
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganometallic rhodium(III) and iridium(III) cyclopentadienyl complexes with curcumin and bisdemethoxycurcumin co-ligands†
Riccardo
Pettinari
*a,
Fabio
Marchetti
b,
Claudio
Pettinari
a,
Francesca
Condello
a,
Agnese
Petrini
a,
Rosario
Scopelliti
c,
Tina
Riedel
c and
Paul J.
Dyson
*c
aSchool of Pharmacy, University of Camerino, via S. Agostino 1, 62032 Camerino MC, Italy. E-mail: riccardo.pettinari@unicam.it; Tel: +39-0737-632338
bSchool of Science and Technology, University of Camerino, via S. Agostino 1, 62032 Camerino MC, Italy
cInstitute of Chemical Sciences and Engineering, Swiss Federal Institute of Technology (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch; Fax: +41-21-6939885; Tel: +41-21-6939854
First published on 9th November 2015
Abstract
A series of half-sandwich cyclopentadienyl rhodium(III) and iridium(III) complexes of the type [Cp*M(curc/bdcurc)Cl] and [Cp*M(curc/bdcurc)(PTA)][SO3CF3], in which Cp* = pentamethylcyclopentadienyl, curcH = curcumin and bdcurcH = bisdemethoxycurcumin as O^O-chelating ligands, and PTA = 1,3,5-triaza-7-phosphaadamantane, is described. The X-ray crystal structures of three of the complexes, i.e. [Cp*Rh(curc)(PTA)][SO3CF3] (5), [Cp*Rh(bdcurc)(PTA)][SO3CF3] (6) and [Cp*Ir(bdcurc)(PTA)][SO3CF3] (8), confirm the expected “piano-stool” geometry. With the exception of 5, the complexes are stable under pseudo-physiological conditions and are moderately cytotoxic to human ovarian carcinoma (A2780 and A2780cisR) cells and also to non-tumorigenic human embryonic kidney (HEK293) cells, but lack the cancer cell selectivity observed for related arene ruthenium(II) complexes.
Introduction
Curcumin is a naturally occurring polyphenol derived from the rhizomes of turmeric (Curcuma longa).1 Curcumin (curcH) is the major component of three curcuminoids that give turmeric its characteristic yellow color. The minor curcuminoid components are demethoxycurcumin (dcurcH) and bisdemethoxycurcumin (bdcurcH) in which one or both –OMe functionalities at the outer phenol rings are removed.Curcumin displays potent activity in vitro and in animal studies, acting as an anti-proliferative, anti-metastatic and antiangiogenic agent.2–5 Numerous studies have also explored the antioxidant, anti-hepatotoxic, anti-hyperlipidemic, antiviral, and anti-Alzheimer's disease effects.6,7 Despite the promising biological effects of curcumin at the preclinical and clinical levels its therapeutic applications are, however, restricted by poor solubility and rapid metabolism resulting in very low bioavailability following oral administration.8 One strategy used to improve the biological (drug-like) properties of curcumin, which avoids covalently modifying the compound,9,10 is to coordinate it to metal complexes.11–13
Ruthenium compounds have emerged, in recent years, as promising alternatives to platinum drugs by displaying specific activities against different cancers and favourable toxicity and clearance properties.14–21 Previously, we have demonstrated that half-sandwich ruthenium-arene complexes with curcumin and bisdemethoxycurcumin and an ancillary chloride ligand show promising activity as anticancer agents.22,23 We have also shown that the replacement of chloride with the 1,3,5-triaza-7-phosphaadamantane ligand (PTA) led to the formation of RAPTA-type complexes with superior solubility properties and also superior cytotoxicities.24 It was found that the presence or absence of peripheral methoxy groups in curcumin and the different arene rings do not strongly influence the biological activity whereas the PTA ligand appears to significantly improve the pharmacological properties of the curcumin-modified arene ruthenium(II) complexes.
Rhodium and iridium complexes have been explored for their anticancer properties with initial efforts being focused on Rh(I) and Ir(I) compounds with a square-planar geometry similar to that of cisplatin.25,26 Recently, more stable cyclopentadienyl Rh(III) and Ir(III) complexes, with higher structural diversity and a higher coordination number (6 versus 4),27,28 have been shown to have highly potent anticancer activity.29–40
Based on the highly promising pharmacological properties of the arene ruthenium(II) complexes incorporating curcumin and bisdemethoxycurcumin co-ligands, combined with the exciting developments on the anticancer properties of Rh(III) and Ir(III) organometallic compounds, we decided to integrate these two areas and prepare and evaluate some pentamethylcyclopentadienyl Rh(III) and Ir(III) compounds containing curcumin-based ligands and, in some cases, PTA. It should be noted that two curcumin complexes of Rh(III) and Ir(III) have been reported,41 but structural data and biological studies were not described.
Results and discussion
Synthesis
The rhodium(III) and iridium(III) pentamethylcyclopentadienyl complexes (1–4) were prepared in high yield (78–93%) from the reaction of the appropriate dimer, [Cp*MCl2]2, (M = Rh or Ir, Cp* = η5-pentamethylcyclopentadienyl) with the appropriate pro-ligand and KOH in methanol (Scheme 1). Analytical and spectral data for 1 and 3 are in accordance with literature data.41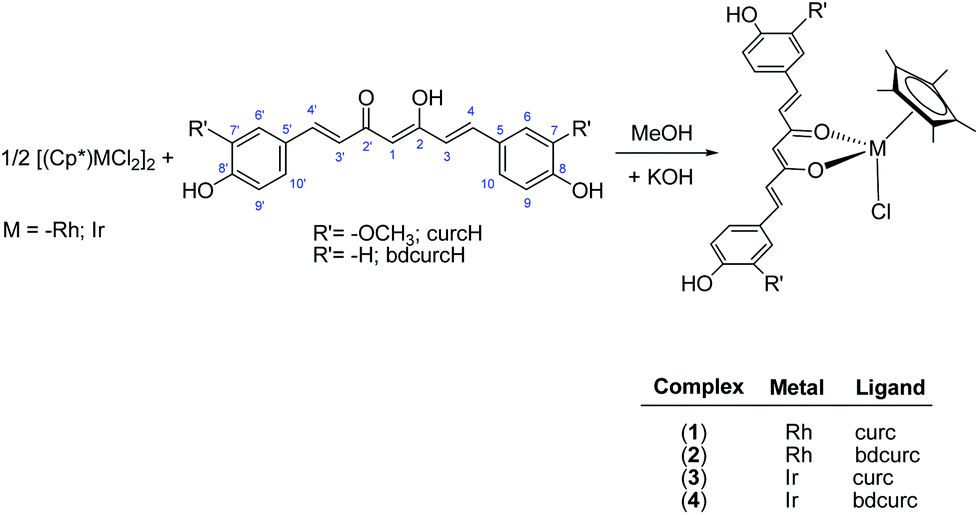 | ||
| Scheme 1 Synthesis of 1–4. | ||
Complexes 1–4 are air stable and soluble in acetone, acetonitrile and DMSO and slightly soluble in alcohols. In addition, the curcumin complexes, 1 and 3, are also soluble in chlorinated solvents. Such different solubilities appear to be due to the different R’ substituents of curcumin ligands (Scheme 1). The IR spectra of 1–4 show the typical ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC) bands of curc and bdcurc at lower wavenumbers than the corresponding bands in the free ligands as a consequence of coordination through both the carbonyl arms to the metal. In the far-IR region, several absorptions were observed in the range 482–445 and 252–242 cm−1. These may be assigned to ν(M–O) and ν(M–Cl) stretches, respectively.42
O, CC) bands of curc and bdcurc at lower wavenumbers than the corresponding bands in the free ligands as a consequence of coordination through both the carbonyl arms to the metal. In the far-IR region, several absorptions were observed in the range 482–445 and 252–242 cm−1. These may be assigned to ν(M–O) and ν(M–Cl) stretches, respectively.42
The NMR spectra of 1–4 corroborate the expected structures containing the bidentate curcumin and bisdemethoxycurcumin ligands. For example, a doublet is observed in the 13C{1H}43 NMR spectra of the rhodium derivatives 1 and 2 around δ 95 (J(103Rh–13C) ≈ 6.9 Hz), whereas in the spectra of iridium derivatives 3 and 4 a singlet is observed. The electrospray ionisation (ESI) mass spectra of 1–4 display peaks corresponding to the species [Cp*M(curc/bdcurc)]+, in which the chloride ligand has dissociated.
The chloride ligand in 1–4 is readily replaced by the water-soluble phosphine 1,3,5-triaza-7-phosphaadamantane (PTA), by treatment of the complexes with AgSO3CF3 in methanol containing PTA, affording [Cp*M(curc/bdcurc)(PTA)][SO3CF3] (M = Rh 5 and 6 and M = Ir 7 and 8), as depicted in Scheme 2. The substitution of the chloride ligand by PTA and the formation of an ionic compound were confirmed by the disappearance of the ν(M–Cl) band in the IR spectra of 5–8. Moreover, a characteristic absorption pattern in the region 1000–1200 cm−1, indicative of a non-coordinated O3SCF3− anion, has also been observed.44 The 1H NMR spectra of 5–8 in CD3CN display the expected signals due to the coordinated Cp*, curc or bdcurc and PTA ligands. The resonances due to the PTA are observed at a lower field with respect to those of uncoordinated PTA, thus confirming coordination to the metal center.45
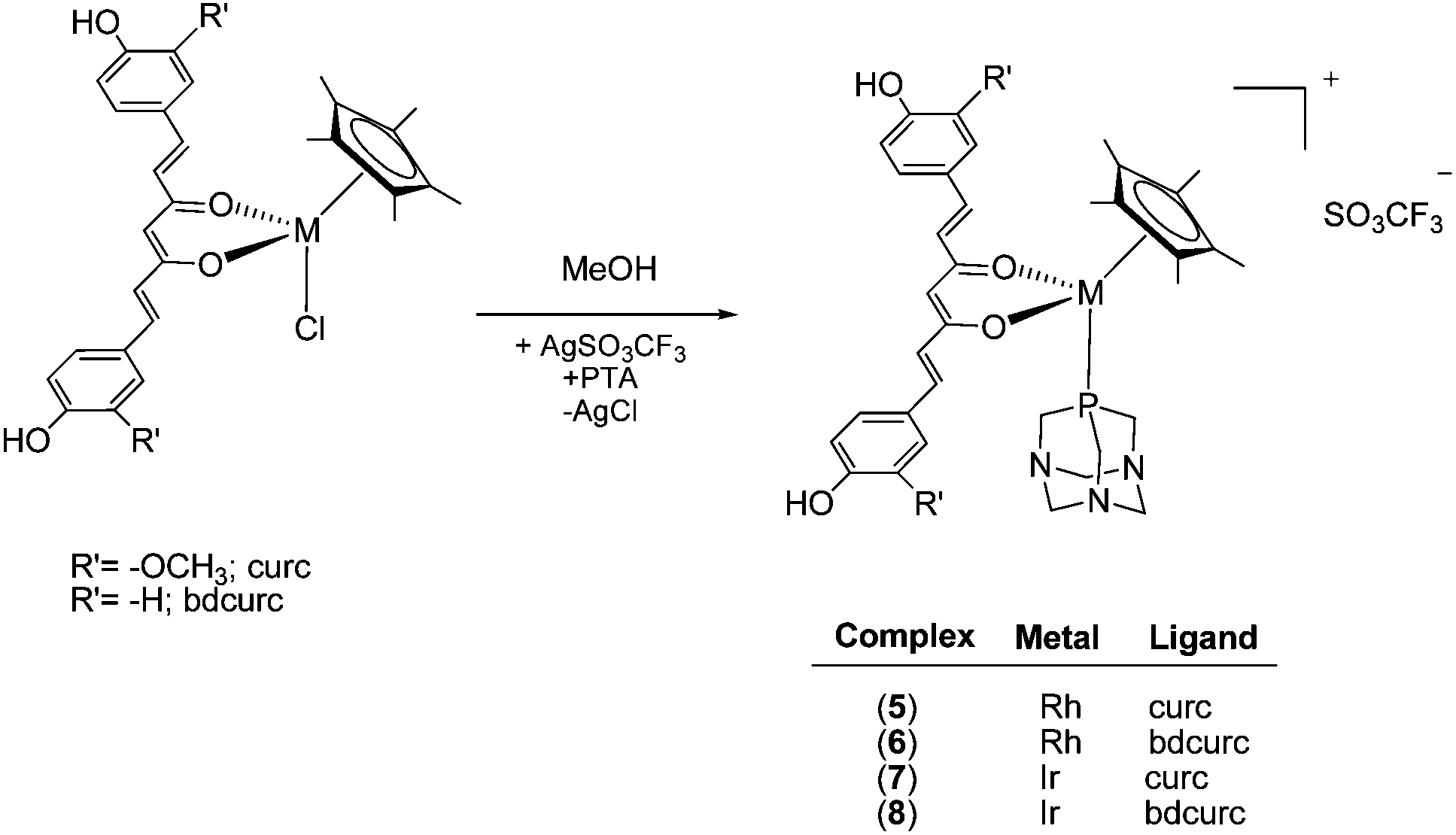 | ||
| Scheme 2 Synthesis of 5–8. | ||
The 31P NMR spectra of rhodium complexes 5 and 6 in CD3CN display a doublet centered at ca. −34 ppm with a 1JP–Rh value of ca. 151 Hz, and the iridium derivatives 7 and 8 comprise a single resonance at −56 ppm, in a range typical of related compounds.46,47
The ESI mass spectra of 5–8 in acetonitrile show two main peak envelopes, that of highest relative intensity corresponding to the fragment [Cp*M(curc/bdcurc)]+, upon dissociation of PTA, the other corresponding to the intact species [Cp*M(curc/bdcurc)(PTA)]+.
Complex 5 in water–DMSO (80–20%) solution is stable, its 31P NMR spectra remaining unchanged after 96 h. The stability of 5 was also determined under pseudo-pharmacological conditions in 5 mM NaCl solution (being a model for the low intracellular chloride concentration in cells) and in 100 mM NaCl solution (approximating to the higher chloride levels in blood). Solutions of the complexes (c = 2.0 mM) in aqueous NaCl (c = 5 mM or 100 mM in D2O containing 20% of [D6]DMSO) were prepared and maintained at 37 °C for 7 days. The decomposition of the complexes was monitored by 1H and 31P NMR spectroscopy.
The 31P{1H} spectrum of 5 displays a doublet at −36.0 ppm (JPRh = 155 Hz), corresponding to the starting species, [Cp*Rh(curc)(PTA)]+ (RhA) (Scheme 3 and Fig. 1).
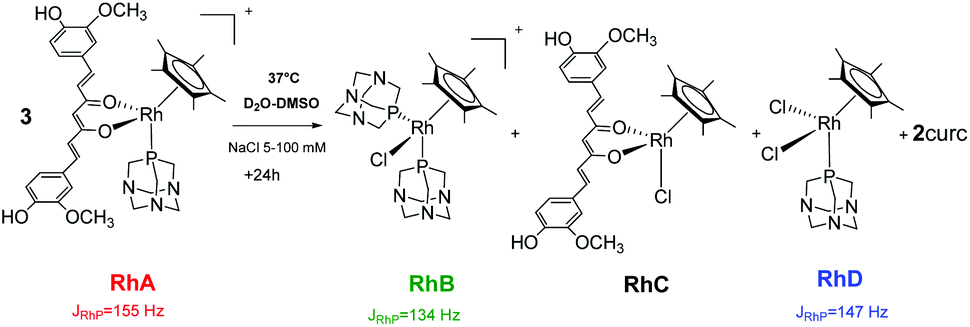 | ||
| Scheme 3 Decomposition of 5 (RhA) into [Cp*Rh(PTA)2Cl]+ (RhB), [Cp*Rh(curc)Cl] (RhC), [Cp*Rh(PTA)Cl2] (RhD) and into curc, under pseudo-pharmacological conditions. | ||
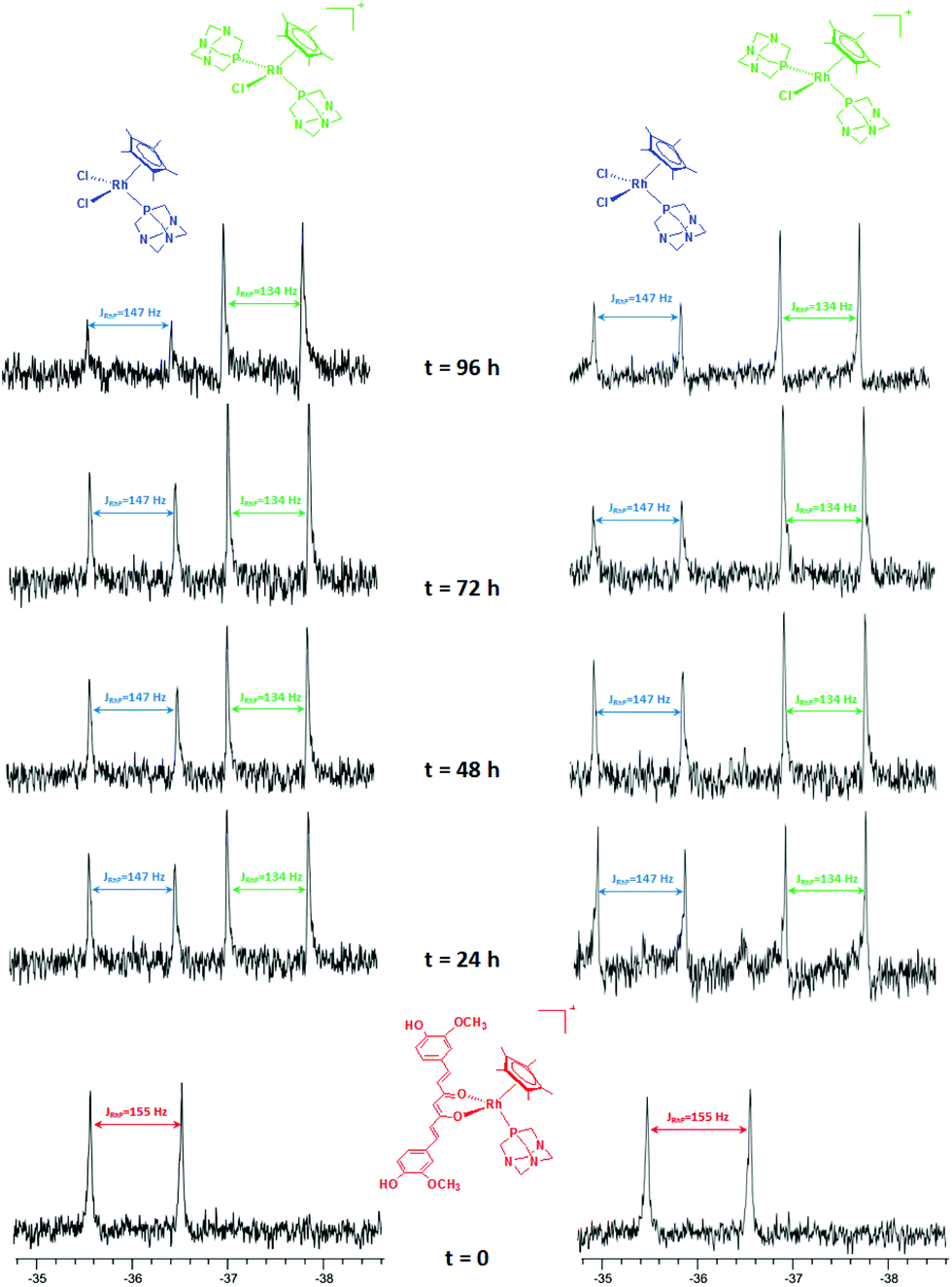 | ||
| Fig. 1 31P NMR spectra of 5 in 5 mM (left) and in 100 mM (right) aqueous NaCl solution. | ||
After 1 day, signals indicative of two new species are observed in both 5 and 100 mM aqueous NaCl solutions. The first doublet in the range −35.7/−36.0 ppm (JPRh = 147 Hz) corresponds to the [Cp*Rh(PTA)2Cl]+ (RhB) species, previously reported by Macchioni and co-workers,48 arising from the release of the curc ligand and its replacement with a chloride and a second PTA from another [Cp*Rh(curc)(PTA)]+ species, which in turn likely affords a third neutral species of the formula [Cp*Rh(curc)Cl] (RhC). The second doublet in the range −37.3/−37.4 ppm (JPRh = 134 Hz) corresponds to the [Cp*Rh(PTA)Cl2] (RhD) species.48
After 96 h an excess of PTA was added and, immediately, the resonance of [Cp*Rh(PTA)Cl2] (RhD) disappeared. Whereas, the intensity of [Cp*Rh(PTA)2Cl]+ (RhB) increased and the resonances of [Cp*Rh(curc)(PTA)]+ (RhA) reappeared, together with a new signal due to [Cp*Rh(PTA)3]+2 (JPRh = 120 Hz).48
The 31P NMR spectrum of the iridium compound 7 in water–DMSO (80–20%) solution showed immediately the formation of the hydrolysis products [Cp*IrCl2(PTA)] (IrC: −63.8 ppm) and [Cp*IrCl(OD2)(PTA)]+ (IrB: −65.7 ppm), in equilibrium with the starting species [Cp*Ir(curc)(PTA)]+ (IrA: −54.0 ppm). The nature of [Cp*IrCl2(PTA)] has been confirmed by adding an excess of NaCl (Fig. 2b), whereas the [Cp*IrCl(OD2)(PTA)]+ species has been hypothesized on the basis of a previous work by Peruzzini.47
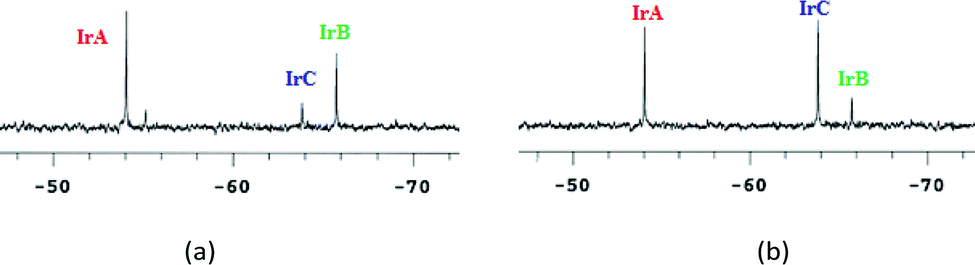 | ||
| Fig. 2 (a) 31P NMR chemical shifts for 7 in D2O–DMSO and (b) after addition of excess NaCl. | ||
The difference in stability of iridium complexes with respect to rhodium analogues has been previously observed for complexes with other ligands such as poly(pyrazolyl)borates.49,50
The 31P NMR spectra of 5 in D2O containing 20% DMSO were recorded at different pD values to determine the pKa value of the coordinated PTA ligand. The chemical shift was plotted against pD (Fig. 3) and the curve fitted using the Henderson–Hasselbalch equation to give a pKa of 3.36 ± 0.02 (0.44 is subtracted to account for the difference between the pH and pD).51
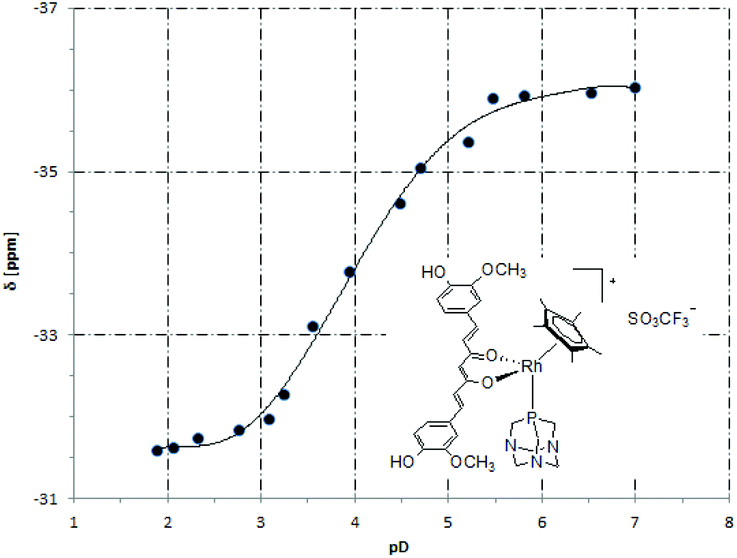 | ||
| Fig. 3 A plot of the 31P NMR chemical shift (δ) versus pD for 5. | ||
The solid-state structures of 5, 6 and 8 were established by X-ray crystallography (see the Experimental section). Their structures are shown in Fig. 4 and relevant bond distances and angles are given in the caption. The three metal complexes display the standard piano-stool geometry. Ligands show an almost planar geometry (the angles between the aromatic rings being 1.3°, 4.6° and 4.0° for compounds 5, 6 and 8, respectively), with a small twist around the central backbone (the calculated dihedral angles for the –CC(O)–C moieties varies from 1.7° in 5 to 8° in 8 to 8.8° in 6). The bond distances and angles around the metal centers (see the caption to Fig. 4) fall in the range of values found in the literature for similar compounds.19 Strong intermolecular hydrogen bonds are observed between the ligands and the anions (CF3SO3−) [O⋯O, 2.716(2) Å, <OHO, 156(3)° in 5; O⋯O, 2.716(4) Å, <OHO, 167° in 6; O⋯O, 2.706(7) Å, <OHO, 167° in 8] and between the ligands and the coordinated PTA of neighboring complexes [O⋯N, 2.810(2) Å, <OHN, 155(3)° in 5; O⋯N, 2.760(4) Å, <OHN, 163° in 6; O⋯N, 2.744(7) Å, <OHN, 163° in 8]. Additional intramolecular OH⋯O interactions are observed in the crystal of 5.
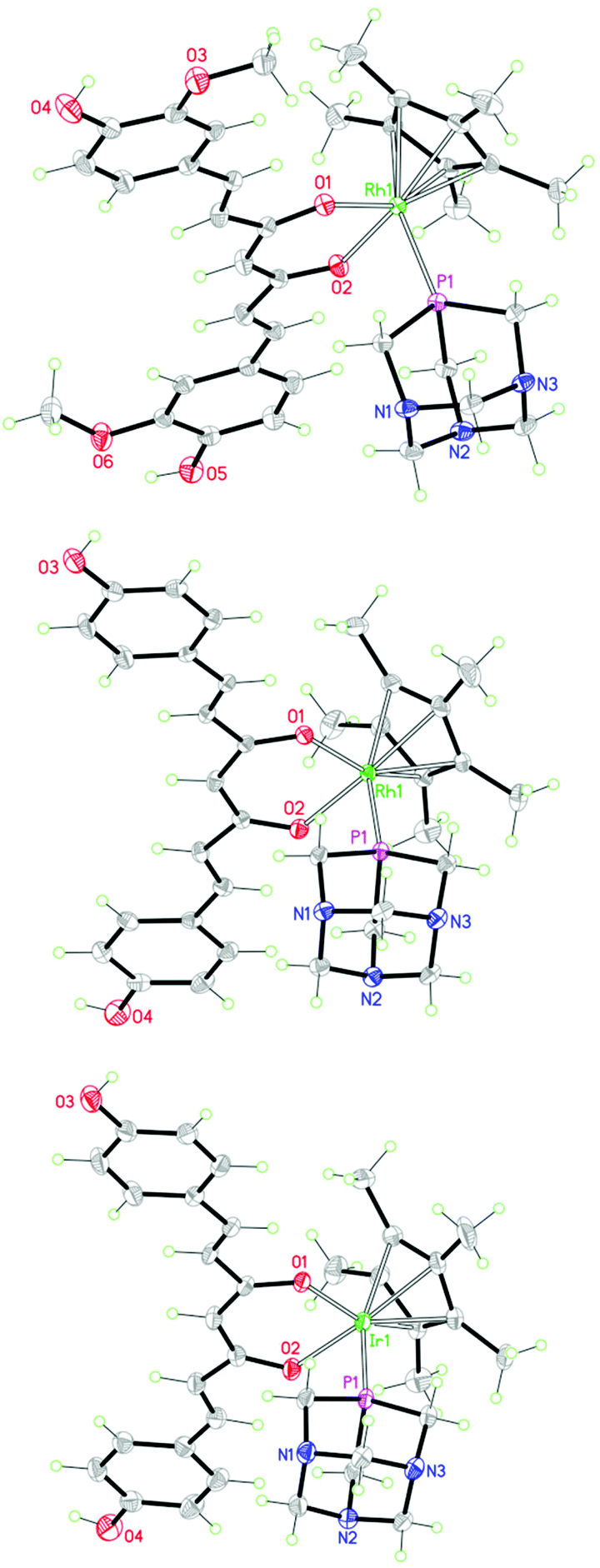 | ||
| Fig. 4 Molecular structures of 5 (top), 6 (middle) and 8 (bottom). The counter-anion has been omitted for clarity. Bond distances (Å) and angles (°): [5] Rh1–O1, 2.107(1); Rh1–O2, 2.091(1); Rh1–P1, 2.314(1); Rh1-η5(centroid), 1.802(1). [6] Rh1–O1, 2.109(2); Rh1–O2, 2.089(2); Rh1–P1, 2.313(1); Rh1-η5(centroid), 1.808(2). [8] Ir1–O1, 2.117(4); Ir1–O2, 2.101(4); Ir1–P1, 2.299(2), Ir1-η5(centroid), 1.810(3). | ||
The antiproliferative effects of 1–8 were investigated in two human ovarian cancer cell lines, A2780 and the cisplatin resistant A2780cisR cell line, as well as in non-tumorigenic human embryonic kidney (HEK293) cells. IC50 values for compounds 1–8 were determined after a 72 hour exposure period using the MTT assay (Table 1 and the Experimental section).
| Compound | A2780 (IC50, μM) | A2780cisR (IC50, μM) | HEK293 (IC50, μM) |
|---|---|---|---|
| curcH | 4.0 ± 0.1 | 3.2 ± 0.2 | 1.1 ± 0.1 |
| bdcurcH | 4.6 ± 0.1 | 4.6 ± 0.2 | 2.2 ± 0.1 |
| 1 | 14.9 ± 2.1 | 12.3 ± 0.3 | 13.7 ± 0.3 |
| 2 | 20.0 ± 2.0 | 18.1 ± 0.2 | 31.8 ± 0.2 |
| 3 | 23.2 ± 0.8 | 14.1 ± 2.5 | 16.3 ± 0.7 |
| 4 | 20.7 ± 5.6 | 23.6 ± 2.3 | 26.1 ± 2.9 |
| 5 | 12.5 ± 0.5 | 16.0 ± 3.0 | 17.2 ± 4.8 |
| 6 | 14.7 ± 2.2 | 17.7 ± 0.2 | 19.0 ± 4.0 |
| 7 | 23.2 ± 2.3 | 39.4 ± 9.4 | 29.4 ± 3.9 |
| 8 | 21.0 ± 1.0 | 33.3 ± 3.6 | 15.6 ± 1.4 |
All the complexes were cytotoxic at moderate micromolar concentrations against both cancer cell lines with IC50 values in the 12–23 μM range for A2780 cells and in the 12–40 μM range for A2780cisR cells, indicating a lack of cross-resistance. In HEK293 cells comparable IC50 values were obtained (14–32 μM) suggesting a lack of cancer cell selectivity.
Previously reported Rh(III) Cp* complexes containing maltol or allomaltol as O^O-chelating ligands were all inactive toward A549, CH1 and SW480 cell lines,52 whereas the lapachol complex shows good activity and selectivity toward the CH1 ovarian carcinoma cell line.53 A cyclometalated rhodium(III) complex shows lower54 or comparable IC50 values toward A2780 cells.39,55 Ir(III) Cp* complexes of the formula [(η5-Cp*)Ir(L^L′)Cl]0/+ containing N^N-(phenanthroline, 2,2′-bipyridine, ethylenediamine) and N^O-(picolinate) chelating ligands, are inactive on A2780 cells with IC50 values >100 μM,31,33 even with modified Cp*-type ligands.31,56 However, varying the chelating ligand32,40,57–59 can lead to compounds with IC50 values considerably higher than cisplatin.
Cytotoxicity data for the A2780 cell line are comparable with the IC50 value obtained for [(p-cymene)Ru(curc)Cl] in the HEK293 cell line, however, the rhodium(III) and iridium(III) complexes are 20–60 times less active than their corresponding ruthenium(II) analogues in the ovarian cancer cell lines.
The rhodium(III) and iridium(III) complexes with chloride 1–4 or PTA 5–8 co-ligands exhibit similar biological activities. This behaviour contrasts to that of the related ruthenium(II) complexes, in which the activity/selectivity of the PTA-containing complexes was superior to the chloride-based complexes.22 However, in keeping with the ruthenium(II) complexes, the nature of the curcuminoid ligand employed, i.e. curcumin and bisdemethoxycurcumin, plays little discernible role. It is not unreasonable to assume that following dissociation of the curcuminoid ligand the rhodium(III)/iridium(III) complexes interact with different biological targets compared to the arene ruthenium(II) derivatives, implying a different mechanism of action.
Conclusions
Compared to platinum- or ruthenium-based anticancer agents, the study of the medicinal properties of rhodium and iridium complexes is poorly developed. Here, we described a series of novel pentamethylcyclopentadienyl rhodium(III) and iridium(III) complexes with curcumin or bisdemethoxycurcumin co-ligands and a chloride or PTA ligand, these complexes being closely related to the previously studied arene ruthenium(II) complexes.24 Unlike related ruthenium(II) complexes with a PTA ligand, which display 100 times higher cytotoxicity towards cancer cells versus healthy cells, the compounds reported here are moderately cytotoxic to both human ovarian cancer cells and non-tumorigenic human embryonic kidney cells. Since it has been shown that all these compounds are able to release their curcuminoid ligands under physiological-like conditions it would suggest that the differences in activity between the pentamethylcyclopentadienyl M(III) (M = Rh/Ir) and arene ruthenium(II) compounds originate from the different metal fragments, which are known to have different binding preferences for certain biological targets.60Experimental section
General procedures
The dimers [Cp*MCl2]2 (M = Rh or Ir) were purchased from Aldrich. Curcumin and bisdemethoxycurcumin were purchased from TCI Europe and were used as received. All other materials were obtained from commercial sources and were used as received. IR spectra were recorded from 4000 to 30 cm−1 on a Perkin-Elmer Frontier Spectrometer FT-IR/FIR instrument. 1H and 13C NMR spectra were recorded on a 400 Mercury Plus Varian instrument operating at room temperature (400 MHz for 1H and 100 MHz for 13C) relative to TMS. Positive and negative ion electrospray ionization mass spectra (ESI-MS) were obtained on a Series 1100 MSI detector HP spectrometer using methanol as the mobile phase. Solutions (3 mg mL−1) for analysis were prepared using reagent-grade methanol. Masses and intensities were compared to those calculated using IsoPro Isotopic Abundance Simulator, version 2.1.28. Melting points are uncorrected and were recorded on a STMP3 Stuart scientific instrument and on a capillary apparatus. Samples for microanalysis were dried in vacuo to constant weight (20 °C, ca. 0.1 Torr) and analysed on a Fisons Instruments 1108 CHNS-O elemental analyzer.Synthesis of complexes 1–8
C), 1505 s, 472 m, 454 w, 242 s ν(Rh–Cl). 1H NMR (CDCl3, 293 K): δ, 1.69 (s, 15H, CH3Cp*), 3.94 (s, 6H, OCH3 of curc), 5.78 (s, 1H, C(1)H of curc), 6.51 (d, 2H, C(3, 3′)H of curc, 3Jtrans = 16 Hz), 6.90 (d, 2H, C(9, 9′)H of curc, 3JaromH–H = 9 Hz), 7.05 (m, 4H, C(6, 6′)H and C(10, 10′)H of curc), 7.57 (d, 2H, C(4, 4′)H of curc, 3Jtrans = 16 Hz). 13C NMR (CDCl3, 293 K): δ, 8.8 (s, CH3Cp*), 56.1 (s, OCH3 of curc), 94.7 (d, CCp*, J(103Rh–13C) = 6.9 Hz), 102.2 (s, C(1) of curc), 109.4 (s, C(6, 6′) of curc), 114.8 (s, C(9, 9′) of curc), 122.5 (s, C(10, 10′) of curc), 126.7 (s, C(5, 5′) of curc), 128.8 (s, C(3, 3′) of curc), 138.4 (s, C(4, 4′) of curc), 146.8 (s, C(7, 7′) of curc), 147.2 (s, C(8, 8′) of curc), 178.7 (s, C(2, 2′)O of curc). ESI-MS (+) CH3OH (m/z, relative intensity %): 605 [100] [Cp*Rh(curc)]+.
C), 1492 s, 478 m, 450 w, 244 s ν(Rh–Cl). 1H NMR (CD3CN, 293 K): δ, 1.63 (s, 15H, CH3Cp*), 5.94 (s, 1H, C(1)H of bdcurc), 6.65 (d, 2H, C(3, 3′)H of bdcurc, 3Jtrans = 16 Hz), 6.88 (m, 4H, C(7, 7′)H and C(9, 9′)H of bdcurc), 7.46 (s, 2H, OH of bdcurc), 7.55 (m, 4H, C(6, 6′)H and C(10, 10′)H of bdcurc), 7.62 (d, 2H, C(4, 4′)H of curc, 3Jtrans = 16 Hz). 13C NMR (CD3CN, 293 K): δ, 9.4 (s, CH3Cp*), 95.5 (d, CCp*, J(103Rh–13C) = 7.0 Hz), 102.3 (s, C(1) of bdcurc), 116.8 (s, C(7, 7′) and C(9, 9′) of bdcurc), 122.4 (s, C(6, 6′ and (s, C(10, 10′) of bdcurc), 126.7 (s, C(5, 5′) of bdcurc), 131.2 (s, C(4, 4′) of bdcurc), 141.1 (s, C(3, 3′) of bdcurc), 146.8 (s, C(8, 8′) of bdcurc), 178.7 (s, C(2, 2′)O of bdcurc). ESI-MS (+) CH3CN (m/z, relative intensity %): 545 [100] [Cp*Rh(bdcurc)]+.
C), 1504 s, 474 m, 461 w, 245 s ν(Ir–Cl). 1H NMR (CDCl3, 293 K): δ, 1.66 (s, 15H, CH3Cp*), 3.93 (s, 6H, OCH3 of curc), 5.50 (s, 1H, C(1)H of curc), 6.45 (d, 2H, C(3, 3′)H of curc, 3Jtrans = 16 Hz), 6.89 (d, 2H, C(9, 9′)H of curc, 3JaromH–H = 8 Hz), 7.05 (m, 4H, C(6, 6′)H and C(10, 10′)H of curc), 7.55 (d, 2H, C(4, 4′)H of curc, 3Jtrans = 16 Hz). 13C NMR (CDCl3, 293 K): δ, 9.0 (s, CH3Cp*), 56.2 (s, OCH3 of curc), 83.8 (s, CCp*), 103.3 (s, C(1) of curc), 109.3 (s, C(6, 6′) of curc), 114.9 (s, C(9, 9′) of curc), 122.5 (s, C(10, 10′) of curc), 126.3 (s, C(5, 5′) of curc), 128.9 (s, C(3, 3′) of curc), 138.3 (s, C(4, 4′) of curc), 147.0 (s, C(7, 7′) of curc), 147.2 (s, C(8, 8′) of curc), 176.5 (s, C(2, 2′)O of curc). ESI-MS (+) CH3OH (m/z, relative intensity %): 695 [100] [Cp*Ir(curc)]+.
C), 1493 s, 1269 s, 1158 s, 482 m, 445 w, 252 s ν(Ir–Cl). 1H NMR (CD3CN, 293 K): δ, 1.63 (s, 15H, CH3Cp*), 5.51 (s, 1H, C(1)H of bdcurc), 6.57 (d, 2H, C(3, 3′)H of bdcurc, 3Jtrans = 16 Hz), 6.87 (m, 4H, C(7, 7′)H and C(9, 9′)H of bdcurc), 7.37 (s, 2H, OH of bdcurc), 7.55 (m, 4H, C(6, 6′)H and C(10, 10′)H of bdcurc), 7.61 (m, 2H, C(4, 4′)H of bdcurc). 13C NMR (CD3CN, 293 K): δ, 8.9 (s, CH3Cp*), 92.7 (s, CCp*), 101.6 (s, C(1) of bdcurc), 116.6 (s, C(7, 7′) and C(9, 9′) of bdcurc), 121.4 (s, C(6, 6′ and (C(10, 10′) of bdcurc), 126.5 (s, C(5, 5′) of bdcurc), 131.0 (s, C(4, 4′) of bdcurc), 141.0 (s, C(3, 3′) of bdcurc), 160.5 (s, C(8, 8′) of bdcurc), 183.8 (s, C(2, 2′)O of bdcurc). ESI-MS (+) CH3CN (m/z, relative intensity %): 635 [100] [Cp*Ir(bdcurc)]+.
C), 1496 s, 1270 s, 1148 s, 1029 s ν(SO3CF3), 570 m ν(Rh–P), 472 m, 454 w, 421 w ν(Rh–P). 1H NMR (CD3CN, 293 K): δ, 1.68 (d, 15H, CH3Cp*, JHP = 3.4 Hz), 3.94 (s, 6H, OCH3 of curc), 4.17 (s, 6H, NCH2N, PTA), 4.49 (s, 6H, PCH2N, PTA), 5.65 (s, 1H, C(1)H of curc), 6.76 (d, 2H, C(3, 3′)H of curc, 3Jtrans = 16 Hz), 6.89 (d, 2H, C(9, 9′)H of curc, 3JaromH–H = 8 Hz), 6.95 (sbr, 2H, OH of curc), 7.16 (dd, 2H, C(10, 10′)H of curc, 3JaromH–H = 8 Hz), 7.28 (s, 2H, C(6, 6′)H of curc), 7.61 (d, 2H, C(4, 4′)H of curc 3Jtrans = 16 Hz). 13C NMR (CD3CN, 293 K): δ, 7.2 (s, CH3Cp*), 47.8 (d, NCH2P, PTA, JCP = 11.1 Hz), 54.8 (s, OCH3 of curc), 71.3 (d, NCH2N, PTA, JCP = 6.9 Hz), 97.9 (d, CCp*, J(103Rh–13C) = 6.8 Hz), 102.7 (s, C(1) of curc), 109.5 (s, C(6, 6′) of curc), 114.0 (s, C(9, 9′) of curc), 121.8 (s, C(10, 10′) of curc), 124.4 (s, C(5, 5′) of curc), 126.9 (s, C(3, 3′) of curc), 139.0 (s, C(4, 4′) of curc), 146.7 (s, C(7, 7′) of curc), 147.4 (s, C(8, 8′) of curc), 179.2 (s, C(2, 2′)O of curc). 31P NMR (CD3CN, 293 K): δ = −34.9 (d, JPRh = 151 Hz, PTA). ESI-MS (+) CH3CN (m/z, relative intensity %): 605 [100] [Cp*Rh(curc)]+, 762 [10] [Cp*Rh(curc)(PTA)]+.
C), 1493 s, 1277 s, 1160 s, 1025 s ν(SO3CF3), 580 m, 571 m ν(Rh–P), 476 s, 450 w, 346 w ν(Rh–P). 1H NMR (CD3CN, 293 K): δ, 1.67 (d, 15H, CH3Cp*, JHP = 3.4 Hz), 4.16 (s, 6H, NCH2N, PTA), 4.48 (s, 6H, PCH2N, PTA), 5.65 (s, 1H, C(1)H of bdcurc), 6.71 (d, 2H, C(3, 3′)H of bdcurc, 3Jtrans = 16 Hz), 6.88 (m, 4H, C(7, 7′)H and C(9, 9′)H of bdcurc), 7.40 (s, 2H, OH of bdcurc), 7.55 (m, 4H, C(6, 6′)H and C(10, 10′)H of bdcurc), 7.62 (d, 2H, C(4, 4′)H of bdcurc, 3Jtrans = 16 Hz). 13C NMR (CD3CN, 293 K): δ, 8.5 (s, CH3Cp*), 48.8 (d, NCH2P, PTA, JCP = 9.9 Hz), 72.5 (d, NCH2N, PTA, JCP = 11.1 Hz), 91.6 (s, CCp*) (d, CCp*, J(103Rh–13C) = 7.0 Hz), 104.0 (s, C(1) of bdcurc), 116.1 (s, C(7, 7′) and C(9, 9′) of bdcurc), 125.4 (s, C(6, 6′) and C(10, 10′) of bdcurc), 127.6 (s, C(5, 5′) of bdcurc), 130.2 (s, C(4, 4′) of bdcurc), 139.9 (s, C(3, 3′) of bdcurc), 159.8 (s, C(8, 8′) of bdcurc), 178.4 (s, C(2, 2′)O of bdcurc). 31P NMR (CD3CN, 293 K): δ = −34.9 (d, JPRh = 151 Hz, PTA). ESI-MS (+) CH3CN (m/z, relative intensity %): 545 [100] [Cp*Rh(bdcurc)]+, 702 [10] [Cp*Rh(bdcurc)(PTA)]+.
C), 1493 s, 1270 s, 1149 s, 1028 s ν(SO3CF3), 575 m ν(Ir–P), 472 m, 453 w, 390 w ν(Ir–P). 1H NMR (CD3CN, 293 K): δ, 1.70 (d, 15H, CH3Cp*, JHP = 1.9 Hz). 3.94 (s, 6H, OCH3 of curc), 4.16 (s, 6H, NCH2N, PTA), 4.48 (m, 6H, PCH2N, PTA), 5.71 (s, 1H, C(1)H of curc), 6.69 (d, 2H, C(3, 3′)H of curc, 3Jtrans = 16 Hz), 6.89 (d, 2H, C(9, 9′)H of curc, 3JaromH–H = 8 Hz)), 7.04 (s, 2H, OH of curc), 7.18 (dd, 2H, C(10, 10′)H of curc, 3JaromH–H = 8 Hz), 7.30 (s, 2H, C(6, 6′)H of curc), 7.63 (d, 2H, C(4, 4′)H of curc, 3Jtrans = 16 Hz). 13C NMR (CDCl3, 293 K): δ, 9.4 (s, CH3Cp*), 49.4 (d, PCH2N, PTA, JCP = 18.8 Hz), 57.1 (s, OCH3 of curc), 73.6 (d, NCH2N, PTA, JCP = 7.1 Hz), 93.5 (s, CCp*), 105.9 (s, C(1) of curc), 111.7 (C(6, 6′) of curc), 116.4 (C(9, 9′) of curc), 124.1 (C(10, 10′) of curc), 125.7 (C(5, 5′) of curc), 129.2 (C(3, 3′) of curc), 141.4 (C(4, 4′) of curc), 149.0 (C(7, 7′) of curc), 149.7 (C(8, 8′) of curc), 179.5 (C(2, 2′)O of curc). 31P NMR (CD3CN, 293 K): δ = −56.1 (s, PTA). ESI-MS (+) CH3CN (m/z, relative intensity %): 695.2 [100] [Cp*Ir(curc)]+, 852 [30] [Cp*Ir(curc)(PTA)]+.
C), 1491 s, 1278 s, 1160 s, 1025 s ν(SO3CF3), 600 m, 577 m ν(Ir–P), 482 s, 456 w, 346 w ν(Ir–P). 1H NMR (CD3CN, 293 K): δ, 1.69 (d, 15H, CH3Cp*, JHP = 2.0 Hz), 4.16 (s, 6H, NCH2N, pta), 4.48 (m, 6H, PCH2N, pta), 5.71 (s, 1H, C(1)H of bdcurc), 6.63 (d, 2H, C(3, 3′)H of bdcurc, 3Jtrans = 16 Hz), 6.88 (m, 4H, C(7, 7′)H and C(9, 9′)H of bdcurc), 7.44 (s, 2H, OH of bdcurc), 7.57 (m, 4H, C(6, 6′)H and C(10, 10′)H of bdcurc), 7.64 (d, 2H, C(4, 4′)H of bdcurc, 3Jtrans = 16 Hz). 13C NMR (CD3CN, 293 K): δ, 8.5 (s, CH3Cp*), 48.1 (d, PCH2N, PTA, JCP = 18.4 Hz), 72.5 (d, NCH2N, PTA, JCP = 7.7 Hz), 92.4 (s, CCp*), 105.0 (s, C(1) of bdcurc), 116.2 (s, C(7, 7′) and C(9, 9′) of bdcurc), 124.4 (s, C(6, 6′) and C(10, 10′) of bdcurc), 127.7 (s, C(5, 5′) of bdcurc), 130.2 (s, C(4, 4′) of bdcurc), 140.1 (s, C(3, 3′) of bdcurc), 159.2 (s, C(8, 8′) of bdcurc), 178.4 (s, C(2, 2′)O of bdcurc). 31P NMR (CD3CN, 293 K): δ = −56.3 (s, PTA). ESI-MS (+) CH3CN (m/z, relative intensity %): 635 [100] [Cp*Ir(bdcurc)]+, 792 [20] [Cp*Ir(bdcurc)(PTA)]+.
X-ray crystal structure determination
Diffraction data were measured at low temperature [100(2) K] using Mo Kα radiation on a Bruker APEX II CCD diffractometer equipped with a kappa geometry goniometer. The datasets were reduced by using EvalCCD61 and then corrected for absorption.62 The solutions and refinements were performed by using SHELX.63 The crystal structures were refined using full-matrix least-squares based on F2 with all non-hydrogen atoms anisotropically defined. Hydrogen atoms were placed in calculated positions by means of the “riding” model. Disorder problems dealing with CF3SO3− were encountered during the last stages of refinement and treated by using the split model (SADI and SIMU cards were applied to retain a reasonable geometry and acceptable ADP).Cell culture and inhibition of cell growth
The human A2780 and A2780cisR ovarian carcinoma and HEK (human embryonic kidney) cells were obtained from the European Collection of Cell Cultures (Salisbury, U.K.). A2780 and A2780cisR cells were grown routinely in RPMI-1640 medium, while HEK cells were grown in DMEM medium, with 10% fetal bovine serum (FBS) and 1% antibiotics at 37 °C and 5% CO2. Cytotoxicity was determined using the MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide). The cells were seeded in 96-well plates as monolayers with 100 μL of cell suspension (approximately 5000 cells) per well and pre-incubated for 24 h in a medium supplemented with 10% FBS. Compounds were prepared as DMSO solutions and then dissolved in the culture medium and serially diluted to the appropriate concentration, to give a final DMSO concentration of 0.5%. 100 μL of the drug solution was added to each well, and the plates were incubated for another 72 h. Subsequently, MTT (5 mg mL−1 solution) was added to the cells and the plates were incubated for a further 2 h. The culture medium was aspirated, and the purple formazan crystals formed by the mitochondrial dehydrogenase activity of vital cells were dissolved in DMSO. The optical density, directly proportional to the number of surviving cells, was quantified at 590 nm using a multiwell plate reader, and the fraction of surviving cells was calculated from the absorbance of untreated control cells. Evaluation is based on means from at least two independent experiments, each comprising triplicates per concentration level.Determination of pKa values
The pH values of NMR samples in D2O were measured at 298 K, directly in the NMR tube, using a 713 pH meter (Metrohm) equipped with an electrode calibrated with buffer solutions at pH values of 4, 7, and 9. The pH values were adjusted with dilute HNO3 and NaOH. The pH titration curves were fitted to the Henderson–Hasselbalch equation using the program Matlab (MathWorks Software) with the assumption that the observed chemical shifts are weighted averages according to the populations of the protonated and deprotonated species. The resonance frequencies change smoothly with pH between the chemical shifts of the charged form HA+, stable in acidic solution, and those of the neutral, deprotonated form A, which is present at a high pH. At any pH, the observed chemical shift is a weighted average (δav) of the two extreme values δ(HA+) and δ(A):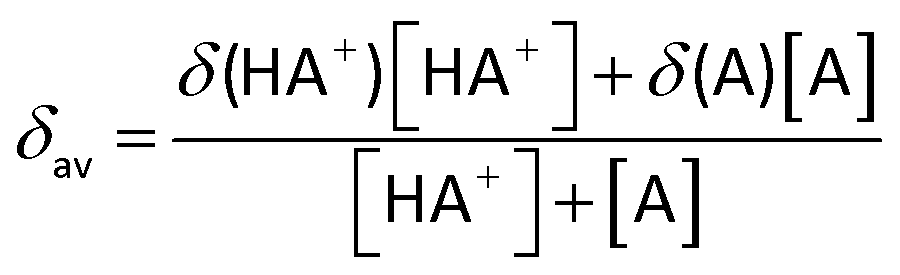
The midpoint of the titration occurs when the concentrations of the acid and its conjugate base are equal: [HA+] = [A], that is, when the pH equals the pKa of the compound. The pH at the midpoint of the curve is corrected by subtracting 0.44 to the pD values since the measurements were made in D2O.64
Acknowledgements
This work was financially supported by the University of Camerino (Fondo di Ateneo per la Ricerca 2011–2012), by MIUR (PRIN 2010-2011; 2010BNZ3F2) and by COST (CM 1302, European Network on Smart Inorganic Polymers).Notes and references
- B. B. Aggarwal, C. Sundaram, N. Malani and H. Ichikawa, Curcumin: the Indian solid gold, in The molecular targets and therapeutic uses of curcumin in health and disease, Springer, USA, 2007, pp. 1–75 Search PubMed.
- A. K. Renfrew, N. S. Bryce and T. W. Hambley, Chem. Sci., 2013, 4, 3731–3739 RSC.
- S. Banerjee, P. Prasad, A. Hussain, I. Khan, P. Kondaiah and A. R. Chakravarty, Chem. Commun., 2012, 48, 7702–7704 RSC.
- M. Sagnou, D. Benaki, C. Triantis, T. Tsotakos, V. Psycharis, C. P. Raptopoulou, I. Pirmettis, M. Papadopoulos and M. Pelecanou, Inorg. Chem., 2011, 50, 1295–1303 CrossRef CAS PubMed.
- G. Bar-Sela, R. Epelbaum and M. Schaffer, Curr. Med. Chem., 2010, 17, 190–197 CrossRef CAS PubMed.
- R. Waranyoupalin, S. Wongnawa, M. Wongnawa, C. Pakawatchai, P. Panichayupakaranant and P. Sherdshoopongse, Cent. Eur. J. Chem., 2009, 7, 388–394 CrossRef CAS.
- A. Goel, A. B. Kunnumakkara and B. B. Aggarwal, Biochem. Pharmacol., 2008, 75, 787–809 CrossRef CAS PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- A. Arezki, G. G. Chabot, L. Quentin, D. Scherman, G. Jaouen and E. Brulé, MedChemComm, 2011, 2, 190–195 RSC.
- A. Arezki, E. Brule and G. Jaouen, Organometallics, 2009, 28, 1606–1609 CrossRef CAS.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- A. Valentini, F. Conforti, A. Crispini, A. De Martino, R. Condello, C. Stellitano, G. Rotilio, M. Ghedini, G. Federici, S. Bernardini and D. Pucci, J. Med. Chem., 2009, 52, 484–491 CrossRef CAS PubMed.
- B. Balaji, B. Balakrishnan, S. Perumalla, A. A. Karande and A. R. Chakravarty, Eur. J. Med. Chem., 2014, 85, 458–467 CrossRef CAS PubMed.
- S. K. Singh and D. S. Pandey, RSC Adv., 2014, 4, 1819–1840 RSC.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- F. Caruso, M. Rossi, A. Benson, C. Opazo, D. Freedman, E. Monti, M. B. Gariboldi, J. Shaulky, F. Marchetti, R. Pettinari and C. Pettinari, J. Med. Chem., 2012, 55, 1072–1081 CrossRef CAS PubMed.
- L. Bonfili, R. Pettinari, M. Cuccioloni, V. Cecarini, M. Mozzicafreddo, M. Angeletti, G. Lupidi, F. Marchetti, C. Pettinari and A. M. Eleuteri, ChemMedChem, 2012, 7, 2010–2020 CrossRef CAS PubMed.
- R. Pettinari, F. Marchetti, F. Condello, C. Pettinari, G. Lupidi, R. Scopelliti, S. Mukhopadhyay, T. Riedel and P. J. Dyson, Organometallics, 2014, 33, 3709–3715 CrossRef CAS.
- T. Giraldi, G. Sava, G. Mestroni, G. Zassinovich and D. Stolfa, Chem.-Biol. Interact., 1978, 22, 231–238 CrossRef CAS PubMed.
- G. Sava, S. Zorzet, L. Perissin, G. Mestroni, G. Zassinovich and A. Bontempi, Inorg. Chim. Acta, 1987, 137, 69–71 CrossRef CAS.
- C. Pettinari, R. Pettinari, M. Fianchini, F. Marchetti, B. W. Skelton and A. H. White, Inorg. Chem., 2005, 44, 7933–7942 CrossRef CAS PubMed.
- C. Pettinari, R. Pettinari, F. Marchetti, A. Macchioni, D. Zuccaccia, B. W. Skelton and A. H. White, Inorg. Chem., 2007, 46, 896–906 CrossRef CAS PubMed.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- M. Gras, B. Therrien, G. Süss-Fink, A. Casini, F. Edafe and P. J. Dyson, J. Organomet. Chem., 2010, 695, 1119–1125 CrossRef CAS.
- Z. Liu, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Organometallics, 2011, 30, 4702–4710 CrossRef CAS.
- Z. Liu, L. Salassa, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Inorg. Chem., 2011, 50, 5777–5783 CrossRef CAS PubMed.
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijnincx, G. J. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS PubMed.
- Y. Geldmacher, K. Splith, I. Kitanovic, H. Alborzinia, S. Can, R. Rubbiani, M. A. Nazif, P. Wefelmeier, A. Prokop, I. Ott, S. Wölfl, I. Neundorf and W. S. Sheldrick, J. Biol. Inorg. Chem., 2012, 17, 631–646 CrossRef CAS PubMed.
- M. A. Nazif, R. Rubbiani, H. Alborzinia, I. Kitanovic, S. Wölfl, I. Ott and W. S. Sheldrick, Dalton Trans., 2012, 41, 5587–5598 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264–3279 RSC.
- R. Payne, P. Govender, B. Therrien, C. M. Clavel, P. J. Dyson and G. S. Smith, J. Organomet. Chem., 2013, 729, 20–27 CrossRef CAS.
- M. U. Raja, J. Tauchman, B. Therrien, G. Süss-Fink, T. Riedel and P. J. Dyson, Inorg. Chim. Acta, 2014, 409, 479–483 CrossRef CAS.
- L. C. Sudding, R. Payne, P. Govender, F. Edafe, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, J. Organomet. Chem., 2014, 774, 79–85 CrossRef CAS.
- V. Novohradsky, Z. Liu, M. Vojtiskova, P. J. Sadler, V. Brabec and J. Kasparkova, Metallomics, 2014, 6, 682–690 RSC.
- F. Kühlwein, K. Polborn and W. Beck, Z. Anorg. Allg. Chem., 1997, 623, 1211–1219 CrossRef.
- Y. Nakamura, K. Isobe, H. Morita, S. Yamazaki and S. Kawaguchi, Inorg. Chem., 1972, 11, 1573–1578 CrossRef CAS.
- Z. Liu, I. Romero-Canelón, A. Habtemariam, G. J. Clarkson and P. J. Sadler, Organometallics, 2014, 33, 5324–5333 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons, Inc., 2008 Search PubMed.
- R. Wanke, P. Smoleński, M. F. C. Guedes Da Silvan, L. M. D. R. S. Martins and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 10158–10168 CrossRef CAS PubMed.
- A. Dorcier, W. H. Ang, S. Bolano, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- M. Erlandsson, V. R. Landaeta, L. Gonsalvi, M. Peruzzini, A. D. Phillips, P. J. Dyson and G. Laurenczy, Eur. J. Inorg. Chem., 2008, 620–627, DOI:10.1002/ejic.200700792.
- S. Bolaño, M. Plaza, J. Bravo, J. Castro, M. Peruzzini, L. Gonsalvi, G. Ciancaleoni and A. Macchioni, Inorg. Chim. Acta, 2010, 363, 509–516 CrossRef.
- R. Pettinari, C. Pettinari, F. Marchetti, M. Monari, E. Mosconi and F. De Angelis, Organometallics, 2013, 32, 3895–3902 CrossRef CAS.
- E. Carmona, A. Cingolani, F. Marchetti, C. Pettinari, R. Pettinari, B. W. Skelton and A. H. White, Organometallics, 2003, 22, 2820–2826 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- M. Schmidlehner, V. Pichler, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, J. Organomet. Chem., 2015, 782, 69–76 CrossRef CAS.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Göschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348–3350 RSC.
- J. Ruiz, V. Rodríguez, N. Cutillas, K. G. Samper, M. Capdevila, Ò. Palacios and A. Espinosa, Dalton Trans., 2012, 41, 12847–12856 RSC.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed.
- V. Novohradsky, L. Zerzankova, J. Stepankova, A. Kisova, H. Kostrhunova, Z. Liu, P. J. Sadler, J. Kasparkova and V. Brabec, Metallomics, 2014, 6, 1491–1501 RSC.
- A. J. Millett, A. Habtemariam, I. Romero-Canelón, G. J. Clarkson and P. J. Sadler, Organometallics, 2015, 34, 2683–2694 CrossRef CAS PubMed.
- Z. Liu, I. Romero-Canelõn, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- J. M. Hearn, I. Romero-Canelón, B. Qamar, Z. Liu, I. Hands-Portman and P. J. Sadler, ACS Chem. Biol., 2013, 8, 1335–1343 CrossRef CAS PubMed.
- A. Casini, F. Edafe, M. Erlandsson, L. Gonsalvi, A. Ciancetta, N. Re, A. Ienco, L. Messori, M. Peruzzini and P. J. Dyson, Dalton Trans., 2010, 39, 5556–5563 RSC.
- A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M. Schreurs, J. Appl. Crystallogr., 2003, 36, 220–229 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Mikkelsen and S. O. Nielsen, J. Phys. Chem., 1960, 64, 632–637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1412115–1412117. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03037d |
| This journal is © The Royal Society of Chemistry 2015 |