
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of molecular oxygen by a molybdenum complex for catalytic oxidation†
Antoine
Dupé‡
a,
Martina E.
Judmaier‡
a,
Ferdinand
Belaj
a,
Klaus
Zangger
b and
Nadia C.
Mösch-Zanetti
*a
aInstitute of Chemistry, University of Graz, Schubertstrasse 1, 8010 Graz, Austria. E-mail: nadia.moesch@uni-graz.at
bInstitute of Chemistry, University of Graz, Heinrichstraße 28/II, 8010 Graz, Austria
First published on 9th November 2015
Abstract
A sterically demanding molybdenum(VI) dioxo complex was found to catalytically activate molecular oxygen and to transfer its oxygen atoms to phosphines. Intermediate peroxo as well as reduced mono-oxo complexes were isolated and fully characterized. Monomeric Mo(IV) monooxo species proved to be of an unusual nature with the coordinated phosphine trans to the oxo group. The reduced molybdenum centers can activate O2 to form a stable Mo(VI) oxo–peroxo complex unambiguously characterized by single crystal X-ray diffraction analysis. NMR experiments demonstrate that both oxygen atoms of the peroxo unit are transferred to an accepting substrate, generating the Mo(IV) intermediate and restarting the catalytic cycle.
Introduction
High-valent molybdenum-oxo complexes are of considerable interest as catalysts for oxidation reactions.1,2 In nature, molybdenum oxotransferases are a broad class of enzymes that transfer an oxygen atom to or from a substrate, using mainly water as a source of oxygen.3 The active site of these enzymes consists of a molybdenum(VI) center substituted by at least one oxo group and one or two molybdopterin ligands.4 In order to mimic the activity of such enzymes, a wide range of model compounds containing a high valent [MoO2]2+ core have been prepared.5 While oxygen atom transfer (OAT) capability of such complexes can be investigated using phosphines or sulfides as oxygen accepting substrates, they are also commonly used as catalysts for alkene epoxidation reactions and have thus found wide applications in industry.6 In the most efficient systems, tert-butyl hydroperoxide or hydrogen peroxide solutions are used as oxidant.1,7 However, following the principles of sustainable chemistry, the use of molecular oxygen as oxidant would be highly desirable; it is abundant, cheap, readily available and ideally does not lead to the formation of side-products.8 Despite these advantages, controlling the reactivity of dioxygen with transition metal centers is a challenge. Although many iron, manganese and copper complexes are known to react with dioxygen,9 only rare examples of molecular oxygen activation by molybdenum have been previously reported,10,11 including one by our group,12 and the reactivity of these oxo–peroxo molybdenum is limited. Few homogeneously molybdenum catalyzed oxidations employing molecular oxygen were reported. A bimetallic molybdenum/copper catalyst was disclosed by Osborn and coworkers for the oxidation of alcohols.13 It represents a “Wacker-type reaction” where the alcohol is oxidized by molybdenum but reoxidation is mediated by the copper co-catalyst and not by molecular oxygen directly. Selected reports can be found in literature where O2 was used in Mo catalyzed oxidation chemistry14 but high catalyst loadings were needed and/or it remains unclear whether autooxidation is occurring.In the course of our ongoing research on high-valent molybdenum-oxo complexes bearing Schiff-base ligands and their reactivity in OAT reactions,15–17 we report herein the synthesis and characterization of a new molybdenum(VI) dioxo complex which can be reduced and subsequently activate dioxygen to form a new molybdenum(VI) oxo–peroxo complex. This activation proceeds via the formation of a five-coordinated molybdenum(IV) mono-oxo complex or a highly unusual trans-[MoO(PMe)3]2+ intermediate which was characterized by NMR spectroscopy and elemental analysis. Furthermore, we demonstrate that the molybdenum(VI) oxo–peroxo complex is also active in OAT reaction and able to transfer both oxygen atoms of the peroxo moiety to an accepting substrate. Benchmark oxidation of tertiary phosphine18 confirms the catalytic activity of the complexes. This reactivity is a new step toward the use of oxygen as oxidant in molybdenum-catalyzed oxidation reactions.
Results and discussion
The molybdenum-dioxo complex [MoO2L2] 1 was synthesized by addition of a solution of the ligand 2,4-di-tert-butyl-6-((tert-butylimino)methyl)phenol (HL)19 in toluene to a solution of 1 equiv. of [MoO2Cl2] in toluene in presence of an excess trimethylamine (Scheme 1). After stirring for 15 h, filtration and washing with cold pentane, 1 was obtained as a orange solid in good yield (64%). The 1H NMR spectrum reveals the formation of a single, symmetric isomer as only one set of resonances for both coordinated ligands is observed. The IR spectrum of 1 exhibits two strong νMo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands at 885 and 912 cm−1 for both the symmetric and asymmetric stretching mode of the cis-[MoO2]2+ fragment, in good accordance with the literature.16,20 Mass spectrometry as well as elemental analysis confirmed the formation of complex 1 as [MoO2L2].
O bands at 885 and 912 cm−1 for both the symmetric and asymmetric stretching mode of the cis-[MoO2]2+ fragment, in good accordance with the literature.16,20 Mass spectrometry as well as elemental analysis confirmed the formation of complex 1 as [MoO2L2].
 | ||
| Scheme 1 Synthesis of Mo–dioxo complex 1. | ||
This structure was confirmed by single crystal X-ray diffraction analysis. Suitable crystals were obtained from concentrated solution of 1 in toluene (Fig. 1). Crystal data and structure refinements are presented in the Experimental section. Complex 1 exhibits a six-coordinate metal center in a distorted octahedral geometry.
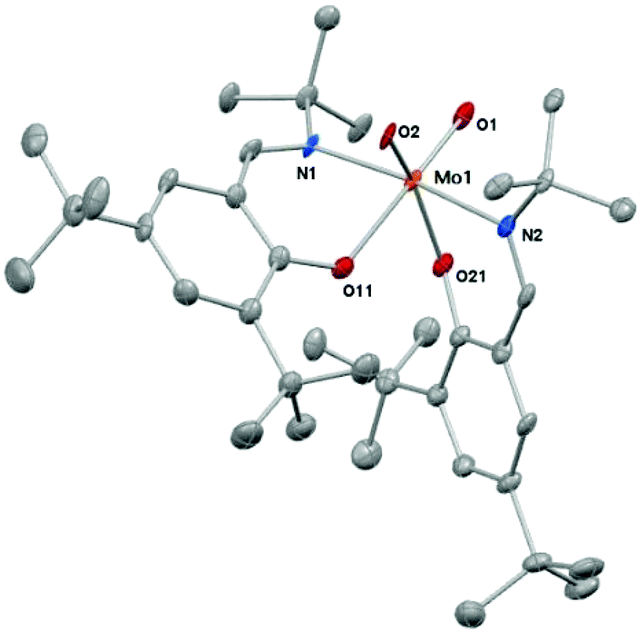 | ||
| Fig. 1 Molecular structure of complex 1. Thermal ellipsoids have been drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bonds lengths (Å) and angles (°): Mo(1)–O(2) 1.709(3); Mo(1)–O(1) 1.720(3); Mo(1)–O(21) 2.037(3); Mo(1)–O(11) 2.105(3); Mo(1)–N(1) 2.187(3); Mo(1)–N(2) 2.228(4); O(11)–C(11) 1.309(5); C(1)–N(1) 1.290(6); N(1)–C(10) 1.518(5); O(21)–C(21) 1.345(5); C(2)–N(2) 1.311(5); N(2)–C(20) 1.528(6); N(1)–Mo(1)–N(2) 171.91(13); O(1)–Mo(1)–O(11) 165.89(13); O(2)–Mo(1)–O(21) 159.04(13); C(11)–O(11)–Mo(1) 124.1(3); C(1)–N(1)–C(10) 116.0(3); C(1)–N(1)–Mo(1) 116.9(3); C(10)–N(1)–Mo(1) 124.8(3); C(21)–O(21)–Mo(1) 131.2(3); C(2)–N(2)–C(20) 116.0(3); C(2)–N(2)–Mo(1) 121.8(3); C(20)–N(2)–Mo(1) 121.7(3). | ||
Reactivity of the dioxo complex 1 in oxygen atom transfer (OAT) reactions was studied using trimethylphosphine and triphenylphosphine as oxygen acceptor substrates, allowing easy monitoring by 31P NMR spectroscopy. Complex 1 was first reacted with an excess (4–5 equiv.) trimethylphosphine in toluene under inert conditions (Scheme 2).
 | ||
| Scheme 2 Synthesis of molybdenum(IV) complex 2. | ||
The reaction was indicated by a quick change of color from deep orange to crimson and 31P NMR measurements reveal the formation of OPMe3 and further reduced molybdenum(IV) complex 2. After evaporation of the solvent and excess PMe3 then re-dissolution of the crude material in cold heptane, OPMe3 is removed via subsequent filtration over a pad of Celite. The pure reduced complex 2 was isolated as a highly sensitive to moisture and air, crimson solid in excellent yield (87%). Interestingly, 1H and 13C NMR spectra revealed the formation of a symmetric compound as only one set of resonances for both coordinate ligands is observed (Table 1). Characteristic signals in the 1H NMR spectrum in benzene-d6 are given at 8.33 ppm (imine proton), 7.42 and 7.19 ppm (aromatic protons). The 31P NMR spectrum of a concentrated solution of 2 in benzene-d6 points to the existence of a single Mo–PMe3 species (−9.47 ppm) further evidenced by the occurrence of a doublet at 0.91 ppm with the integration of nine protons in the 1H NMR spectrum.
| Compd. | Ar–CHN | Ar–H | Ar–H |
|---|---|---|---|
| 1 | 8.33 | 7.66 | 7.15 |
| 2 | 8.32 | 7.40 | 7.16 |
| 3 | 8.21 | 7.47 | 7.24 |
| 4 | 8.63, 8.55 | 7.60, 7.49 | 7.19, 7.19 |
The symmetric coordination in complex 2 has not been described in the literature up to now. In such type of OAT reactions, either the formation of a dimeric Mo(V)–O–Mo(V) species is observed, or the free coordination site is captured by a phosphine in cis position to the [MoO]2+ metal core, forming complexes of the type [MoO(PR3)L2]12,15,21 or [MoO(OPR3)L2].22 Such complexes render the geometry around the metal center non-symmetric and therefore lead to two sets of resonances for both coordinate ligands in NMR analysis. Our results point to the symmetric coordination of the PMe3 molecule in trans position to the MoO moiety. Although an equilibrium between a trans and cis species is in principle feasible, this is very unlikely as we observe narrow peaks in NMR spectroscopy. Furthermore, low temperature 31P NMR spectroscopy in toluene-d8 at −30 °C shows only one signal (see ESI†). This unusual arrangement is presumably caused by the high steric demand of the ligand. Two isomers are conceivable with the phosphine and the oxo groups trans to each other, namely the trans-N,N isomer depicted in Scheme 2 and the trans-N,O isomer, but the latter seems sterically too congested to exist.
ESI-MS measurements and elemental analysis corroborates the formation of a monomeric [MoO(PMe3)L2] complex. Information about the molecular size of the complex were obtained from 2D DOSY diffusion measurements on a sample containing complex 2 and free ligand in benzene-d6. For the free ligand (HL), a diffusion coefficient D = −8.805 (log(m2 s−1)) corresponding to 1.57 × 10−9 m2 s−1 was found, while complex 2 diffuses with a D = −8.878 (log(m2 s−1)) corresponding to 1.32 × 10−9 m2 s−1. Based on the Stokes–Einstein equation, the relative hydrodynamic radii of two components are related to the diffusion coefficients by D1/D2 = r2/r1. Therefore, the experimental diffusions coefficients of complex 2 and free ligand correspond to relative hydrodynamic radii of 1.18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Such an increase of 18% of the diameter of the complex relative to HL can only be explained by a monomer. In addition, the DOSY analysis of a sample containing the dioxo complex 1 and the reduced complex 2 showed similar diffusion for both complexes (see ESI†).
:1. Such an increase of 18% of the diameter of the complex relative to HL can only be explained by a monomer. In addition, the DOSY analysis of a sample containing the dioxo complex 1 and the reduced complex 2 showed similar diffusion for both complexes (see ESI†).
The OAT reaction of complex 1 with 2 equiv. PPh3 in benzene-d6 at room temperature as well results in a color change from deep orange to purple. After 5 h under stirring, an NMR sample was taken. 1H and 13C NMR spectra exhibited again one set of resonances for both coordinate ligands, indicating formation of a new symmetric compound [MoOL2] 3. Characteristic signals in 1H NMR in benzene-d6 (Table 1) are given at 8.21 ppm (imine proton), 7.47 and 7.24 ppm (aromatic protons). 31P NMR confirmed formation of OPPh3 (signal at 24.78 ppm) but showed that the other equivalent of PPh3 does not coordinate the molybdenum center and thus remains free (signal at 5.41 ppm). Evaporation of the solvent led to a highly air sensitive purple powder which contains 3, OPPh3 and PPh3. Removal of OPPh3 was performed by filtration of a heptane solution of the mixture over dry Celite, which however did not remove residual PPh3. Thus, sublimation of residual PPh3 at 100 °C was attempted to isolate 3, but only lead to decomposition of the complex. Nevertheless, its sensitivity towards air as well as 1H and 31P NMR spectroscopy lead us to believe that the monooxo [MoOL2] complex 3 is formed in situ rather than a Mo(V) dimer or a Mo(IV) phosphino species. The monomeric structure of 3 was confirmed by DOSY analysis, with a diffusion coefficient similar to that obtained for complex 2.
Although complexes with the [MoO(O2)]2+ or [MoO(O2)2]2+ cores are relatively abundant in the literature, as complexes thereof are available from the reaction of [MoO3] and the appropriate ligand in aqueous H2O2,23 the activation of molecular oxygen by molybdenum compounds is scarce in the literature. As previously described by our group, re-oxidation of the molybdenum(IV) complex type [MoO(PMe3)L2] with molecular oxygen either results in the formation of [MoO2]2+ or [MoO(O2)]2+ complexes,12,15 depending on the steric demand in the ligand backbone. When benzene-d6 solutions of 2 and 3 were exposed to dry molecular oxygen, color change from crimson to orange for complex 2 and from purple to orange for 3 occurred. 1H and 13C NMR spectra indicate the formation of the same, non-symmetric compound in both reactions, as two sets of resonances for both coordinated ligands are observed (e.g. in the 1H NMR spectra 2 × Ar–CHN at 8.55 and 8.63 ppm, Table 1). Such behavior is in good accordance to the literature regarding the formation of a [MoO(O2)]-type complex, as the coordination of the O2 molecule in cis position to the [MoO]2+ renders the geometry around the metal center non-symmetric.16 It is noteworthy that this [MoO(O2)L2] complex 4 can be synthesized directly by reacting 1 with a 5-fold excess phosphine under O2 atmosphere. After purification, 4 was isolated as an orange solid in 93% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of the oxo–peroxo Mo(VI) complex 4. | ||
Single crystals suitable for X-ray diffraction analyses of complex 4 were obtained from concentrated THF solutions layered with pentane at room temperature. Crystal data and structure refinements are presented in the Experimental section. The structure reveals a hepta-coordinated metal center in a distorted pentagonal bi-pyramidal geometry (Fig. 2). The metal center is ligated by one terminal oxygen atom, a η2-peroxo moiety and two bidentate Schiff base ligands. The terminal oxygen atom is found to be in cis position to the η2-peroxo moiety. All these features as well as bonds lengths and angles values are in good accordance with similar [MoO(O2)]2+ complexes reported previously in the literature.10,12,24
 | ||
| Fig. 2 Molecular structure of complex 4. Thermal ellipsoids have been drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–O1 1.6909(10); Mo1–O2 1.9487(10); Mo1–O3 1.9447(10); Mo1–O11 2.0494(9); Mo1–O31 2.0269(9); Mo1–N1 2.2548(11); Mo1–N2 2.2162(11); O2–O3 1.4332(13); O1–Mo1–O11 170.37(4); O2–Mo1–O31 155.69(4); O3–Mo1–O31 153.96(4); N1–Mo1–N3 165.72(4); O1–Mo1–O2 99.17(5); O1–Mo1–O3 101.82(5); O1–Mo1–O31 91.99(4); O1–Mo1–N1 91.57(4). | ||
The re-oxidation of the reduced [MoO(PMe3)L2] complex 2 with molecular O2, yielding the [MoO(O2)L2] complex 4 (Scheme 3), provided an interesting information. The 31P NMR spectra measured directly after exposition of 2 to O2 showed that the phosphine attached to the Mo is released as a free phosphine when 4 is formed. The same NMR sample, maintained under O2 atmosphere was submitted again for analysis after 24 h. The 31P NMR spectra revealed that the equivalent of free PMe3 formed during the re-oxidation had been converted to OPMe3. Thus, when 2 reacted with O2 as presented in Scheme 3, substitution of the phosphine by O2 occurred and only subsequently PMe3 was oxidized to OPMe3 by the complex 4 (see Fig. S3 in ESI†). These observations are noteworthy as phosphine is usually strongly coordinated, so that prior oxidation is required for its displacement. Here, the steric demand of the ligand and the trans-effect resulting from the presence of the oxo group leads to a weakly coordinated PMe3 rendering the system reactive. Such labilization of phosphine ligands due to a trans-effect was already observed with other Mo complexes.25 The fact that the free PMe3 equivalent could be oxidized under O2 atmosphere in presence of 4 lead us to investigate the role of the oxo–peroxo complex as possible catalyst for the oxidation of phosphine using O2, as described thereafter. In order to exclude auto oxidation of PMe3 and to prove the reactivity of the oxo–peroxo complex in OAT, the reaction of 4 with 2 equiv. PMe3 in exclusion of O2 was investigated (Scheme 4). Monitoring the reaction by 1H and 31P NMR spectroscopy revealed that the oxo–peroxo complex 4 is able to oxidize tertiary phosphines, but more importantly that both atoms of the peroxo group are involved in this transfer. Although the reaction does not go to completion, 1H NMR spectra show formation of a new molybdenum species and OPMe3 in a 1:2 ratio, along with unreacted PMe3 and complex 4. As shown in Fig. 3, after t = 5 h approx. 25% conversion is observed and after t = 30 h 50%. This Mo intermediate exhibits a single set of signal very similar to that of the Mo(IV) monooxo complex 2 (e.g. signals at 8.34, 7.40 and 7.21 ppm). The small differences between the 1H NMR signals of the observed Mo intermediate and 2 can be explained by differences in the coordination of the phosphine. In the intermediate, PMe3 is only weakly interacting with Mo but is not fully coordinated. This explanation is supported by the 1H NMR signal of the weakly interacting PMe3 (0.86 ppm, JP–C = 4.2 Hz), which is different from free PMe3 (0.81 ppm, JP–C < 2 Hz) and from the fully bound PMe3 in 2 (0.91 ppm, JP–C = 7 Hz). During the course of the reaction, no formation of a plausible Mo(VI) dioxo intermediate (complex 1) was observed, indicating that the transfer of the first oxygen atom from the peroxo unit is the rate determining step of the process, while transfer of the second oxygen atom is either concerted or occurs much faster. This was corroborated by kinetic experiments using UV-Vis spectroscopy. The OAT reaction from complex 1 with a 100-fold excess PMe3 (pseudo-first order conditions) to form the reduced complex 2 was found to have a rate constant k = 0.003 s−1. The corresponding reaction of complex 4 with 100 equiv. PMe3 and the same concentration for the complex and phosphine was found to be orders of magnitude slower as hardly any reaction occurred in the observed timeframe (see ESI†).
 | ||
| Scheme 4 OAT reaction of the oxo–peroxo complex 4 in exclusion of O2 in benzene-d6. | ||
 | ||
| Fig. 3 1H NMR spectra in benzene-d6 at t = 5 h (left) and t = 30 h (right) of the reaction of molybdenum oxo–peroxo complex 4 with 2 equiv. PMe3 under O2 exclusion. | ||
As both the dioxo and oxo–peroxo complexes are active in OAT reaction, catalytic oxidation of PMe3 was investigated. In the literature, the molybdenum complexes which activate O2 could not catalytically oxidize phosphines. [MoO(O2)(CN)4][PPh4]2 is capable of oxidizing 3 mol PPh3 per 2 mol molybdenum in the presence of molecular O2,10 but no further oxidation of PPh3 was obtained due to conproportionation of the dioxo Mo(VI) and the monoxo Mo(IV) intermediates. Although PMe3 is easily oxidized without any metal in presence of water and air, the stability of PMe3 against dry O2 in dry benzene-d6 solutions was confirmed by running a blank experiment (see ESI†). For the catalytic run, a solution of 1 (10 mg, 14 μmol) in benzene-d6 was prepared and placed under O2 atmosphere. Then 100 equivalents PMe3 were added and the reaction was left to stir for 24 h (Scheme 5). The reaction was monitored by 1H and 31P NMR analysis. Integration of PMe3 and OPMe3 signals after 24 h showed the conversion of 19 equivalents PMe3 to OPMe3. After evaporation of the solvent and unreacted phosphine, mass balance confirmed formation of OPMe3 besides the residue of the catalyst. Control experiments in the presence of sodium molybdate or [MoO2(acac)2] did not lead to formation of OPMe3. The reactivity of 1 is limited by decomposition of the complex, but is a successful progress toward the use of molecular oxygen as sole oxidant in catalytic OAT reactions.
 | ||
| Scheme 5 Conversion of PMe3 to OPMe3 catalyzed by 1 using O2. | ||
Conclusion
We were able to prepare a new molybdenum(VI) dioxo complex 1 which is active in oxygen atom transfer reaction to tertiary phosphines, allowing the isolation of the unusual trans-[MoO(PMe3)L2] complex 2. The activation of molecular oxygen by the reduced species yields the oxo–peroxo complex [MoO(O2)L2] 4 in excellent yield. 4 is also active in OAT and NMR spectroscopy studies show that both atoms of the peroxo group are transferred to phosphine. Hence, the molybdenum(VI) dioxo complex 1 exhibits promising results as catalyst for oxidations using O2 as the oxidant.Experimental section
All reactions have been carried out under nitrogen using standard Schlenk or glovebox techniques. [MoO2Cl2] was purchased from Sigma-Aldrich and used in the Glovebox without further purification. The Schiff base ligand was synthesized according to previously published literature.19 Solvents were purified via a Pure-Solv MD-4-EN solvent purification system from Innovative Technology, Inc. The 1H and 13C NMR spectra were recorded on a Bruker Optics Instrument 300 MHz. Peaks are denoted as singlet (s), doublet (d), doublet of doublets (dd), triplet (t) and multiplet (m), Ar denotes aromatic protons. Chemical shifts are reported in ppm and are referenced using the residual solvent peak. To obtain self-diffusion coefficients we used two-dimensional diffusion ordered spectroscopy (DOSY).26 The employed pulse sequence was a bipolar pulse pair longitudinal eddy current delay (BPP-LED) sequence, using 32 scans per increment, 60 ms diffusion delay time, 1 ms gradient pulses and variation of the gradient strength in 32 increments, linearly varied between 2 and 95% of maximum (which is 53.5 G cm−1). DOSY analysis was performed using the Bruker DOSY package of TopSpin 3.1. All DOSY measurements were carried out at 300 K on a Bruker Avance III 500 MHz NMR spectrometer using a 5 mm TXI probe with z-axis gradients. IR spectra were measured as solid samples on a Bruker Alpha P Diamond FTIR-ATR spectrometer or as liquid samples in benzene on a Bruker FT-MIR matrix MF in situ spectrometer using a glass fiber optic probe. ESI-MS spectra were recorded in acetonitrile on an Agilent 1100 Series LCMSD (SL type). Elemental analyses were carried out using a Heraeus Vario Elementar automatic analyzer at the Institute of Inorganic Chemistry at the University of Technology in Graz.[MoO2L2] (1)
Ligand HL (500 mg, 1.7 mmol, 2 equiv.) was dissolved in 5 ml of dry toluene and slowly added to a suspension of [MoO2Cl2] (180 mg, 0.9 mmol, 1 equiv.) in 5 mL of dry toluene under inert conditions. Dry triethylamine (0.26 mL, 1.9 mmol, 2.2 equiv.) was then added. The formation of the complex was immediately indicated by a quick change of color from pale yellow to deep red. To ensure complete complex formation, the solution was stirred 5 h at room temperature. The mixture was then filtered with a cannula and the solvent was removed in vacuo. The residue was washed thrice with 10 ml of cold pentane to afford complex 1 as an orange solid in 64% yield (407 mg). Single crystals suitable for X-ray diffraction analyses were obtained by slow evaporation from concentrated toluene solution at room temperature.1 H NMR (300 MHz, benzene-d6, ppm) δ = 8.33 (s, 1H, Ar–CHN), 7.66 (d, 4JH–H = 2.7 Hz, 1H, Ar–H), 7.15 (d, 4JH–H = 2.7 Hz, 1H, Ar–H), 1.56 (s, 9H, NC(CH3)3), 1.33 (s, 9H, Ar–C(CH3)3), 1.30 (s, 9H, Ar–C(CH3)3).
13 C NMR (75 MHz, benzene-d6, ppm) δ = 167.48 (Ar–CHN), 164.31 (Ar–O), 139.32 (Ar), 139.00 (Ar), 131.03 (Ar–H), 129.28 (Ar–H), 123.39 (Ar), 64.96 (NC(CH3)3), 35.28 (Ar–C(CH3)3), 34.18 (Ar–C(CH3)3), 31.59 (Ar–C(CH3)3), 31.24 (N(CH3)3), 29.57 (Ar–C(CH3)3).
IR (ATR, cm−1): ν = 1613 (m, CN), 1587 (m, CN), 1534 (m), 1387 (m), 1255 (m), 1169 (s), 913 (s, MoO), 883 (s, MoO), 836 (m), 745 (m), 533 (m).
Anal. Calcd for MoO4N2C38H60: C, 64.75; H, 8.58; N, 3.97. Found: C, 64.29; H, 8.42; N, 3.73%.
[MoO(PMe3)L2] (2)
[MoO2L2] (100 mg, 0.14 mmol, 1 equiv.) was dissolved in dry toluene and excess PMe3 (70 μL, 0.7 mmol, 5 equiv.) was added. The OAT was indicated by a quick change of color from orange to dark red. After stirring for 3 hours, the solvent and excess PMe3 were removed under reduced pressure. The obtained material was re-dissolved in cold dry heptane (5 mL) and filtered over a pad of Celite. After evaporation of the solvent, complex 2 was obtained as dark red solid. Yield: 95 mg (87%).1 H NMR (300 MHz, benzene-d6, ppm) δ = 8.32 (s, 2H, Ar–CHN), 7.40 (d, 4JH–H = 2.6 Hz, 2H, Ar–H), 7.16 (m, 2H, Ar–H), 1.65 (s, 18H, N–C(CH3)3), 1.34 (s, 18H, Ar–C(CH3)3), 1.31 (s, 18H, Ar–C(CH3)3), 0.91 (d, JP–H = 7 Hz, 9H, OPMe3).
13 C NMR (75 MHz, benzene-d6, ppm) δ = 164.57 (Ar–CHN), 162.96 (Ar–O), 138.78 (Ar), 136.58 (Ar), 131.55 (Ar–H), 128.90 (Ar–H), 122.59 (Ar), 64.47 (N–C(CH3)3), 35.67 (Ar–C(CH3)3), 34.05 (Ar–C(CH3)3), 32.07 (Ar–C(CH3)3), 31.81 (N–C(CH3)3), 30.60 (Ar–C(CH3)3), 17.55 (d, JP–C = 17.9 Hz, Mo–P(CH3)3).
31 P NMR (121 MHz, benzene-d6, 298 K, ppm) δ −9.47 (Mo − P(CH3)3).
IR (FT-IR, benzene, cm−1): ν 1605 (m, CN), 1542 (m), 1460 (s), 1436 (s), 1396 (s), 1362 (m), 1307 (s), 1256 (s), 1165 (s), 932 (s, MoO), 836 (m).
ESI-MS (50 V) m/z (%): 690.3 (100) [M − HP(CH3)3].
Anal. Calcd for MoO3N2PC41H69: C, 64.38; H, 9.09; N, 3.66. Found: C, 64.25; H, 9.23; N, 3.38%.
[MoOL2] (3)
[MoO2L2] (100 mg, 0.14 mmol, 1 equiv.) was dissolved in dry toluene and an excess PPh3 (74 mg, 0.28 mmol, 2 equiv.) was added. The OAT was indicated by a quick change of color from orange to purple. After stirring for 15 hours, the solvent was removed under reduced pressure. The obtained material was re-dissolved in cold dry heptane (5 mL) and filtered over a pad of celite. After evaporation of the heptane, [MoOL2] was obtained as a purple solid in 60% yield, with partial decomposition to the free ligand and traces of triphenylposphine.1 H NMR (300 MHz, benzene-d6, ppm) δ = 8.21 (s, 2H, Ar–CHN), 7.47 (d, 4JH–H = 2.6 Hz, 2H, Ar–H), 7.24 (d, 4JH–H = 2.6 Hz, 2H, Ar–H), 1.61 (s, 18H, N–C(CH3)3), 1.34 (s, 18H, Ar–C(CH3)3), 1.33 (s, 18H, Ar–C(CH3)3).
13 C NMR (75 MHz, benzene-d6, ppm) δ = 165.66 (Ar–CHN), 163.68 (Ar–O), 139.01 (Ar), 138.52 (Ar), 132.05 (Ar–H), 129.07 (Ar–H), 119.06 (Ar), 65.97 (N–C(CH3)3), 35.80 (Ar–C(CH3)3), 34.27 (Ar–C(CH3)3), 31.69 (Ar–C(CH3)3), 30.83 (N–C(CH3)3), 30.14 (Ar–C(CH3)3).
[MoO(O2)L2] (4)
[MoO2L2] (100 mg, 0.14 mmol, 1 equiv.) was dissolved in dry benzene (5 mL) followed by the addition of excess PMe3 (53 mg, 70 μL, 0.70 mmol, 5 equiv.). The OAT was indicated by a quick change of color from orange to dark red. The solution was stirred for 30 minutes, followed by exposure of this solution to dry molecular oxygen for 5 minutes. The reaction solution quickly changed the color from dark violet to orange. The mixture was left to stir for 30 minutes and the solvent was removed in vacuo. The product was dissolved in heptane (5 mL) and twice filtered over a pad of Celite. [MoO(O2)L2] was obtained as an orange solid. Yield: 87 mg (93%). For elemental analyses, the complex was dissolved in minimum amount of CH2Cl2 and again filtered over a pad of Celite. Single crystals suitable for X-ray diffraction analyses were obtained from concentrated THF solutions layered with pentane.1 H NMR (300 MHz, benzene-d6, ppm) δ = 8.63 (s, 1H, Ar–CHN), 8.55 (s, 1H, Ar–CHN), 7.60 (d, 4JH–H = 2.4 Hz, 1H, Ar–H), 7.49 (d, 4JH–H = 2.4 Hz, 1H, Ar–H), 7.19 (d, 4JH–H = 2.4 Hz, 2H, Ar–H), 1.87 (s, 9H, NC(CH3)3), 1.74 (s, 9H, NC(CH3)3), 1.41 (s, 9H, Ar–C(CH3)3), 1.27 (s, 9H, Ar–C(CH3)3), 1.23 (s, 9H, Ar–C(CH3)3), 1.09 (s, 9H, Ar–C(CH3)3).
13 C NMR (75 MHz, benzene-d6, ppm) δ = 166.36 (overlapping signals, 2 × Ar–CHN), 161.74 (Ar–O), 159.65 (Ar–O), 140.22 (Ar), 139.79 (Ar), 138.97 (Ar), 137.78 (Ar), 131.83 (Ar–H), 130.86 (Ar–H), 130.16 (Ar–H), 130.37 (Ar–H), 124.45 (Ar), 122.63 (Ar), 67.96 (NC(CH3)3), 67.49 (NC(CH3)3), 35.44 (Ar–C(CH3)3), 34.77 (Ar–C(CH3)3), 34.13 (Ar–C(CH3)3), 34.06 (Ar–C(CH3)3), 32.78 (NC(CH3)3), 31.60 (NC(CH3)3), 31.54 (2 × Ar–C(CH3)3), 30.25 (Ar–C(CH3)3), 29.91 (Ar–C(CH3)3).
IR (ATR, cm−1): ν 1606 (s, CN), 1541 (m), 1255 (s), 1168 (s), 931 (s, MoO), 838 (s), 777 (s), 747 (m), 542 (s, Mo–O).
Anal. Calcd for MoO5N2C38H60·0.23 CH2Cl2: C, 62.00; H, 8.23; N, 3.78. Found: C, 62.18; H, 8.44; N, 3.90%.
Oxygen atom transfer reactivity of [MoO(O2)L2] (4)
[MoO(O2)L2] (25 mg, 0.035 mmol, 1 equiv.) was dissolved in dry benzene-d6 under inert conditions in a Young NMR tube. 2 equivalent PMe3 (8 μL, 0.07 mmol) was added using a micropipette. The OAT reaction was monitored by 1H and 31P NMR spectroscopy.Catalytic oxidation of trimethylphosphine
[MoO2L2] (complex 1, 10 mg, 14 μmol) was placed in a Schlenk flask in the glovebox. The Schlenk flask was evacuated then refilled with dry O2. Dry benzene-d6 (2 mL) and trimethylphosphine (108 mg, 0.15 μL, 1.4 mmol, 100 equiv.) were added and the reaction was left to stir under O2 atmosphere for 24 h. The reaction was monitored by 1H and 31P NMR at t = 4 h, 20 h and 24 h. After removal of the solvent, the mass balance was calculated and the yield of OPMe3 confirmed by integration of the signals in the 1H NMR spectrum at t = 24 h (yield = 19%). To confirm the stability of PMe3 under O2 atmosphere, a blank experiment was performed. A Schlenk flask was evacuated then refilled with dry O2. Dry benzene-d6 (2 mL) and trimethylphosphine (108 mg, 0.15 μL, 1.4 mmol, 100 equiv.) were added and the reaction was left to stir under O2 atmosphere for 24 h. The reaction was monitored by 1H and 31P NMR at t = 4 h, 20 h and 24 h. at t = 24 h, a signal for OPMe3 could be observed in the 1H NMR (1% yield) but no signal was observed in 31P NMR. After 24 h, the solvent and free PMe3 were evaporated, no solid OPMe3 could be isolated.OAT kinetic study
In the glovebox, a solution of 7 mg of complex 1 in 5 mL toluene (2 × 10−3 mol L−1) and a solution of 15 mg PMe3 in 1 mL toluene (0.2 mol L−1) were prepared. Using a micropipette, 100 μL of the solution containing the complex were placed in a quartz cuvette and 2 mL toluene were added. Then 100 μL of the solution containing PMe3 were added and the cuvette was sealed with a Teflon stopper and parafilm. The sample was removed from the glovebox and the measurement immediately started. The reaction was monitored by the dissapearance of the signal at 430 nm, at room temperature using a Cary50 Conc. UV-Vis spectrophotometer under PC control using the kinetics program “Scan” included with the instrument software. The same procedure was followed preparing a solution of 7 mg of complex 4 in 5 mL toluene (2 × 10−3 mol L−1).Structure determination
For X-ray structure analyses the crystals were mounted onto the tip of glass fiber and data collection was performed at 100 K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) with a BRUKER-AXS SMART APEX II diffractometer equipped with a CCD detector. Essential details of the crystal-data and structure refinements for compounds 1 and 4 are summarized in Table 2. Crystallographic data for the structures of compounds 1 and 4 have been deposited with the Cambridge Crystallographic Data Center [CCDC 1413968 for 1, CCDC 1413969 for 4].| 1 | 4 | |
|---|---|---|
| Empirical formula | C38H60MoN2O4 | 2MoO5N2C38H60·xC4H8O·(1 − x)C5H12; x = 0.786(6) |
| Formula weight | 704.82 | 1513.75 |
| Crystal description | Plate, red | Block, red |
| Crystal size (mm) | 0.27 × 0.17 × 0.05 mm | 0.32 × 0.25 × 0.25 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, C2/c |
| Unit cell dimensions | ||
| a (Å) | 10.3863(4) | 24.3085(13) |
| b (Å) | 11.4627(5) | 15.8102(8) |
| c (Å) | 33.1945(13) | 20.8035(11) |
| α (°) | 90 | 90 |
| β (°) | 98.6499(17) | 94.112(2) |
| γ (°) | 90 | 90 |
| Volume (Å3) | 3907.0(3) | 7974.7(7) |
| Z, calculated density (g cm−3) | 4, 1.198 | 4, 1.261 |
| F(000) | 1504 | 3233.9 |
| Linear absorption coefficient μ (mm−1) | 0.373 | 0.373 |
| Absorption correction | Semi-empirical from equiv. | Semi-empirical from equiv. |
| Temperature (K) | 100 | 100 |
| Wavelength (Mo Kα) (Å) | 0.71073 | 0.71073 |
| Theta range for data collection (°) | 2.46 to 28.45 | 1.79 to 30.00 |
| Limiting indices | −12 ≤ h ≤ 12 | −34 ≤ h ≤ 29 |
| −5 ≤ k ≤ 14 | −22 ≤ k ≤ 22 | |
| −40 ≤ l ≤ 40 | −29 ≤ l ≤ 29 | |
| Reflections collected/unique | 26791/7661 |
46647/11648 |
| Reflections with I > 2σ(I) | 5550 | 10882 |
| R(int), R(sigma) | 0.0549, 0.0723 | 0.0187, 0.0148 |
| Completeness to theta max. | 0.999 | 0.999 |
| Refinement method | Full matrix least squares on F2 | Full matrix least squares on F2 |
| Data/restraints/parameters | 7661/432/0 | 11648/486/12 |
| Goodness-of-fit on F2 | 1.107 | 1.100 |
| Final R1 wR2 [I > 2σ(I)] | R 1 = 0.0585 | R 1 = 0.0277 |
| wR2 = 0.1401 | wR2 = 0.0689 | |
| R indices (all data) | R 1 = 0.0869 | R 1 = 0.0304 |
| wR2 = 0.1507 | wR2 = 0.0709 | |
| Largest diff. peak and hole (e Å−3) | 1.484 and −2.767 | 1.283 and −0.599 |
N double bond were put at the external bisectors of the C–C–X angles at C–H distances of 0.95 Å and common isotropic displacement parameters were refined for these H atoms of the same ligand. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same tert-butyl group and idealized geometries with tetrahedral angles, enabling rotation around the C–C bonds, and C–H distances of 0.98 Å.
Acknowledgements
The authors gratefully acknowledge financial support from the Austrian Science Fund (FWF): project number P26264 and from NAWI Graz.Notes and references
- M. Amini, M. M. Haghdoost and M. Bagherzadeh, Coord. Chem. Rev., 2013, 257, 1093 CrossRef CAS.
- M. Schubert, J. Leppin, K. Wehming, D. Schollmeyer, K. Heinze and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 2494 CrossRef CAS PubMed; R. Sanz and M. Pedrosa, Curr. Org. Synth., 2009, 6, 239 CrossRef.
- R. Hille, Dalton Trans., 2013, 42, 3029 RSC; M. J. Romão, Dalton Trans., 2009, 4053 RSC; R. H. Holm, Coord. Chem. Rev., 1990, 100, 183 CrossRef CAS; R. Hille, Trends Biochem. Sci., 2002, 27, 360 CrossRef PubMed.
- F. J. Hine, A. J. Taylor and C. D. Garner, Coord. Chem. Rev., 2010, 254, 1570 CrossRef CAS; P. Basu and S. J. N. Burgmayer, Coord. Chem. Rev., 2011, 255, 1016 CrossRef PubMed; R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963 CrossRef PubMed.
- J. H. Enemark, J. J. A. Cooney, J.-J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175 CrossRef CAS PubMed; H. Sugimoto and H. Tsukube, Chem. Soc. Rev., 2008, 37, 2609 RSC; C. Schulzke, Eur. J. Inorg. Chem., 2011, 1189 CrossRef; R. H. Holm, E. I. Solomon, A. Majumdar and A. Tenderholt, Coord. Chem. Rev., 2011, 255, 993 CrossRef; S. J. N. Burgmayer, M. Kim, R. Petit, A. Rothkopf, A. Kim, S. Bel Hamdounia, Y. Hou, A. Somogyi, D. Habel-Rodriguez, A. Williams and M. L. Kirk, J. Inorg. Biochem., 2007, 101, 1601 CrossRef PubMed; J. M. Berg and R. H. Holm, J. Am. Chem. Soc., 1985, 107, 925 CrossRef; A. Majumdar and S. Sarkar, Coord. Chem. Rev., 2011, 255, 1039 CrossRef.
- J. Kollar, US3351635, 1967 RSC; M. N. Sheng and J. G. Zajacek, GB1136923, 1965 RSC; J.-M. Brégeault, Dalton Trans., 2003, 3289 RSC; S. Shylesh, M. Jia and W. R. Thiel, Eur. J. Inorg. Chem., 2010, 4395 CrossRef CAS.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552 CAS.
- I. W. C. E. Arends and R. A. Sheldon, in Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 147–185 Search PubMed; W. B. Tolman, Activation of small molecules. Organometallic and bioinorganic perspectives, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006 Search PubMed.
- J. Serrano-Plana, I. Garcia-Bosch, A. Company and M. Costas, Acc. Chem. Res., 2015, 48, 2397 CrossRef CAS PubMed; W. Nam, Acc. Chem. Res., 2015, 48, 2415 CrossRef PubMed; E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef PubMed; K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef PubMed; P. Comba, Y.-M. Lee, W. Nam and A. Waleska, Chem. Commun., 2014, 50, 412 RSC; K. E. Dalle, T. Gruene, S. Dechert, S. Demeshko and F. Meyer, J. Am. Chem. Soc., 2014, 136, 7428 CrossRef PubMed; P. Haack and C. Limberg, Angew. Chem., Int. Ed., 2014, 53, 4282 CrossRef PubMed; C. E. MacBeth, A. P. Golombek, V. G. Young Jr., C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938 CrossRef PubMed; E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570 CrossRef.
- H. Arzoumanian, J. F. Petrignani, M. Pierrot, F. Ridouane and J. Sanchez, Inorg. Chem., 1988, 27, 3377 CrossRef CAS.
- J. Tachibana, T. Imamura and Y. Sasaki, J. Chem. Soc., Chem. Commun., 1993, 1436 RSC; M. Minato, D.-Y. Zhou, K.-i. Sumiura, Y. Oshima, S. Mine, T. Ito, M. Kakeya, K. Hoshino, T. Asaeda, T. Nakada and K. Osakada, Organometallics, 2012, 31, 4941 CrossRef CAS.
- G. Lyashenko, G. Saischek, A. Pal, R. Herbst-Irmer and N. C. Mösch-Zanetti, Chem. Commun., 2007, 701 RSC.
- C. Y. Lorber, S. P. Smidt and J. A. Osborn, Eur. J. Inorg. Chem., 2000, 655 CrossRef CAS.
- M. A. Katkar, S. N. Rao and H. D. Juneja, RSC Adv., 2012, 2, 8071 RSC; S. N. Rao, N. Kathale, N. N. Rao and K. N. Munshi, Inorg. Chim. Acta, 2007, 360, 4010 CrossRef CAS; W. A. Herrmann, G. M. Lobmaier, T. Priermeier, M. R. Mattner and B. Scharbert, J. Mol. Catal. A: Chem., 1997, 117, 455 CrossRef.
- G. Lyashenko, G. Saischek, M. E. Judmaier, M. Volpe, J. Baumgartner, F. Belaj, V. Jancik, R. Herbst-Irmer and N. C. Mösch-Zanetti, Dalton Trans., 2009, 5655 RSC.
- M. E. Judmaier, C. Holzer, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 9956 CrossRef CAS PubMed.
- T. Arumuganathan, M. Volpe, B. Harum, D. Wurm, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 150 CrossRef CAS PubMed; M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 1440 CrossRef PubMed; J. A. Schachner, P. Traar, C. Sala, M. Melcher, B. N. Harum, A. F. Sax, M. Volpe, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 7642 CrossRef PubMed; M. E. Judmaier, C. H. Sala, F. Belaj, M. Volpe and N. C. Mösch-Zanetti, New J. Chem., 2013, 37, 2139 RSC; N. C. Mösch-Zanetti, D. Wurm, M. Volpe, G. Lyashenko, B. Harum, F. Belaj and J. Baumgartner, Inorg. Chem., 2010, 49, 8914 CrossRef PubMed.
- S. K. Sharma, P. S. May, M. B. Jones, S. Lense, K. I. Hardcastle and C. E. MacBeth, Chem. Commun., 2011, 47, 1827 RSC; W.-D. Wang and J. H. Espenson, Inorg. Chem., 2001, 40, 1323 CrossRef CAS PubMed; B. G. Jacobi, D. S. Laitar, L. Pu, M. F. Wargocki, A. G. DiPasquale, K. C. Fortner, S. M. Schuck and S. N. Brown, Inorg. Chem., 2002, 41, 4815 CrossRef PubMed.
- G. Alesso, M. Sanz, M. E. G. Mosquera and T. Cuenca, Eur. J. Inorg. Chem., 2008, 4638 CrossRef CAS.
- J. Liimatainen, A. Lehtonen and R. Sillanpää, Polyhedron, 2000, 19, 1133 CrossRef CAS.
- K. Most, J. Hoßbach, D. Vidović, J. Magull and N. C. Mösch-Zanetti, Adv. Synth. Catal., 2005, 347, 463 CrossRef CAS; K. Heinze and A. Fischer, Eur. J. Inorg. Chem., 2007, 1020 CrossRef; K. Hüttinger, C. Förster, T. Bund, D. Hinderberger and K. Heinze, Inorg. Chem., 2012, 51, 4180 CrossRef PubMed.
- B. L. Tran and C. J. Carrano, Inorg. Chem., 2007, 46, 5429 CrossRef CAS PubMed; A. J. Millar, C. J. Doonan, P. D. Smith, V. N. Nemykin, P. Basu and C. G. Young, Chemistry, 2005, 11, 3255 CrossRef PubMed; V. N. Nemykin and P. Basu, Inorg. Chem., 2005, 44, 7494 CrossRef PubMed.
- W. R. Thiel and T. Priermeier, Angew. Chem., Int. Ed. Engl., 1995, 34, 1737 CrossRef CAS; J.-Y. Piquemal, S. Halut and J.-M. Brégeault, Angew. Chem., Int. Ed., 1998, 37, 1146 CrossRef; M. Herbert, F. Montilla, E. Álvarez and A. Galindo, Dalton Trans., 2012, 41, 6942 RSC; M. Bagherzadeh, M. Zare, V. Amani, A. Ellern and L. Keith Woo, Polyhedron, 2013, 53, 223 CrossRef.
- J. A. Brito, M. Gomez, G. Muller, H. Teruel, J.-C. Clinet, E. Dußach and M. A. Maestro, Eur. J. Inorg. Chem., 2004, 4278 CrossRef CAS.
- J. Leppin, C. Förster and K. Heinze, Inorg. Chem., 2014, 53, 1039 CrossRef CAS PubMed.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1993, 115, 4291 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray crystallographic data, characterization data. CCDC 1413968 for 1 and 1413969 for 4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02931g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |