
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dinuclear copper(I) complexes with N-heterocyclic thione and selone ligands: synthesis, characterization, and electrochemical studies†
Martin M.
Kimani
a,
David
Watts
a,
Leigh A.
Graham
b,
Daniel
Rabinovich
b,
Glenn P. A.
Yap
c and
Julia L.
Brumaghim
*a
aDepartment of Chemistry, Clemson University, Clemson, SC 29634-0973, USA. E-mail: brumagh@clemson.edu
bDepartment of Chemistry, The University of North Carolina at Charlotte, 9201 University City Boulevard, Charlotte, NC 28223, USA
cDepartment of Chemistry and Biochemistry, The University of Delaware, Newark, DE 19716, USA
First published on 18th August 2015
Abstract
The synthesis, characterization, and structures of a series of homoleptic and heteroleptic copper(I) complexes supported by N-heterocyclic chalcogenone ligands is reported herein. The quasi-reversible Cu(II/I) reduction potentials of these copper complexes with monodentate (dmit or dmise) and/or bidentate (BmmMe, BsemMe, BmeMe, BseeMe) chalcogenone ligands are highly dependent upon the nature and number of the donor groups and can be tuned over a 470 mV range (−369 to 102 mV). Copper–selone complexes have more negative Cu(II/I) reduction potentials relative to their thione analogs by an average of 137 mV, and increasing the number of methylene units linking the heterocyclic rings in the bidentate ligands results in more negative reduction potentials for their copper complexes. This ability to tune the copper reduction potentials over a wide range has potential applications in synthetic and industrial catalysis as well as the understanding of important biological processes such as electron transfer in blue copper proteins and respiration.
Introduction
The chemistry of monodentate and bidentate sulfur and selenium Lewis donor ligands towards soft and borderline metals has recently received much attention due to their potential applications in catalysis,1,2 the preparation of radiopharmaceuticals,3 and in supramolecular, bioinorganic, organometallic, and coordination chemistry.4,5 Thus, great strides have been made in understanding the coordination chemistry of bis(mercaptoimidazolyl)borate (BmR) and bis(mercaptoimidazolyl)methane (BmmR) ligands, first pioneered by Parkin6,7 and Williams,8 respectively. In contrast, the reactivity of the corresponding selenium analogs, the bis(selenoimidazolyl)borates (BseR),9,10 bis(selenoimidazolyl)methanes (BsemR),1,11 and related derivatives,5 remains markedly underdeveloped.We are interested in the coordination chemistry of the aforementioned bidentate neutral ligands as well as that of the closely related bis(mercaptoimidazolyl)ethanes (BmeR) and bis(selenoimidazolyl)ethanes (BseeR) with copper(I) to understand the fundamentals of the copper–sulfur and copper–selenium interactions and their effect on Cu(I)/Cu(II) redox potentials. The high propensity for sulfur- and selenium-containing ligands to bridge metal centers also results in diverse coordination frameworks12 and these groups are also potential synthons for the formation of heterocyclic carbenes via potassium metal reduction.13 There is also increased interest in copper chalcogenolates as single-source precursors in the synthesis of semiconductor materials via metal organic chemical vapor deposition.14
Although coordination complexes of the BmmMe ligand with rhenium(I),3 iron(II),15 cobalt(II),11 rhodium(I),1,16 iridium(I),17 nickel(II),11 silver(I),18,19 gold(I/III),19 zinc(II),20 tin(II),21 lead(II)22,23 and antimony(III)8 have been isolated, it is rather surprising that only one report of copper(I) derivatives has been published,19 particularly given the reported affinity of copper for sulfur- and selenium-containing ligands.24
In this work, we report the synthesis and crystal structures of a series of dinuclear, three- and four-coordinate copper(I) complexes with the aim of understanding the effect of the methylene linkers and chalcogenone donor groups on the redox potentials of the Cu(I)/Cu(II) couple. These reduction potentials are highly dependent upon S/Se ligand coordination and can be tuned in a wide potential range using a variety of monodentate and bidentate thione and selone ligands. Such redox tuning has practical applications ranging from understanding biological processes such as electron transfer in blue copper proteins and respiration,25 to industrial and synthetic applications in catalysis.2,26 Homoleptic and heteroleptic copper(I) complexes bearing monodentate (dmit or dmise) or bidentate (BmmMe, BsemMe, BmeMe, BseeMe) chalcogenone ligands (Fig. 1) have been synthesized and characterized using elemental analysis, infrared (IR) and multinuclear (1H, 13C, 19F, 77Se) NMR spectroscopies, single-crystal X-ray diffraction, electrospray ionization mass spectrometry, and cyclic voltammetry.
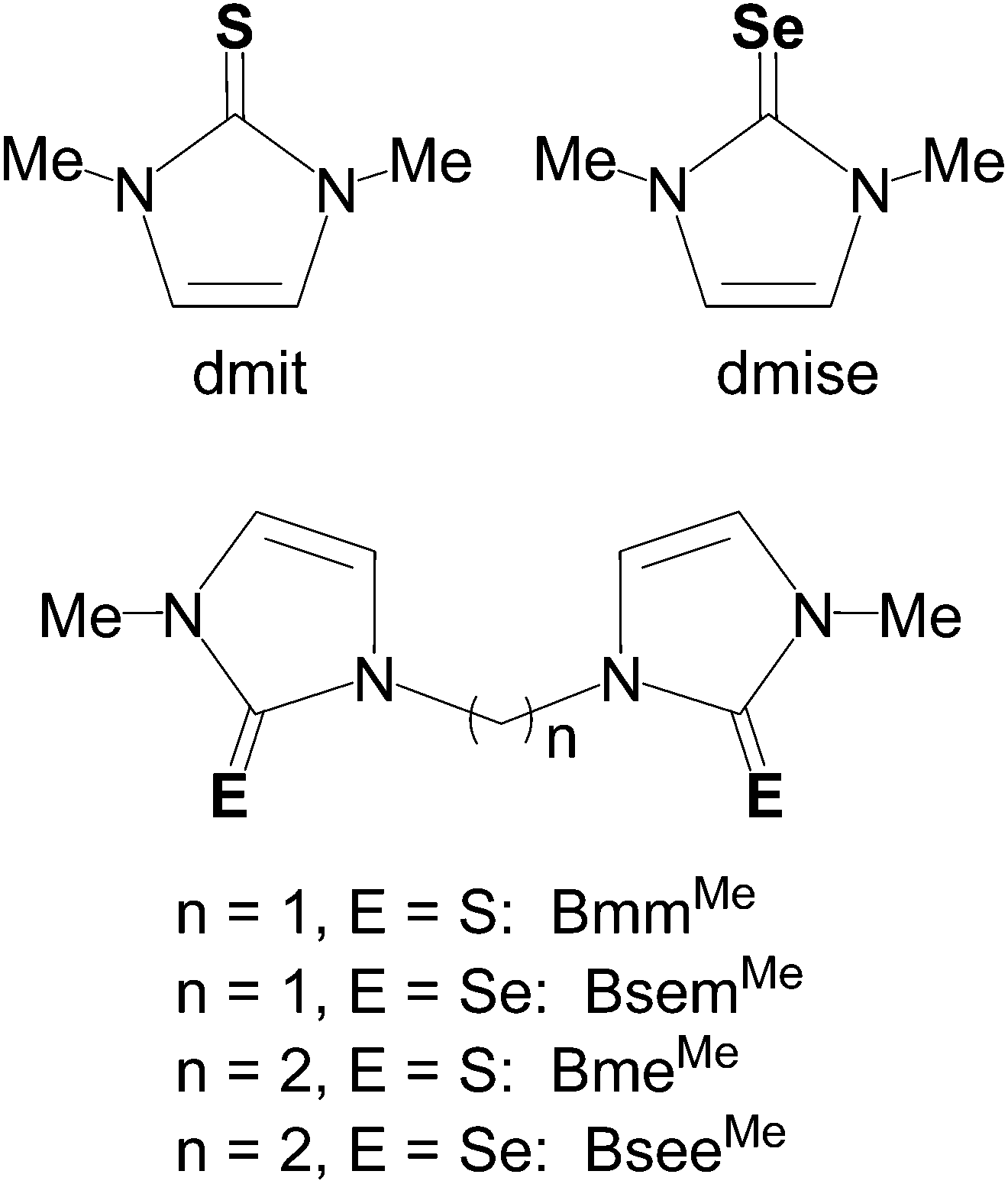 | ||
| Fig. 1 Chalcogenone ligands used in this study. | ||
Results and discussion
Synthesis of dinuclear copper(I) thione and selone complexes
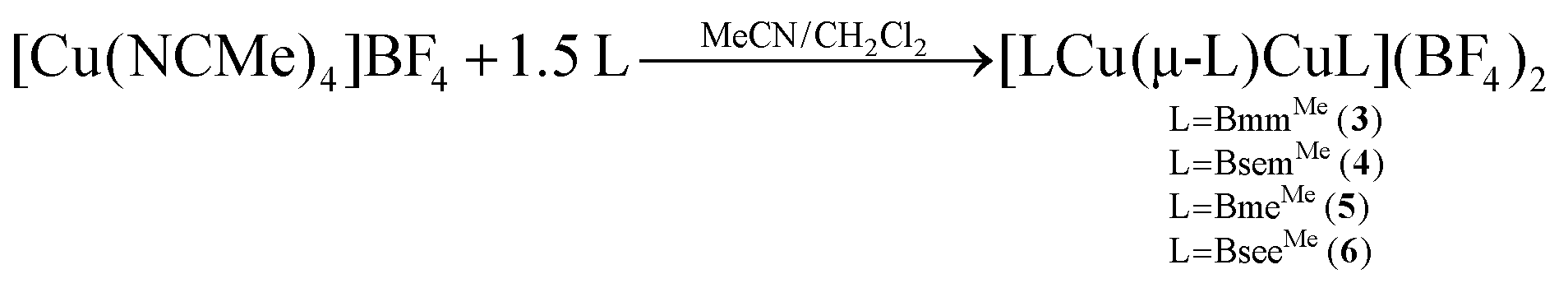
Homoleptic dinuclear copper complexes were synthesized via the reaction of [Cu(NCMe)4]BF4 with the appropriate amount of N,N′-dimethylimidazole thione (dmit) or N,N′-dimethylimidazole selone (dmise) in acetonitrile (eqn (1)) or bis(mercaptoimidazolyl)methane (BmmMe), bis(selenoimidazolyl)methane (BsemMe), bis(mercaptoimidazolyl)ethane (BmeMe), and bis(selenoimidazolyl)ethane (BseeMe) in a mixed-solvent system of acetonitrile and dichloromethane (eqn (2)). | (1) |
 | (2) |
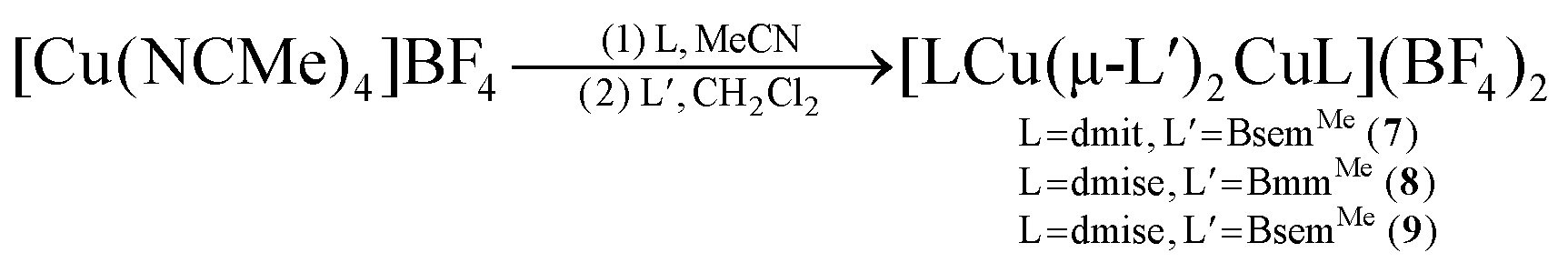 | (3) |
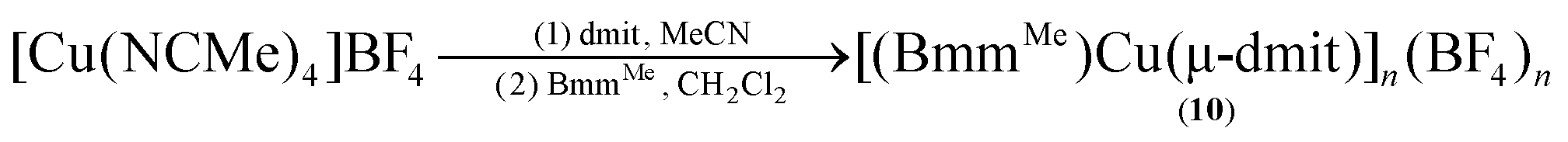 | (4) |
In turn, heteroleptic dinuclear complexes of copper(I) were synthesized via a convenient two-step, one-pot synthesis by treating equimolar amounts of [Cu(NCMe)4]BF4 and dmit or dmise in acetonitrile, followed by cannula addition of BmmMe or BsemMe in dichloromethane (eqn (3)). Similarly, treating equimolar amounts of [Cu(NCMe)4]BF4 and dmit in acetonitrile followed by addition of one molar equivalent of BmmMe in dichloromethane afforded a polynuclear copper(I) complex (eqn (4)).
Structural analyses of dinuclear copper complexes
The molecular structures of several complexes have been obtained using X-ray crystallography. More specifically, single crystals suitable for X-ray diffraction studies were obtained for [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2·CH3CN (2), [(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3), [(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4), [(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5), [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7), [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9), and [(BmmMe)Cu(μ-dmit)]n(BF4)n (10).The X-ray crystal structure of [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2·CH3CN (2), is shown in Fig. 2, and selected bond lengths (Å) and angles (°) are given in Table 1. The structural unit of [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2 is made up of two copper(I) centers, with the Se atom of the dimethylimidazole selone (dmise) ligands bridging the two copper atoms, forming a bent CuSeCu core. Each copper atom is further bonded to two dmise ligands and thus each copper adopts a distorted trigonal planar geometry. The average of the four Cu–Se distances involving terminal dmise ligands (2.35 Å) is shorter than those involving the bridging dmise ligand (2.42 Å) but is slightly longer than those in the monomeric copper selone complexes (∼2.30 Å) reported by Kimani et al.27 In a similar vein, these values are comparable to those observed in the three-coordinate copper selone complexes Cu(dmise)2X, (X = Cl, Br, I)28 and the diphosphine selenide derivative [Cu3I3{Ph2P(Se)–(CH2)3–P(Se)Ph2}2]n.29
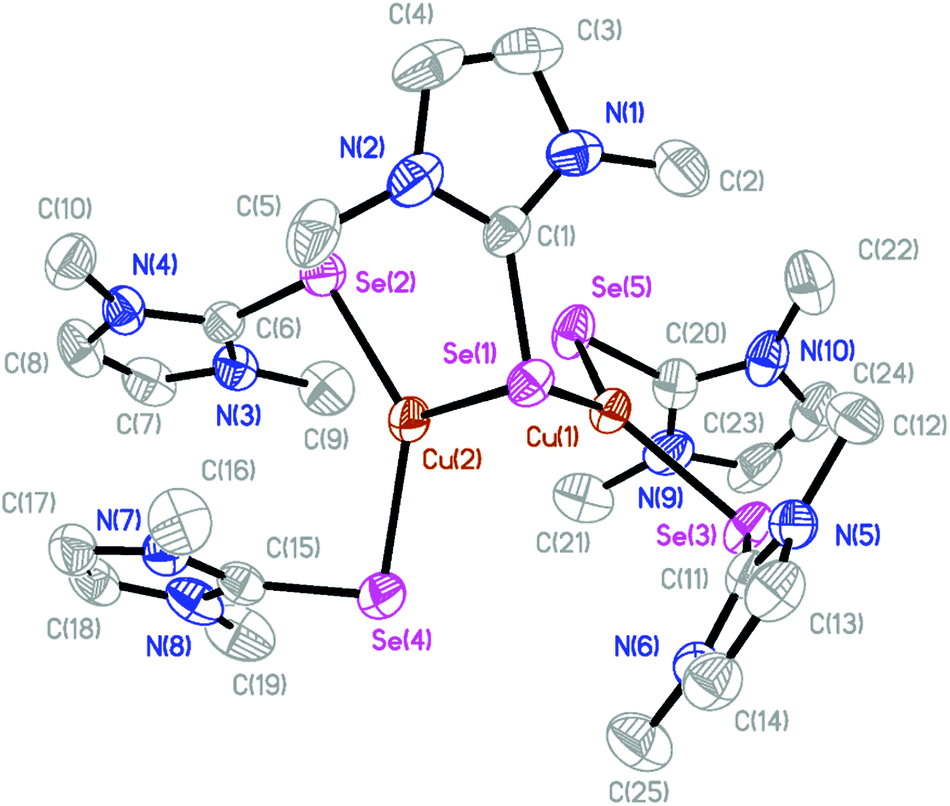 | ||
| Fig. 2 The crystal structure diagram of the cation in [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2·CH3CN (2) showing 50% probability ellipsoids. Hydrogen atoms, counterions, and the solvent molecules are omitted for clarity. | ||
| Cu(1)–Se(1) | 2.3986(9) | Se(5)–Cu(1)–Se(3) | 118.37(4) |
| Cu(2)–Se(1) | 2.4382(10) | Se(5)–Cu(1)–Se(1) | 128.11(4) |
| Cu(1)–Se(3) | 2.3460(10) | Se(3)–Cu(1)–Se(1) | 113.34(3) |
| Cu(1)–Se(5) | 2.3377(9) | Se(2)–Cu(1)–Se(4) | 133.26(4) |
| Cu(2)–Se(2) | 2.3458(11) | Se(2)–Cu(1)–Se(1) | 111.91(4) |
| Cu(2)–Se(4) | 2.3592(12) | Se(4)–Cu(1)–Se(1) | 112.68(4) |
| Cu(1)–Cu(2) | 2.6326(11) |
The molecular structures of the isostructural complexes [(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3) and [(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4) are shown in Fig. 3 and 4, with selected bond lengths and angles given in Tables 2 and 3, respectively. The dinuclear complexes feature two terminal and one bridging bis(chalcogenone) ligands, forming “butterfly” shape [Cu2E2] cores (E = S, Se). Each copper(I) ion adopts a distorted tetrahedral geometry, with angles ranging from 96.45 to 123.86° for 3 and from 100.50 to 123.36° for 4. The Cu⋯Cu distances (2.96 and 2.97 Å for 3 and 4, respectively), significantly longer than twice the covalent radius of copper(I) (2.34 Å), precludes the existence of a copper–copper bonding interaction in these complexes. As expected, the terminal Cu–S and Cu–Se bond distances in 3 and 4 (averages 2.29 and 2.42 Å, respectively) and shorter than those involving the corresponding values involving bridging ligands (averages 2.44 and 2.52 Å, respectively).
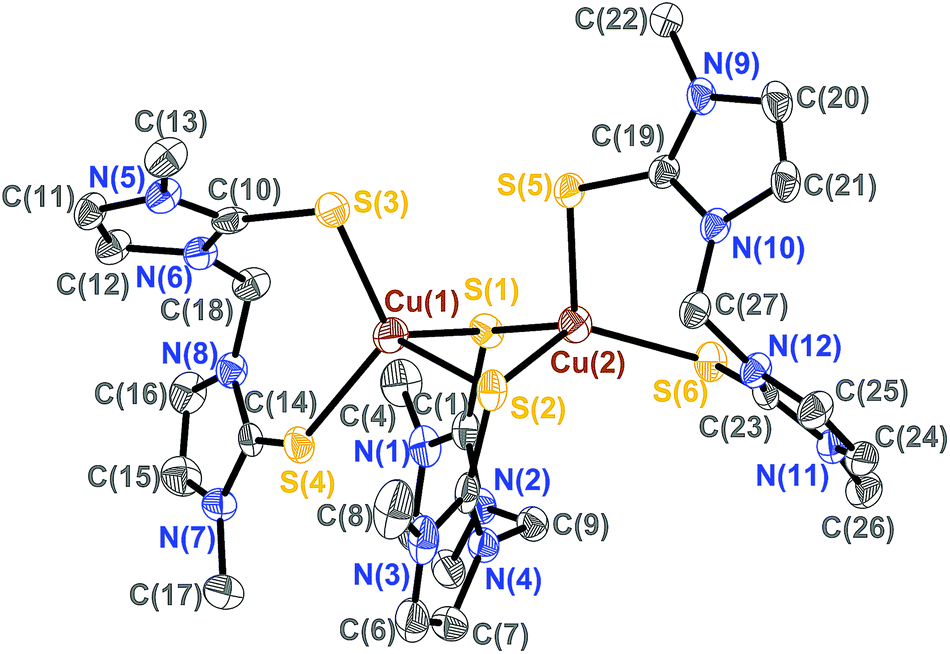 | ||
| Fig. 3 The crystal structure diagram of the cation in [(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3) showing 50% probability ellipsoids. Hydrogen atoms and counterions are omitted for clarity. | ||
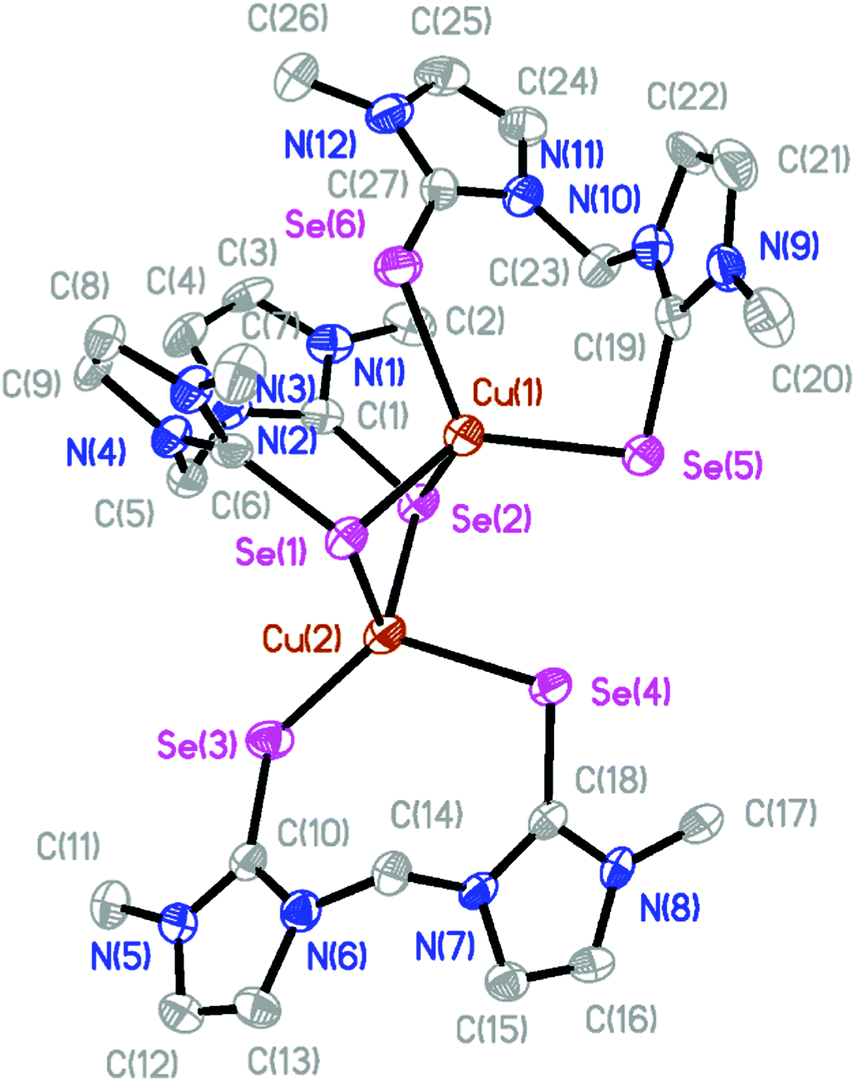 | ||
| Fig. 4 The crystal structure diagram of the cation in [(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4) showing 50% probability ellipsoids. Hydrogen atoms, counterions, and solvent molecules are omitted for clarity. | ||
| Cu(1)–S(1) | 2.6675(17) | S(1)–Cu(1)–S(2) | 96.45(4) |
| Cu(1)–S(2) | 2.3338(15) | S(1)–Cu(1)–S(3) | 113.78(6) |
| Cu(1)–S(3) | 2.2710(16) | S(1)–Cu(1)–S(4) | 105.57(5) |
| Cu(1)–S(4) | 2.3067(15) | S(2)–Cu(1)–S(3) | 118.32(5) |
| Cu(2)–S(1) | 2.3006(15) | S(2)–Cu(1)–S(4) | 103.97(6) |
| Cu(2)–S(2) | 2.4706(16) | S(3)–Cu(1)–S(4) | 116.26(5) |
| Cu(2)–S(5) | 2.2964(16) | S(1)–Cu(2)–S(2) | 103.03(5) |
| Cu(2)–S(6) | 2.3033(14) | S(1)–Cu(2)–S(5) | 107.51(5) |
| Cu(1)–Cu(2) | 2.9741(13) | S(1)–Cu(2)–S(6) | 112.87(5) |
| S(2)–Cu(2)–S(5) | 96.51(5) | ||
| S(2)–Cu(2)–S(6) | 123.86(5) | ||
| S(5)–Cu(2)–S(6) | 111.07(5) |
| Cu(1)–Se(1) | 2.5128(12) | Se(1)–Cu(1)–Se(2) | 100.50(4) |
| Cu(1)–Se(2) | 2.5617(13) | Se(1)–Cu(1)–Se(5) | 115.76(4) |
| Cu(1)–Se(5) | 2.4221(11) | Se(1)–Cu(1)–Se(6) | 100.21(4) |
| Cu(1)–Se(6) | 2.4315(12) | Se(2)–Cu(1)–Se(5) | 110.93(4) |
| Cu(2)–Se(1) | 2.5073(12) | Se(2)–Cu(1)–Se(6) | 113.12(5) |
| Cu(2)–Se(2) | 2.4981(12) | Se(5)–Cu(1)–Se(6) | 115.10(5) |
| Cu(2)–Se(3) | 2.4091(15) | Se(1)–Cu(2)–Se(2) | 102.43(4) |
| Cu(2)–Se(4) | 2.4267(11) | Se(1)–Cu(2)–Se(3) | 122.52(4) |
| Cu(1)–Cu(2) | 2.9616(18) | Se(1)–Cu(2)–Se(4) | 95.13(4) |
| Se(2)–Cu(2)–Se(3) | 107.50(4) | ||
| Se(2)–Cu(2)–Se(4) | 102.71(4) | ||
| Se(3)–Cu(2)–Se(4) | 123.36(4) |
The centrosymmetric copper complex [(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5) (Fig. 5) exhibits two copper(I) centers, each arranged in a distorted trigonal planar geometry arising from the coordination of a terminal bidentate BmeMe ligand and one of the thione moieties from a bridging bis(monodentate) BmeMe ligand. As summarized in Table 4, the sum of angles around each copper center is 354.91° and the average C–S bond distance is 2.29 Å.
 | ||
| Fig. 5 Crystal structure diagram of the cation in [(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5) displaying 50% probability density ellipsoids. Hydrogen atoms and counterions are omitted for clarity. | ||
| Cu(1)–S(4) | 2.2871(16) |
| Cu(1)–S(3) | 2.3030(16) |
| Cu(1)–S(2) | 2.2900(14) |
| S(4)–Cu(1)–S(2) | 122.49(5) |
| S(4)–Cu(1)–S(3) | 114.70(6) |
| S(3)–Cu(1)–S(2) | 117.72(5) |
The molecular structures of [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7) and [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9) are shown in Fig. 6, with selected bond length and angles for the isostructural complexes given in Table 5. The two dinuclear complexes are centrosymmetric and exhibit rhombic Cu2Se2 cores, with all the bis(selone) ligands exhibiting the unusual bridging monodentate:bidentate (μ–κ1:κ2) coordination mode. Each copper center is coordinated to a terminal dmit or dmise ligand and three selone moieties from BsemMe ligands (one terminal and two bridging), with an overall distorted tetrahedral geometry in each case. The angles surrounding the copper centers in the two complexes are very similar, ranging from 95.38 to 118.61° for 7 and from 94.97 to 118.58° for 9. The Cu⋯Cu distances (2.73 and 2.74 Å for 7 and 9, respectively) are slightly shorter than the sum of the van der Waals radii of copper, suggesting the presence of weak Cu–Cu interactions. The average lengths of the bridging Cu–Se bonds derived from BsemMe ligands (2.52 and 2.51 Å for 7 and 9, respectively) are longer than the average terminal Cu–Se bond lengths associated with the same ligands (2.42 Å for both complexes).
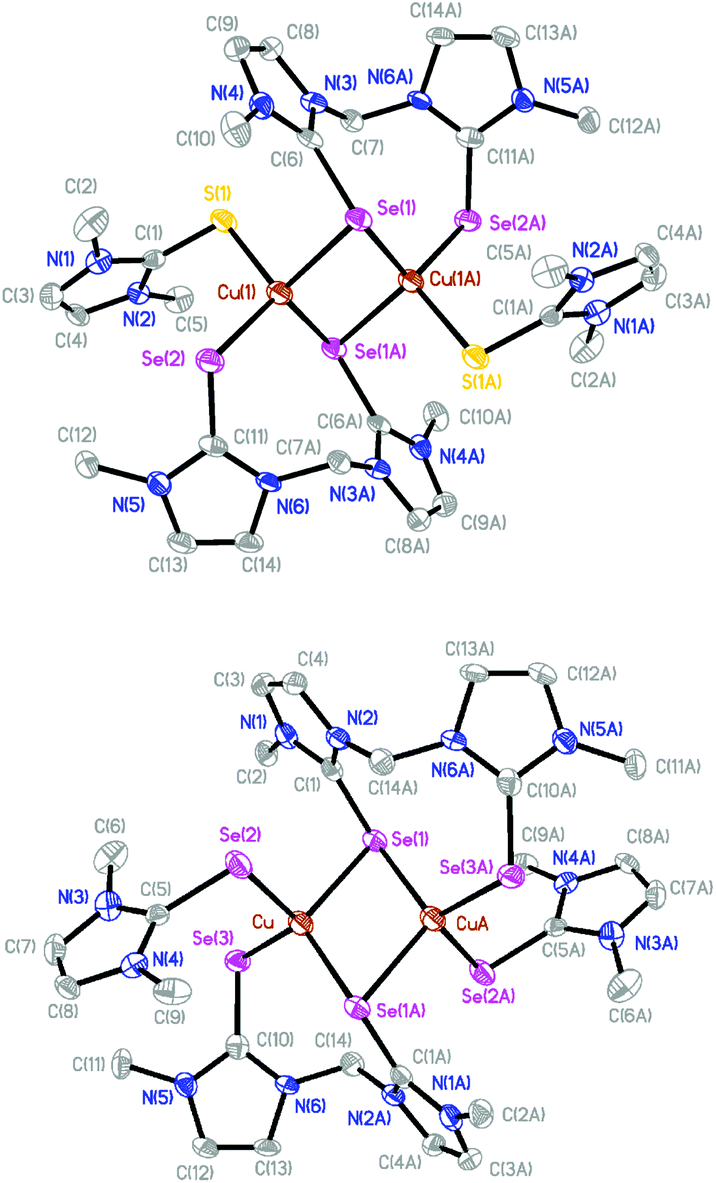 | ||
| Fig. 6 Crystal structure diagrams of the cations in [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7, top) and [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9, bottom) showing 50% probability ellipsoids. Hydrogen atoms and counterions are omitted for clarity. | ||
| 7 | 9 | ||
|---|---|---|---|
| Cu(1)–S(1) | 2.3455(16) | Cu–Se(1) | 2.5349(12) |
| Cu(1)–Se(2) | 2.4222(12) | Cu–Se(A1) | 2.4950(13) |
| Cu(1)–Se(1A) | 2.5013(11) | Cu–Se(2) | 2.4583(13) |
| Cu(1)–Se(1) | 2.5328(11) | Cu–Se(3) | 2.4238(14) |
| Se(1)–Cu(1A) | 2.5013(11) | Cu(A)–Se(1) | 2.4950(13) |
| Cu(1)–Cu(1A) | 2.7297(19) | Cu–Cu(A) | 2.739(2) |
| S(1)–Cu(1)–Se(2) | 116.36(6) | Se(1)–Cu–Se(2) | 105.02(5) |
| S(1)–Cu(1)–Se(1A) | 95.38(5) | Se(1)–Cu–Se(3) | 107.35(5) |
| Se(2)–Cu(1)–Se(1A) | 118.61(4) | Se(1)–Cu–Se(A1) | 114.02(5) |
| S(1)–Cu(1)–Se(1) | 105.58(5) | Se(2)–Cu–Se(3) | 115.95(5) |
| Se(2)–Cu(1)–Se(1) | 105.96(4) | Se(2)–Cu–Se(A1) | 94.97(5) |
| Se(1A)–Cu(1)–Se(1) | 114.33(4) | Se(3)–Cu–Se(A1) | 118.58(4) |
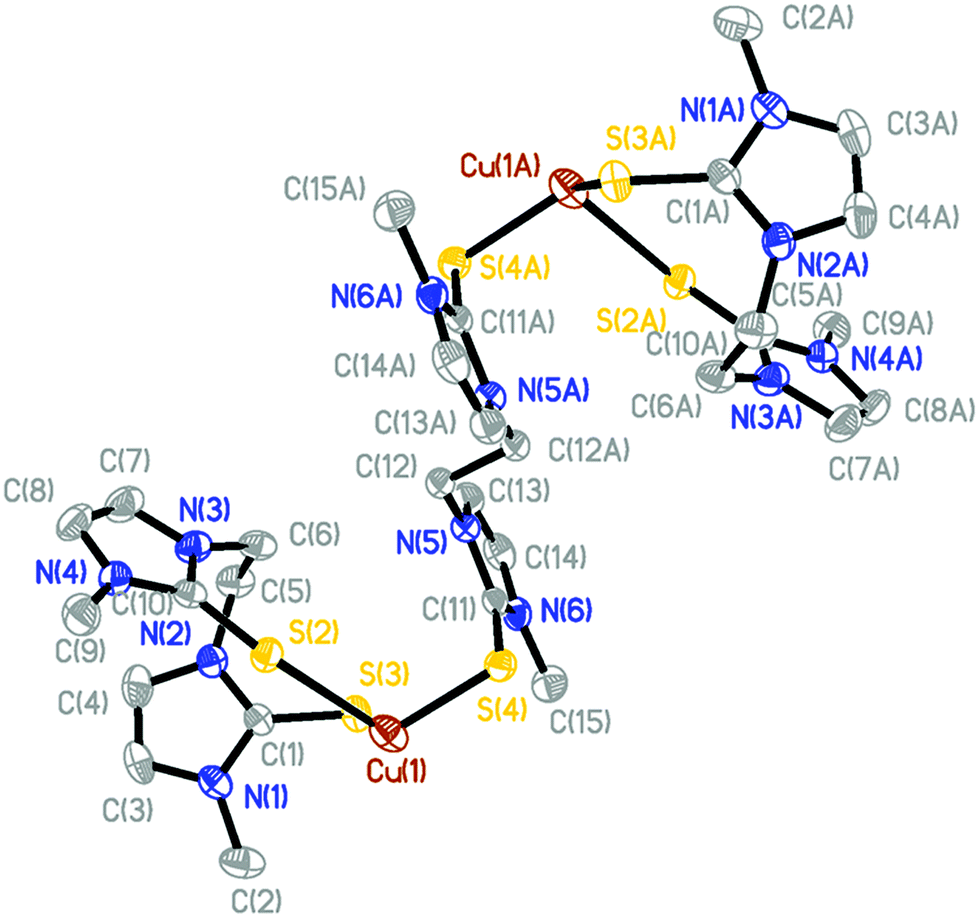
The X-ray structure of [(BmmMe)Cu(μ-dmit)]n(BF4)n (10), unlike all the ones described above, reveals the formation of a coordination polymer in which an infinite chain of four-coordinate copper(I) centers are bound to two terminal sulfur atoms from a bidentate BmmMe ligand and two sulfur atoms from bridging dmit ligands (Fig. 7 and ESI, Fig. S1†). The geometry around Cu(1) is best described as distorted tetrahedral, with S–Cu–S angles ranging from 95.06° to 123.18°, and average Cu–S bond lengths of 2.36 Å (Table 6).
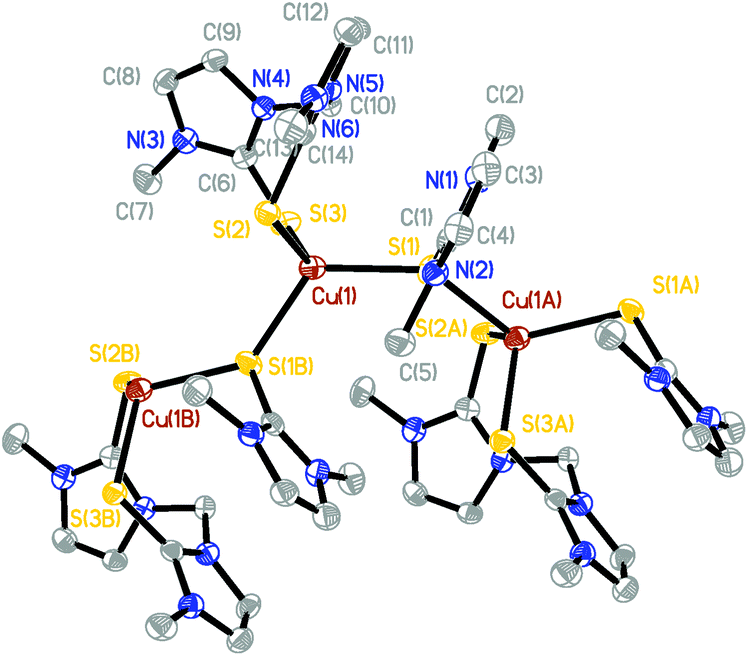 | ||
| Fig. 7 Crystal structure diagram of the cationic portion of [(BmmMe)Cu(μ-dmit)]n(BF4)n (10) showing three repeating units of the coordination polymer and 50% probability ellipsoids. Hydrogen atoms and counterions are omitted for clarity. | ||
| Cu(1)–S(1) | 2.3689(10) | S(1)–Cu(1)–S(2) | 110.51(5) |
| Cu(1)–S(2) | 2.3748(10) | S(1A)–Cu(1)–S(2) | 95.06(4) |
| Cu(1)–S(3) | 2.3347(10) | S(1A)–Cu(1)–S(1) | 123.18(2) |
| Cu(1)–S(1A) | 2.3520(10) | S(3)–Cu(1)–S(1) | 105.05(4) |
| Cu(1A)–S(1) | 2.3520(10) | S(3)–Cu(1)–S(1A) | 105.05(4) |
| S(1)–C(1) | 1.718(3) | S(3)–Cu(1)–S(2) | 117.58(3) |
| S(2)–C(6) | 1.698(3) | C(1)–S(1)–Cu(1) | 104.53(11) |
| S(3)–C(14) | 1.694(3) | C(6)–S(2)–Cu(1) | 99.25(11) |
Although the number of reported N-heterocyclic thione and selone complexes of copper(I) is limited, further comparison of the metrical parameters observed in the structures described above can be made. The tetrahedrally coordinated dinuclear copper selone complexes 4, 7, and 9 have average terminal Cu–Se bond lengths of 2.43 Å, longer than the average terminal Cu–Se bond distances of 2.30 Å for [(TpmR)Cu(dmise)][BF4] (R = H, Me, iPr), 2.33 Å for Tp*Cu(dmise),27 and an average of 2.41 Å for [Cu(C11H14Se2)2][BF4],30 but shorter than the 2.49 Å in [Cu(1,10-phen)2(C5H10N2Se)][2ClO4].31 The Se–C bonds in 2, in the range of 1.85–1.88 Å, are slightly lengthened relative to those in uncoordinated dmise (1.89 Å).32
In a similar vein, the copper thione complexes 3, 5, 7 and 10 have an average terminal Cu–S bond distance of 2.34 Å, longer than the corresponding terminal bond distances observed in most previously reported copper thione and thiolate complexes, including [(TpmR)Cu(dmit)]BF4 (2.20 Å; R = H, Me), Tp*Cu(dmit),27 [Cu(diditme)2Cl] (2.23 Å),33 Cu3(BmMe)3 (∼2.28 Å), (BmMe)Cu(PPh3) (2.28 Å),34 but somewhat shorter than those in [Cu(PPh3)2(bzimH2)Cl] (2.38 Å),35 [CuCl(1κS-imzSH)(PPh3)2] (2.36 Å),36 and significantly shorter than in [Cu(HB(3,5-iPrPz)3(SMeIm)] (2.45 Å).37 The S–C bond lengths in complexes 6, 8, and 10 (in the range 1.694–1.704 Å), are slightly lengthened relative to those in uncoordinated dmit (1.68 Å),38 and 1-methyl-4-imidazoline-2-thione (1.68 Å).39
NMR spectroscopy of dinuclear copper thione and selone complexes
The dinuclear copper complexes were characterized by 1H, 13C{1H}, 77Se{1H}, and 19F{1H} NMR spectroscopy. In the 1H NMR spectra of dmit, dmise, BmmMe, BsemMe, BmeMe, and BseeMe the olefinic CH protons on the heterocyclic ring are shifted downfield by δ 0.2 to 0.5 from its position in the free ligand upon copper coordination. This same downfield shift was observed by Rabinovich et al. for [Pb2(BmmMe)5](ClO4)4,40 Gardinier et al. for [Ag(mbit)2]+ complexes,41 and Kimani et al. for [(TpmR)Cu(L)]+ derivatives (R = H, Me, iPr; L = dmit, dmise).27 The 13C{1H} NMR resonances for the complexed and uncomplexed thione and selone and thione ligands are given in Table 7. Substantial shifting of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S/CSe resonances of the dmit, dmise, BmmMe, BsemMe carbon atoms are observed upon copper complexation relative to the free ligands. Coordination of the thiones and selones via the sulfur and selenium atoms results in upfield shifts of δ 5–8 ppm for both the CS and CSe carbons, in agreement with previous reports.34,42
S/CSe resonances of the dmit, dmise, BmmMe, BsemMe carbon atoms are observed upon copper complexation relative to the free ligands. Coordination of the thiones and selones via the sulfur and selenium atoms results in upfield shifts of δ 5–8 ppm for both the CS and CSe carbons, in agreement with previous reports.34,42
| Ligand or complex |
CS (dmise) |
CSe |
77Se |
|---|---|---|---|
| t = terminal, b = bridging. | |||
| dmit | 162.4t | ||
| dmise | 155.6t | −6 | |
| BmmMe | 163.7b | ||
| BsemMe | 157.0b | 16 | |
| BmeMe | 162.3b | ||
| BseeMe | 155.6b | 22 | |
| [Cu2(dmit)5](BF4)2 (1) | 157.3t | ||
| [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2 (2) | 147.2 | — | |
| [(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3) | 158.0b | ||
| [(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4) | 149.7b | −28 | |
| [(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5) | 155.2 | ||
| [Cu2(BseeMe)3](BF4)2 (6) | 148.0b | −43 | |
| [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7) | 157.7t | 151.6b | −24 |
| [(dmise)Cu(μ-BmmMe)2Cu(dmise)](BF4)2 (8) | 158.8b | 149.3t | |
| [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9) | 149.0t, 151.3b | −26 | |
| [(BmmMe)Cu(μ-dmit)]n(BF4)n (10) | 156.6t, 158.4b | ||
77Se{1H} NMR spectroscopy studies revealed upfield shifts for the selenium resonances in the copper complexes relative to those of unbound BsemMe and BseeMe. The 77Se{1H} NMR signal for complex 2 could not be obtained, whereas all the complexes with BsemMe and BseeMe ligands exhibited upfield selenium resonance shifts of ∼40 ppm upon coordination to copper. This upfield shift of the 77Se{1H} NMR resonance upon copper binding is direct evidence that the BsemMe and BseeMe ligands bind to copper in a bidentate fashion via the selenium atoms.
Electrochemical studies of the dinuclear copper complexes
Cyclic voltammetry studies of the chalcogenones and their dinuclear copper complexes were conducted to determine the influence of the methylene linkers on the redox potential of the chalcogenone ligands and the change in Cu(II/I) reduction potential upon coordination of the chalcogenone ligands to copper. All the uncoordinated chalcogenone ligands exhibit chemically reversible and quasi-reversible electrochemical behavior, with the selone ligands having more negative reduction potentials relative to the analogous thione ligands (Fig. 8 and Table 8). The unbound bidentate ethylene-bridged ligands (BmeMe and BseeMe) have larger peak separations between the oxidized and reduced products relative to the methylene-bridged ligands (BmmMe and BsemMe), suggesting faster electron transfer in the latter.43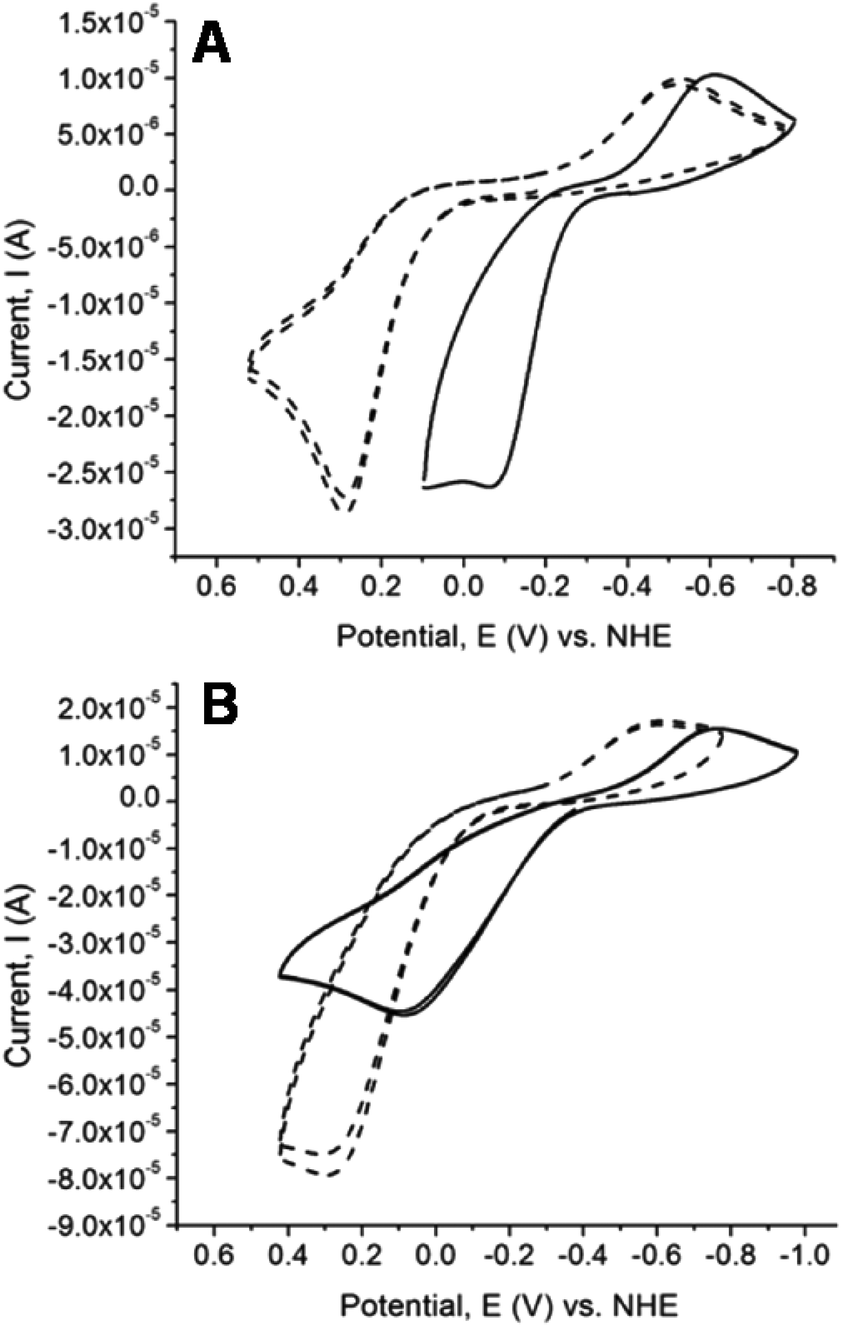 | ||
| Fig. 8 Cyclic voltammetry (CV) scan for (A) BmmMe (dashed lines) and BsemMe (solid lines), (B) BmeMe (dashed lines) and BseeMe (solid lines). All data were collected with 1 mM complex in acetonitrile. | ||
| Ligand | E pa | E pc | ΔE | E 1/2 |
|---|---|---|---|---|
| dmit | 424 | −761 | 1158 | −167 |
| dmise | 39 | −773 | 812 | −367 |
| BmmMe | 289 | −525 | 814 | −118 |
| BsemMe | −53 | −613 | 560 | −333 |
| BmeMe | 292 | −587 | 879 | −148 |
| BseeMe | 83 | −768 | 851 | −342 |
| Cu(II/I) | Cu(I/0) | |||||||
|---|---|---|---|---|---|---|---|---|
| Complex | E pa | E pc | ΔE | E 1/2 | E pa | E pc | ΔE | E 1/2 |
| [Cu2(dmit)5](BF4)2 (1) | 147 | −565 | 712 | −210 | −747 | −1129 | 382 | −938 |
| [Cu2(dmise)5](BF4)2 (2) | −101 | −603 | 502 | −352 | −724 | −1107 | 383 | −920 |
| [Cu2(BmmMe)3](BF4)2 (3) | 120 | −500 | 620 | −180 | −742 | −1298 | 556 | −1020 |
| [Cu2(BsemMe)3](BF4)2 (4) | −37 | −575 | 538 | −306 | −796 | −1336 | 540 | −1066 |
| [Cu2(BmeMe)3](BF4)2 (5) | 228 | −634 | 862 | −203 | −816 | −1299 | 483 | −1058 |
| [Cu2(BseeMe)3](BF4)2 (6) | −131 | −606 | 475 | −369 | −936 | −1152 | 216 | −1044 |
| [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7) | 192, −6 | −44, −478 | 225, 439 | 74, −242 | −710 | −1119 | 409 | −915 |
| [(dmise)Cu(μ-BmmMe)2Cu(dmise)](BF4)2 (8) | 174, −23 | 31, −608 | 149, 585 | 102, −315 | −671 | −1107 | 436 | −889 |
| [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9) | −68 | −645 | 577 | −356 | −774 | −1231 | 457 | −1003 |
| [(BmmMe)Cu(μ-dmit)]n(BF4)n (10) | 147 | −535 | 682 | −195 | −791 | −1222 | 431 | −1007 |
The reduction potentials of the unbound selone ligands are: dmise −367 mV < BseeMe (−342 mV) < BsemMe (−333 mV). The analogous thione ligands follow the same trend: dmit (−169 mV) < BmeMe (−148 mV) < BmmMe (−118 mV), versus normal hydrogen electrode (NHE; Table 8). The reduction potentials of the free bidentate chalcogenones indicate that increasing the length of the linker from methylene to ethylene results in more negative reduction potentials.
The Cu(II/I)and Cu(I/0) redox potentials of the complexes versus NHE are given in Table 8. The cyclic voltamograms (CV) of the copper complexes 1, 2, 3, 4, 5, 6, 9, and 10 exhibit two, one-electron redox potential waves belonging to the Cu(II/I)and Cu(I/0) couples, with the exception of complexes 7 and 8 which exhibit three, one-electron redox potential waves. The Cu(I/0) redox couple commences at potentials more than −1000 mV vs. NHE and after switching the scan direction at potentials close to 750 mV, Cu(0) is stripped off the electrode (Fig. 9). All the dinuclear copper thione and selone complexes exhibit one-electron Cu(II/I) oxidation and reduction waves with large ΔE values, indicating that these redox processes are not fully reversible (ESI, Fig. S2†).
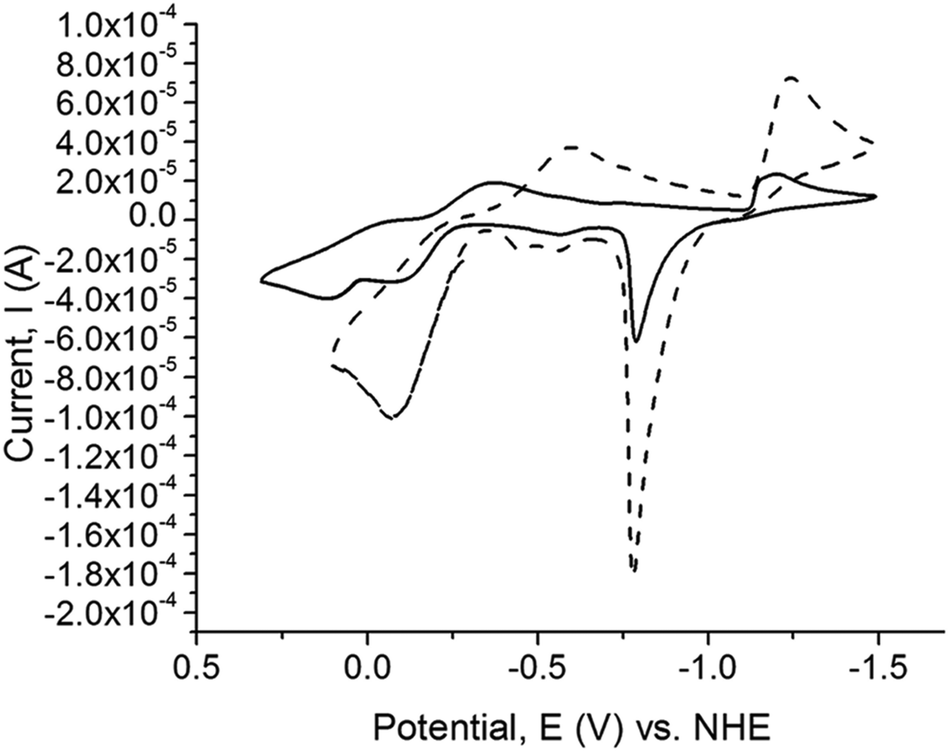 | ||
| Fig. 9 Cyclic voltammetry (CV) scans for [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7) (solid line) and [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9) (dashed line). All data were collected with 1 mM compound in acetonitrile with n-butylammonium phosphate as the supporting electrolyte (0.1 M) at a scan rate of 100 mV s−1. | ||
Upon examination of the reduction potentials for the copper complexes 1, 2, 3, 4, 5, and 6, it is clear that the selone-containing complexes exhibit more negative Cu(II/I) reduction potentials relative to the analogous thione complexes regardless of whether the thione and selone ligands are bridging. A similar trend was reported by Kimani et al. for the electrochemistry of only monodentate [TpmRCu(X)]+ complexes (X = dmise or dmit).27
Interestingly, increasing the length of the linker in the bidentate ligands from methylene to ethylene results in lower Cu(II/I) reduction potentials for [Cu2(BseeMe)3](BF4)2 (6) (−369 mV) compared to [Cu2(BsemMe)3](BF4)2 (4) (−306 mV), and the same trend is observed for the thione complex [Cu2(BmeMe)3](BF4)2 (5) (−203 mV) relative to [Cu2(BmmMe)3](BF4)2 (3) (−180 mV). The dinuclear copper complex 9 with both BsemMe and dmise ligands has a lower reduction potential of (−356 mV) relative to complex 10 which has both BmmMe and dmit ligands (−195 mV; Table 8).
The heterogeneous dinuclear complex [(dmit)2Cu2(BsemMe)2](BF4)2 (7) (ESI, Fig. S2I†) exhibits two different reduction and oxidation potentials for the Cu(II/I) couple, whereas [(dmise)2Cu2(BmmMe)2](BF4)2 (8) (ESI, Fig. S2H†) exhibits three oxidation and reduction waves. One reduction and oxidation wave in the dinuclear copper complex 8 likely corresponds to the reduction potential of the bidentate BmmMe ligand (E1/2 = −51 mV), whereas the remaining two waves correspond to Cu(II/I) reduction potentials, similar to those observed for complex 7. These two different Cu(II/I) reduction potentials are only observed for the dinuclear copper complexes with mixed thione and selone ligands, and effect which has not been previously reported for copper complexes (Table 8).
The unbound dmit and dmise ligands have more negative reduction potentials than the bidentate chalcogenones (BmmMe, BsemMe, BmeMe and BseeMe). The reduction potentials from the bidentate chalcogenones indicate that increasing the length of the linker from methylene to ethylene results in more negative reduction potentials. All the synthesized copper–selone complexes have more negative Cu(II/I) reduction potentials relative to the analogous copper–thione complexes. The copper–selone complexes stabilize the Cu(II) oxidation state more effectively than the copper–thione complexes by an average of 144 mV, consistent with previously observed results.27,28
Notably, the Cu(II/I) reduction potential of the dinuclear copper chalcogenone complexes 1 to 10 can be tuned in a 470 mV window from 102 mV to −369 mV by simply changing the nature of the chalcogen donor and the denticity of thione and selone ligands. This ability to tune the copper redox potentials could have potential applications in copper-based catalysis. Compared to naturally occurring cupredoxins with a Cu(II/I) reduction potential range of 90 to 670 mV,44 the synthesized copper chalcogenone complexes have significantly more negative Cu(II/I) reduction potentials.
Conclusions
Dinuclear homoleptic and heteroleptic copper(I) complexes with monodentate and bidentate chalcogenone ligands have been synthesized and characterized, and the electrochemistry of the resulting species has been investigated and compared. Treating the copper(I) starting material [Cu(NCMe)4]BF4 with bidentate (BmmMe, BsemMe, BmeMe, BseeMe) and monodentate chalcogenone ligands (dmit and dmise) results in the formation of dinuclear copper complexes (1, 2, 3, 4, 5, and 6). The dinuclear copper complexes adopt either trigonal or tetrahedral geometries with both terminal and bridging thione or selone ligands. The heteroleptic dinuclear copper complexes [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7) and [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9) adopt distorted tetrahedral geometry where each copper is coordinated to three selenium atoms from BsemMe ligands and one sulfur atom from dmit for 7 and one selenium atom from dmise for 9. Interestingly, the mixed ligand complex 10 consists of infinite chains of tetrahedrally coordinated Cu(I) ions bound to two sulfur atoms from a BmmMe ligand and a bridging sulfur atom from a dmit ligand.The copper selone complexes 2, 4, 6, and 9 have more negative Cu(II/I) reduction potentials relative to their sulfur analogs (1, 3, 5, and 10), and increasing the length of the methylene linker in the bidentate chalcogenone ligands results in more negative reduction potentials for their copper complexes. This study provides detailed comparative coordination chemistry of thiones and selones with copper and its effect on the Cu(II/I) reduction potentials. Simply changing the chalcogens and denticity of the thione and selone ligands results in Cu(II/I) reduction potentials of the synthesized copper chalcogenone complexes that can be tuned in a range of 471 mV, a difference that would have significant effects in redox-mediated reactions.
Experimental section
Materials
The synthesis and manipulation of all copper complexes was performed under an inert atmosphere of argon or nitrogen using standard Schlenk techniques. Acetonitrile, methanol, and ether were purified using standard procedures and freshly distilled under argon atmosphere prior to use. N,N′-Dimethylimidazole thione (dmit), N,N′-dimethylimidazole selone (dmise),45 [Cu(NCMe)4][BF4],46 bis(mercaptoimidazolyl)methane (BmmMe), bis(selonoimidazolyl)methane (BsemMe), bis(mercaptoimidazolyl)ethane (BmeMe), and bis(selonoimidazolyl)ethane (BseeMe)47 were synthesized according to published procedures. The following reagents were used as received: selenium powder (VWR), sulfur powder (VWR), cuprous oxide (stabilized, Aldrich), 1-methylimidazole (VWR), iodomethane (VWR), and dibromomethane (Alfa Aesar).Instrumentation
1H, 13C{1H}, 77Se{1H} and 19F{1H} spectra were obtained on Bruker-AVANCE 300 and 500 MHz NMR spectrometers. 1H and 13C{1H} NMR chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS) and referenced to solvent. 19F{1H} NMR spectra were externally referenced to CCl3F (δ 0 ppm).48 The 77Se{1H} NMR chemical shifts were obtained in CDCl3 and externally referenced to diphenyl diselenide (δ 461 ppm),49 and reported relative to dimethyl selenide (δ 0 ppm). All 77Se NMR chemical shifts are reported in Table 7.Electrochemical experiments were performed with a BAS 100B potentiostat. A three-compartment cell was used with an Ag/AgCl reference electrode, Pt counter electrode, and a glassy carbon working electrode. Freshly-distilled acetonitrile was used as the solvent with tetra-n-butylammonium phosphate as the supporting electrolyte (0.1 M). Solutions containing 1 mM analyte were deaerated for 2 min by vigorous nitrogen purge. The measured potentials were corrected for junction potentials relative to ferrocenium/ferrocene (0.586 mV vs. Ag/AgCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50) and adjusted from Ag/AgCl to NHE (−0.197 V (ref. 51)) All E1/2 values were calculated from (Epa + Epc)/2 at a scan rate of 100 mV s−1, and ΔE = Epa − Epc.
50) and adjusted from Ag/AgCl to NHE (−0.197 V (ref. 51)) All E1/2 values were calculated from (Epa + Epc)/2 at a scan rate of 100 mV s−1, and ΔE = Epa − Epc.
Infrared spectra were obtained using Nujol mulls on KBr salt plates with a Magna 550 IR spectrometer. Abbreviations used in the description of vibrational data are as follows: vs, very strong; s, strong; m, medium; w, weak; b, broad. Electrospray ionization mass spectrometry (ESI-MS) was conducted using a QSTAR XL Hybrid MS/MS System from Applied Biosystems via direct injection of sample (0.05 mL min−1 flow rate) into a Turbo Ionspray ionization source. Samples were run under positive mode, with ionspray voltage of 5500 V, and TOF scan mode. MALDI-TOF-MS was conducted on a Bruker Microflex. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile was used as a matrix for co-crystallization of the copper complex characterized. All the peak envelopes matched their calculated isotopic distributions. Melting points were determined using a Barnstead Electrothermal 9100 apparatus in silicon-grease-sealed glass capillary tubes. Absorption spectra were collected using a Varian Cary-50 Bio spectrophotometer in quartz cuvettes with a path length of 1 cm. Elemental analysis (EA) was performed using PerkinElmer Series II CHNS/O Analyzer 2400.
[Cu2(dmit)5](BF4)2 (1)
Dmit (322 mg, 2.5 mmol) was dissolved in acetonitrile (30 mL) and cannula transferred to a solution of [Cu(NCMe)4]BF4 (312 mg, 1 mmol) in acetonitrile (20 mL). The reaction was stirred at room temperature for 3 h, and the solvent volume was reduced in vacuo to about 5 mL. The product was precipitated with diethyl ether (10 mL) to afford an off-white solid that was dried in vacuo. Yield: 74% (350 mg, 0.371 mmol). Mp = 132 °C. NMR (CD3CN): 1H δ 3.63 (s, 6 H, CH3), 6.99 (s, 2 H, CH); 13C{1H} δ 35.1 (CH3), 120.4 (CH), 157.3 (CS). IR (cm−1): 521 s, 672 vs, 724 vs, 746 vs, 801 s, 1047 b, 1175 vs, 1236 vs, 1284 v, 1378 s, 1464 vs, 1569 vs, 1684 w, 2276 s, 2304 s, 2723 w, 2859 b, 3118 w, 3142 w. MALDI-TOF-MS: 319.51 [Cu(dmit)2]+. Anal. Calc. for C25H40Cu2N10S5B2F8: C, 31.89; N, 14.87; H, 4.28. Found: C, 31.80; N, 14.56; H, 4.23%.
[(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2 (2)
Complex 2 was prepared following the procedure for 1 except dmise (437 mg, 2.5 mmol) was used in place of dmit. Yield: 85% (496 mg, 0.425 mmol). Mp = 126 °C. NMR (CD3CN): 1H δ 3.69 (s, 6 H, CH3), 7.16 (s, 2 H, CH); 13C{1H} δ 37.1 (CH3), 121.6 (CH), 147.2 (CSe); 19F{1H} δ −151.62, −151.63. IR (cm−1): 521 s, 624 w, 660 s, 744 s, 933 s, 1021 b, 1238 s, 1285 s, 1378 s, 1457 s, 1570 s, 1818 w, 2252 w, 2276 vs, 2304 vs, 2918 b, 3139 w, 3172 w, 3230 w. MALDI-TOF-MS: 415.07 [Cu(dmise)2]+. Anal. Calc. for C25H40Cu2N10Se5B2F8: C, 25.53; N, 11.91; H, 3.43. Found: C, 25.42; N, 11.73; H, 3.45%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3)
Tetrahydrofuran (5 mL) was added to a 4-dram vial containing a mixture of [Cu(NCMe)4]BF4 (131 mg, 0.416 mmol) and BmmMe (150 mg, 0.624 mmol), resulting in the immediate formation of a white solid suspended in a colorless solution. After stirring the suspension for 18 h, the product was isolated by filtration and dried in vacuo for 2 h. Yield: 88% (187 mg, 0.183 mmol). Mp = 128 °C. NMR (d6-DMSO): 1H δ 3.43 (s, 18 H, CH3), 6.59 (s, 6 H, CH2), 7.38 (s, 6 H, imidazole H), 7.62 (s, 6 H, imidazole H); 13C δ 35.9 (q, 1JC–H = 142, 6 C, CH3), 56.3 (t, 1JC–H = 158, 3 C, CH2), 118.8 (dd, 1JC–H = 201, 2JC–H = 11, 6 C, imidazole C), 121.1 (dd, 1JC–H = 200, 2JC–H = 9, 6 C, imidazole C), 155.8 (s, 6 C, CS); 19F{1H} δ −151.57, −151.63. UV-vis (CH3CN): 274 nm. IR (cm−1): 3173 m, 3140 m, 3119 m, 3027 w, 2903 w, 2943 m, 1575 s, 1467 vs, 1401 vs, 1378 s, 1317 m, 1286 w, 1249 s, 1215 s, 1166 m, 1064 vs, 788 m, 762 s, 742 s, 724 m, 701 w, 655 w, 521 m, 470 w. Anal. Calc. for C27H36B2Cu2F8N12S6: C, 31.74; H, 3.55; N, 16.45. Found: C, 31.93; H, 3.56; N, 16.32%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4)
BsemMe (215 mg, 0.75 mmol) was dissolved in dichloromethane (20 mL) before being cannula transferred to a solution of [Cu(NCMe)4]BF4 (160 mg, 0.5 mmol) in acetonitrile (10 mL). The reaction mixture was stirred at room temperature for 3 h. The solvent volume in the reaction mixture was then reduced to about 5 mL and the product was precipitated with diethyl ether. The growth of single crystals for X-ray analysis was performed from slow vapor diffusion of diethyl ether into acetonitrile solution. Yield: 45% (262 mg, 0.225 mmol). Mp = 139 °C. NMR (d6-DMSO): 1H δ 3.54 (s, 6 H, CH3), 6.82 (s, 2 H, CH2), 7.33 (d, JHH = 2.0, 2 H, CH), 7.59 (d, JHH = 2.0, 2 H, CH); 13C{1H} δ 37.5 (CH3), 59.7 (CH2), 121.3 (CH), 123.4 (CH), 149.7 (CSe); 19F{1H} δ −151.59, −151.63. UV-vis (CH3CN): 292 nm. IR (cm−1): 460 s, 473 w, 521 vs, 604 w, 655 s, 697 s, 731 vs, 779 w, 790 s, 1059 b, 1207 s, 1234 s, 1249 s, 1318 s, 1378 s, 1464 vs, 1575 vs, 1676 vs, 2727 b, 3145 w. Mass spectrum (ESI-MS): m/z 1216.59 [Cu2(BsemMe)3(BF4)]+, 882.68 [Cu2(BsemMe)2(BF4)]+, 796.67 [Cu2(BsemMe)2]2+, 398.83 [Cu(BsemMe)]+. Anal. Calc. for C29H39Cu2N13Se6B2F8: C, 25.91; N, 13.55; H, 2.92. Found: C, 25.98; N, 13.12; H, 3.04%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5)
Complex 5 was prepared following the procedure for 4 except BmeMe (191 mg, 0.75 mmol) was used in place of BsemMe. Yield: 47% (252 mg, 0.236 mmol). Mp = 230 °C. NMR (d6-DMSO): 1H δ 3.52 (s, 6 H, CH3), 4.63 (s, 4 H, CH2), 7.18 (d, 2 H, CH), 7.30 (d, 2 H, CH); 13C{1H} δ 35.6 (CH3), 45.7 (CH2), 119.7 (CH), 120.9 (CH), 155.2 (CS); 19F{1H} δ −148.31, −148.35. UV-vis (CH3CN): 273 nm. IR (cm−1): 501 w, 522 s, 622 w, 670 s, 680 s, 720 vs, 736 vs, 1059 vs, 1137 w, 1197 s, 1227 s, 1247 vs, 1287 w, 1378 vs, 1415 vs, 1466 vs, 1570 vs, 1694 w, 2927 b, 3137 w. Anal. Calc. for C30H42Cu2N12S6B2F8: C, 33.87; N, 15.50; H, 3.98. Found: C, 29.88; N, 13.68; H, 3.45%.
[Cu2(BseeMe)3](BF4)2 (6)
Complex 6 was prepared following the procedure for 4 except that BseeMe (223 mg, 0.75 mmol) was used in place of BsemMe. Yield: 30% (174 mg, 0.153 mmol). Mp = 270 °C. NMR (d6-DMSO): 1H δ 3.58 (s, 6 H, CH3), 4.73 (s, 4 H, CH2), 7.33 (d, 2 H, CH), 7.47 (d, 2 H, CH); 13C{1H} δ 39.7 (CH3), 47.5 (CH2), 121.5 (CH), 122.8 (CH), 148.0 (CSe); 19F{1H} δ −148.10, −148.16. UV-vis (CH3CN): 288 nm. IR (cm−1): 522 s, 666 vs, 724 vs, 738 vs, 747 vs, 800 w, 930 w, 1057 vs, 1128 vs, 1183 vs, 1223 s, 1246 vs, 1287 w, 1378 vs, 1409 vs, 1467 vs, 1569 vs, 2854 vs, 2919 b, 3114 w, 3146 w, 3173 w. Anal. Calc. for C30H42Cu2N12Se6B2F8: C, 26.79; N, 12.49; H, 3.15. Found: C, 26.97; N, 12.48; H, 3.12%.
[(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7)
Dmit (129 mg, 1 mmol) was dissolved in acetonitrile (20 mL) and cannula transferred to a solution of [Cu(NCMe)4]BF4 (312 mg, 1 mmol) in acetonitrile (10 mL). The reaction was stirred at room temperature for 3 h, resulting in the formation of a yellow solution. To this reaction mixture was cannula added BsemMe (336 mg, 1 mmol) in dichloromethane (10 mL) and stirred overnight. The solvent volume was reduced in vacuo to about 3 mL and the product was precipitated with diethyl ether to afford an off-white solid, which was dried in vacuo. Yield: 38% (427 mg, 0.378 mmol). Mp = 209 °C. NMR (CD3CN): 1H δ 3.60 (s, 6 H, CH3), 3.62 (s, 6 H, CH3), 6.65 (s, 2 H, CH2), 6.98 (s, 2 H, CH), 7.23 (d, JHH = 2.5, 2 H, CH), 7.38 (d, JHH = 2.0, 2 H, CH); 13C{1H} δ 35.8 (CH3), 37.8 (CH3), 60.5 (CH2), 120.0 (CH), 121.0 (CH), 123.3 (CH), 151.6 [CSe (BsemMe)], 157.7 [CS (dmit)]; 19F{1H} δ −151.52, −151.57. UV-vis (CH3CN): 274 nm. IR (cm−1): 508 s, 521 s, 611 s, 640 s, 650 s, 657 s, 676 s, 723 vs, 746 vs, 790 vs, 839 s, 867 s, 1033 b, 1145 s, 1177 s, 1207 s, 1229 s, 1249 s, 1290 s, 1321 s, 1372 s, 1395 s, 1465 s, 1571 vs, 1602 s, 1673 s, 2920 b, 3088 s. Mass spectrum (ESI-MS): m/z 732.73 [Cu(BsemMe)2]+, 526.85 [(dmit)Cu(BsemMe)]+, 398.82 [Cu(BsemMe)]+, 318.97 [Cu(dmit)2]+, 190.95 [Cu(dmit)]+. Anal. Calc. for C28H40Cu2N12Se4S2B2F8: C, 27.44; N, 13.72; H, 3.29. Found: C, 27.28; N, 13.60; H, 3.27%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(dmise)Cu(μ-BmmMe)2Cu(dmise)](BF4)2 (8)
Complex 8 was prepared following the same procedure for 7 except that dmise (176 mg, 1 mmol) was used in place of dmit and BmmMe (242 mg, 1 mmol) was used in place of BsemMe. Yield: 30% (347 mg, 0.302 mmol). Mp = 174 °C. NMR (CD3CN): 1H δ 3.52 (s, 6 H, CH3, BmmMe), 3.69 (s, 6 H, CH3, dmise), 6.48 (s, 2 H, CH2), 7.04 (d, JHH = 3.0, 2 H, CH), 7.14 (s, 2 H, CH, dmise), 7.26 (d, JHH = 3.0, 2 H, CH); 13C{1H} δ 35.9 (CH3), 37.7 (CH3), 57.5 (CH2), 118.9 (CH), 121.4 (CH), 122.5 (CH), 149.3 [CSe (dmise)], 158.8 [CS (BmmMe)]; 19F{1H} δ −151.48, −151.53. UV-vis (CH3CN): 269 nm. IR (cm−1): 521 s, 672 vs, 725 vs, 741 vs, 761 vs, 796 vs, 848 s, 983 s, 1033 b, 1217 vs, 1234 vs, 1250 vs, 1287 s, 1314 s, 1376 vs, 1401 vs, 1429 s, 1464 b, 1571 vs, 1699 b, 2851 b, 3141 s, 3171 s. Anal. Calc. for C28H40Cu2N12Se2S4B2F8: C, 29.72; N, 14.85; H, 3.56. Found: C, 29.60; N, 14.61; H, 3.53%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9)
Complex 9 was prepared following the same procedure for 7 except that dmise (176 mg, 1 mmol) was used instead of dmit. Yield: 46% (558 mg, 0.456 mmol). Mp = 193 °C. NMR (CD3CN): 1H δ 3.62 (s, 6 H, CH3), 3.68 (s, 6 H, CH3), 6.68 (s, 2 H, CH2), 7.14 (s, 2 H, CH), 7.24 (d, JHH = 2.0, 2 H, CH), 7.40 (d, JHH = 2.0, 2 H, CH); 13C{1H} δ 36.5 (CH3), 37.8 (CH3), 60.5 (CH2), 121.1 (CH), 122.0 (CH), 123.7 (CH), 149.0 [CSe (dmise)], 151.3 [CSe (BsemMe)]; 19F{1H} δ −151.56, −151.61. UV-vis (CH3CN): 278 nm. IR (cm−1): 521 s, 623 s, 650 s, 658 s, 724 s, 745 s, 791 s, 837 s, 1055 b, 1176 s, 1207 s, 1230 vs, 1248 s, 1287 s, 1320 s, 1378 b, 1464 vs, 1571 vs, 1673 s, 2925 b, 3132 b. Mass spectrum (ESI-MS): m/z 732.76 [Cu(BsemMe)2]+, 572.81 [(dmise)Cu(BsemMe)]+, 398.83 [Cu(BsemMe)]+, 239.02 [Cu(dmise)]+. Anal. Calc. for C28H40Cu2N12Se6B2F8: C, 25.49; N, 12.74; H, 3.06. Found: C, 24.85; N, 12.48; H, 3.00%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
[(BmmMe)Cu(μ-dmit)]n(BF4)n (10)
Complex 10 was prepared following the same procedure for 7 except that BmmMe (242 mg, 1 mmol) was used instead of BsemMe. Yield: 34% (354 mg, 0.335 mmol). Mp = 159 °C. NMR (CD3CN): 1H δ 3.52 (s, 6 H, CH3, BmmMe), 3.62 (s, 6 H, CH3, dmit), 6.49 (s, 2 H, CH2), 7.00 (s, 2 H, CH), 7.06 (d, JHH = 2.5, 2 H, CH), 7.25 (d, JHH = 2.5, 2 H, CH); 13C{1H} δ 35.9 (CH3), 57.5 (CH2), 119.0 (CH), 120.3 (CH), 121.5 (CH), 156.6 [CS (dmit)], 158.4 [CS (BmmMe)]; 19F{1H} δ −151.30, −151.35. UV-vis (CH3CN): 268 nm. IR (cm−1): 503 s, 521 s, 603 s, 633 s, 670 vs, 729 vs, 760 s, 782 s, 848 s, 1032 b, 1174 s, 1234 vs, 1286 s, 1395 vs, 1464 vs, 1572 vs, 1684 b, 2250 s, 2725 s, 2921 b, 3140 b. Anal. Calc. for C28H40Cu2N12S6B2F8: C, 32.41; N, 16.20; H, 3.88. Found: C, 32.55; N, 16.15; H, 3.97%. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex.
X-ray data collection and structural determination
Single crystals grown from vapor diffusion were mounted on a glass filament with silicon grease and immediately cooled to 168 K in a cold nitrogen gas stream. Single crystals suitable for X-ray analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of [(dmise)2Cu(μ-dmise)Cu(dmise)2](BF4)2 (2), [(BmmMe)Cu(μ-BmmMe)Cu(BmmMe)](BF4)2 (3), [(BsemMe)Cu(μ-BsemMe)Cu(BsemMe)](BF4)2 (4), [(BmeMe)Cu(μ-BmeMe)Cu(BmeMe)](BF4)2 (5), [(dmit)Cu(μ-BsemMe)2Cu(dmit)](BF4)2 (7), [(dmise)Cu(μ-BsemMe)2Cu(dmise)](BF4)2 (9), and [(BmmMe)Cu(μ-dmit)]n(BF4)n (10). Intensity data were collected using a Rigaku Mercury CCD detector and an AFC8S diffractometer. The space group P21/c for 9 was determined from the observed systematic absences. No symmetry higher than triclinic was observed for 2, 4, 5, 7, and 10 and assignment of the centrosymmetric space group option, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , provided chemically reasonable refinement results. Data reduction including the application of Lorentz and polarization (Lp) effects and absorption corrections used the CrystalClear program.52 The structures were solved by direct methods and subsequent Fourier difference techniques, and refined anisotropically, by full-matrix least squares, on F2 using SHELXTL 6.10.53 In the final cycle of least squares, independent anisotropic displacement factors were refined for the non-hydrogen atoms and the methyl hydrogen atoms were fixed in “idealized” positions with C–H = 0.96 Å. Their isotropic displacement parameters were set equal to 1.5 times Ueq of the attached carbon atom.
, provided chemically reasonable refinement results. Data reduction including the application of Lorentz and polarization (Lp) effects and absorption corrections used the CrystalClear program.52 The structures were solved by direct methods and subsequent Fourier difference techniques, and refined anisotropically, by full-matrix least squares, on F2 using SHELXTL 6.10.53 In the final cycle of least squares, independent anisotropic displacement factors were refined for the non-hydrogen atoms and the methyl hydrogen atoms were fixed in “idealized” positions with C–H = 0.96 Å. Their isotropic displacement parameters were set equal to 1.5 times Ueq of the attached carbon atom.
For complex 2, the largest peak in the final Fourier difference map (1.08 e A−3) was located 0.83 Å from Se(4) and the lowest peak (−0.81 e A−3) was located at a distance of 0.86 Å from Se(4). The largest peak for complex 4 in the final Fourier difference map (0.82 e A−3) was located 0.08 Å from Se(4) and the lowest peak (−0.79 e A−3) was located at a distance of 0.77 Å from Se(5). The largest peak for 7 in the final Fourier difference map (1.10 e A−3) was located 1.23 Å from N(5) and the lowest peak (−0.78 e A−3) was located at a distance of 0.88 Å from Se(1). The largest peak for 9 in the final Fourier difference map (1.16 e A−3) was located 1.19 Å from H(6C) and the lowest peak (−0.74 e A−3) was located at a distance of 0.92 Å from Se(1). The largest peak for 10 in the final Fourier difference map (0.42 e A−3) was located 1.73 Å from S(1), and the lowest peak (−0.42 e A−3) was located at a distance of 0.76 Å from Cu(1).
For complex 3, a suitable crystal was mounted using viscous oil onto a plastic mesh, and cooled to the data collection temperature. Data were collected on a Bruker-AXS APEX CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The systematic absences in the diffraction data were consistent with Pna21 and Pnma. The absence of a molecular mirror or inversion point, and the observed occupancy, Z = 4, were consistent with Pna21, the noncentrosymmetric option. The Flack parameter refined to zero, indicating that the true hand of the data was determined. This data set was treated with absorption corrections based on redundant multiscan data. The structures were solved using direct methods and refined with full-matrix, least-squares procedures on F2. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were treated as idealized contributions. Scattering factors are contained in the SHELXTL 6.12 program library.54 Final refinement parameters for the structures of 2, 3, 4, 5, 7, 9, and 10 are provided in Tables 9 and 10.
| 2 | 4 | 3 | 5 | |
|---|---|---|---|---|
| a R 1 = [∑||Fo| − |Fc||]/∑|Fo|; wR2 = {[∑w[(Fo)2 − (Fc)2]2}1/2. | ||||
| Chemical formula | C27H43B2Cu2F8N11Se5 | C29H39B2Cu2F8N13Se6 | C27H36B2Cu2F8N12Se6 | C33H46Cu2N13S6B2F8 |
| F.W. (g mol−1) | 1217.22 | 1344.19 | 1021.74 | 1437.29 |
| Space group |
P |
P |
Pna2(1) |
P |
| Crystal system | Triclinic | Triclinic | Orthorhombic | Triclinic |
| a, Å | 11.712(2) | 11.972(2) | 14.997(7) | 10.368(2) |
| b, Å | 14.126(3) | 14.325(3) | 15.362(7) | 10.699(2) |
| c, Å | 14.800(3) | 15.568(3) | 17.487(8) | 10.804(2) |
| α, ° | 87.32(3) | 89.58(3) | 90 | 98.29(3) |
| β, ° | 73.78(3) | 77.29(3) | 90 | 116.81(3) |
| γ, ° | 71.01(3) | 68.69(3) | 90 | 91.25(3) |
| V, Å3 | 2220.5(8) | 2418.7(8) | 4029(3) | 1053.4(4) |
| Z | 2 | 2 | 4 | 2 |
| D cal, mg m−3 | 1.821 | 1.846 | 1.685 | 1.677 |
| Indices (min) | [−14, −17, −18] | [−14, −17, 0] | [−19, −20, −23] | [−12, −11, −13] |
| (max) | [14, 17, 18] | [14, 17, 19] | [19, 19, 22] | [12, 11, 13] |
| Parameters | 508 | 548 | 520 | 274 |
| F(000) | 1184 | 1296 | 2072 | 542 |
| μ, mm−1 | 5.124 | 5.462 | 1.444 | 1.384 |
| 2θ range, ° | 3.19–26.38 | 2.94–26.34 | 1.76–28.24 | 3.09–26.30 |
| Collected reflections | 18943 |
9716 | 40237 |
9129 |
| Unique reflections | 8943 | 9716 | 9227 | 9129 |
| Final R (obs. data)a, R1 | 0.0461 | 0.0470 | 0.0527 | 0.0553 |
| wR2 | 0.1125 | 0.1116 | 0.1267 | 0.1363 |
| Final R (all data), R1 | 0.0616 | 0.0666 | 0.0682 | 0.0553 |
| wR2 | 0.1263 | 0.1276 | 0.1371 | 0.1581 |
| Goodness of fit (S) | 1.117 | 1.062 | 1.006 | 1.046 |
| Largest diff. peak | 1.081 | 0.817 | 1.107 | 0.929 |
| Largest diff. hole | −0.813 | −0.792 | −1.468 | −0.880 |
| 9 | 7 | 10 | |
|---|---|---|---|
| a R 1 = [∑||Fo| − |Fc||]/∑|Fo|; wR2 = {[∑w[(Fo)2 − (Fc)2]2}1/2. | |||
| Chemical formula | C28H40Cu2N12Se6B2F8 | C28H40Cu2N12S2Se4B2F8 | C28H40Cu2N12S6B2F8 |
| F.W. (g mol−1) | 1319.18 | 1225.38 | 1037.78 |
| Space group |
P |
P |
P21/c |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| a, Å | 8.21868(16) | 8.1987(16) | 9.4763(19) |
| b, Å | 11.247(2) | 11.198(2) | 27.970(6) |
| c, Å | 12.904(3) | 12.935(3) | 7. 8016(16) |
| α, ° | 66.67(3) | 65.68(3) | 90 |
| β, ° | 84.64(3) | 84.17(3) | 99. 89 (3) |
| γ, ° | 77.72(3) | 77.75(3) | 90 |
| V, Å3 | 1066.1(4) | 1057.5(4) | 2037.1(7) |
| Z | 1 | 1 | 2 |
| D cal, Mg m−3 | 2.055 | 1.924 | 1.692 |
| Indices (min) | [−10, −14, 16] | [−12, −21, −24] | [−11, −34, −7] |
| (max) | [9, 14, 11] | [11, 21, 26] | [11, 34, 9] |
| Parameters | 266 | 267 | 266 |
| F(000) | 636 | 600 | 1056 |
| μ, mm−1 | 6.194 | 4.622 | 1.429 |
| 2θ range, ° | 3.12–26.75 | 2.95–26.35 | 2.18–26.31 |
| Collected reflections | 9066 | 8161 | 16881 |
| Unique reflections | 4435 | 4221 | 4096 |
| Final R (obs. data)a, R1 | 0.0503 | 0.0455 | 0.0440 |
| wR2 | 0.1120 | 0.1049 | 0.0984 |
| Final R (all data), R1 | 0.0796 | 0.0658 | 0.0591 |
| wR2 | 0.1319 | 0.1182 | 0.1074 |
| Goodness of fit (S) | 1.093 | 1.100 | 1.089 |
| Largest diff. peak | 1.158 | 1.097 | 0.416 |
| Largest diff. hole | −0.736 | −0.778 | −0.424 |
Acknowledgements
J.L.B. thanks the National Science Foundation for a CAREER Award (CHE-0545138) and additional financial support from grant CHE-1213912. D.R. also acknowledges financial support from the National Science Foundation (CHE-0911407) and The University of North Carolina at Charlotte. M.M.K. thanks the Clemson University Chemistry Department for a graduate fellowship. We thank Carolyn E. Quarles for performing the ESI-MS experiments.References
- W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612–5620 RSC.
- H. R. Kim, I. G. Jung, K. Yoo, K. Jang, E. S. Lee, J. Yun and S. U. Son, Chem. Commun., 2010, 46, 758–760 RSC.
- L. Maria, C. Moura, A. Paulo, I. C. Santos and I. Santos, J. Organomet. Chem., 2006, 691, 4773–4778 CrossRef CAS PubMed.
- G. Parkin, New J. Chem., 2007, 31, 1996–2014 RSC.
- G. Roy, P. N. Jayaram and G. Mugesh, Chem. – Asian J., 2013, 8, 1910–1921 CrossRef CAS PubMed.
- C. Kimblin, B. M. Bridgewater, D. G. Churchill, T. Hascall and G. Parkin, Inorg. Chem., 2000, 39, 4240–4243 CrossRef CAS.
- C. Kimblin, T. Hascall and G. Parkin, Inorg. Chem., 1997, 36, 5680–5681 CrossRef CAS.
- D. J. Williams, D. VanDerveer, R. L. Jones and D. S. Menaldino, Inorg. Chim. Acta, 1989, 165, 173–178 CrossRef CAS.
- V. K. Landry, D. Bucella, K. Pang and G. Parkin, Dalton Trans., 2007, 866–870 RSC.
- V. K. Landry and G. Parkin, Polyhedron, 2007, 26, 4751–4757 CrossRef CAS PubMed.
- W.-G. Jia, Y.-B. Huang, Y.-J. Lin, G.-L. Wang and G.-X. Jin, Eur. J. Inorg. Chem., 2008, 4063–4073 CrossRef CAS PubMed.
- M. O. Awaleh, A. Badia and F. Brisse, Inorg. Chem., 2007, 46, 3185–3191 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- J. Cheon, J. Arnold, K. M. Yu and E. D. Bourret, Chem. Mater., 1995, 7, 2273–2276 CrossRef CAS.
- S. Meyer, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chim. Acta, 2010, 363, 3088–3092 CrossRef CAS PubMed.
- I. R. Crossley, A. F. Hill, E. R. Humphrey and M. K. Smith, Organometallics, 2006, 25, 2242–2247 CrossRef CAS.
- W.-G. Jia, Y.-B. Huang and G.-X. Jin, J. Organomet. Chem., 2009, 694, 3376–3380 CrossRef CAS PubMed.
- R. M. Silva, M. D. Smith and J. R. Gardinier, Inorg. Chem., 2006, 45, 2132–2142 CrossRef CAS PubMed.
- M. T. Aroz, M. C. Gimeno, M. Kulcsar, A. Laguna and V. Lippolis, Eur. J. Inorg. Chem., 2011, 2884–2894 CrossRef CAS PubMed.
- J. Nunn, I. Zahedi, G. Bauer, M. F. Haddow, S. N. A. Halim, A. Pérez-Redondo and G. R. Owen, Inorg. Chim. Acta, 2011, 365, 462–468 CrossRef CAS PubMed.
- F. Bigoli, P. Deplano, F. A. Devillanova, V. Lippolis, M. L. Mercuri, M. A. Pellinghelli and E. F. Trogu, Inorg. Chim. Acta, 1998, 267, 115–121 CrossRef CAS.
- T. A. Pinder, D. VanDerveer and D. Rabinovich, Inorg. Chem. Commun., 2007, 10, 1381–1384 CrossRef CAS PubMed.
- D. J. Williams, A. Shilatifard, D. VanDeveer, L. A. Lipscomb and R. L. Jones, Inorg. Chim. Acta, 1992, 202, 53–57 CrossRef CAS.
- J. Choudhury, P. Sinha, S. Prabhakar, M. Vairamani and S. Roy, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 2943–2955 CrossRef CAS PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y. G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–117 CrossRef CAS PubMed.
- E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 78–88 CrossRef CAS PubMed.
- M. M. Kimani, J. L. Brumaghim and D. VanDerveer, Inorg. Chem., 2010, 49, 9200–9211 CrossRef CAS PubMed.
- M. M. Kimani, J. L. Brumaghim and C. A. Bayse, Dalton Trans., 2011, 40, 3711–3723 RSC.
- R. Balamurugan, M. Palaniandavar and R. S. Gopalan, Inorg. Chem., 2001, 40, 2246–2255 CrossRef CAS PubMed.
- D. G. Booth, W. Levason, J. J. Quirk, G. Reid and S. M. Smith, J. Chem. Soc., Dalton Trans., 1997, 3493–3500 RSC.
- A. J. Blake, V. Lippolis, T. Pivetta and G. Verani, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, m364–m367 CAS.
- D. J. Williams, M. R. Fawcettbrown, R. R. Raye, D. Vanderveer, Y. T. Pang, R. L. Jones and K. L. Bergbauer, Heteroat. Chem., 1993, 4, 409–414 CrossRef CAS PubMed.
- F. A. Devillanova, G. Verani, L. P. Battaglia and B. A. Corradi, Transition Met. Chem., 1980, 5, 362–364 CrossRef CAS.
- A. Beheshti, W. Clegg, V. Nobakht, M. P. Mehr and L. Russo, Dalton Trans., 2008, 6641–6646 RSC.
- P. Aslanidis, S. K. Hadjikakou, P. Karagiannidis and P. J. Cox, Inorg. Chim. Acta, 1998, 271, 243–247 CrossRef CAS.
- T. S. Lobana, R. Sharma and R. J. Butcher, Z. Anorg. Allg. Chem., 2008, 634, 1785–1790 CrossRef CAS PubMed.
- L. Basumallick, S. D. George, D. W. Randall, B. Hedman, K. O. Hodgson, K. Fujisawa and E. I. Solomon, Inorg. Chim. Acta, 2002, 337, 357–365 CrossRef CAS.
- D. W. Tomlin, D. P. Campbell, P. A. Fleitz and W. W. Adams, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1153–1154 Search PubMed.
- E. S. Raper, J. R. Creighton, R. E. Oughtred and I. W. Nowell, Acta Crystallogr., Sect. B: Struct. Commun., 1983, 39, 355–360 CrossRef.
- T. A. Pinder, D. VanDerveer and D. Rabinovich, Inorg. Chem. Commun., 2007, 10, 1381–1384 CrossRef CAS PubMed.
- R. M. Silva, M. D. Smith and J. R. Gardinier, Inorg. Chem., 2006, 45, 2132–2142 CrossRef CAS PubMed.
- T. S. Lobana and A. Castineiras, Polyhedron, 2002, 21, 1603–1611 CrossRef CAS.
- D. H. Lee, L. Y. Q. Hatcher, M. A. Vance, R. Sarangi, A. E. Milligan, A. A. N. Sarjeant, C. D. Incarvito, A. L. Rheingold, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2007, 46, 6056–6068 CrossRef CAS PubMed.
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef CAS PubMed.
- G. Roy, D. Das and G. Mugesh, Inorg. Chim. Acta, 2007, 360, 303–316 CrossRef CAS PubMed.
- G. J. Kubas, Inorg. Synth., 1990, 28, 68–70 CrossRef CAS PubMed.
- W. G. Jia, Y. B. Huang, Y. J. Lin and G. X. Jin, Dalton Trans., 2008, 5612–5620 RSC.
- R. K. Harris and B. E. Mann, NMR and the Periodic Table, Academic Press, London, 1978, p. 99 Search PubMed.
- J. D. Odom, W. H. Dawson and P. D. Ellis, J. Am. Chem. Soc., 1979, 101, 5815–5823 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- H. M. Koepp, H. Wendt and H. Z. Strehlow, Electrochemistry, 1960, 64, 483–491 CAS.
- T. H. Dunning, J. Chem. Phys., 1971, 55, 716–723 CrossRef CAS PubMed.
- G. M. Sheldrick, Bruker Analytical X-ray Systems Inc., Madison, WI, 2000.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal packing diagram of 10 (Fig. S1), cyclic voltammograms of complexes 1–10 (Fig. S2). CCDC 1405705–1405711. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02232k |
| This journal is © The Royal Society of Chemistry 2015 |