.-P. Costes and R. Sessoli, Chem. Commun., 2014, 50, 1648 RSC;
(b) F. Gao, L. Cui, Y. Song, Y.-Z. Li and J.-L. Zuo, Inorg. Chem., 2014, 53, 562 CrossRef CAS PubMed;
(c) J. J. Baldoví, S. C. Serra, J. M. Clemente-Juan, E. Coronado, A. G. Arin and A. Palii, Inorg. Chem., 2012, 51, 12565 CrossRef PubMed;
(d) G. Rajaraman, S. K. Singh, T. Gupta and M. Shanmugam, Chem. Commun., 2014, 50, 15513–15516 RSC.
-
(a) P.-H. Lin, T. J. Burchell, R. Clerac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848 CrossRef CAS PubMed;
(b) G.-F. Xu, Q.-L. Wang, P. Gamez, Y. Ma, R. Cl_erac, J. Tang, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem. Commun., 2010, 46, 1506 RSC;
(c) K. A. Thiakou, V. Bekiari, C. P. Raptopoulou, V. Psycharis, P. Lianos and S. P. Perlepes, Polyhedron, 2006, 25, 2869 CrossRef CAS PubMed;
(d) S.-S. Bao, L.-F. Ma, Y. Wang, L. Fang, C.-J. Zhu, Y.-Z. Li and L.-M. Zheng, Chem. – Eur. J., 2007, 13, 2333 CrossRef CAS PubMed;
(e) S. J. Liu, J.-P. Zhao, W.-C. Song, S.-D. Han, Z.-Y. Liu and X.-H. Bu, Inorg. Chem., 2013, 52, 2103 CrossRef CAS PubMed.
-
(a) I. J. Hewitt, Y. Lan, C. E. Anson, J. Luzon, R. Sessoli and A. K. Powell, Chem. Commun., 2009, 6765 RSC;
(b) J. Tang, I. J. Hewitt, T. N. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729 CrossRef CAS PubMed;
(c) M. U. Anwar, S. S. Tandon, L. N. Dawe, F. Habib, M. Murugesu and L. K. Thompson, Inorg. Chem., 2012, 51, 1028 CrossRef CAS PubMed.
-
(a) B.-Q. Ma, D.-S. Zhang, S. Gao, T.-Z. Jin, C.-H. Yan and G.-X. Xu, Angew. Chem., Int. Ed., 2000, 39, 3644 CrossRef CAS;
(b) Y. Gao, G.-F. Xu, Z. L. Hao, J. Tang and Z. Liu, Inorg. Chem., 2009, 48, 11495 CrossRef CAS PubMed;
(c) Y. Bi, X.-T. Wang, W. Liao, X. Wang, R. Deng, H. Zhang and S. Gao, Inorg. Chem., 2009, 48, 11743 CrossRef CAS PubMed;
(d) N. M. Randell, M. U. Anwar, M. W. Drover, L. N. Dawe and L. K. Thompson, Inorg. Chem., 2013, 52, 6731 CrossRef CAS PubMed;
(e) S.-D. Han, X.-H. Miao, S.-J. Liu and X.-H. Bu, Inorg. Chem. Front., 2014, 1, 549 RSC;
(f) F. C. Liu, Y.-F. Zeng, J.-P. Zhao, B. W. Hu, X. Hu, J. Ribasc and X. H. Bu, Dalton Trans., 2009, 2074 RSC.
-
(a) R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530 CrossRef CAS PubMed;
(b) D. T. Thielemann, A. T. Wagner, Y. Lan, C. E. Anson, M. T. Gamer, A. K. Powell and P. W. Roesky, Dalton Trans., 2013, 42, 14794 RSC;
(c) M. T. Gamer, Y. Lan, P. W. Roesky, A. K. Powell and R. Clérac, Inorg. Chem., 2008, 47, 6581 CrossRef CAS PubMed;
(d) J.-B. Peng, X.-J. Kong, Y.-P. Ren, L.-S. Long, R.-B. Huang and L.-S. Zheng, Inorg. Chem., 2012, 51, 2186 CrossRef CAS PubMed;
(e) H. Tian, L. Zhao, H. Lin, J. Tang and G. Li, Chem. – Eur. J., 2013, 19, 13235 CrossRef CAS PubMed.
-
(a) D.-S. Zhang, B.-Q. Ma, T.-Z. Jin, S. Gao, C.-H. Yan and T. C. W. Mak, New J. Chem., 2000, 24, 61 RSC;
(b) N. Mahe, O. Guillou, C. Daiguebonne, Y. G_erault, A. Caneschi, C. Sangregorio, C. J. Y. Hane-Ching, P. E. Car and T. Roisnel, Inorg. Chem., 2005, 44, 7743 CrossRef CAS PubMed;
(c) B. Hussain, D. Savard, T. J. Burchell, W. Wernsdorfer and M. Murugesu, Chem. Commun., 2009, 1100 RSC;
(d) S. Xue, L. Zhao, Y. N. Guo, P. Zhang and J. K. Tang, Chem. Commun., 2012, 48, 8946 RSC;
(e) L. Ungur, S. K. Langley, T. N. Hooper, B. Moubaraki, E. K. Brechin, K. S. Murray and L. F. Chibotaru, J. Am. Chem. Soc., 2012, 134, 18554 CrossRef CAS PubMed.
- X.-J. Zheng, L.-P. Jin and S. Gao, Inorg. Chem., 2004, 43, 1600 CrossRef CAS PubMed.
-
(a) T. Kajiwara, H. Wu, T. Ito, N. Iki and S. Miyano, Angew. Chem., Int. Ed., 2004, 43, 1832 CrossRef CAS PubMed;
(b) V. Chandrasekhar, P. Bag and E. Colacio, Inorg. Chem., 2013, 52, 4562–4570 CrossRef CAS PubMed.
-
(a) G. Xu, Z.-M. Wang, Z. He, Z. Lü, C.-S. Liao and C.-H. Yan, Inorg. Chem., 2002, 41, 6802 CrossRef CAS PubMed;
(b) G.-F. Xu, P. Gamez, S. J. Teat and J. Tang, Dalton Trans., 2010, 39, 4353 RSC.
-
(a) K. Manseki and S. Yanagida, Chem. Commun., 2007, 1242 RSC;
(b) L. G. Westin, M. Kritikos and A. Caneschi, Chem. Commun., 2003, 1012 RSC;
(c) X. Yang, R. A. Jones and M. J. Wiester, Dalton Trans., 2004, 1787 RSC;
(d) A. Kornienko, T. J. Emge, G. A. Kumar, R. E. Riman and J. G. Brennan, J. Am. Chem. Soc., 2005, 127, 3501 CrossRef CAS PubMed.
-
(a) P. C. Andrews, T. Beck, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and P. W. Roesky, Dalton Trans., 2007, 5651 RSC;
(b) R. Wang, H. D. Selby, H. Liu, M. D. Carducci, T. Jin, Z. Zheng, J. W. Anthis and R. J. Staples, Inorg. Chem., 2002, 41, 278 CrossRef CAS PubMed.
-
(a) V. Chandrasekhar, B. M. Pandian, R. Azhakar, J. J. Vittal and R. Clérac, Inorg. Chem., 2007, 46, 5140 CrossRef CAS PubMed;
(b) V. Chandrasekhar, B. M. Pandian, R. Boomishankar, A. Steiner, J. J. Vittal, A. Houri and R. Clérac, Inorg. Chem., 2008, 47, 4918 CrossRef CAS PubMed;
(c) V. Chandrasekhar, P. Bag, W. Kroener, K. Gieb and P. Müller, Inorg. Chem., 2013, 52, 13078 CrossRef CAS PubMed;
(d) V. Chandrasekhar, S. Das, A. Dey, S. Hossain, F. Lloret and E. Pardo, Eur. J. Inorg. Chem., 2013, 4506 CrossRef CAS PubMed;
(e) V. Chandrasekhar, S. Das, A. Dey, S. Hossain, S. Kundu and E. Colacio, Eur. J. Inorg. Chem., 2014, 397 CrossRef CAS PubMed;
(f) C. Meseguer, S. T.-Padilla, M. M. Hänninen, R. Navarrete, A. J. Mota, M. Evangelisti, J. Ruiz and E. Colacio, Inorg. Chem., 2014, 53, 12092–12099 CrossRef CAS PubMed;
(g) A. Deb, T. T. Boron, M. Itou, Y. Sakurai, T. Mallah, V. L. Pecoraro and J. E. Penner-Hahn, J. Am. Chem. Soc., 2014, 136, 4889 CrossRef CAS PubMed;
(h) C. M. Zaleski, J. W. Kampf, T. Mallah, M. L. Kirk and V. L. Pecoraro, Inorg. Chem., 2007, 46, 1954 CrossRef CAS PubMed;
(i) J. Goura, R. Guillaume, E. Rivière and V. Chandrasekhar, Inorg. Chem., 2014, 53, 7815 CrossRef CAS PubMed;
(j) P. Bag, A. Chakraborty, G. Rogez and V. Chandrasekhar, Inorg. Chem., 2014, 53, 6524 CrossRef CAS PubMed.
-
(a) S. Das, S. Hossain, A. Dey, S. Biswas, J.-P. Sutter and V. Chandrasekhar, Inorg. Chem., 2014, 53, 5020–5028 CrossRef CAS PubMed;
(b) V. Chandrasekhar, P. Bag and E. Colacio, Inorg. Chem., 2013, 52, 4562 CrossRef CAS PubMed;
(c) S. Das, S. Hossain, A. Dey, S. Biswas, J.-P. Sutter and V. Chandrasekhar, Inorg. Chem., 2014, 53, 5020–5028 CrossRef CAS PubMed;
(d) T. Fukuda, N. Shigeyoshi, T. Yamamura and N. Ishikawa, Inorg. Chem., 2014, 53, 9080 CrossRef CAS PubMed;
(e) J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003 CrossRef CAS PubMed;
(f) F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242 CrossRef CAS PubMed;
(g) V. E. Campbell, H. Bolvin, E. Rivière, R. Guillot, W. Wernsdorfer and T. Mallah, Inorg. Chem., 2014, 53, 2598 CrossRef CAS PubMed.
- V. Chandrasekhar, S. Hossain, S. Das, S. Biswas and J.-P. Sutter, Inorg. Chem., 2013, 52, 6346 CrossRef CAS PubMed.
- S. Das, A. Dey, S. Biswas, E. Colacio and V. Chandrasekhar, Inorg. Chem., 2014, 53, 3417 CrossRef CAS PubMed.
-
(a)
H. Lueken, Magnetochemie, Teubner, Stuttgart, Germany, 1999 Search PubMed;
(b) M. Speldrich, H. Schilder, H. Lueken and P. Kögerler, Isr. J. Chem., 2011, 51, 215–227 CrossRef CAS PubMed;
(c) J. van Leusen, M. Speldrich, H. Schilder and P. Kögerler, Coord. Chem. Rev., 2015, 289–290, 137–148 CrossRef CAS PubMed.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS PubMed.
-
(a)
Vogel's Textbook of Practical Organic Chemistry, ed. B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, ELBS and Longman, London, 5th edn, 1989 Search PubMed;
(b) D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed;
(c) X. Zeng, D. Coquiére, A. Alenda, E. Garrier, T. Prangé, Y. Li, O. Reinaud and I. Jabin, Chem. – Eur. J., 2006, 12, 6393–6402 CrossRef CAS PubMed.
-
(a)
SMART & SAINT Software Reference manuals, Version 6.45, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2003 Search PubMed;
(b)
G. M. Sheldrick, SADABS, a software for empirical absorption correction, Ver. 2.05, University of Göttingen, Göttingen, Germany, 2002 Search PubMed;
(c)
SHELXTL Reference Manual, Ver. 6.1, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2000 Search PubMed;
(d)
G. M. Sheldrick, SHELXTL, Ver. 6.12, Bruker AXS Inc., Madison, WI, 2001 Search PubMed;
(e)
G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(f) P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef;
(g) A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, c34 Search PubMed;
(h)
K. Bradenburg, Diamond, Ver. 3.1eM, Crystal Impact GbR, Bonn, Germany, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Molecular structures of 2 and 3 (Fig. S1–S2), list of bond lengths and bond angles (Tables S1–S2), Cole–Cole plot at zero dc field (Fig. S3), χ′mvs. T plot at 3000 G (Fig. S4), χ′′mvs. T at 3000 G (Fig. S5), τ vs. T−1 (Fig. S6), PXRD (Fig. S6–S8), reported Ln5 complexes (Fig. S9) and TGA (Fig. S10–S12). CCDC 1031235–1031237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03060a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence