DOI:
10.1039/C5DT02851E
(Paper)
Dalton Trans., 2015,
44, 16325-16331
Preparation of enantiomerically pure open calcocene and strontocene complexes and their application in ring opening polymerizations of rac-lactide†
Received
25th July 2015
, Accepted 15th August 2015
First published on 24th August 2015
Abstract
The synthesis of C2 symmetric enantiomerically pure open Ca and Sr metallocenes, [(η5-pdl*)2Ca(thf)] (1) and [(η5-pdl*)2Sr(thf)2] (2) (pdl* = dimethylnopadienyl) is described and these complexes were fully characterized. The solid state structures confirm that the pdl* ligands coordinate exclusively with the less sterically demanding site to the Ca and Sr atoms. These complexes are active catalysts for the controlled ring opening polymerization (ROP) of rac-lactide to give heterotactically enriched polylactides (PL) with narrow polydispersities (PDI = 1.29–1.31) and without adding further activators.
Introduction
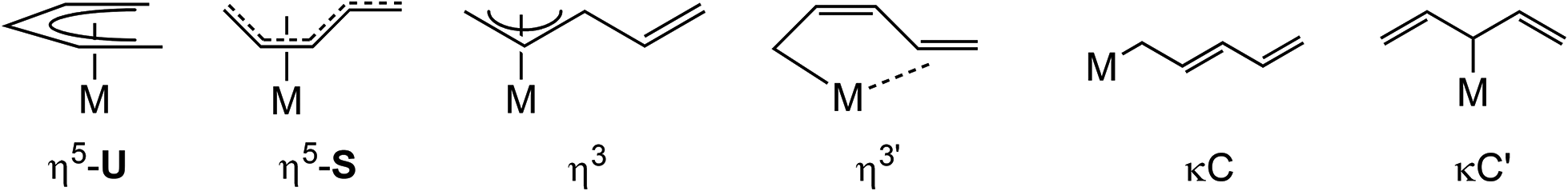
Since the discovery of ferrocene, Cp2Fe, organometallic chemistry has been dominated by the cyclopentadienyl (Cp) ligand; a large number of cyclopentadienyl complexes with tailored steric and electronic properties have been developed1–5 for various applications ranging from material science and small molecule activation to (enantioselective) catalysis. In contrast, the chemistry of the “open” analogues, the pentadienyl (pdl) ligands, and their corresponding metal complexes has received significantly less attention and the investigations have mainly focused on d-transition metals,6–10 whereas complexes of the rare earth,11–21 actinide21–25 and main group metals26–38 have been surprisingly scarce. The diverging developments of these ligand types is surprising, considering that Cp and pdl ligands are indeed related, but pdl derivatives can also adopt different hapticities (Chart 1) and exhibit in its η5-U coordination better π-donor and δ-acceptor capabilities, which allow the stabilization of low oxidation states such as Hf(II) and Zr(II).6–10
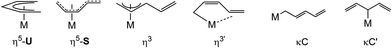 |
| | Chart 1 Different hapticity modes associated with pdl ligands. | |
There are only scattered reports of pdl complexes acting as catalysts in polymerization processes.12,20,39 Inspired by these observations, we recently initiated a research program on sterically demanding and chiral pentadienyl ligands and their coordination chemistry.35–38,40 In the course of these investigations we also set out to close currently existing gaps in the periodic table, to prepare pentadienyl complexes of metals such as the heavier alkaline earths, (Ca, Sr and Ba)37 that are currently less well explored, and to establish new reactivity patterns.
In this contribution the synthesis of the first chiral open metallocenes of Ca and Sr derived from the natural product (1R)-(−)myrtenal is reported, together with their application in the ring-opening polymerization (ROP) of rac-lactide. Ca complexes have become interesting catalysts for various catalytic applications including polymerizations of olefins (styrene) and polar monomers such as lactones and lactides.41 Lactides are interesting renewable precursors for biodegradable and biocompatible polymers and the resulting polylactides show interesting physical and mechanical properties.42–46 While enantiomerically pure polylactide melts at 180 °C, stereocomplex derivatives formed by an equivalent mixture of poly(L-lactide) and poly(D-lactide) melt at 230 °C.47,48 Furthermore the rate of degradation can be modulated by the chain stereochemistry.49–51 Several catalyst systems for the ROP of cyclic esters, based on magnesium, zinc, calcium, aluminum, lanthanides, tin, group 4 metals, germanium, indium and iron, have been reported.42,44–46,52–58
Results and discussion
Complex synthesis and characterization
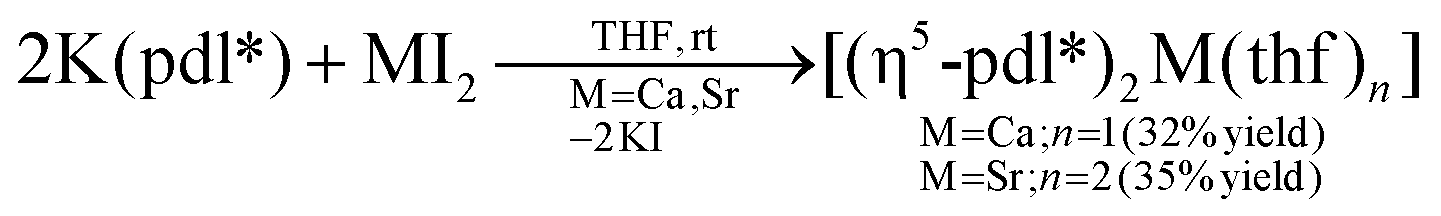
Potassium dimethylnopadienide, K(pdl*) (2 equiv.), reacts cleanly with MI2 (M = Ca and Sr) in THF to give the respective open metallocenes, [(η5-pdl*)2Ca(thf)] (1) and [(η5-pdl*)2Sr(thf)2] (2). These complexes are very air- and moisture-sensitive, but can be isolated after crystallization in moderate yields. They are readily soluble in aromatic and aliphatic hydrocarbons and THF.
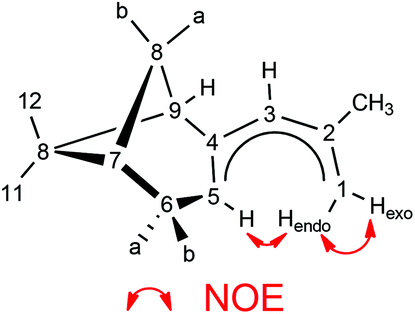
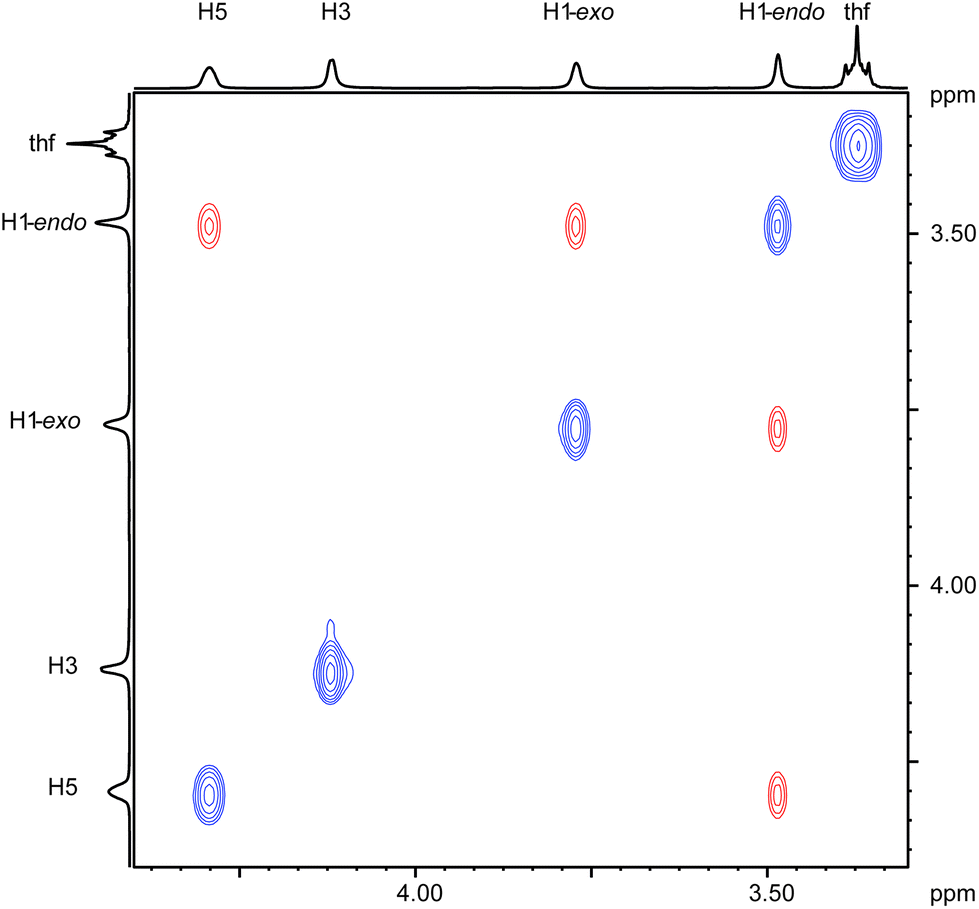
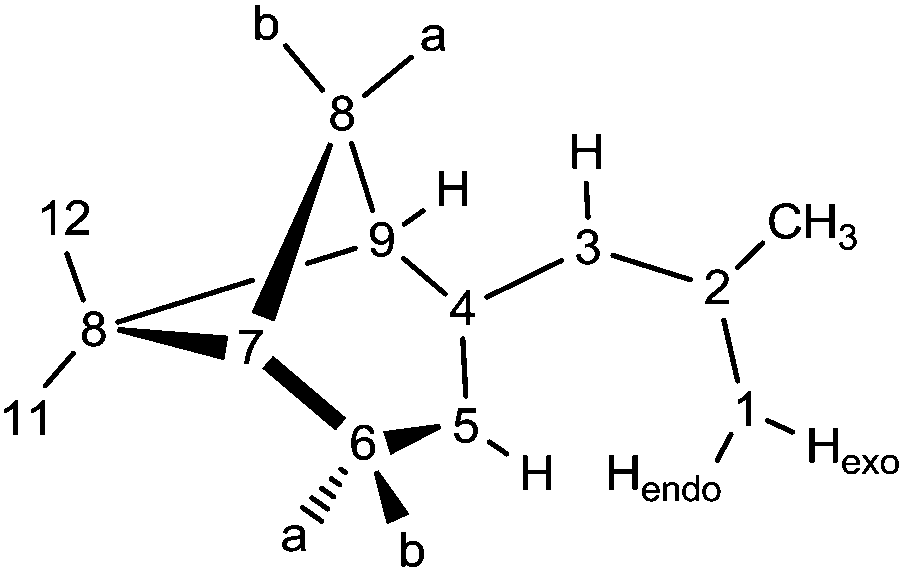
Various spectroscopic techniques, elemental analyses and X-ray diffraction were employed for the characterization of these compounds (see Experimental section for details). In solution the open metallocenes 1 and 2 adopt a C2 symmetric structure, in which the pentadienyl ligands display an η5-U conformation. NOESY spectroscopy shows a strong NOE between the H atoms H1-endo and H5 (Chart 2 and Fig. 1).
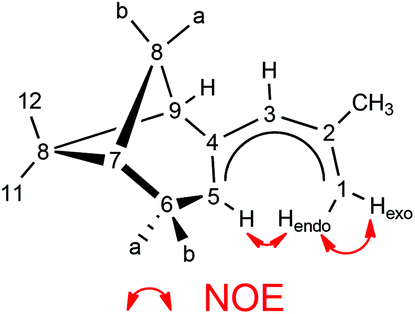 |
| | Chart 2 | |
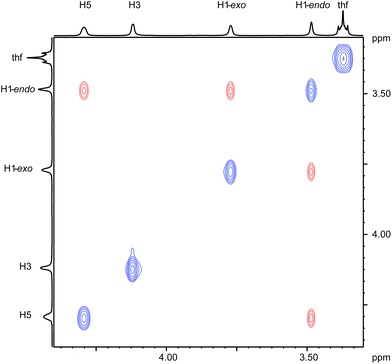 |
| | Fig. 1
1H–1H NOESY NMR spectrum of the pentadienyl moiety in complex 1 recorded in C6D6 at ambient temperature. | |
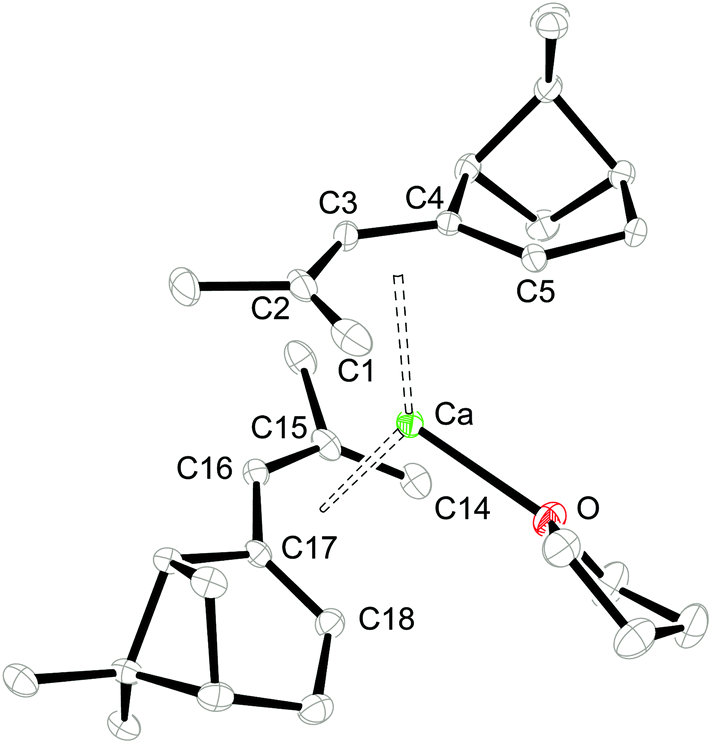
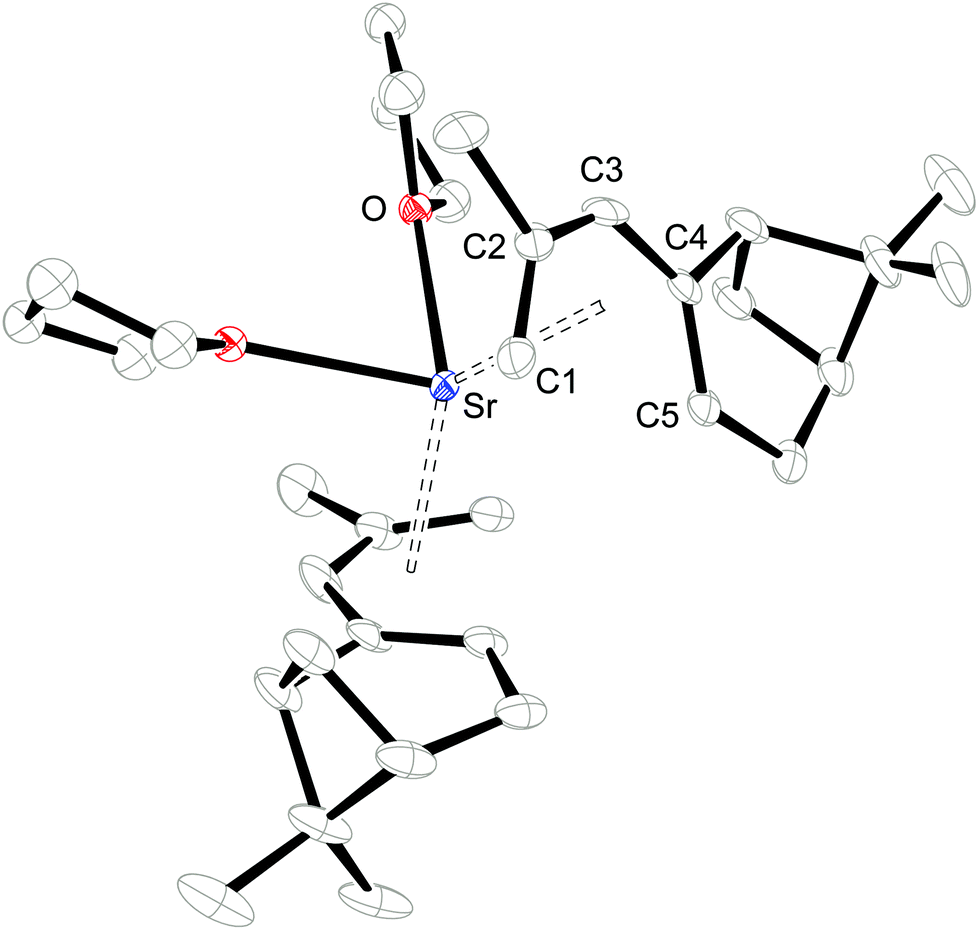
Single crystals of 1 and 2 were grown from THF/hexane mixtures at −30 °C (Table 1) and their molecular structures are shown in Fig. 2 and 3. Attempts to crystallize these compounds from pure hexane solutions only lead to amorphous precipitates, but on addition of even small amounts of THF, crystallization is readily induced. Complex 1 crystallizes in the orthorhombic space group P212121, whereas 2 crystallizes in the trigonal space group P3121 with one uncoordinated THF molecule in the crystal lattice. Selected bond distances and angles can be found in Table 2.
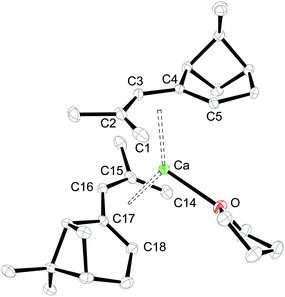 |
| | Fig. 2 ORTEP of 1 with thermal displacement parameters drawn at 30% probability. Hydrogen atoms were omitted for clarity. | |
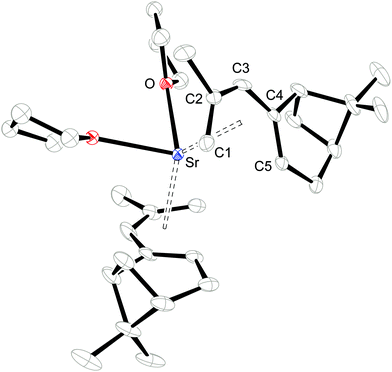 |
| | Fig. 3 ORTEP of 2 with thermal displacement parameters drawn at 30% probability. Hydrogen atoms were omitted for clarity. The coordinated THF is disordered over two positions, but only one of these is shown. | |
Table 1 Crystallographic data
| Compound reference |
1
|
2·(thf) |
| Chemical formula |
C30H46CaO |
C38H62O3Sr |
| Formula mass |
462.75 |
654.50 |
| Crystal system |
Orthorhombic |
Trigonal |
|
a/Å |
13.5295(3) |
14.1418(5) |
|
b/Å |
13.7062(3) |
14.1418(5) |
|
c/Å |
15.0026(3) |
15.6028(5) |
|
α/° |
90.00 |
90.00 |
|
β/° |
90.00 |
90.00 |
|
γ/° |
90.00 |
120.00 |
| Unit cell volume/Å3 |
2782.07(10) |
2702.37(16) |
| Temperature/K |
100(2) |
100(2) |
| Space group |
P212121 |
P3121 |
| No. of formula units per unit cell, Z |
4 |
3 |
| Radiation type |
Mo Kα |
Mo Kα |
| Absorption coefficient, μ/mm−1 |
0.244 |
1.529 |
| No. of reflections measured |
119![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 013 013 |
67826 |
| No. of independent reflections |
5682 |
3696 |
|
R
int
|
0.0554 |
0.0657 |
| Final R1 values (I > 2σ(I)) |
0.0336 |
0.0403 |
| Final wR(F2) values (I > 2σ(I)) |
0.0811 |
0.0894 |
| Final R1 values (all data) |
0.0371 |
0.0453 |
| Final wR(F2) values (all data) |
0.0831 |
0.0919 |
| Goodness of fit on F2 |
1.050 |
1.052 |
| Flack parameter |
0.01(3) |
−0.005(10) |
| CCDC |
1415170
|
1415171
|
Table 2 Selected bond distances (Å) and angles (°)
| Compound |
1
|
2
|
|
pdlcentroid is the centroid of the pentadienyl ligand.
The coordinate THF ligand is disordered over two positions.
α is defined as the angle formed by the two dienyl planes.
Conformational angle of the pdl* with respect to each other (as defined in Chart 3).
|
| C1/C14–C2/C15 |
1.360(3); 1.375(3) |
1.369(6) |
| C2/C15–C3/C16 |
1.428(3); 1.421(3) |
1.417(6) |
| C3/C16–C4/C17 |
1.431(2); 1.439(2) |
1.440(5) |
| C4/C17–C5/C18 |
1.372(2); 1.371(3) |
1.366(5) |
| C1/C14⋯C5/C18 |
3.205; 3.235 |
3.246 |
| M–C1/C14 |
2.728(2); 2.713(2) |
2.918(4) |
| M–C2/C15 |
2.6896(18); 2.6531(18) |
2.904(4) |
| M–C3/C16 |
2.6577(19); 2.6217(18) |
2.862(4) |
| M–C4/C17 |
2.7426(16); 2.7422(17) |
2.942(3) |
| M–C5/C18 |
2.8009(18); 2.9181(19) |
2.958(3) |
| M–C (Å, range) |
2.9181(19)–2.6217(18) |
2.958(3)–2.862(4) |
| M–C (Å, average) |
2.727 ± 0.085 |
2.917 ± 0.037 |
| M–pdl*plane |
2.261; 2.265 |
2.426 |
| M–pdl*centroida (Å, average) |
2.222 |
2.485 |
| M–O (Å) |
2.3400(13) |
2.528(4), 2.604(5)b |
| pdl*centroid–M–pdl*centroida (°) |
135.6 |
133.3 |
|
α
|
25.3 |
67.9 |
|
χ
|
28.4 |
50.0 |
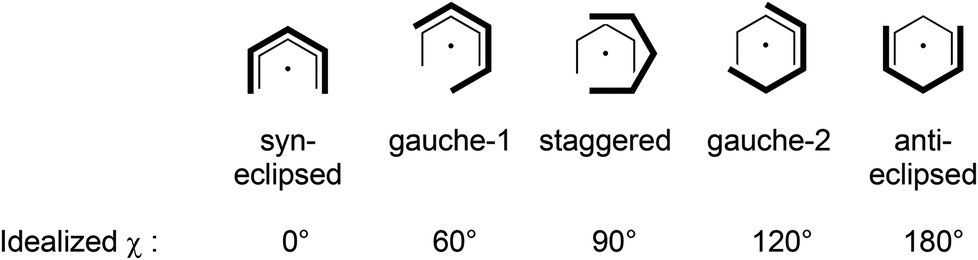
Consistent with the solution studies, the pdl* ligands in 1 and 2 also adopt an η5-U conformation in the solid state; and, as in previous studies on open metallocenes of the transition metals, the pdl* systems coordinate site-selectively with the less sterically demanding face (syn to the CH2-bridge of the bicyclic ligand framework) directed toward the metal atom. Complex 1 shows approximate (non-crystallographic) C2 symmetry, in which the C2-axis runs between the atoms Ca and O. The coordinated THF molecule resides in the usual position at the open edge of the two pdl* ligands. In contrast, 2 exhibits crystallographically imposed C2 symmetry with two coordinated THF ligands, which are located at the rear of the pdl* systems. Therefore the pentadienyls adopt different orientations with respect to each other (Chart 3). In the Ca compound 1 the arrangement of the ligands is better described by a syn-eclipsed conformation (χ = 28.4°), whereas for the Sr system 2 a gauche-1 arrangement is realized (χ = 50.0°). Compared to other enantiomerically pure open metallocenes derived from the pdl* framework or related ligands, these values are the smallest observed so far.35–38,40
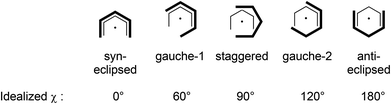 |
| | Chart 3 Conformational of two pentadienyl ligands. | |
Because of the predominantly ionic bonding in these complexes, the various arrangements are probably enforced by the minimization of inter-ligand repulsions. The C–C bond distances feature a distinct short-long-long-short pattern, whereby the shortest M–C bond distance is formed to the central carbon atom (C3-position) of the pdl* ligand, and they become progressively longer from C2/C4 to C1/C5. However, the M–C4/C5 bond distances are significantly longer than the corresponding M–C1/C2 distances, which is presumably a consequence of the more sterically demanding bicyclic framework. Furthermore, the average M–C distances are 2.727 ± 0.085 Å and 2.917 ± 0.037 Å for 1 and 2, respectively. This correlates well with the difference between the ionic radii of heptacoordinate Ca2+ (1.06 Å) and octacoordinate Sr2+ (1.26 Å).59 Another notable aspect is the comparison of pdl* to the sterically more encumbered pdl′ ligand (pdl′ = 2,4-(Me3C)2C5H5), for which only mono THF-adducts [(η5-pdl′)2M(thf)] (M = Ca and Sr) are isolated.37
Polymerization of rac-lactide
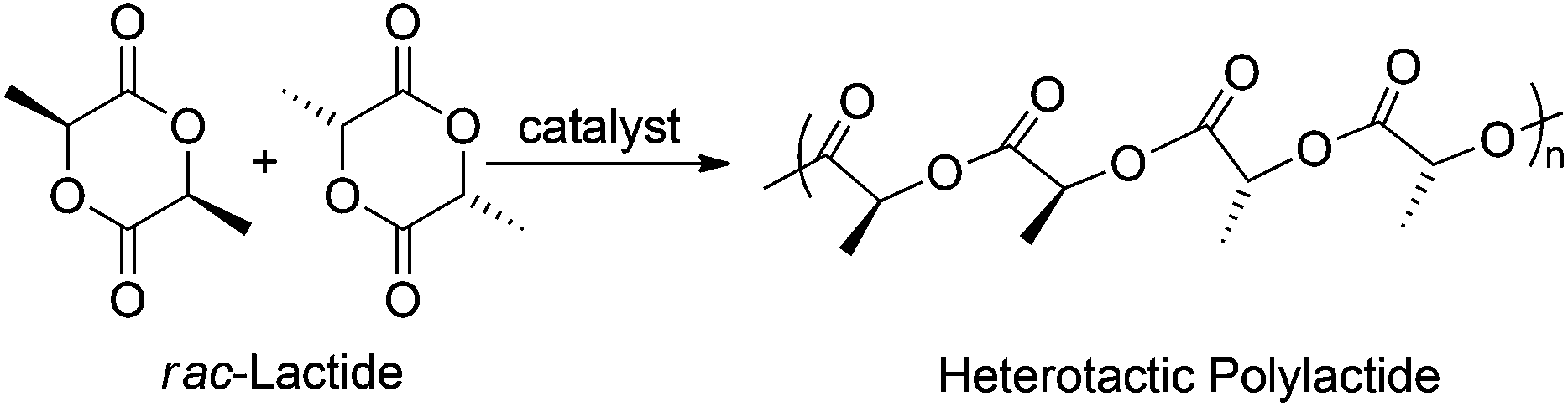
With complexes 1 and 2 in hand we set out to investigate their reactivity in the heterotactic ring opening polymerization (ROP) of rac-lactide (rac-LA) (Chart 4).
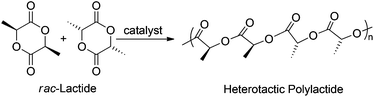 |
| | Chart 4 Heterotactic polylactide formation. | |
Complete conversion of 100 equiv. of lactide ([rac-LA] = 0.5 mol L−1) can be achieved within 12 h at 50 °C. No activators need to be added and a switch in solvent polarity (toluene vs. THF) has no influence on the reaction rate or polymer structure. This suggests that THF is not competing for monomer at the active site of the catalyst. The ROP of rac-lactide catalyzed by 1 and 2 proceeds slower than for allyl rare earth complexes such as [(η3-allyl)3M(dioxane)]x (M = Y, La, Sm) or [(nacnac)M(η3-allyl)3)] (M = La, Y, Sm, Nd; nacnac = HC(MeCNC6H3iPr2-2,6)2),60 but whereas in the latter cases atactic polymers were isolated, the open metallocenes 1 and 2 furnish heterotactic-enriched polylactides with Pr of 0.61–0.63. In addition the experimental Mn values (Mn,exp) are close to the calculated values Mn,calcd, giving rise to a narrow polydispersity (Mw/Mn = 1.29–1.31; Table 3). It should also be noted that the selectivities and activities of 1 and 2 are virtually identical, suggesting that the difference in ionic radii has no influence on the polymerization process. Group 2 complexes bearing either alkoxido, amido or BH4 groups usually initiate faster than complexes 1 and 2. Very reactive complexes in this context are [{HB(3-tBupz)3}CaN(SiMe3)2] or [{HB(3-tBupz)3}Ca(O-2,6-iPr2C6H3)] that polymerizes rac-lactide rapidly at ambient temperature with up to >90% heterolactic polylactide, but significantly broader polydispersities Mw/Mn = 1.6–1.7.61 Other examples also include [(nacnac)M(BH4)(thf)2] (M = Ca and Sr) for which only atactic polymers were obtained and the Mw/Mn increase significantly as a function of conversion approaching values of 1.7–1.8.62 However, polydispersities in the range of 1.4 to 1.8 indicate side reactions such as transesterfications.62
Table 3 Polymerization of rac-lactide catalyzed by open metallocenes 1 and 2a
| Entry |
Cat. |
Solvent |
Conv. (%) |
M
n,expb (kg mol−1) |
M
n,calcc (kg mol−1) |
M
w/Mnb |
P
rd (%) |
|
Conditions: [precat.]/LA (mol/mol) = 1/100; polymerization time, 12 h; solvent, 5 mL; [LA] = 0.5 mol L−1; temperature, 50 °C.
Measured by GPC (using polystyrene standards in THF).
Calculated by ([LA]/[precat.]) × 144 × X (X = conv.).
P
r is the probability of forming an r-dyad by insertion and is determined from the methine region of the homonuclear decoupled 1H NMR spectrum in CDCl3 at 25 °C.
|
| 1 |
1
|
Toluene |
98 |
14.2 |
14.1 |
1.29 |
0.62 |
| 2 |
2
|
Toluene |
100 |
14.6 |
14.4 |
1.31 |
0.60 |
| 3 |
1
|
THF |
97 |
14.1 |
14.0 |
1.30 |
0.63 |
| 4 |
2
|
THF |
98 |
14.3 |
14.1 |
1.29 |
0.61 |
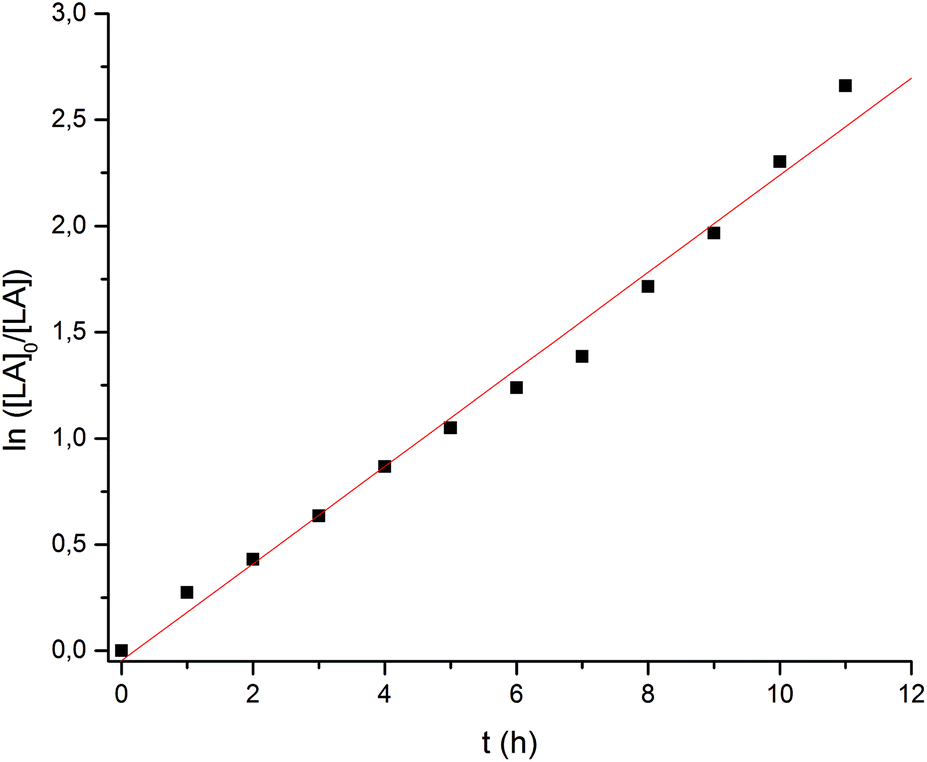
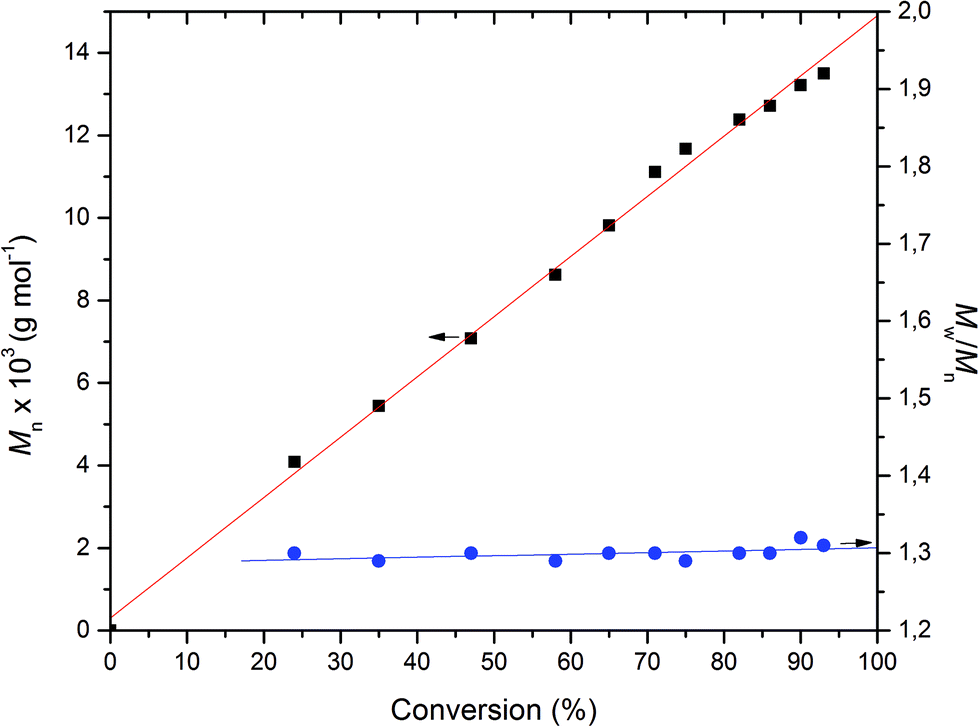
A more detailed analysis was then undertaken for the Ca catalyst 1 to show that it acts as a single-site catalyst performing a controlled polymerization of rac-lactide. First order kinetics with respect to rac-LA concentration was established (Fig. 4) and the Mn,exp values increase linearly with the monomer conversion, whereas the Mw/Mn values remain in the range of 1.28–1.31 independent of conversion suggesting that no side reactions occur (Fig. 5).
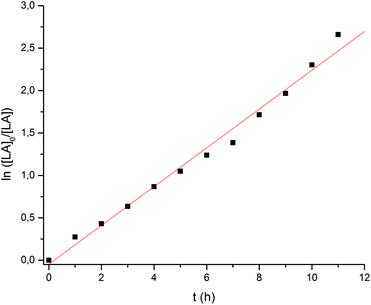 |
| | Fig. 4 Ln([LA]0/[LA]) vs. time plot for the ROP of rac-LA initiated by Ca complex. Conditions: precat./LA (mol/mol) = 1/100, [LA]0 = 0.5 mol L−1, solvent = THF, T = 50 °C. kobs = 0.189 h−1 (or 5.25 × 10−5 s−1). | |
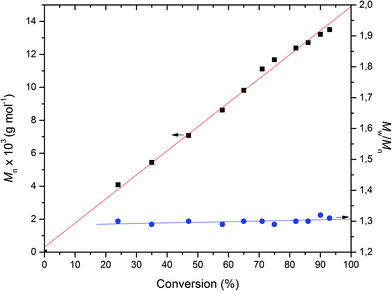 |
| | Fig. 5
M
n and Mw/Mnvs. conversion plots for the ROP of rac-LA initiated by Ca complex. Conditions: precat./LA (mol/mol) = 1/100, [LA]0 = 0.5 mol L−1, solvent = THF, T = 50 °C. | |
Conclusion
The first enenatiomerically pure open metallocenes of group 2 metals were successfully isolated and fully characterized. In the Ca and Sr complexes 1 and 2, the pentadienyl ligands adopt an η5-U conformation in solution and in the solid state. It was further established that the pdl* ligands coordinate exclusively from the sterically less demanding site of the bicycle (that is, syn to the CH2-bridge). However, in contrast to the sterically more demanding pdl′ system, the number of coordinated THF ligands varies between Ca and Sr, suggesting that pdl* is slightly less sterically demanding than the corresponding pdl′ derivative. Complexes 1 and 2 are active catalysts in the ROP of rac-lactide without the necessity to add any activators. During the polymerization process Mn,exp increases linearly with conversion, yielding heterotactic-enriched polymers with narrow polydispersities (PDI = 1.29–1.31). Further investigations regarding catalytic applications of pentadienyl complexes are ongoing and will be reported in due course.
Experimental
General considerations
Synthetic and spectroscopic manipulations were performed under an atmosphere of purified nitrogen, either in a Schlenk apparatus or in a glovebox. Solvents were dried and deoxygenated either by distillation under a nitrogen atmosphere from sodium benzophenone ketyl (THF) or by an MBraun GmbH solvent purification system (all other solvents). NMR data were recorded on a Bruker DPX 200 spectrometer at ambient temperature unless stated otherwise. The chemical shifts were referenced to the residual solvent signal (δH 7.16 for benzene, 7.26 for chloroform, 3.58 ppm for α-H of THF) for the 1H NMR spectra and the solvent signal (δC 128.06 for benzene, 67.21 ppm for α-C of THF) for the 13C{1H} NMR spectra. Elemental analyses were performed by combustion and gas chromatographic analysis with an Elementar varioMICRO instrument. Despite several attempts, the carbon values for complexes 1–2 were lower than expected because of incomplete combustion and the extreme sensitivity of these compounds to air and moisture. Nevertheless, low carbon values were uncommon for alkylcyclopentadienyl complexes of the heavy alkaline-earth metals, e.g.ref. 63 and 64 and literature cited therein, and ref. 65. Racemic lactide (rac-LA) was recrystallized twice from dry toluene and then sublimed under vacuum prior to use.
Synthesis
Preparation of [(η5-pdl*)2Ca(thf)].
A suspension of CaI2 (206 mg, 0.70 mmol) in THF (10 mL) was stirred for 10 min. To this saturated solution a solution of K(pdl*) (300 mg, 1.40 mmol) in THF (10 mL) was added. The mixture was stirred for 30 min at ambient temperature. The red to brown solution was separated from a pale yellow precipitate by filtration and the solvent was reduced under dynamic vacuum to 1 mL. Orange to yellow single crystals grew from a concentrated hexane solution at −30 °C (103 mg, 0.22 mmol, 32%). Anal. Calcd for C30H46CaO: C, 77.86; H, 10.02. Found: C, 77.20; H, 10.39. 1H NMR (400 MHz, ambient temperature, C6D6): δ = 4.29 (s, 2 H, 5), 4.12 (“s”, 2 H, 3), 3.77 (s, 2 H, 1-exo), 3.48 (s, 2 H, 1-endo), 3.41–3.34 (m, 4 H, THF), 2.62–2.53 (m, 4, H, 6b and 8b), 2.49–2.40 (m, 2 H, 6a), 2.23 (“d”, 4 H, J = 5.27 Hz, 7 and 9), 2.13 (s, 6 H, 13), 1.59 (d, 2 H, J = 9.03 Hz, 8a), 1.36 (s, 6 H, 12), 1.22–1.18 (m, 4 H, THF), 1.17 (s, 6 H, 11) ppm. 13C NMR (100 MHz, ambient temperature, C6D6): δ = 154.0 (C, 4), 149.6 (C, 2), 89.2 (CH, 5), 89.1 (CH, 3), 77.1 (CH2, 1), 69.1 (CH2, THF), 52.8 (CH, 9), 41.6 (CH, 7), 38.8 (C, 10), 33.4 (CH2, 8), 32.4 (CH2, 6), 28.8 (CH3, 13), 26.8 (CH3, 12), 25.2 (CH2, THF), 21.6 (CH3, 11) ppm.
Preparation of [(η5-pdl*)2Sr(thf)2].
To a suspension of SrI2 (239 mg, 0.70 mmol) in THF (10 mL) was added a solution of K(pdl*) (300 mg, 1.40 mmol) in THF (10 mL). The brown to red suspension was stirred for 30 min at room temperature. Afterwards the solution was separated from a pale yellow precipitate by filtration and the solvent was reduced under dynamic vacuum to 1 mL. Single crystals grew from a concentrated hexane solution at −30 °C (134 mg, 0.13 mmol, 33%). Anal. Calcd for C34H54SrO2: C, 70.12; H, 9.35. Found: C, 69.26; H, 9.58. 1H NMR (400 MHz, ambient temperature, C6D6): δ = 4.52 (“bs”, 1 H, 5), 3.98–3.83 (m, 2 H, 1-exo and 3), 3.78 (“bs”, 1 H, 1-endo), 3.52–3.45 (m, 8 H, THF), 2.88–2.64 (m, 2 H, 6), 2.53–2.43 (m, 1 H, 8b), 2.28–2.21 (m, 1 H, 7), 2.17–2.09 (m, 1 H, 9), 2.02 (s, 3 H, 13), 1.52 (brd, 1 H, J = 8.53 Hz, 8a), 1.38 (brs, 3 H, 12), 1.37–1.31 (m, 8 H, thf),1.22 (brs, 3 H, 11) ppm. 13C NMR (100 MHz, ambient temperature, C6D6): δ = 152.8 (C, 4), 147.2 (C, 2), 89.2 (CH, 5), 86.6 (CH, 3), 78.9 (CH2, 1), 68.6 (CH2, THF), 52.9 (CH, 9), 41.8 (CH, 7), 39.3 (C, 10), 32.3 (CH2, 8), 32.2 (CH2, 6), 28.6 (CH3, 13), 26.9 (CH3, 12), 25.5 (CH2, THF), 21.6 (CH3, 11) ppm.
General procedure for the polymerization of rac-lactide
In a glovebox, a rac-lactide (rac-LA) (0.360 g, 2.5 mmol), toluene or THF (4.0 mL), and complex (typically 0.025 mmol, in 1.0 mL of toluene or THF) were added sequentially into a Schlenk flask with stirring. The flask containing the reaction mixture was subsequently placed in an oil bath and stirred for 12 h at 50 °C. The polymerization was quenched by the addition of cold acidified methanol (20 mL, 10% HCl). The precipitated polylactide was collected, washed with cold methanol several times, and dried in vacuum at 50 °C overnight.
Crystallographic details
Single crystals of each compound were examined under inert oil. Data were recorded at 100(2) K on Oxford Diffraction diffractometers using monochromated MoKα radiation (Table 1). Absorption corrections were performed on the basis of multi-scans. The structures were refined anisotropically using the SHELXL-97 program.66 Hydrogen atoms at the 1-, 3- and 5-positions of the pdl* ligands were refined freely (but for 2 with distance restraints); methyl groups were refined as idealised rigid groups allowed to rotate but not tip. Other hydrogen atoms were refined using a riding model starting from calculated positions. Absolute configurations were confirmed unambiguously by the Flack parameters. Special features and exceptions: The coordinated THF in 2 was disordered and was refined on two positions. The assignment of the oxygen position for the uncoordinated THF, which is disordered over a twofold axis, was not straightforward. The U values are not convincing, but other permutations of O and C led to very short H⋯H contacts. In any case, this molecule may be more seriously disordered and its geometry should be interpreted with caution. Attempts to refine the structure in space group P31 (in the hopes that the THF molecules might be ordered) were unsuccessful; the disorder remained.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (WA 2513/2, WA 2513/6, WA 2513/7) (M.D.W.) and the National Natural Science Foundation of China (Grant No. 21472013, 21172022) (G.Z.).
Notes and references
- C. Janiak and H. Schumann, Adv. Organomet. Chem., 1991, 33, 291–393 CrossRef CAS.
- J. Okuda, Top. Curr. Chem., 1992, 160, 97–145 CrossRef CAS.
- R. L. Halterman, Chem. Rev., 1992, 92, 965–994 CrossRef CAS.
- A. Glöckner, H. Bauer, M. Maekawa, T. Bannenberg, C. G. Daniliuc, P. G. Jones, Y. Sun, H. Sitzmann, M. Tamm and M. D. Walter, Dalton Trans., 2012, 41, 6614–6624 RSC.
- H. Bauer, A. Glöckner, A. C. Tagne Kuate, S. Schäfer, Y. Sun, M. Freytag, M. Tamm, M. D. Walter and H. Sitzmann, Dalton Trans., 2014, 43, 15818–15828 RSC.
- R. D. Ernst, Struct. Bonding, 1984, 57, 1–53 CrossRef CAS.
- R. D. Ernst, Acc. Chem. Res., 1985, 18, 56–62 CrossRef CAS.
- R. D. Ernst, Chem. Rev., 1988, 88, 1255–1291 CrossRef CAS.
- R. D. Ernst, Comments Inorg. Chem., 1999, 21, 285–325 CrossRef CAS PubMed.
- L. Stahl and R. D. Ernst, Adv. Organomet. Chem., 2008, 55, 137–199 CrossRef CAS.
- R. D. Ernst and T. H. Cymbaluk, Organometallics, 1982, 1, 708–713 CrossRef CAS.
- J. Sieler, A. Simon, K. Peters, R. Taube and M. Geitner, J. Organomet. Chem., 1989, 362, 297–293 CrossRef CAS.
- H. Schumann and A. Dietrich, J. Organomet. Chem., 1991, 401, C33–C36 CrossRef CAS.
- W. Q. Weng, K. Kunze, A. M. Arif and R. D. Ernst, Organometallics, 1991, 10, 3643–3647 CrossRef CAS.
- K. Kunze, A. M. Arif and R. D. Ernst, Bull. Soc. Chim. Fr., 1993, 130, 708–711 CAS.
- M. B. Zielinski, D. K. Drummond, P. S. Iyer, J. T. Leman and W. J. Evans, Organometallics, 1995, 14, 3724–3731 CrossRef CAS.
- J. Wang, C. Zheng, G. Canseco-Melchor, D. J. Hilby, J. A. Maguire and N. S. Hosmane, Organometallics, 2007, 26, 577–585 CrossRef CAS.
- M. R. Kunze, J. Sieler and R. Taube, Z. Anorg. Allg. Chem., 2008, 634, 1045–1050 CrossRef CAS PubMed.
- M. R. Kunze, D. Steinborn, K. Merzeriler, C. Wagner, J. Sieler and R. Taube, Z. Anorg. Allg. Chem., 2007, 633, 1451–1463 CrossRef CAS PubMed.
- M. R. Kunze and R. Taube, Z. Anorg. Allg. Chem., 2010, 636, 2454–2461 CrossRef CAS PubMed.
- B. M. Day, N. F. Chilton and R. A. Layfield, Dalton Trans., 2015, 44, 7109–7113 RSC.
- T. H. Cymbaluk, J. Z. Liu and R. D. Ernst, J. Organomet. Chem., 1983, 255, 311–315 CrossRef CAS.
- D. Baudry, E. Bulot and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1988, 1369–1370 RSC.
- D. Baudry, E. Bulot and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1989, 1316–1317 RSC.
- D. Baudry, E. Bulot, P. Charpin, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1989, 371, 163–174 CrossRef CAS.
- S. A. Solomon, F. M. Bickelhaupt, R. A. Layfield, M. Nilsson, J. Poater and M. Sola, Chem. Commun., 2011, 47, 6162–6164 RSC.
- B. M. Day, J. Clayden and R. A. Layfield, Organometallics, 2013, 32, 4448–4451 CrossRef CAS.
- B. M. Day, J. J. W. McDouall, J. Clayden and R. A. Layfield, Organometallics, 2015, 34, 2348–2355 CrossRef CAS.
- J. S. Overby and T. P. Hanusa, Angew. Chem., Int. Ed. Engl., 1994, 33, 2191–2193 CrossRef PubMed.
- J. Klein and S. Glily, Tetrahedron, 1971, 27, 3477–3494 CrossRef CAS.
- H. Yasuda, Y. Ohnuma, A. Nakamura, Y. Kai, N. Yasuoka and N. Kasai, Bull. Chem. Soc. Jpn., 1980, 53, 1101–1111 CrossRef CAS.
- H. Yasuda, T. Nishi, K. Lee and A. Nakamura, Organometallics, 1983, 2, 21–27 CrossRef CAS.
- H. Yasuda and A. Nakamura, J. Organomet. Chem., 1985, 285, 15–29 CrossRef CAS.
- H. Yasuda, T. Nishi, S. Miyanaga and A. Nakamura, Organometallics, 1985, 4, 359–367 CrossRef CAS.
- A. C. Fecker, A. Glöckner, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Organometallics, 2013, 32, 874–884 CrossRef CAS.
- A. C. Fecker, B.-F. Craciun, M. Freytag, P. G. Jones and M. D. Walter, Organometallics, 2014, 33, 3792–3803 CrossRef CAS.
- M. Reiners, A. C. Fecker, M. Freytag, P. G. Jones and M. D. Walter, Dalton Trans., 2014, 43, 6614–6617 RSC.
- A. C. Fecker, B.-F. Craciun, P. Schweyen, M. Freytag, P. G. Jones and M. D. Walter, Organometallics, 2015, 34, 146–158 CrossRef CAS.
- J. W. Freeman, D. R. Wilson, R. D. Ernst, P. D. Smith, D. D. Klendworth and M. P. McDaniel, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 2063–2075 CrossRef CAS PubMed.
- M. Reiners, D. Baabe, P. Schweyen, M. Freytag, P. G. Jones and M. D. Walter, Inorg. Chim. Acta, 2014, 422, 167–180 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305–313 CrossRef CAS PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Tsuji and Y. Ikada, Polymer, 1999, 40, 6699–6708 CrossRef CAS.
- M. S. Reeve, S. P. McCarthy, M. J. Downey and R. A. Gross, Macromolecules, 1994, 27, 825–831 CrossRef CAS.
- J.-R. Sarasua, R. E. Prud'homme, M. Wisniewski, A. Le Borgne and N. Spassky, Macromolecules, 1998, 31, 3895–3905 CrossRef CAS.
- N. Nomura, J. Hasegawa and R. Ishii, Macromolecules, 2009, 42, 4907–4909 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- A. K. Sutar, T. Maharana, S. Dutta, C.-T. Chen and C.-C. Lin, Chem. Soc. Rev., 2010, 39, 1724–1746 RSC.
- C. A. Wheaton and P. G. Hayes, Comments Inorg. Chem., 2011, 32, 127–162 CrossRef CAS PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- N. Zhao, G. Hou, X. Deng, G. Zi and M. D. Walter, Dalton Trans., 2014, 43, 8261–8272 RSC.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2005, 24, 3792–3799 CrossRef.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC.
- R. A. Collins, J. Unruangsri and P. Mountford, Dalton Trans., 2013, 42, 759–769 RSC.
- M. J. Harvey and T. P. Hanusa, Organometallics, 2000, 19, 1556 CrossRef CAS.
- M. J. McCormick, S. C. Sockwell, T. P. Hanusa and J. C. Huffman, J. Am. Chem. Soc., 1992, 114, 3393 CrossRef.
- M. J. Harvey, T. P. Hanusa and V. G. Young Jr., J. Organomet. Chem., 2001, 626, 43 CrossRef CAS.
-
(a)
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structure from Diffraction Data, University of Göttingen, Göttingen, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence