
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkoxide coordination of iron(III) protoporphyrin IX by antimalarial quinoline methanols: a key interaction observed in the solid-state and solution†
Johandie
Gildenhuys
a,
Chandre J.
Sammy
a,
Ronel
Müller
a,
Victor A.
Streltsov
b,
Tanya
le Roex
a,
David
Kuter
*a and
Katherine A.
de Villiers
 *a
*a
aStellenbosch University, Department of Chemistry and Polymer Science, Private Bag X1, Matieland, 7602, Stellenbosch, South Africa. E-mail: kdev@sun.ac.za; Fax: +27-21-808-3360; Tel: +27-21-808-2741
bManufacturing Flagship, Commonwealth Scientific and Industrial Research Organization, Parkville, Victoria 3050, Australia
First published on 26th August 2015
Abstract
The quinoline methanol antimalarial drug mefloquine is a structural analogue of the Cinchona alkaloids, quinine and quinidine. We have elucidated the single crystal X-ray diffraction structure of the complexes formed between racemic erythro mefloquine and ferriprotoporphyrin IX (Fe(III)PPIX) and show that alkoxide coordination is a key interaction in the solid-state. Mass spectrometry confirms the existence of coordination complexes of quinine, quinidine and mefloquine to Fe(III)PPIX in acetonitrile. The length of the iron(III)–O bond in the quinine and quinidine complexes as determined by Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy unequivocally confirms that coordination of the quinoline methanol compounds to Fe(III)PPIX occurs in non-aqueous aprotic solution via their benzylic alkoxide functional group. UV-visible spectrophotometric titrations of the low-spin bis-pyridyl-Fe(III)PPIX complex with each of the quinoline methanol compounds results in the displacement of a single pyridine molecule and subsequent formation of a six-coordinate pyridine–Fe(III)PPIX-drug complex. We propose that formation of the drug–Fe(III)PPIX coordination complexes is favoured in a non-aqueous environment, such as that found in lipid bodies or membranes in the malaria parasite, and that their existence may contribute to the mechanism of haemozoin inhibition or other toxicity effects that lead ultimately to parasite death. In either case, coordination is a key interaction to be considered in the design of novel antimalarial drug candidates.
Introduction
According to the World Health Organization (WHO), almost half of the world's population remains at risk of contracting malaria,1 a parasitic disease caused by Plasmodium species. While prevention and control measures have curbed the spread of the disease to the point that the WHO has observed a decrease in mortality rates in recent years, resistance to both historically-relevant quinoline treatments and the newer artemisinin-based combination therapies remains a motivating factor to develop novel antimalarial chemotherapies.1 Efforts to do so typically fall into two categories, the first being synthesis of libraries of derivatives of a particular scaffold. This may be informed by activity data collected for earlier generations of derivatives, however is frequently guided by availability of starting materials and the particular methodological expertise within a research group. The alternative approach relies on understanding both the drug target and, in the case of malaria chemotherapy, the antimalarial pharmacophore. In this regard, limited definitive structural data detailing antimalarial drug interactions with proposed targets has to date precluded such rational drug design.Haemozoin formation is a biocrystallisation process exploited by the malaria parasite P. falciparum to rid itself of toxic ferriprotoporphyrin IX (Fe(III)PPIX) released during haemoglobin digestion.2 Fe(III)PPIX is widely considered to be the target of the clinically-relevant 4-aminoquinoline and quinoline methanol antimalarial drugs,3–8 however the mechanism of quinoline antimalarial drug action remains unclear. Hypotheses in the literature include complexation between the drug and the haemozoin precursor (namely Fe(III)PPIX) in solution,5,9–12 direct interaction between the drug and haemozoin,13,14 or between drug–Fe(III)PPIX complexes and haemozoin.15,16 We recently reported a kinetic model describing the inhibition of β-haematin (synthetic haemozoin) formation, which incorporates aspects of these different hypotheses.17 At low drug concentration, adsorption of a monolayer of drug to the surface of the β-haematin crystal is presumed to slow its rate of further growth, while at higher concentrations of drug, the irreversible precipitation of Fe(III)PPIX as its drug complex was proposed as a competing process. Insight into the specific interactions between drug and target for either of these processes is limited. By means of a computational docking study, Leiserowitz and co-workers suggested that adsorption of quinoline antimalarial drugs is most likely to occur on the fastest-growing crystal face of β-haematin, and they propose several intermolecular interactions between surface Fe(III)PPIX molecules and quinoline antimalarial drugs (including chloroquine (CQ), amodiaquine (AQ), quinine (QN) and mefloquine (MQ)) that appear favourable.13 The literature concerning the structure and stoichiometry of Fe(III)PPIX-drug complexes in solution is extensive and has been recently reviewed.18,19 In the case of the quinoline methanol compounds specifically, debate has centred around the structure of their coordination complexes with urohaemin20 and Fe(III)PPIX.9,21–23 Recently, we determined the first single crystal X-ray diffraction (SCD) structures of the coordination complexes formed between Fe(III)PPIX and two active antimalarial compounds, the Cinchona alkaloids QN and quinidine (QD, see Fig. 1a).24 A three-point binding mode, which had been predicted using computation,25 was confirmed. The SCD structures provide indisputable evidence that coordination takes place via the deprotonated benzylic alcohol (alkoxide) functional group of both QN and QD, at least in the solid-state. As expected, π-stacking between the porphyrin and quinoline aryl systems of Fe(III)PPIX and the alkaloids respectively, is also observed. Finally, a charge-assisted intramolecular hydrogen bond formed between the protonated tertiary amine of the quinuclidine moiety of the drug molecules and the deprotonated propionate side chain of Fe(III)PPIX is a feature in both coordination complexes. Interestingly, QD is known to be more active than QN.26,27 The computational investigation resolved that the strain energy within these complexes following formation of the intramolecular hydrogen bond was less in the case of QD compared to QN.25 The authors proposed that the absolute stereochemistry of C8 and C9 in QD afforded the required geometry within its coordination complex with Fe(III)PPIX more readily than QN.
 | ||
| Fig. 1 (a) The molecular structures of quinine (QN: 8S,9R) and quinidine (QD: 8R,9S), two naturally-occurring Cinchona alkaloids. (b) The molecular structures of the erythro isomers of mefloquine (MQ). These are designated (−)-MQ and (+)-MQ, and have 11R,12S and 11S,12R configurations, respectively.43 | ||
In the current work, we report the first SCD structure of racemic MQ complexed with Fe(III)PPIX, and a second SCD structure of QD-Fe(III)PPIX, both as acetonitrile solvates. These structures are complementary to that of QN-Fe(III)PPIX reported previously,24 which is also a solvate of acetonitrile. We show that the geometries within the two MQ-Fe(III)PPIX complexes are analogous to those observed in QN- and QD-Fe(III)PPIX, and suggest that this may account for the observed difference in activity between the (+) and (−) erythro MQ isomers (see Fig. 1b).28 Furthermore, we present Electrospray Ionization Mass Spectrometry (ESI-MS), Extended X-ray Absorption Fine Structure (EXAFS) and UV-visible spectroscopic evidence that strongly supports the existence of these alkoxide-bound coordination complexes in non-aqueous solution. We propose that the interactions reported in this structural study may be of relevance in the lipid environments of the malaria parasite's digestive vacuole where haemozoin formation,29–34 and most likely drug action as a direct consequence,35 have recently been considered to take place.
Experimental
Materials and chemicals
All chemicals and organic solvents used in this study were purchased from Sigma-Aldrich. Antimalarial drugs were purchased as salts, namely QN hemisulfate monohydrate, QD sulfate dihydrate, MQ hydrochloride and CQ diphosphate. Haemin (Cl–Fe(III)PPIX) and haematin (H2O–Fe(III)PPIX) were both of porcine origin. Free base forms of QN and QD were prepared by precipitating the respective salt from aqueous solution using concentrated NaOH, followed by freeze drying. Free base MQ was obtained by precipitating its HCl salt from methanol using concentrated NaOH, followed by centrifugation and drying over phosphorous pentoxide. Dimethyl sulfoxide (DMSO) and acetonitrile were reagent grade or better and were used without further purification.Single crystal X-ray diffraction
Mass spectroscopy
Samples of the mother liquors from the crystallisation experiments were freshly prepared. Measurements were performed using a Waters Synapt G2 mass spectrometer equipped with an electrospray ionization (ESI) source in positive ion mode. Aliquots were delivered via direct injection.X-ray absorption studies
EXAFS spectra were recorded of the single crystals of the QD-Fe(III)PPIX and QN-Fe(III)PPIX complexes previously-reported,24 as well as those of the MQ-Fe(III)PPIX complex reported in the current work. Solutions that contained higher concentrations of both Fe(III)PPIX and drugs compared to the crystallisation experiments, but with both individual components present in their original ratios, were also investigated. The target concentration of Fe(III)PPIX (∼10 mM) represents an 8 to 11 fold increase in concentration relative to that used in the crystallisation experiments. In practice however, owing to the poor solubility of haemin in acetonitrile, this could not be easily attained, especially for QN- and MQ-Fe(III)PPIX. The raw EXAFS scans (χ(k)) were collected at the X-ray absorption spectroscopy (XAS) beamline (1.9 T Wiggler) at the Australian Synchrotron in Melbourne. Crystals were placed between two Kapton tapes with as much of the mother liquor removed as possible and mounted on a Teflon cell. All liquid samples were prepared immediately prior to the EXAFS experiments being conducted, and solutions were filtered through a PTFE filter disk (0.45 μm pore size) to remove undissolved haemin and to obtain homogeneous samples for analysis. These were then injected into Teflon cells and flash frozen in liquid nitrogen. A cryogenic temperature (18 K) was maintained for the duration of the measurements to limit thermal disorder and avoid sample degradation. For each sample, at least three iron Kα-edge (E0 = 7112 eV) scans were recorded in fluorescence mode up to k = 12 Å−1 using a 100-element Ge detector (cooled by liquid N2). Data collections were made from several locations within the samples to determine the extent of sample homogeneity and to limit sample damage due to continuous exposure to the X-ray beam.Replicate data sets for each sample were merged using the XFIT AVERAGE program.36 Data were processed using ATHENA (v0.8.061), and later fitted to a proposed model using ARTEMIS (v0.8.014).37–39 Within this suite of programs, the AUTOBK algorithm was applied for background removal and normalization.40 In the case of QD-Fe(III)PPIX, both the solid-state and frozen solution EXAFS spectra could be simultaneously refined, owing to the similarity between the two data sets. For QN- and MQ-Fe(III)PPIX on the other hand, the EXAFS data sets of the solid-state and frozen solution samples were refined independently. A previously-built model of the porphyrin core of Fe(III)PPIX41 served as the basis for the structural fitting in each case. The SCD structures reported previously24 and in the current work were used to identify non-hydrogen atoms (within 5.0 Å of the absorbing Fe atom) belonging to either the coordinated quinoline methanol drug (designated as axial atoms) or the neighbouring π-stacked complex (designated as atoms outside of the complex). Of the axial atoms, the alkoxide oxygen atom, the benzylic carbon atom and the quinoline and quinuclidinium/piperidinium carbon atoms immediately adjacent to this were explicitly refined. All other C and N atoms (both axial and outside of the complex) were refined as one of five arbitrary atom types (Cp1, Cp2, Cp3, Cp4 or Cn1), and assigned initial Fe–X distances in the range 3.5 to 5.0 Å. For each Fe(III)PPIX-drug complex, fitting of experimental data to the model required inclusion of different numbers of each atom type (see Results and discussion for further details). The initial distances, together with the associated Debye–Waller factors, were allowed to refine during the fitting process.
UV-visible spectrophotometric titrations
 | (1) |
The association of pyridine with Fe(III)PPIX was measured by adding aliquots of pyridine to a fresh sample of the same Fe(III)PPIX working solution described above and recording the spectrum after each addition. A total volume of 290 μL pyridine (approximately 10% (v/v)) was added. Data were corrected for dilution and analysed in HypSpec according to the equilibria described by eqn (2) and (3) in order to obtain equilibrium constants β2 and β3. To minimise variables in the refinement process, the spectrum of Fe(III)PPIX (M) was constrained. Spectra of the monopyridyl- and bis-pyridyl-Fe(III)PPIX complexes (MPy and M(Py)2, respectively) were allowed to freely refine.
 | (2) |
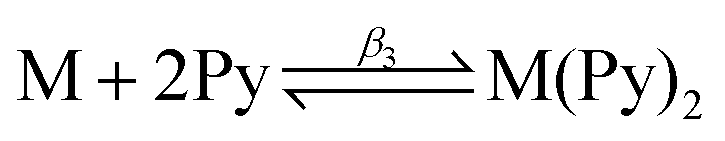 | (3) |
 | (4) |
Results and discussion
Solid-state structures of quinoline methanol complexes of Fe(III)PPIX
We have shown previously that QN and QD form coordination complexes with Fe(III)PPIX in the solid-state via their benzylic alcohol functional groups.24 Somewhat surprisingly, the μ-propionato dimer of Fe(III)PPIX that constitutes haemozoin and β-haematin was recently crystallised from a DMSO solution in the presence of the antimalarial CQ (as its free base).17 It was therefore of interest to determine whether, in the presence of CQ free base, the formation of the coordination complexes of the quinoline methanol antimalarial drugs (Fig. 1) with Fe(III)PPIX would be favoured over the formation of the μ-propionato dimer. Single crystals were obtained from acetonitrile solutions of Fe(III)PPIX, which contained CQ free base and either QD or MQ salts. In both cases, the coordination complex, namely QD-Fe(III)PPIX or MQ-Fe(III)PPIX, was preferentially formed over the μ-propionato dimer. Interestingly, there is no evidence of CQ in either of the structures.The MQ salt used in this study was obtained as a racemate of erythro enantiomers. The absolute configurations of these (−) and (+) isomers have been determined as (11R,12S) and (11S,12R), respectively.43 It is not unexpected therefore, that the X-ray diffraction structure of MQ-Fe(III)PPIX includes two unique complexes in the asymmetric unit (Fig. S1†). These are designated as I and II for clarity (Fig. 2), and include the (−) and (+) isomers of MQ, respectively.
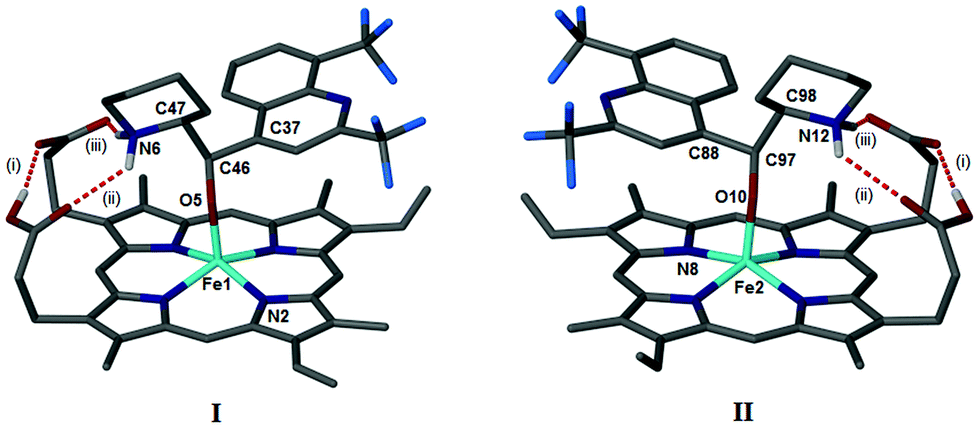 | ||
| Fig. 2 The asymmetric unit of the MQ-Fe(III)PPIX structure includes two unique complexes, I and II, which have been reoriented relative to one another for clarity such that their enantiomeric relationship is evident. Complex I includes the (−) erythro isomer of MQ, while complex II includes the (+) erythro isomer. The absolute configurations of the two stereochemical centres are (C11R,C12S) in I and (C11S,C12R) in II. Position 11 corresponds to C46 and C97 in the SCD structures of I and II, respectively, while position 12 corresponds to C47 and C98 in the SCD structures of I and II. Selected atom labels have been included for later reference. Intramolecular hydrogen bonds are shown as red dashed lines and labelled as (i), (ii), and (iii). Solvent molecules and non-relevant H atoms have been removed for clarity. Atom colour labelling: grey – C, cyan – Fe, dark blue – N, light blue – F, red – O and white – H. | ||
As observed previously for QN-Fe(III)PPIX and QD-Fe(III)PPIX,24 coordination of MQ to the metal centre of Fe(III)PPIX occurs via the deprotonated benzylic alcohol functional group. The Fe–O bond lengths in I and II are 1.899(2) and 1.892(2) Å respectively, which are comparable to other alkoxide-bound species.24,44–47 π-Stacking is also a feature within and between MQ-Fe(III)PPIX complexes. This is best appreciated when viewing the unit cell down the crystallographic b-axis as shown in Fig. S2.† π-Stacking between the unligated faces of Fe(III)PPIX molecules in adjacent complexes is observed, resulting in dimers consisting of either two molecules of I or two molecules of II. The distance between the mean porphyrin planes is 3.81 and 3.70 Å in dimers of I and II respectively, with the Fe(III) centres offset relative to one another. Dimers of I align to form rows parallel to the b-axis, as do dimers of II. These rows pack together in an alternating fashion to form sheets in the bc plane, which stack together to form the three dimensional structure (Fig. S3†). In addition to ligand-metal coordination and π-stacking, intramolecular hydrogen bonding is also a prominent feature within each complex. The first of three such hydrogen bonds exists between the propionic acid and propionate side chains of Fe(III)PPIX (labelled (i) in Fig. 2). The second and third (labelled (ii) and (iii), respectively, in Fig. 2) occur in turn between each of these groups and the diprotic piperidinium nitrogen atom of MQ. The salt bridge formed in the third instance is reminiscent of that originally predicted25 and later observed24 in the QN-Fe(III)PPIX and QD-Fe(III)PPIX complexes. This network of intramolecular hydrogen bonds is shown in the ESI (Fig. S4†), and the hydrogen bond geometries are reported in Table S3.† An interesting observation is the absence of intermolecular hydrogen bonds between the propionate and propionic acid side chains of neighbouring complexes as were observed in the QN-Fe(III)PPIX and QD-Fe(III)PPIX complexes. Rather, disordered solvent molecules (acetonitrile and water, since no precautions were taken to ensure that the acetonitrile was thoroughly dry before use) occupy spaces between adjacent complexes.
An earlier computational study examined the influence of the charge-assisted intramolecular hydrogen bond (salt bridge) on the resultant geometries of the QN- and QD-Fe(III)PPIX complexes.25 The investigation focused on the energies of each complex as two dihedral angles, designated φ and ψ (where φ refers to the axial Fe–O bond dihedral and ψ refers to the dihedral about the C8–C9 bond in the coordinated drug), were varied relative to one another. Of the two complexes, QD-Fe(III)PPIX was found to incur the lowest strain energy. It was argued that the absolute configuration (at C8 and C9) of QD, the more active isomer,27 confers a greater degree of geometrical preorganization, which in turn facilitates the formation of the salt bridge between the drug and the porphyrin. Parallels can be drawn between the erythro isomers of MQ and the Cinchona alkaloids QN and QD. Importantly, the two chiral centres in QN and QD that are considered relevant for their activity (C8 and C9)48 are preserved in MQ (C11 and C12). Furthermore, it has been shown that the (+) erythro isomer of MQ is almost twice as active as the (−) erythro isomer.28 With the present elucidation of the SCD structures of MQ-Fe(III)PPIX, it was possible to compare the geometries of I and II to their analogous alkaloid complex and explore whether a similar trend exists between their structures and the reported activities of the unbound MQ stereoisomers. It was not possible during the current study to extend the original Hyperchem calculations25 to examine the strain energy surfaces for each of the hydrogen-bonded MQ-Fe(III)PPIX complexes. Rather, a qualitative analysis was carried out. The values of the dihedral angles φ and ψ in I and II were compared to those in QN-Fe(III)PPIX and QD-Fe(III)PPIX, respectively (Table 1). In general, close agreement is observed. An overlay of the observed MQ dihedral angles with the strain energy surfaces previously computed for the QN- and QD-Fe(III)PPIX complexes provides visual appreciation (Fig. S5†). It would appear that the complex induced by the less active (−)-MQ isomer (I) adopts a conformation that is higher in strain energy than the complex induced by the more active (+)-MQ isomer (II). This is consistent with what was observed for the QN- and QD-Fe(III)PPIX complexes, and suggests that the (+) erythro isomer of MQ may be structurally preorganised in a similar manner as QD. The φ/ψ strain energy analysis suggests that the difference in energy between the two complexes is between 2 and 3 kcal mol−1. This energy barrier is relatively small, and given that the crystallisation procedure involved heating the mother liquor at 50 °C for 20 minutes, and 37 °C for a further 24 hours, both complexes would be accessible in solution, though in differing proportions. The fact that I and II appear in the crystal structure in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio suggests that additional factors (e.g. crystallisation energy, entropy) may have strongly influenced the composition of the crystal, however such an investigation is not within the scope of the current work.
:1 ratio suggests that additional factors (e.g. crystallisation energy, entropy) may have strongly influenced the composition of the crystal, however such an investigation is not within the scope of the current work.
In addition to the SCD structure of MQ-Fe(III)PPIX presented above, a new structure of QD-Fe(III)PPIX has been obtained under the same conditions. The molecular structure of this complex is in good agreement with that of the species recovered from a 3:7 (v/v) DMSO-methanol solution.24 The asymmetric unit of the acetonitrile solvate includes two unique complexes (labelled III and IV), and the three-point binding mode between the drug and Fe(III)PPIX that is discussed above for MQ is observed in both (Fig. S6†). The complexes π-stack in the solid-state via their unligated porphyrin faces, and the iron centres are laterally-shifted relative to one another. Close packing is assisted by a series of intermolecular hydrogen bonds between the porphyrin propionate and propionic acid groups, as well as between the suspected propionic acid group and the quinoline nitrogen atom of adjacent complexes of IV (Table S4†). As before, the presence of a charge-assisted intramolecular hydrogen bond between the protonated quinuclidine nitrogen atom of QD and the porphyrin propionate side chain reduces conformational freedom within each coordination complex. Notably, the crystallisation medium does not appear to strongly influence the magnitudes of the φ and ψ dihedral angles. These differ by no more than six degrees compared to those measured in the complex recovered from DMSO-methanol (Table 1 and Table S5†).24
Coordination of quinoline methanol antimalarials to Fe(III)PPIX in solution
While the SCD structures conclusively demonstrate coordination of the benzylic alcohol (as an alkoxide) moiety of quinoline methanol antimalarial drugs to the iron(III) centre of Fe(III)PPIX in the solid-state, evidence for this binding mode in solution is more contested.9,21–23 Since single crystals of QN-, QD- and MQ-Fe(III)PPIX were recovered from acetonitrile, a solvent routinely used in mass spectrometry, this provided a serendipitous opportunity to investigate whether these coordination complexes persist in the mother liquors. As expected, peaks arising from the individual components that comprised the samples, namely haemin, MQ, QN, QD and CQ, were observed (Table S6†). More importantly, peaks corresponding to the coordination complexes of all species were also detected, confirming the existence of these complexes in non-aqueous solution. The MQ-Fe(III)PPIX complex is observed at m/z 994.2946 (Fig. S7†), which corresponds to [M + H]+ (calculated m/z = 994.2940). M is considered to be the species observed by SCD, i.e. the neutral coordination complex formed between the zwitterionic drug and a Fe(III)PPIX molecule with one ionised propionate group. Subsequent protonation (via the medium) could occur at this latter site, or alternatively at the quinoline nitrogen atom of the drug when feasible. Similarly, the QN- and QD-Fe(III)PPIX complexes are observed at m/z values of 940.3632 (Fig. S8†) and 940.3629 (Fig. S9†), respectively, which are in very good agreement with the calculated monoisotopic mass of the [M + H]+ species (m/z = 940.3611). Previously, Alumasa et al. reported similar MS data for a QN-Fe(III)PPIX adduct precipitated from basic aqueous solution and redissolved in acetonitrile for analysis.49 Taking this and other spectroscopic data into account, the authors proposed that the complex present in aqueous solution involves non-covalent coordination of QN free base to the unligated iron(III) centre of a fully ionized Fe(III)PPIX molecule via the benzylic alcohol group of the drug. The structure is notably different to the coordination complex observed by SCD, in which coordination is facilitated by the deprotonated alkoxide of the QN zwitterion.24MS alone is not able to distinguish between the two above-mentioned structural interpretations, and consequently we undertook an EXAFS spectroscopic investigation to probe the local environment around the iron(III) centre. This technique has been successfully applied in various investigations of Fe(III)PPIX50–53 and haemozoin (β-haematin),54 as well as synthetic analogues of the latter.55–58 Knowing that the Fe–O bond length of a bound alkoxide (1.816(2)–1.866(4) Å)24,44–47 is considerably shorter than that of a bound alcohol (2.112(8)–2.160(8) Å),59,60 we were particularly interested to apply EXAFS spectroscopy to study the length of Fe–O bonds in the complexes present in non-aqueous solution, with the aim of differentiating between the structure implicated by SCD (alkoxide-bound) and that proposed by Roepe and co-workers (alcohol-bound).49 EXAFS spectra were therefore recorded of the frozen solutions of each complex. To facilitate analysis, we further recorded spectra of the corresponding solid-state samples. Atoms comprising the porphyrin core (Cα, Cβ, Cmeso and N), selected non-hydrogen axial atoms from the Cinchona alkaloids (C4′, C8, C9 and O(H) in Fig. 1a) or MQ (C4, C11, C12 and O(H) in Fig. 1b), and the iron(III) atom of a neighbouring π-stacked complex were identified, and their distances from the absorbing iron(III) centre refined explicitly. As outlined in the experimental methods, arbitrary C and N atoms at longer distances (3.5–5.0 Å) were used as descriptors for additional atoms arising from either the axially-bound quinoline methanol drug or the porphyrin of a neighbouring π-stacked complex. In the case of QD-Fe(III)PPIX, there was a high degree of congruency between the experimental spectra obtained for the crystals and mother liquor. To increase the number of independent points, the model was refined to both data sets simultaneously. On the other hand, differences in the spectra of QN-Fe(III)PPIX in the solid-state and in solution necessitated their independent refinement. The EXAFS spectrum for MQ-Fe(III)PPIX in solution was noisy, most likely owing to a lower concentration of Fe(III)PPIX solubilised in acetonitrile in the presence of MQ, and a reliable fit could not be achieved. Fitting was possible however, for the solid-state spectrum. In addition to those atoms refined explicitly, 17, 18 and 25 additional atoms were required to yield acceptable fits of the experimental spectra of QD-, QN- and MQ-Fe(III)PPIX, respectively (see Table 2 for refinement statistics). Best fits of the experimental spectra are presented in R-space (Fig. 3) and k-space (Fig. S10†), while fitted distances of all atoms relative to the absorbing iron(III) centre are given in Table S7.† The comparative distances determined by SCD of all explicitly-refined atoms relative to the absorbing iron(III) centre are given in Table S8,† while tentative assignments of the additional C and N atoms for each complex are presented in Table S9.† To aid appreciation of the goodness of fit of each EXAFS structure (for solid-state samples), an overlay of all atoms at their refined distances is shown relative to the respective SCD structure in Fig. S11.†
 | ||
| Fig. 3 Experimental and fitted Fourier transformed (R-space) EXAFS spectra. (a) The best fit of the model (grey line) to the experimental spectra of QD-Fe(III)PPIX in the solid-state (black solid line) and frozen solution (black dashed line). Spectra were fitted over the range 2.0 ≤ k ≤ 10.0 Å−1 and 0.0 ≤ R ≤ 5.0 Å. (b) The best fit of the model (grey line) to the experimental spectrum of a crystalline sample of MQ-Fe(III)PPIX (black solid line). Spectra were fitted over the range 2.0 ≤ k ≤ 10.0 Å−1 and 0.0 ≤ R ≤ 5.0 Å. (c) The best fit of the model (grey line) to the experimental spectrum of a crystalline sample of QN-Fe(III)PPIX (black solid line). Spectra were fitted over the range 2.0 ≤ k ≤ 10.0 Å−1 and 1.0 ≤ R ≤ 5.0 Å. (d) The best fit of the model (grey line) to the experimental spectrum of the frozen solution of QN-Fe(III)PPIX (black dashed line). Spectra were fitted over the range 2.0 ≤ k ≤ 9.4 Å−1 and 0.0 ≤ R ≤ 5.0 Å. All spectra (a to d) were fitted with k3-weighting in R-space at the final step. | ||
| QD-Fe(III)PPIXa | QN-Fe(III)PPIX | MQ-Fe(III)PPIXd | ||
|---|---|---|---|---|
| Solid-state | Solution | |||
| a EXAFS data collected from crystalline and solution samples were fitted simultaneously. b Parameters pertain to the solid-state sample. c Parameters pertain to the frozen solution sample. d Only data obtained from the crystalline sample of MQ-Fe(III)PPIX were successfully fit. | ||||
| n ind | 45.03 | 20.03 | 23.11 | 25.00 |
| n var | 19 | 14 | 15 | 16 |
| R-factor | 0.033b | 0.022 | 0.044 | 0.021 |
| 0.029c | ||||
| Reduced χ2 | 1.38 | 0.91 | 1.62 | 1.63 |
| S 0 2 | 0.89(9) | 0.81(9) | 1.00(10) | 0.65(6) |
| ΔE0 (eV) | −0.3(1.4)b | 0.1(2.0) | 2.1(1.6) | 0.7(1.5) |
| 1.3(1.3)c | ||||
The Fe–O bond length refined to 1.86(2) and 1.86(3) Å for QD- and QN-Fe(III)PPIX, respectively, in the solid-state structures determined by EXAFS (Table 3). These values compare favourably to the corresponding bond lengths observed in the SCD structures (1.862(2) and 1.866(4) Å, respectively).24 While a less favourable agreement is observed for MQ-Fe(III)PPIX, the fitted EXAFS Fe–O distance (1.85(2) Å) is within three standard deviations of the SCD equivalent (1.899(2) Å and 1.892(2) Å for I and II respectively). These observations confirm the coordination of the alkoxide species in all cases in the solid-state, and demonstrate the sensitivity of the EXAFS technique to provide reliable structural information regarding the first coordination shell of Fe(III)PPIX. For the mother liquors of QN- and QD-Fe(III)PPIX, Fe–O bond lengths from fitted EXAFS spectra confirm the presence of a coordinated alkoxide species, with values in very good agreement with the corresponding SCD structures (Table 3). The data is particularly resolute in the case of QD-Fe(III)PPIX, so much so that the solution EXAFS spectrum could be simultaneously refined together with the solid-state spectrum. Despite a lower precision in the fit for QN-Fe(III)PPIX in solution, the Fe–O bond length (1.85(3) Å) is within one standard deviation of the value resolved by SCD.
| QD-Fe(III)PPIX | QN-Fe(III)PPIX | MQ-Fe(III)PPIX | |||
|---|---|---|---|---|---|
| Solid-state | Solution | I | II | ||
| Fe–OSCD | 1.862(2) | 1.866(4) | 1.899(2) | 1.892(2) | |
| Fe–OEXAFS | 1.86(2) | 1.86(3) | 1.85(3) | 1.85(2) | |
Previously, changes in the UV-visible spectrum of Fe(III)PPIX in acetonitrile following titration with QN24 were shown to be consistent with those arising from a QN-Fe(III)PPIX coordination complex prepared in benzene.9 Coordination in the latter case was presumed to occur via the quinuclidine N atom. Based on SCD evidence however, the solution structure was reinterpreted, and coordination of QN to Fe(III)PPIX was proposed to occur in the same manner via the alkoxide.24 Indeed, in the current study, EXAFS spectroscopy has confirmed this to be the case. In order to obtain a quantitative measure of the coordinating ability of each drug to Fe(III)PPIX in acetonitrile, association constants for complexation (β1) were determined from spectrophotometric titrations (Table 4). The trend in binding strength reported for the three quinoline methanol antimalarial drugs to Fe(III)PPIX in 40% (v/v) aqueous DMSO5,26 is also observed in acetonitrile, with QD displaying the strongest interaction and MQ the weakest. However, the absolute values of the association constants are between five and twenty-four fold larger in acetonitrile. To further probe the relative stability of the coordination interaction observed in solution between the three drugs and Fe(III)PPIX, we considered the effect of a competing ligand, namely pyridine. This compound readily coordinates to free Fe(III)PPIX in solution to form a peach-coloured low-spin bis-pyridyl-Fe(III)PPIX complex.61,62 The latter species has a characteristic UV-visible spectrum identified by a sharp Soret peak at 405 nm and a distinctive spectroscopic envelope between 510 and 580 nm. The stepwise coordination of pyridine to Fe(III)PPIX in acetonitrile was monitored spectrophotometrically (Fig. S12,† a and b), and the resultant equilibrium constants for the processes described by eqn (2) and (3) were determined as 1.76 ± 0.07 (logβ2) and 2.83 ± 0.03 (logβ3), respectively. These represent a 10–17 fold enhancement over values obtained in aqueous DMSO.63 Having confirmed that pyridine indeed behaves as a coordinating ligand in acetonitrile, its ability to displace each quinoline methanol drug from the respective drug–Fe(III)PPIX complex was probed through competition titrations (Fig. S12, c–h†). These were conducted in two different ways. In the first case, increasing concentrations of pyridine were added to acetonitrile solutions of preformed drug–Fe(III)PPIX complexes. The alternative approach involved the titration of drug into an acetonitrile solution containing the preformed bis-pyridyl-Fe(III)PPIX complex. Both methods were expected to yield the same overall results, however differences in the fitted equilibrium constants were observed. These were attributed to the significant changes in solvent composition arising from the addition of relatively large volumes of neat pyridine required in the first method. Since the solvent composition of the titrant (drug solution) used in the second approach was the same as that of the working solution (see Experimental section for details), the results obtained are considered more reliable, and are therefore presented here. For comparative purposes however, the results obtained using the first approach are presented in the ESI (Table S10†).
The spectrophotometric titration data of the bis-pyridyl-Fe(III)PPIX complex, M(Py)2, with each of the quinoline methanol compounds could not be successfully fit using the speciation equilibria described by eqn (1)–(3) only. Rather, incorporation of an additional equilibrium process involving the formation of a unique six-coordinate Fe(III)PPIX species ligated by the drug and a single pyridine molecule (PyMD) was essential (see eqn (4)). In the fitting procedure, spectra of all Fe(III)PPIX species were summed together in varying ratios (that were governed by β values) to reproduce the experimental data at each wavelength. The spectrum of M, as well as those of MD and M(Py)2, were obtained from initial and endpoints, respectively, of the spectrophotometric titrations discussed above. The spectrum of MPy was predicted following analysis of the titration data for Fe(III)PPIX and pyridine. These four spectra together with their respective association constants (β1, β2 and β3) were constrained during the refinement of the data obtained from titration of M(Py)2 with drug. This left only two unknown parameters, namely the spectrum of PyMD and the association constant β4 (Table 4), which were optimized to reproduce the experimental data. The predicted UV-visible spectrum of PyMD is unique, and can thus be distinguished from all other Fe(III)PPIX species that may be present in the experiment (Fig. 4). In the Soret band region of PyMD, there is generally a decrease in the intensity of the peak relative to the bis-pyridyl-Fe(III)PPIX complex. The shoulder feature at shorter wavelength is somewhat more pronounced than in the case of M(Py)2, however is significantly less intense compared to MPy (see Fig. 4a). Compared to the spectra of the three MD species, the Soret band of PyMD in each case is redshifted by approximately 10 nm and displays a somewhat less intense shoulder feature at shorter wavelength. In the longer wavelength region (450–700 nm), the characteristic charge-transfer band at 600 nm in the spectra of MD is retained in the spectra of PyMD, however is consistently observed at a reduced intensity. Furthermore, the spectroscopic envelope that is characteristic of M(Py)2 is also observed in each case of PyMD, although this varies with intensity for the three different drugs. This feature is most prominent in the spectrum of PyMD arising from the MQ species, and least evident in the spectrum arising from the QD species.
 | ||
| Fig. 4 UV-visible spectra of Fe(III)PPIX species (10 μM) in acetonitrile solution following spectrophotometric titration. (a) The initial spectrum of Fe(III)PPIX (black solid line) and predicted spectra of mono- and bis-pyridyl-Fe(III)PPIX complexes (grey solid and grey dashed lines, respectively). (b–d) Spectra of five-coordinate drug–Fe(III)PPIX complexes (black solid lines) and predicted spectra of six-coordinate pyridine–Fe(III)PPIX–drug complexes (grey solid lines). In each case, the longer wavelength region has been expanded, and the spectrum of the bis-pyridyl–Fe(III)PPIX species included (black dashed line) for comparison. The drugs present in each case are quinidine (b), quinine (c) and mefloquine (d). | ||
The displacement of a single pyridine ligand from M(Py)2 by the quinoline methanol drug to form the observed PyMD species is given by eqn (5a), and the respective step-wise equilibrium constant by eqn (5b) (see the ESI† for the derivation). The association of pyridine with the five-coordinate drug–Fe(III)PPIX complex (MD) to yield PyMD is described by eqn (6a), and the equilibrium constant K2 is obtained from eqn (6b) (see the ESI† for the derivation).
 | (5a) |
| logK1 = logβ4 − logβ3 | (5b) |
 | (6a) |
| logK2 = logβ4 − logβ1 | (6b) |
The initial intention behind the spectrophotometric titrations was to probe the ability of pyridine to act as a competing ligand for the coordination site occupied by the alkoxide drug moiety in each MD complex. It is evident from the spectroscopic data and extracted equilibrium constants however, that this does not occur in the acetonitrile system. The large positive values determined for logK1 (Table 4) instead support the formation of a six-coordinate PyMD species over either MPy or M(Py)2. The values follow the trend observed for logβ1, which describe the formation of the three MD complexes, however they are approximately one order of magnitude weaker. The difference can be attributed to the necessary dissociation of one pyridine molecule from M(Py)2 before the drug is able to coordinate. The PyMD species presumably contains a single pyridine molecule bound to the unligated face of the five-coordinate drug–Fe(III)PPIX complexes. Indeed, the strength of this ligation by pyridine (described by eqn (6a)) is the same at the 95% confidence level as that found for the association of one pyridine molecule to Fe(III)PPIX in acetonitrile (logβ2 reported above). The moderate values of logK2 for this process (Table 4) are essentially the same irrespective of the drug present.
The UV-visible spectroscopic data above conclusively demonstrate that pyridine is unable to displace the coordinated quinoline methanol antimalarial drugs, at least in acetonitrile. This is not unexpected given a previous report, which showed that in the absence of proton competition (such as in acetonitrile), coordination to Fe(III)PPIX is dependent on the pKa of the ligand.63 The pKa of pyridine (5.2363) is lower than that of an alkoxide (predicted value for QN and QD = 13.8964), and hence coordination is expected to be stronger in the case of the latter. Indeed, a recent computational study has shown the alkoxide-iron(III) porphine interaction to be particularly favourable compared to coordination by neutral ligands.65 These current findings may have important implications for detergent- and lipid-based pyridine hemochrome assays that rely on the bis-coordination of free Fe(III)PPIX by pyridine to quantitate and evaluate the ability of compounds to inhibit the formation of β-haematin.17,62,66 We have shown in non-aqueous solution, at least for quinoline methanol antimalarial drugs, that the major Fe(III)PPIX species that exists in the presence of pyridine is not the anticipated bis-pyridyl complex, but rather the six-coordinate pyridine–Fe(III)PPIX-drug complex. The latter species has a unique UV-visible spectrum, which bears some resemblance to that of the low-spin bis-pyridyl–Fe(III)PPIX complex in the longer wavelength region. At first glance, this may be mistaken as an indication of the presence of the bis-pyridyl-Fe(III)PPIX species. If this were the case, the reduced intensity of the Soret band would be suggestive of a lower concentration of free Fe(III)PPIX. This in turn would result in a prospective inhibitor displaying a lower than actual activity. Follow-up studies using techniques such as electron paramagnetic resonance and nuclear magnetic resonance spectroscopy would most certainly be necessary in order to confirm the existence and structure of the six-coordinate PyMD species, however are beyond the scope of the current investigation.67,68
We have suggested previously, that owing to its absolute stereochemistry at C8 and C9, QD may be structurally preorganised compared to QN and therefore possess an inherent predisposition to form a complex with Fe(III)PPIX.25 The interaction is primarily facilitated by coordination, which we have shown occurs in both the solid-state and non-aqueous solution via the alkoxide zwitterionic form of the quinoline methanol antimalarial drug. An early study by Behere and Goff demonstrated that an aliphatic amine is able to promote the formation of an alkoxide–iron(III) porphyrin complex in aprotic solution by enabling proton transfer from an alcohol.21 Given however, that MQ hydrochloride and QD sulfate dihydrate were used in the current study, the aliphatic amine would not have been able to facilitate the proton transfer necessary for alkoxide formation in either case. While the quinoline nitrogen atom may have served as a basic site in the case of QD, this would not have been a viable base in the case of MQ owing to the steric hinderance of the –CF3 substituents on the quinoline ring of MQ. The only component in both experimental solutions to possess the free aliphatic amine functional group necessary to facilitate alkoxide formation would have been CQ free base. Almost three decades ago, Blauer reported reversible changes with pH in the circular dichroism spectra of Fe(III)PPIX in the presence of QD in aqueous solution. He attributed the changes to an ionisable functional group that would be of uttermost importance in the formation of the optically-active complexes, and considered the tertiary amine of the quinuclidine ring the most likely candidate.23 Following coordination, structural preorganisation almost certainly promotes the subsequent formation of the intramolecular charge-assisted hydrogen bond. As observed for each complex in the solid-state, the tertiary amine of the quinuclidine ring (QD and QN) and the secondary amine of the piperidine ring (MQ) are directly involved in this important interaction. Unfortunately, it has not been possible to confirm the presence of this hydrogen bond in the solution-state coordination complexes investigated in the current study.
Conclusions
The data reported herein provide insight into the speciation and drug–Fe(III)PPIX interactions that may be relevant in non-aqueous environments within the digestive vacuole of the malaria parasite, such as lipid bodies or membranes. The determination of the structure of MQ-Fe(III)PPIX supports the three point binding model that is evident in the solid-state structures of the Cinchona alkaloid complexes of Fe(III)PPIX, and we have been able to reconcile the differences in activity of the erythro isomers of MQ as a result. Previously, the difference in activity of (+) and (−) erythro MQ has been attributed to the orientation of the benzylic alcohol and terminal amine functional groups and their subsequent hydrogen bonding capacity relative to cellular constituents.43 The current study implicates Fe(III)PPIX as a feasible drug target, and infers that differences in activity are directly related to the ability of a compound to complex free Fe(III)PPIX. We have provided conclusive evidence that the latter process is facilitated by coordination of the quinoline methanol benzylic alcohol (alkoxide) and further involves intramolecular hydrogen bonding of the terminal amine. MS, EXAFS and UV-visible spectroscopic data confirm the coordination of the alkoxide species in non-aqueous solution, and while the existence of the intramolecular hydrogen bond that presumably forms following coordination has not been established in solution, we strongly suspect that it may be present. Having recovered single crystals of the MQ- and QD-Fe(III)PPIX complexes from solutions that contained CQ free base, which in the absence of MQ or QD is observed to promote μ-propionato (haemozoin) dimer formation, it is tempting to suggest that complex formation is the mechanism of haemozoin inhibition in the parasite. The evidence is however, insufficient. While drug–Fe(III)PPIX complex formation may contribute to the mechanism of inhibition, it is also plausible that the coordination complexes may be involved in subsequent toxicity effects, which ultimately lead to parasite death. In either case, the interactions identified in this study may yet prove to be important for the rational design of novel antimalarials.Acknowledgements
This material is based upon work supported in part by the National Research Foundation (NRF) of South Africa under the Thuthuka Programme (KdV, Grant no. 76320 and TlR, Grant no. 69103). Beamtime at the Australian Synchrotron was made possible following a successful application by VS (Ref AS131/XAS/5839). Travel by JG to and from the Australian Synchrotron to participate in the EXAFS experiments was made possible by a travel grant under the NRF Research Equipment Programme (Grant no. 87336). We gratefully acknowledge Professor Timothy Egan at the University of Cape Town for use of HypSpec software. Any opinions, findings, or conclusions expressed in this material are those of the authors and do not necessarily reflect the views of the NRF.Notes and references
- WHO, World malaria report, World Health Organisation, 2014 Search PubMed.
- T. J. Egan, J. M. Combrink, J. Egan, G. R. Hearne, H. M. Marques, S. Ntenteni, B. T. Sewell, P. J. Smith, D. Taylor, D. A. van Schalkwyk and J. C. Walden, Biochem. J., 2002, 365, 343–347 CrossRef CAS.
- A. C. Chou, R. Chevli and C. D. Fitch, Biochemistry, 1980, 19, 1543–1549 CrossRef CAS.
- A. F. G. Slater and A. Cerami, Nature, 1992, 355, 167–169 CrossRef CAS PubMed.
- T. J. Egan, W. W. Mavuso, D. C. Ross and H. M. Marques, J. Inorg. Biochem., 1997, 68, 137–145 CrossRef CAS.
- A. Dorn, S. R. Vippagunta, H. Matile, C. Jaquet, J. L. Vennerstrom and R. G. Ridley, Biochem. Pharmacol., 1998, 55, 727–736 CrossRef CAS.
- T. J. Egan, D. C. Ross and P. A. Adams, FEBS Lett., 1994, 352, 54–57 CrossRef CAS.
- C. D. Fitch, Life Sci., 2004, 74, 1957–1972 CrossRef CAS PubMed.
- D. C. Warhurst, Biochem. Pharmacol., 1981, 30, 3323–3327 CrossRef CAS.
- G. Blauer, Biochem. Int., 1988, 17, 729–734 CAS.
- Y. Sugioka and M. Suzuki, Biochim. Biophys. Acta, 1991, 1074, 19–24 CrossRef CAS.
- P. A. Adams, P. A. M. Berman, T. J. Egan, P. J. Marsh and J. Silver, J. Inorg. Biochem., 1996, 63, 69–77 CrossRef CAS.
- R. Buller, M. L. Peterson, Ö. Almarsson and L. Leiserowitz, Cryst. Growth Des., 2002, 2, 553–562 CAS.
- I. Solomonov, M. Osipova, Y. Feldman, C. Baehtz, K. Kjaer, I. K. Robinson, G. T. Webster, D. McNaughton, B. R. Wood, I. Weissbuch and L. Leiserowitz, J. Am. Chem. Soc., 2007, 129, 2615–2627 CrossRef CAS PubMed.
- D. J. Sullivan, Jr., I. Y. Gluzman, D. G. Russel and D. E. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11865–11870 CrossRef.
- D. J. Sullivan, Jr., H. Matile, R. G. Ridley and D. E. Goldberg, J. Biol. Chem., 1998, 273, 31103–31107 CrossRef PubMed.
- J. Gildenhuys, T. Le Roex, T. J. Egan and K. A. de Villiers, J. Am. Chem. Soc., 2013, 135, 1037–1047 CrossRef CAS PubMed.
- T. J. Egan, J. Inorg. Biochem., 2006, 100, 916–926 CrossRef CAS PubMed.
- K. A. de Villiers and T. J. Egan, in Handbook of Porphyrin Science, ed. G. C. Ferreira, K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co. Pte. Ltd., Singapore, 2014, vol. 27, pp. 211–254 Search PubMed.
- I. Constantinidis and J. D. Satterlee, J. Am. Chem. Soc., 1988, 110, 927–932 CrossRef CAS.
- D. V. Behere and H. M. Goff, J. Am. Chem. Soc., 1984, 106, 4945–4950 CrossRef CAS.
- G. Blauer, Arch. Biochem. Biophys., 1986, 251, 306–314 CrossRef CAS.
- G. Blauer, Arch. Biochem. Biophys., 1986, 251, 315–322 CrossRef CAS.
- K. A. de Villiers, J. Gildenhuys and T. le Roex, ACS Chem. Biol., 2012, 7, 666–671 CrossRef CAS PubMed.
- K. A. de Villiers, H. M. Marques and T. J. Egan, J. Inorg. Biochem., 2008, 102, 1660–1667 CrossRef CAS PubMed.
- T. J. Egan, E. Hempelmann and W. W. Mavuso, J. Inorg. Biochem., 1999, 73, 101–107 CrossRef CAS.
- D. C. Warhurst, J. C. Craig, I. S. Adagu, D. J. Meyer and S. Y. Lee, Malar. J., 2003, 2, 26 CrossRef PubMed.
- J. M. Karle, R. Olmeda, L. Gerena and W. K. Milhous, Exp. Parasitol., 1993, 76, 345–351 CrossRef CAS PubMed.
- T. J. Egan, J. Y.-J. Chen, K. A. de Villiers, T. E. Mabotha, K. J. Naidoo, K. K. Ncokazi, S. J. Langford, D. McNaughton, S. Pandiancherri and B. R. Wood, FEBS Lett., 2006, 580, 5105–5110 CrossRef CAS PubMed.
- J. M. Pisciotta, I. Coppens, A. K. Tripathi, P. F. Scholl, J. Shuman, S. Bajad, V. Shulaev and D. J. Sullivan, Jr., Biochem. J., 2007, 402, 197–204 CrossRef CAS PubMed.
- A. N. Hoang, K. K. Ncokazi, K. A. de Villiers, D. W. Wright and T. J. Egan, Dalton Trans., 2010, 39, 1235–1244 RSC.
- A. N. Hoang, R. D. Sandlin, A. Omar, T. J. Egan and D. W. Wright, Biochemistry, 2010, 49, 10107–10116 CrossRef CAS PubMed.
- S. Kapishnikov, T. Berthing, L. Hviid, M. Dierolf, A. Menzel, F. Pfeiffer, J. Als-Nielsen and L. Leiserowitz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11184–11187 CrossRef CAS PubMed.
- S. Kapishnikov, A. Weiner, E. Shimoni, P. Guttmann, G. Schneider, N. Dahan-Pasternak, R. Dzikowski, L. Leiserowitz and M. Elbaum, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11188–11193 CrossRef CAS PubMed.
- J. M. Pisciotta and D. J. Sullivan, Jr., Parasitol. Int., 2008, 57, 89–96 CrossRef CAS PubMed.
- P. J. Ellis and H. C. Freeman, J. Synchrotron Radiat., 1995, 2, 190–195 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. Ravel and M. Newville, Phys. Scr., T, 2005, 115, 1007–1010 CrossRef.
- M. Newville, P. Livins, Y. Yacoby, J. J. Rehr and E. A. Stern, Phys. Rev. B: Condens. Matter, 1993, 47, 14126–14131 CrossRef CAS.
- V. Streltsov, R. Dilanian, N. Klonis, E. Hanssen, H. Quiney and L. Tilley, in 10th Conference of the Asian Crystallographic Association, Busan, Korea, 2010.
- HypSpec, Protonic Software, Leeds, England Search PubMed.
- J. M. Karle and I. L. Karle, Antimicrob. Agents Chemother., 2002, 46, 1529–1534 CrossRef CAS.
- J. L. Hoard, M. J. Hamor, T. A. Hamor and W. S. Caughey, J. Am. Chem. Soc., 1965, 87, 2312–2319 CrossRef CAS.
- C. Lecomte, D. L. Chadwick, P. Coppens and E. D. Stevens, Inorg. Chem., 1983, 22, 2982–2992 CrossRef CAS.
- K. Hatano and T. Uno, Bull. Chem. Soc. Jpn., 1990, 63, 1825–1827 CrossRef CAS.
- M. R. Johnson, W. K. Seok, W. Ma, C. Slebodnick, K. M. Wilcoxen and J. A. Ibers, J. Org. Chem., 1996, 61, 3298–3303 CrossRef CAS.
- J. M. Karle, I. L. Karle, L. Gerena and W. K. Milhous, Antimicrob. Agents Chemother., 1992, 36, 1538–1544 CrossRef CAS.
- J. N. Alumasa, A. P. Gorka, L. B. Casabianca, E. Comstock, A. C. de Dios and P. D. Roepe, J. Inorg. Biochem., 2011, 105, 467–475 CrossRef CAS PubMed.
- N. Binsted, R. W. Strange and S. S. Hasnain, Biochemistry, 1992, 31, 12117–12125 CrossRef CAS.
- D. Kuter, V. Streltsov, N. Davydova, G. A. Venter, K. J. Naidoo and T. J. Egan, Inorg. Chem., 2014, 53, 10811–10824 CrossRef CAS PubMed.
- K. Dziedzik-Kocurek, J. Stanek and K. Burda, Hyperfine Interact., 2008, 185, 87–93 CrossRef.
- K. Dziedzik-Kocurek, D. Okla and J. Stanek, Nukleonika, 2013, 58, 7–11 Search PubMed.
- A. F. G. Slater, W. J. Swiggard, B. R. Orton, W. D. Flitter, D. E. Goldberg, A. Cerami and G. B. Henderson, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 325–329 CrossRef CAS.
- M. S. Walczak, K. Lawniczac-Jablonska, A. Sienkiewicz, I. N. Demchenko, E. Piskorska, G. Chatain and D. S. Bohle, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 238, 32–38 CrossRef CAS PubMed.
- M. S. Walczak, K. Lawniczak-Jablonska, A. Sienkiewicz, M. T. Klepka, L. Suarez, A. J. Kosar, M.-J. Bellemare and D. S. Bohle, J. Non-Cryst. Solids, 2010, 356, 1908–1913 CrossRef CAS PubMed.
- M. S. Walczak, K. Lawniczak-Jablonska, A. Wolska, A. Sienkiewicz, L. Suarez, A. J. Kosar and D. S. Bohle, J. Phys. Chem. B, 2011, 115, 1145–1150 CrossRef CAS PubMed.
- M. S. Walczak, K. Lawniczak-Jablonska, A. Wolska, M. Sikora, A. Sienkiewicz, L. Suarez, A. J. Kosar, M.-J. Bellemare and D. S. Bohle, J. Phys. Chem. B, 2011, 115, 4419–4426 CrossRef CAS PubMed.
- F. W. B. Einstein and A. C. Willis, Inorg. Chem., 1978, 17, 3040–3045 CrossRef CAS.
- P. Gans, G. Buisson, E. Duee, J. R. Regnard and J. C. Marchon, J. Chem. Soc., Chem. Commun., 1979, 393–395 RSC.
- J. H. A. Akoyunoglou, H. S. Olcott and W. D. Brown, Biochemistry, 1963, 2, 1033–1041 CrossRef CAS.
- K. K. Ncokazi and T. J. Egan, Anal. Biochem., 2005, 338, 306–319 CrossRef CAS PubMed.
- D. Kuter, K. Chibale and T. J. Egan, J. Inorg. Biochem., 2011, 105, 684–692 CrossRef CAS PubMed.
- MarvinSketch, ChemAxon Ltd., Budapest, 2011 Search PubMed.
- M. C. Durrant, Dalton Trans., 2014, 43, 9754–9765 RSC.
- R. D. Sandlin, M. D. Carter, P. J. Lee, J. M. Auschwitz, S. E. Leed, J. D. Johnson and D. W. Wright, Antimicrob. Agents Chemother., 2011, 55, 3363–3369 CrossRef CAS PubMed.
- F. A. Walker, Coord. Chem. Rev., 1999, 185–186, 471–534 CrossRef CAS.
- F. A. Walker, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 5, pp. 81–183 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SCD data tables and additional figures, as well as MS, EXAFS and UV-visible spectroscopic data tables and spectra are presented. The SCD structural data for MQ-Fe(III)PPIX and QD-Fe(III)PPIX determined in the current work have been deposited with the Cambridge Crystallographic Data Centre. CCDC 1057567 and 1057568. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02671g |
| This journal is © The Royal Society of Chemistry 2015 |