
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural diversity of bimetallic rhodium and iridium half sandwich dithiolato complexes†
Phillip S.
Nejman
,
Brian
Morton-Fernandez
,
David J.
Moulding
,
Kasun S.
Athukorala Arachchige
,
David B.
Cordes
,
Alexandra M. Z.
Slawin
,
Petr
Kilian
and
J. Derek
Woollins
*
University of St Andrews, EaStCHEM School of Chemistry, St Andrews, Fife KY16 9ST, UK. E-mail: jdw3@st-and.ac.uk
First published on 27th August 2015
Abstract
The synthesis of a range of rhodium(III) and iridium(III) half sandwich complexes with aryl dithiolato ligands of varying geometry and flexibility are reported. These include dinuclear [Cp*M(S–R–S)]2 complexes 3b and 4b, M = Rh, Ir; S–R–S = naphthalene-1,8-dithiolate (b) and four dinuclear complexes bearing bridging dithiolate ligands [(Cp*M)2(μ2-Cl)(μ2-S–R–S)]Cl 3c, 4c, 5b, 6b, M = Rh, Ir; S–R–S = naphthalene-1,8-dithiolate (b) or acenaphthene-5,6-dithiolate (c). The introduction of a less rigid biphenyl dithiolate backbone resulted in the tetranuclear dicationic complex [(Cp*Rh)4(S-R-S)3]Cl2 (3d), S–R–S = biphenyl-2,2′-dithiolate (d) with dithiolate ligands in two different bridging modes. All new complexes were fully characterised by multinuclear NMR, IR, Raman and MS spectroscopy and single crystal X-ray diffraction.
Introduction
The coordination of S,S bidentate ligands remains an important area of chemistry. Complexes bearing this type of ligand have a number of industrial applications including catalysts in vulcanisation1,2 and lubricant additives.3 These complexes also show a range of electrochemical properties.4,5 In addition S,S donors can support unusual magnetic properties6,7 and are important in biological systems.8 As part of our interest in the properties of sulfur donor systems we have investigated a series of dithiolate ligands containing polyaromatic backbones.The coordination chemistry of these types of ligands has seen little study compared to other dithiolates such as benzene-1,2-dithiolate or ethane-1,2-dithiolate. The notable exceptions to this being a series of publications by Teo in the late 1970s and early 1980s on the oxidative addition of tetrathionaphthalene (TTN), tetrachlorotetrathionaphthalene (TCTTN) and tetrathiotetracene (TTT) (Fig. 1) to a variety of low valent metal centres.9–16
 | ||
| Fig. 1 Structurally related naphthalene ligands. | ||
Compounds in which two metal centres are bridged by two sulfur atoms are of particular relevance to the work presented here as they give rise to M2S2 centres. Examples of these include the tetra iron species [{(CO)3Fe}2(TTN){Fe(CO)3}2] and the polymeric nickel [{Ni}2(TTN)]n and cobalt [{(CO)2Co}2(TTN)]n systems.10 Another example of a complex bearing this type of ligand is the unusual trinuclear nickel(II) species [Ni3(PPh3)3(S2C10Cl6)3], which was obtained by the oxidative addition of hexachlorodithionaphthalene (HCDTN) to [Ni(cod)2] (cod = 1,5-cyclooctadiene) in the presence of triphenylphosphine.17 This trinuclear structure is in contrast to the mononuclear square planar compounds [M(PPh3)2(HCDTN)] (M = Pd or Pt) obtained by reaction of [Pd(PPh3)3] or [Pt(PPh3)4] with the same ligand.17 There have also been examples of oligomeric, dimeric and monomeric zinc complexes with no co-ligands, with pyridine or with neocuproin, respectively, of sterically crowded and electron poor naphthalene-1,8-dithiolate derivatives.18
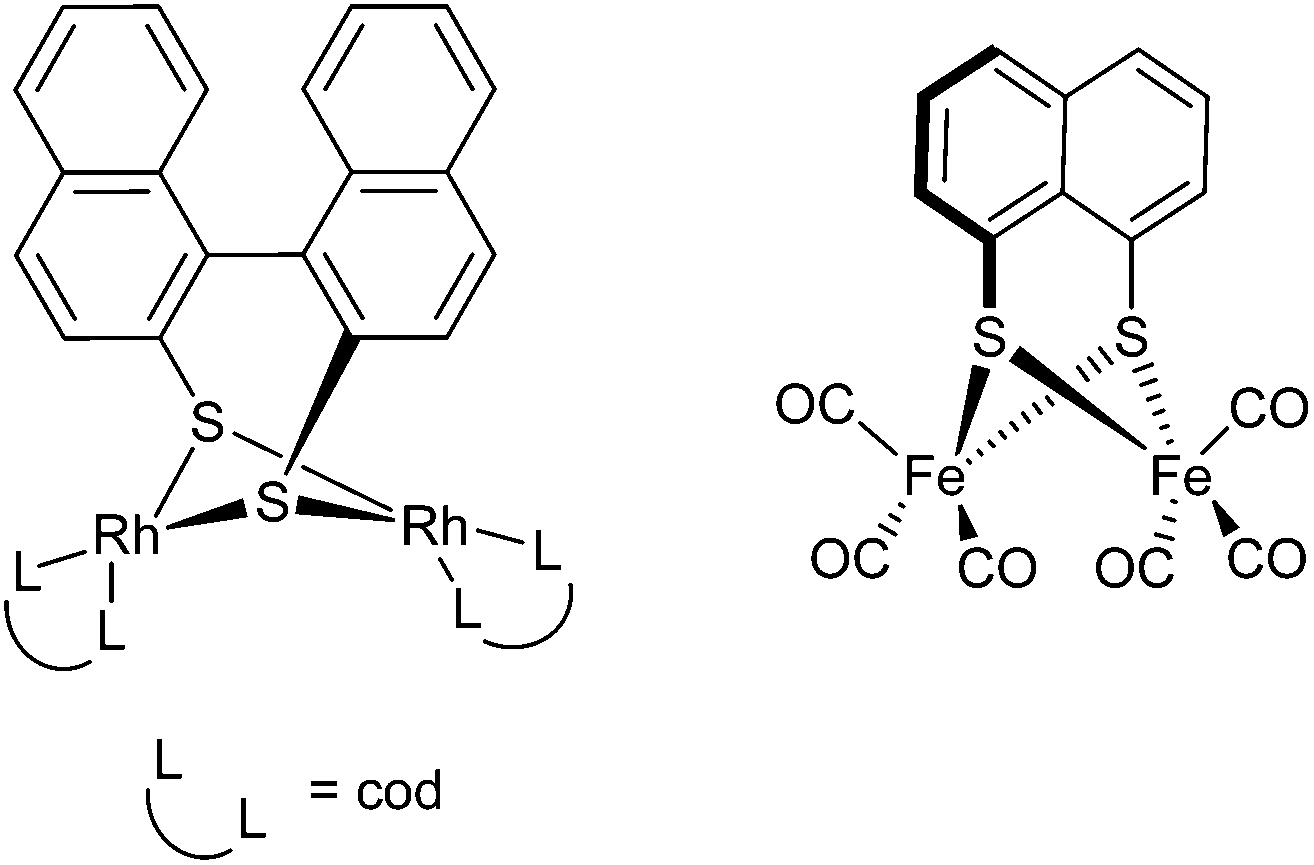
A number of complexes containing the 1,1′-binaphthalene-2,2′-dithiolate ligand have been prepared from metathesis reactions, for example, by reaction of the dithiol with [Rh(μ-OMe)(cod)]2 to give a dinuclear complex with a bridging disulfide ligand (Fig. 3).19 In many of these reactions the purpose has been to develop complexes for catalytic polymerisation reactions such as the regioselective hydroformylation of styrene. Complexes containing the ligand 4,4′-biphenanthrene-3,3′-dithiolate have also been shown to react with carbon monoxide to give interesting dinuclear tetracarbonyl complexes and with PR3 (R = Ph, C6H11, OC6H4(o-tBu)) to give mixed ligand di- and tetranuclear complexes.20,21 Ruiz and co-workers have also produced a palladium dimer complex using the mixed thiol and thio-ether ligand. The dimer was shown to convert to a monomer on addition of triphenylphosphine.22 More recently there has been interest in using naphthalene-1,8-dithiolate and 1,1′-biphenyl-2,2′-dithiolate iron complexes as electron transfer catalysts designed to mimic iron hydrogenases (Fig. 3).23–28
The coordination chemistry of the structurally related acenaphthene-5,6-dithiolate motif is less well documented with only one example of a complex incorporating this type of ligand out with our research. Topf and co-workers have used the acenaphthene backbone as a linker between a 1,2-diimine unit and a dithiolate binding site.29 The iron carbonyl complex bearing this ligand was shown to have potential as a multielectron transfer photosensitiser for artificial photosynthesis and as a bio-inspired photoredox catalyst.29 Apart from electron transfer mimics there has been little study on complexes bearing the 1,1′-biphenyl-2,2′-dithiolate ligand. A derivatised version of dibenzo[c,e]-1,2-dithiin has been bound to copper30 with a molybdenum complex also known.31,32
Herein we describe the synthesis of a series of rhodium(III) and iridium(III) half sandwich dithiolato complexes. The complexes have been characterised, principally by multinuclear NMR spectroscopy and single crystal X-ray diffraction. Tuning of the reaction conditions allowed investigation into the different binding modes of the dithiolate ligands.
Results and discussion
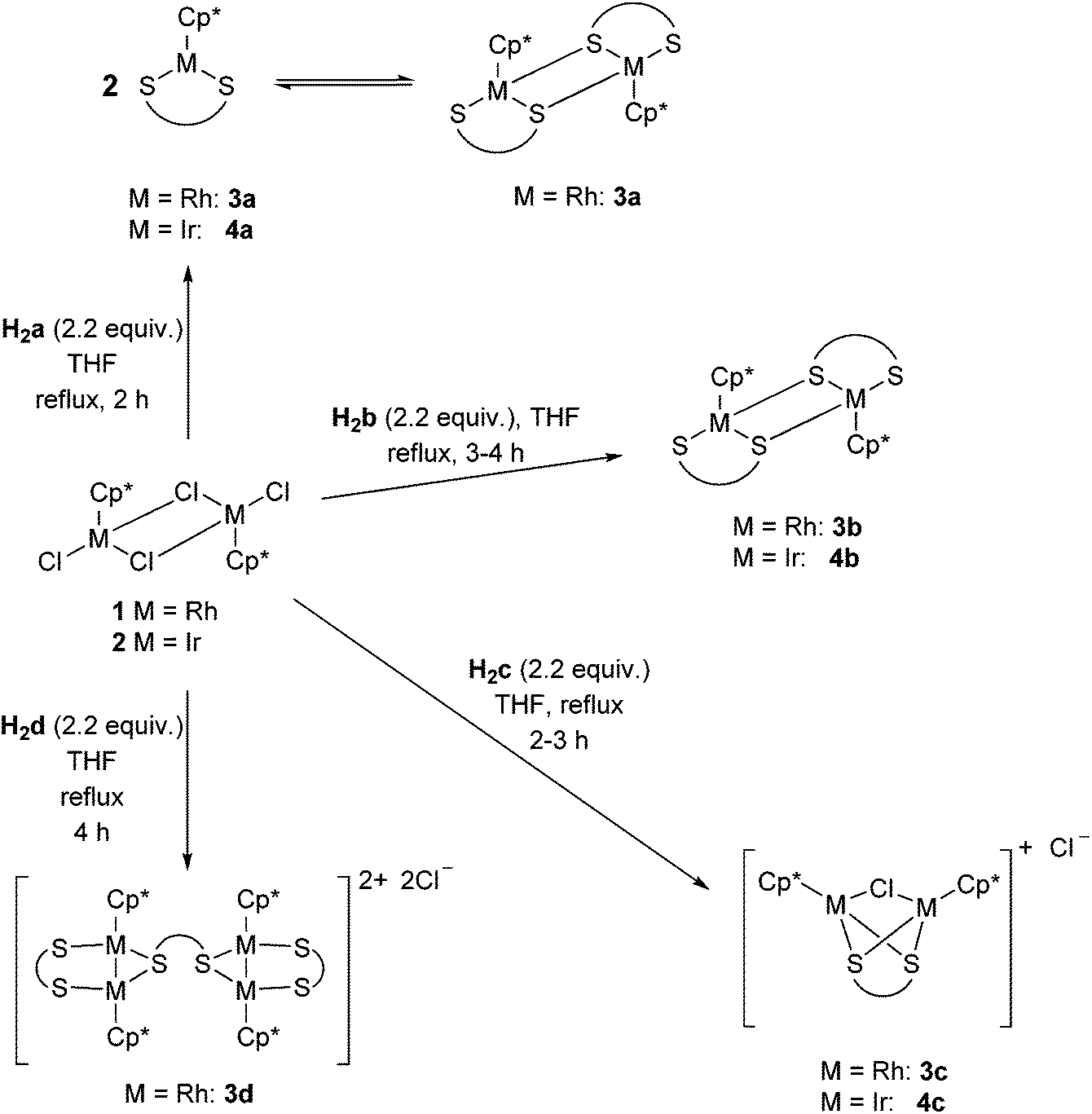
The diprotic proligands benzene-1,2-dithiol (H2a), naphthalene-1,8-dithiol (H2b), acenaphthene-5,6-dithiol (H2c) and 1,1′-biphenyl-2,2′-dithiol (H2d) (Fig. 2) were prepared following literature procedures.33–37 The half sandwich complexes 1 and 2 were also prepared following literature procedures.38 The syntheses of complexes 3a–d and 4a–c are shown in Scheme 1. The metathesis of the chloride ligands in 1 and 2 with the dithiolato ligands proceeds smoothly in refluxing THF with elimination of HCl. New complexes 3b–d and 4b were isolated in 40–84% yields. However, the iridium complex 4c was obtained in only a 2% yield using this method. Improved yields for the formation of 3c (83%) and 4c (98%) were obtained using a different method discussed below and outlined in Scheme 2. In all cases purification was performed by column chromatography on silica using either dichloromethane or a dichloromethane/methanol (or ethanol) mixture. | ||
| Fig. 2 Dithiol proligands studied in this work. | ||
 | ||
| Fig. 3 Examples of catalysts bearing dithiolato ligands [asymmetric hydroformylation (left) and electron transfer catalyst (right)]. | ||
 | ||
| Scheme 1 Reaction scheme for the preparation of complexes 3a–d and 4a–c. | ||
 | ||
| Scheme 2 Alternative reaction scheme for the preparation of 3c and 4c and the formation of 5b and 6b. | ||
In the work by Xi and co-workers 3a and 4a were prepared by the addition of a methanol solution of 1 or 2 to a methanol solution containing H2a and sodium methoxide at room temperature for 6 hours (3a) or 15 hours (4a).39 We found that heating 1 or 2 with proligand H2a in THF under reflux for 2 hours, followed by purification as above, resulted in comparable yields to those previously reported.
Since the 1H, 13C{1H} NMR and structural data for 3a and 4a were reported earlier, we have limited our discussion to complexes 3b–d, 4b/c, 5b and 6b. The 1H NMR data (CDCl3) for complexes 3b and 4b show the Cp* signal shifted upfield (Δδ = 0.45 (3b), 0.36 (4b) ppm) compared to precursors 1 and 2, respectively, and six distinct aromatic signals from the naphthalene backbone (8.14–7.09 ppm). Both of these dimeric complexes proved to be stable in solution over a period of several days as the 1H NMR spectrum showed no additional peaks which would correspond to the monomer after this time. This is in contrast to 3a, which exists in both the mono and dimeric form in solution, and 4a, which is a stable 16 electron species showing no dimeric form in solution.39 The 13C{1H} NMR data (CDCl3) mirror the proton NMR spectra for 3b and 4b with 1JC–Rh coupling (5.7 Hz) observed in complex 3b for the quaternary Cp* carbons. APCI mass spectra show both the [M + H]+ and [½M + H]+ signals for the complexes 3b and 4b, with purity of these two complexes confirmed by means of elemental analysis. Accurate elemental analysis was also obtained for 3a and 4a to show our synthetic method also resulted in pure material.
Despite the proligands H2b and H2c being closely related (both sterically and electronically), reactions with the latter gave complexes of different connectivity. Thus, the reaction of 1 or 2 with H2c produced a set of cationic complexes, 3c and 4c, where the dithiolate acts as a bridging ligand replacing two of the chloride ligands. One bridging chloride remains whilst the final chloride acts as the counter ion. A similar outcome was observed when the biphenyl ligand (H2d) was used, namely the tetranuclear complex 3d. In this case the bridging chloride was replaced with another biphenyl ligand, which, due to its rotational flexibility allowed two of the binuclear moieties to be joined. The 1H NMR data (CDCl3) showed the expected upfield shift of the Cp* signal, consistent with coordination of the thiolate ligand. For 3d two Cp* signals (2 × 30H) were observed which is likely due to the restricted rotation around the aryl–aryl bond of the bridging biphenyl ligand. The aromatic backbone of 3c and 4c showed two signals in the range of 8.37–7.31 ppm, whilst in 3d multiplets were observed due to overlapping signals. The 13C{1H} NMR spectra of 3c/d and 4c (CDCl3) again mirror the proton NMR spectra with marginally larger carbon–rhodium coupling observed (1JC–Rh = 6.5–7.4 Hz) for 3c and 3d than in our previous complex 3b. The [M − Cl]+ (3c, 4c) and [M − 2Cl]2+ (3d) fragments were observed in the ES mass spectra, purity was confirmed by elemental analysis for 3c and 4c.
Given the similarity between proligands H2b and H2c, further investigation into forming the dimeric analogues to 3b and 4b (with completely displaced Cl ligands) was performed. We followed the procedure employed by Xi and co-workers using sodium methoxide in methanol at room temperature, however, this still resulted in the formation of complexes 3c and 4c (Scheme 2). Further attempts at higher temperatures (refluxing for between 7 and 48 hours) were carried out to try and drive the reaction forward to the dimeric complex. In all cases the cationic complexes 3c and 4c were the only products observed by 1H NMR spectroscopy. The conditions shown in Scheme 2 gave the highest yields for 3c and 4c of 83% and 98% respectively. In light of this attempts at selectively forming the complex containing a bridging dithiolate ligand were made with H2b (Scheme 2). From these reactions we isolated both 5b and the dimeric complex 3b in a 58% and 14% yield, respectively, as well as 6b (75%) and the dimeric complex 4b (7%). The 1H and 13C{1H} NMR spectra of the new complexes 5b and 6b match the proposed structure with further confirmation obtained by means of elemental analysis and ES mass spectrometry.
X-ray crystallography
The crystal structures of complexes 3b–d, 4b–c, 5b and 6b are shown in Fig. 4 and 5 with selected structural parameters shown in Tables 1 and 2. The X-ray analyses show that in every example the metal centre adopts a piano stool geometry. Analysis of the single crystal structures of 5b and 6b (which are analogous to 3b and 4b) is not included due to the disorder in the Cp* rings/solvent. We include the data only to confirm the connectivity of the complexes. | ||
| Fig. 4 Crystal structures of 3b (Top left), 4b (Top right), 3c (Bottom left) and 4c (Bottom right). Water molecules and chloride counter ions from 3c and 4c are omitted for clarity. Hydrogen atoms are omitted from all structures for clarity. | ||
 | ||
| Fig. 5 Crystal structures of 3d (Top), 5b (Bottom left) and 6b (Bottom right). The Cp* rings of 6b have been wire framed for clarity. Solvent molecules and chloride counter ions from 5b and 6b are omitted for clarity. Hydrogen atoms are omitted from all structures for clarity. | ||
| 3b | 3c | 4b | 4c | |
|---|---|---|---|---|
| a Splay angle = [(S(1)–C(1)–C(10))+(C(1)–C(10)–C(9))+(C(10)–C(9)–S(9))] − 360. | ||||
| M1–S1 | 2.343(2) | 2.3739(6) | 2.3221(7) | 2.3844(8) |
| M1–S9 | 2.373(2) | 2.3708(7) | 2.3541(7) | 2.3937(8) |
| M1–S1′ | 2.425(2) | — | 2.3927(7) | — |
| M1–Cl1 | — | 2.4754(6) | — | 2.4790(8) |
| M2–S1 | — | 2.3820(7) | — | 2.3855(9) |
| M2–S9 | — | 2.3718(6) | — | 2.3864(7) |
| M2–Cl2 | — | 2.4786(7) | — | 2.4761(8) |
| S1–M1–S9 | 87.56(7) | 80.08(2) | 88.05(2) | 78.60(3) |
| S1–M1–S1′ | 81.71(7) | — | 79.70(2) | — |
| S1–M1–Cl1 | — | 80.81(2) | — | 78.21(3) |
| S1–M2–S9 | — | 79.90(2) | — | 78.72(3) |
| S1–M2–Cl1 | — | 80.58(2) | — | 78.25(3) |
| S9–M1–S1′ | 79.39(6) | — | 78.94(2) | — |
| S9–M1–Cl1 | — | 80.96(2) | — | 78.27(2) |
| S9–M2–Cl1 | — | 80.88(2) | — | 78.46(2) |
| Splay anglea | 20.8(6) | 15.7(2) | 21.4(2) | 14.5(3) |
| S1–C1⋯C9–S9 | −11.3(4) | 2.7(1) | −11.1(2) | 2.6(2) |
| C1–C10–C5–C6 | 179.9(6) | 178.4(2) | 179.4(3) | 179.8(3) |
| C9–C10–C5–C4 | −175.4(6) | 179.1(2) | 175.1(3) | 178.4(3) |
| 3d | |
|---|---|
| M1–S1 | 2.366(2) |
| M1–S8 | 2.336(2) |
| M1–S41 | 2.466(2) |
| M2–S1 | 2.343(2) |
| M2–S8 | 2.383(2) |
| M2–S41 | 2.449(2) |
| S1–M1–S8 | 80.09(5) |
| S1–M1–S41 | 74.67(5) |
| S1–M2–S8 | 79.58(5) |
| S1–M2–S41 | 75.41(5) |
| S8–M1–S41 | 79.16(5) |
| S8–M2–S41 | 78.61(5) |
| C1–C2–C7–C8 | −28(1) |
| C41–C42–C42′–C41′ | 69.2(9) |
In the case of 3b and 4b the coordination sphere of the metal is completed by S1 acting as a μ3-bridging atom resulting in a dimeric 18e complex. The M1–S1 bond lengths [3b 2.343(2) Å; 4b 2.3221(7) Å] and M1–S9 bond lengths [3b 2.373(2) Å; 4b 2.3541(7) Å] are comparable to other compounds of this type.40–42 The M1–S1′ bond length is marginally longer than the other M–S bonds in both 3b and 4b as one of the sulfur atoms forms another dative bond. The M1–S1, M1–S9 and M1–S1′ bond lengths in 3b and 4b are related to M1′–S1′, M1′–S9′ and M1′–S1 through a crystallographically imposed centre of symmetry. The peri S⋯S distance has increased compared to the pro-ligand H2b [2.951(2) Å]37 for 3b [3.264(3) Å] and 4b [3.250(1) Å] as the metal centre bridges the peri positions. All the non-Cp* angles around the metal centre are reduced to less than 90° ranging from 79.40(7)°−87.57(7)° in 3b and 78.94(2)°−88.05(2)° in 4b. This is a result of the naphthalene backbone restricting the position of the sulfur atoms meaning a more idealised geometry is unattainable. In this instance the splay angles are both large and positive [3b 20.7(7)°, 4b 21.4(2)°] showing the effect of the metal forcing the sulfur atoms apart. Both 3b and 4b have comparable S1–C1⋯C9–S9 torsion angles (≈11°) and show distinct buckling of the central naphthalene ring system.
The metal–sulfur bond lengths in the ionic complexes 3c [2.3708(7)–2.3820(7) Å] and 4c [2.383(1)–2.394(1) Å] are similar to other complexes bearing bridging dithiolate ligands.43 The bridging metal–chlorine bond lengths are slightly elongated compared to the starting complexes 1 and 2.44,45 All the non-Cp* angles around the metal are reduced to less than 90° [3c 79.90(7)–80.96(7)° and 4c 78.17(4)–78.56(4)°] and show less variation compared to 3b and 4b. The splay angles are both positive, 3c 15.7(2)°; 4c 14.6(3)°, as the sulfur atoms bridge the two metal centres. Less strain on the backbone is observed in 3c and 4c with the S1–C1⋯C9–S9 torsion and central C1–C10–C5–C6 and C9–C10–C5–C4 ring torsions lower than those seen previously.
The ionic tetranuclear complex 3d contains two distinct Rh–S bond lengths from the terminal and bridging ligands. The terminal ligand has Rh–S bond lengths ranging from 2.336(2)–2.383(2) Å which are similar to those we have previously observed in 3c. However the bridging ligand shows Rh–S bond lengths in the range of 2.449(2)–2.466(2) Å which are similar to lengths observed in 3b. Unlike the other charged complexes presented here the non-Cp* angles around the metal centre show a wide range [74.67(5)°–80.09(5)°]. This is likely due to the steric demands of the biphenyl backbone preventing the sulfur atom (S41) from adopting a more idealised position. The aryl–aryl torsion on the terminal ligand is 69.2(9)° which is similar to that observed for the pro-ligand.37 In contrast the aryl–aryl torsion of the bridging ligand is −28(1)° as the ligand chelates the two metal centres.
Conclusion
We have prepared and characterised a series of Rh(III) and Ir(III) η5-Cp* half sandwich complexes by chloride ligand replacement reactions of [Cp*RhCl2]2 and [Cp*IrCl2]2 with a series of dithiols with aromatic backbones. This work demonstrates the utility and versatility of these sulfur ligands in organometallic complexes. The ligands have shown remarkable variety in the type of complexes formed. A subtle change in the organic backbone (naphthalene to acenaphthene) resulted in a profound difference in the structure of the complex formed. In addition the introduction of rotationally free backbone produced yet another type of structure. Single crystal X-ray diffraction confirmed these three distinct complex classes; such a variety is achieved through the utilisation of κ1 and κ2 bonding of the sulfur donor atoms and via chelating and bridging coordination modes of the dithiolate ligands.Experimental
General
Unless otherwise stated all manipulations were performed under an oxygen-free nitrogen atmosphere using standard Schlenk techniques and glassware. Solvents were collected from an MBraun Solvent Purification System or dried and stored according to common procedures.46 [Cp*RhCl2]2 and [Cp*IrCl2]2 were prepared following a literature procedure which is included in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 The synthesis of [Cp*Rh(o-C6H4S2)]2 and [Cp*Ir(o-C6H4S2)] using our method based on literature preparation39 is reported in the ESI.† The disulfide precursors were made according to literature methods.34–36 Proligands H2b–d were prepared following a modified literature procedure,37,47H2a was prepared according to literature.33,48 IR and Raman spectra were collected on a Perkin Elmer 2000 NIR/Raman Fourier Transform spectrometer with a dipole pumped NdYAG near-IR excitation laser. 1H and 13C NMR spectra were obtained on either a Bruker Avance III 500 spectrometer or a JEOL GSX Delta 270 with δH & δC relative to TMS, residual solvent peaks (CDCl3; δH 7.26, δC 77.2 ppm) were used for calibration. All measurements were performed at 25 °C with shifts reported in ppm; pt has been used to denote a pseudo triplet. Electrospray (ES+) mass spectra were carried out by the University of St Andrews Mass Spectrometry service and Atmospheric Pressure Chemical Ionisation (APCI+) by the EPSRC National Mass Spectrometry Service, Swansea. Elemental analyses were performed by Stephen Boyer at the London Metropolitan University.
38 The synthesis of [Cp*Rh(o-C6H4S2)]2 and [Cp*Ir(o-C6H4S2)] using our method based on literature preparation39 is reported in the ESI.† The disulfide precursors were made according to literature methods.34–36 Proligands H2b–d were prepared following a modified literature procedure,37,47H2a was prepared according to literature.33,48 IR and Raman spectra were collected on a Perkin Elmer 2000 NIR/Raman Fourier Transform spectrometer with a dipole pumped NdYAG near-IR excitation laser. 1H and 13C NMR spectra were obtained on either a Bruker Avance III 500 spectrometer or a JEOL GSX Delta 270 with δH & δC relative to TMS, residual solvent peaks (CDCl3; δH 7.26, δC 77.2 ppm) were used for calibration. All measurements were performed at 25 °C with shifts reported in ppm; pt has been used to denote a pseudo triplet. Electrospray (ES+) mass spectra were carried out by the University of St Andrews Mass Spectrometry service and Atmospheric Pressure Chemical Ionisation (APCI+) by the EPSRC National Mass Spectrometry Service, Swansea. Elemental analyses were performed by Stephen Boyer at the London Metropolitan University.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.78 (2 H, d, 3JHH = 7.2 Hz, Ar–), 7.70 (2 H, d, 3JHH = 7.8 Hz, Ar–), 7.50 (2 H, d, 3JHH = 7.8 Hz, Ar–), 7.17 (2 H, pt, 3JHH = 7.4 Hz, Ar–), 7.14 (2 H, pt, 3JHH = 7.5 Hz, Ar–), 1.17 (30 H, s, C–C
), 7.78 (2 H, d, 3JHH = 7.2 Hz, Ar–), 7.70 (2 H, d, 3JHH = 7.8 Hz, Ar–), 7.50 (2 H, d, 3JHH = 7.8 Hz, Ar–), 7.17 (2 H, pt, 3JHH = 7.4 Hz, Ar–), 7.14 (2 H, pt, 3JHH = 7.5 Hz, Ar–), 1.17 (30 H, s, C–C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ). 13C NMR (125 MHz, CDCl3): δ = 141.1 (Cq, Ar–
). 13C NMR (125 MHz, CDCl3): δ = 141.1 (Cq, Ar–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ), 136.1 (Cq, Ar–), 135.5 (Cq, Ar–), 132.0 (CH, Ar–), 130.5 (Cq, Ar–), 129.3 (CH, Ar–), 129.2 (CH, Ar–), 124.9 (CH, Ar–), 124.7 (CH, Ar–), 123.3 (CH, Ar–), 96.5 (Cq, d, 1JCRh = 5.7 Hz, –CH3), 8.0 (C–H3). HRMS (APCI+): m/z Calcd for C40H43S4Rh2: 857.0352, found: 857.0359 (M + H, 25%); Calcd for C20H22S2Rh: 429.0217, found 429.0215 (½M + H, 100).
), 7.76 (2 H, dd, 3JHH = 7.2 Hz, 4JHH = 1.2 Hz, Ar–), 7.73 (2 H, d, 3JHH = 8.1 Hz, Ar–), 7.50 (2 H, d, 3JHH = 8.1 Hz, Ar–), 7.12 (2 H, pt, 3JHH = 7.6 Hz, Ar–), 7.09 (2 H, pt, 3JHH = 7.6 Hz, Ar–), 1.22 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 137.6 (Cq, Ar–), 135.7 (Cq, Ar–), 130.2 (CH, Ar–), 129.2 (CH, Ar–), 128.4 (CH, Ar–), 127.4 (Cq, Ar–), 125.0 (CH, Ar–), 124.0 (CH, Ar–), 123.5 (Cq, Ar–), 123.3 (CH, Ar–), 91.1 (Cq, –CH3), 7.7 (C–H3). HRMS (APCI+): m/z Calcd for C40H43S4Ir2: 1035.1475, found 1035.1479 (M + H, 20%); Calcd for C20H22S2Ir: 519.0792, found 519.0781 (½M + H, 100).
), 136.1 (Cq, Ar–), 135.5 (Cq, Ar–), 132.0 (CH, Ar–), 130.5 (Cq, Ar–), 129.3 (CH, Ar–), 129.2 (CH, Ar–), 124.9 (CH, Ar–), 124.7 (CH, Ar–), 123.3 (CH, Ar–), 96.5 (Cq, d, 1JCRh = 5.7 Hz, –CH3), 8.0 (C–H3). HRMS (APCI+): m/z Calcd for C40H43S4Rh2: 857.0352, found: 857.0359 (M + H, 25%); Calcd for C20H22S2Rh: 429.0217, found 429.0215 (½M + H, 100).
), 7.76 (2 H, dd, 3JHH = 7.2 Hz, 4JHH = 1.2 Hz, Ar–), 7.73 (2 H, d, 3JHH = 8.1 Hz, Ar–), 7.50 (2 H, d, 3JHH = 8.1 Hz, Ar–), 7.12 (2 H, pt, 3JHH = 7.6 Hz, Ar–), 7.09 (2 H, pt, 3JHH = 7.6 Hz, Ar–), 1.22 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 137.6 (Cq, Ar–), 135.7 (Cq, Ar–), 130.2 (CH, Ar–), 129.2 (CH, Ar–), 128.4 (CH, Ar–), 127.4 (Cq, Ar–), 125.0 (CH, Ar–), 124.0 (CH, Ar–), 123.5 (Cq, Ar–), 123.3 (CH, Ar–), 91.1 (Cq, –CH3), 7.7 (C–H3). HRMS (APCI+): m/z Calcd for C40H43S4Ir2: 1035.1475, found 1035.1479 (M + H, 20%); Calcd for C20H22S2Ir: 519.0792, found 519.0781 (½M + H, 100).
Method 2: A MeOH (25 mL) solution containing [Cp*RhCl2]2 (100 mg, 0.16 mmol), H2c (70 mg, 0.32 mmol) and NaOMe (17 mg, 0.32 mmol) was stirred at room temperature O/N. The solvent was removed and the crude product purified by column chromatography (silica/CH2Cl2:MeOH (9:1)). 3c was obtained (101 mg, 0.13 mmol, 83%). Crystals suitable for X-ray work were obtained from CH2Cl2/ether. Anal. calcd for C32H38Cl2Rh2S2 (761.99 g mol−1): C, 50.34; H, 5.01. Found: C, 50.12; H, 4.71. IR (KBr): νmax/cm−1 2918m (νC–H), 1591m, 1444s, 1376s, 1353s, 1024s, 733m. Raman (glass capillary): νmax/cm−1 3048w (νAr–H), 2919s (νC–H), 1592s, 1407s, 588s (νC–S), 460s, 430s, 416s, 269w (νRh–Cl). 1H NMR (500 MHz, CDCl3): δ = 8.37 (2 H, d, 3JHH = 7.2 Hz, Ar–), 7.45 (2 H, d, 3JHH = 7.2 Hz, Ar–), 3.37 (4 H, s, C![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) –C), 1.23 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 150.0 (Cq, Ar–), 139.8 (Cq, Ar–), 132.2 (CH, Ar–), 128.0 (Cq, Ar–), 124.5 (Cq, Ar–), 120.1 (CH, Ar–), 97.0 (Cq, d, 1JCRh = 7.4 Hz, –CH3), 30.8 (H2–H2), 8.2 (C–H3). MS (ES+): m/z 727.02 (M − Cl, 100%).
–C), 1.23 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 150.0 (Cq, Ar–), 139.8 (Cq, Ar–), 132.2 (CH, Ar–), 128.0 (Cq, Ar–), 124.5 (Cq, Ar–), 120.1 (CH, Ar–), 97.0 (Cq, d, 1JCRh = 7.4 Hz, –CH3), 30.8 (H2–H2), 8.2 (C–H3). MS (ES+): m/z 727.02 (M − Cl, 100%).
Method 2: This was prepared as per method 2 complex 3c using [Cp*IrCl2]2 (150 mg, 0.18 mmol), H2c (79 mg, 0.36 mmol) and NaOMe (20 mg, 0.36 mmol). 4c was obtained (174 mg, 0.18 mmol, 98%). Crystals suitable for X-ray work were obtained from CH2Cl2/ether. Anal. calcd for C32H38Cl2Ir2S2 (942.10 g mol−1): C, 40.75; H, 4.07. Found: C, 40.67; H, 4.12. IR (KBr): νmax/cm−1 3132m (νAr–H), 2918m (νC–H), 1592m, 1452s, 1355s, 1214m, 1030s, 860m. Raman (glass capillary): νmax/cm−1 2920s (νC–H), 1593m, 1408s, 1344m, 584m (νC–S), 430s. 1H NMR (500 MHz, CDCl3): δ = 8.27 (2 H, d, 3JHH = 7.1 Hz, Ar–), 7.31 (2 H, d, 3JHH = 7.1 Hz, Ar–), 3.21 (4 H, s, C–C), 1.25 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 148.7 (Cq, Ar–), 140.0 (Cq, Ar–), 129.3 (CH, Ar–), 128.0 (Cq, Ar–), 122.2 (Cq, Ar–), 120.3 (CH, Ar–), 90.0 (Cq, –CH3), 30.8 (H2–H2), 8.0 (C–H3). HRMS (ES+): m/z Calcd for C22H23ClS2Ir: 907.1362, found 907.1316 (M − Cl, 100%).
:EtOH (9:1)) to afford 3d as an orange solid (155 mg, 0.096 mmol, 40%). Crystals suitable for X-ray work were obtained from CH2Cl2/ether. IR (KBr): νmax/cm−1 3047w (νAr–H), 2917w (νC–H), 1452s, 1376m, 1021s, 754s, 495w. Raman (glass capillary): νmax/cm−1 3054m (νAr–H), 2916s (νC–H), 1582s, 1426m, 1300m, 1041s, 437m, 415s. 1H NMR (500 MHz, CDCl3): δ = 8.32 (2 H, dd, 3JHH = 7.89, 4JHH = 1.24 Hz, Ar–), 8.07 (2 H, dd, 3JHH = 7.83, 4JHH = 1.24 Hz, Ar–), 8.02–7.97 (2 H, m, Ar–), 7.74 (4 H, t, 3JHH = 8.60 Hz, Ar–), 7.53–7.47 (4 H, m, Ar–), 7.46–7.26 (10 H, m, Ar–), 1.20 (30 H, s, C–C), 1.17 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 138.5 (Cq, Ar–), 138.4 (Cq, Ar–), 137.4 (CH, Ar–), 137.1 (CH, Ar–), 136.7 (CH, Ar–), 135.9 (Cq, Ar–), 132.8 (CH, Ar–), 132.7 (CH, Ar–), 131.7 (CH, Ar–), 130.4 (CH, Ar–), 128.9 (CH, Ar–), 128.5 (CH, Ar–), 128.4 (CH, Ar–), 128.3 (CH, Ar–), 126.3 (CH, Ar–), 126.0 (CH, Ar–), 125.2 (Cq, Ar–), 124.8 (CH, Ar–), 98.6 (Cq, d, 1JCRh = 6.7 Hz, –CH3), 98.4 (Cq, d, 1JCRh = 6.5 Hz, –CH3), 8.7 (C–H3), 8.6 (C–H3). MS (ES+): m/z 909.06 (M − C32H37Rh2S2, 100%), 455.04 (M − C54H60Rh3S4, 20).
:MeOH (9:1)) to afford 5b as a red solid (102 mg, 0.13 mmol, 58%). Crystals suitable for X-ray work were obtained from CH2Cl2/ether. Anal. calcd for C30H36Cl2Rh2S2 (735.97 g mol−1): C, 48.91; H, 4.93. Found: C, 48.83; H, 5.04. IR (KBr): νmax/cm−1 2979w (νC–H), 2918m (νC–H), 1625m, 1493s, 1450s, 1377s, 1079m, 1023s, 832s, 769m. Raman (glass capillary): νmax/cm−1 3065w (νAr–H), 2919s (νC–H), 1546s, 894m, 589m (νC–S), 460m, 430s, 322m 1H NMR (500 MHz, CDCl3): δ = 8.44 (2 H, dd, 3JHH = 7.2 Hz, 4JHH = 1.2 Hz, Ar–), 8.13 (2 H, dd, 3JHH = 8.2 Hz, 1.1 Hz, Ar–), 7.59 (2 H, dd, 3JHH = 8.2, 7.2 Hz, Ar–), 1.24 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 135.0 (Cq, Ar–), 131.8 (CH, Ar–), 131.3 (CH, Ar–), 129.3 (Cq, Ar–), 128.9 (Cq, Ar–), 125.9 (CH, Ar–), 97.3 (Cq, d, 1JCRh = 7.6 Hz, –CH3), 8.3 (C–H3). MS (ES+): m/z 701.00 (M − Cl, 100%).
:MeOH (9:1)). 6b was obtained as a red/orange solid (124 mg, 0.13 mmol, 75%). Crystals suitable for X-ray work were obtained from CH2Cl2/ether. Anal. calcd for C30H36Cl2Ir2S2 (916.08 g mol−1): C, 39.30; H, 3.96. Found: C, 39.35; H, 4.08. IR (KBr): νmax/cm−1 2978w (νC–H), 2918m (νC–H), 1626m, 1490m, 1452s, 1381s, 1030s, 831s, 768s. Raman (glass capillary): νmax/cm−1 3056w (νAr–H), 2921s (νC–H), 1547s, 1426m, 893s, 588m (νC–S), 549m, 443s, 425s. 1H NMR (400 MHz, CDCl3): δ = 8.39 (2 H, dd, 3JHH = 7.2 Hz, 4JHH = 1.2 Hz, Ar–), 8.11 (2 H, dd, 3JHH = 8.2 Hz, 4JHH = 1.1 Hz, Ar–), 7.50 (2 H, dd, 3JHH = 8.2, 7.2 Hz, Ar–), 1.29 (30 H, s, C–C). 13C NMR (125 MHz, CDCl3): δ = 135.2 (Cq, Ar–), 130.6 (CH, Ar–), 129.5 (Cq, Ar–), 128.6 (CH, Ar–), 126.4 (CH, Ar–), 90.3 (Cq, –CH3), 8.1 (C–H3) MS (ES+): m/z 880.99 (M − Cl, 100%).
Crystal structure analysis
Tables 3 and 4 list the details of data collections and refinements. Data for 3c was collected using a Rigaku FRX (Mo-K, confocal optic) equipped with a Dectris P200 detector at −100 °C; for 4b using a Rigaku Saturn70 at −148 °C and for 3b, 3d, 4c, 5b and 6b using a Rigaku FRX (Mo-K, confocal optic) equipped with a Dectris P200 detector at −180 °C. Intensities were corrected for Lorentz polarization, and absorption. Structures were solved by direct methods and refined by full-matrix least-squares against F2 (SHELXL).49 Hydrogen atoms were assigned riding isotropic displacement parameters and constrained to idealised geometries. Non-hydrogen atoms were refined anisotropically. In the structures of 5b and 6b there is disorder within the Cp* and solvent molecules. Numerous crystallisations were attempted without success, this data represents the best obtained and is used to confirm the connectivity of the complexes only. CCDC no. 1410515–1410521.| 3b | 3c | 3d | |
|---|---|---|---|
| Empirical formula | C40H42Rh2S2 | C32H40Cl2ORh2S2 | C78H88Cl6Rh4S6 |
| M | 856.82 | 781.50 | 1842.25 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P2(1)/n | P2(1)/c | Fdd2 |
| a [Å] | 9.912(4) | 13.9932(11) | 29.217(4) |
| b [Å] | 16.351(7) | 13.8243(10) | 44.750(7) |
| c [Å] | 10.915(5) | 16.3045(11) | 11.2618(17) |
| α [°] | 90 | 90 | 90 |
| β [°] | 90.662(12) | 101.8010(18) | 90 |
| γ [°] | 90 | 90 | 90 |
| V [Å3] | 1768.9(13) | 3087.4(4) | 14724(4) |
| Z | 2 | 4 | 8 |
| ρ calcd [g cm−3] | 1.609 | 1.681 | 1.662 |
| μ [cm−1] | 11.957 | 13.999 | 13.109 |
| Measured refln. | 12656 |
37059 |
16519 |
| Unique refln. | 3278 | 5682 | 6472 |
| R [I > 2σ(I)] | 0.0665 | 0.0200 | 0.0299 |
| wR | 0.1653 | 0.0563 | 0.0866 |
| 4b | 4c | |
|---|---|---|
| Empirical formula | C40H42Ir2S2 | C32H40Cl2Ir2OS2 |
| M | 1035.45 | 960.13 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P2(1)/n | P2(1)/c |
| a [Å] | 9.8117(9) | 14.1227(16) |
| b [Å] | 16.2184(13) | 13.8101(14) |
| c [Å] | 10.8329(8) | 16.3080(18) |
| α [°] | 90 | 90 |
| β [°] | 90.330(6) | 101.803(2) |
| γ [°] | 90 | 90 |
| V [Å3] | 1723.8(2) | 3113.4(6) |
| Z | 2 | 4 |
| ρ calcd [g cm−3] | 1.995 | 2.048 |
| μ [cm−1] | 80.054 | 88.943 |
| Measured refln | 12983 |
41759 |
| Unique refln. | 3030 | 5686 |
| R [I>2σ(I)] | 0.0149 | 0.0171 |
| wR | 0.0334 | 0.0378 |
References
- J. A. McCleverty, N. Spencer, N. A. Bailey and S. L. Shackleton, J. Chem. Soc., Dalton Trans., 1980, 1939–1944 RSC
.
- J. A. McCleverty, N. J. Morrison, N. Spencer, C. C. Ashworth, N. A. Bailey, M. R. Johnson, J. M. A. Smith, B. A. Tabbiner and C. R. Taylor, J. Chem. Soc., Dalton Trans., 1980, 1945–1957 RSC
- J. R. Phillips, J. C. Poat, A. M. Z. Slawin, D. J. Williams, P. T. Wood and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1995, 2369–2375 RSC
- A. M. Bond and R. L. Martin, Coord. Chem. Rev., 1984, 54, 23–98 CrossRef CAS
-
R. Eisenberg, in Prog. Inorg. Chem, John Wiley & Sons, Inc, 2007, pp. 295–369 Search PubMed
- F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. L. McInnes, R. E. P. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976–6980 CrossRef CAS PubMed
- C.-L. Zhou, Z.-M. Wang, B.-W. Wang and S. Gao, Polyhedron, 2011, 30, 3279–3283 CrossRef CAS
-
J. D. Woollins, in Biological Interactions Of Sulfur Compounds, ed. S. C. Mitchell, Taylor & Francis, London, 1996, pp. 1–19 Search PubMed
- B. K. Teo, F. Wudl, J. H. Marshall and A. Kruger, J. Am. Chem. Soc., 1977, 99, 2349–2350 CrossRef CAS
- B. K. Teo, F. Wudl, J. J. Hauser and A. Kruger, J. Am. Chem. Soc., 1977, 99, 4862–4863 CrossRef CAS
- B. K. Teo and P. A. Snyder-Robinson, Inorg. Chem., 1978, 17, 3489–3497 CrossRef CAS
- B. K. Teo and P. A. Snyder-Robinson, Inorg. Chem., 1979, 18, 1490–1495 CrossRef CAS
- B. K. Teo and P. A. Snyder-Robinson, J. Chem. Soc., Chem. Commun., 1979, 255–256 RSC
- B. K. Teo and P. A. Snyder-Robinson, Inorg. Chem., 1981, 20, 4235–4239 CrossRef CAS
- B. K. Teo, V. Bakirtzis and P. A. Snyder-Robinson, J. Am. Chem. Soc., 1983, 105, 6330–6332 CrossRef CAS
- B. K. Teo and P. A. Snyder-Robinson, Inorg. Chem., 1984, 23, 32–39 CrossRef CAS
- W. P. Bosman and H. G. M. van der Linden, J. Chem. Soc., Chem. Commun., 1977, 714–715 RSC
- M. Tesmer and H. Vahrenkamp, Eur. J. Inorg. Chem., 2001, 1183–1188 CrossRef CAS
- C. Claver, S. Castillon, N. Ruiz, G. Delogu, D. Fabbri and S. Gladiali, J. Chem. Soc., Chem. Commun., 1993, 1833–1834 RSC
- N. Ruiz, A. Aaliti, J. Forniés-Cámer, A. Ruiz, C. Claver, C. J. Cardin, D. Fabbri and S. Gladiali, J. Organomet. Chem., 1997, 545–546, 79–87 CrossRef CAS
- N. Ruiz, S. Castillon, A. Ruiz, C. Claver, A. Aaliti, A. Alvarez-Larena, J. F. Piniella and G. Germain, J. Chem. Soc., Dalton Trans., 1996, 969–973 RSC
- N. Ruiz, I. del Río, J. L. Jiménez, C. Claver, J. Forniés-Cámer, C. C. J. Cardin and S. Gladiali, J. Mol. Catal. A: Chem., 1999, 143, 171–180 CrossRef CAS
- R. J. Wright, C. Lim and T. D. Tilley, Chem. – Eur. J., 2009, 15, 8518–8525 CrossRef CAS PubMed
- A. P. S. Samuel, D. T. Co, C. L. Stern and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 8813–8815 CrossRef CAS PubMed
- P. Li, S. Amirjalayer, F. Hartl, M. Lutz, B. d. Bruin, R. Becker, S. Woutersen and J. N. H. Reek, Inorg. Chem., 2014, 53, 5373–5383 CrossRef CAS PubMed
- J. Ballmann, S. Dechert, S. Demeshko and F. Meyer, Eur. J. Inorg. Chem., 2009, 3219–3225 CrossRef CAS
- K. Charreteur, M. Kdider, J.-F. Capon, F. Gloaguen, Y. F. Pétillon, P. Schollhammer and J. Talarmin, Inorg. Chem., 2010, 49, 2496–2501 CrossRef CAS PubMed
- A. Albers, S. Demeshko, S. Dechert, C. T. Saouma, J. M. Mayer and F. Meyer, J. Am. Chem. Soc., 2014, 136, 3946–3954 CrossRef CAS PubMed
- C. Topf, U. Monkowius and G. Knör, Inorg. Chem. Commun., 2012, 21, 147–150 CrossRef CAS PubMed
- M. R. Malachowski, M. Adams, N. Elia, A. L. Rheingold and R. S. Kelly, J. Chem. Soc., Dalton Trans., 1999, 2177–2182 RSC
- R. L. McNaughton, A. A. Tipton, N. D. Rubie, R. R. Conry and M. L. Kirk, Inorg. Chem., 2000, 39, 5697–5706 CrossRef CAS PubMed
- R. Conry and A. A. Tipton, J. Biol. Inorg. Chem., 2001, 6, 359–366 CrossRef CAS PubMed
- J. Zhao, Z. Wei, X. Zeng and X. Liu, Dalton Trans., 2012, 41, 11125–11133 RSC
- A. J. Ashe, J. W. Kampf and P. M. Savla, Heteroat. Chem., 1994, 5, 113–119 CrossRef CAS
- C. G. M. Benson, C. M. Schofield, R. A. M. Randall, L. Wakefield, F. R. Knight, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2013, 427–437 CrossRef CAS
- S. Cossu, G. Delogu, D. Fabbri and P. Maglioli, Org. Prep. Proced. Int., 1991, 23, 455–457 CrossRef CAS
- P. S. Nejman, B. Morton-Fernandez, N. Black, D. B. Cordes, A. M. Z. Slawin, P. Kilian and J. D. Woollins, J. Organomet. Chem., 2015, 776, 7–16 CrossRef CAS
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS
- R. Xi, M. Abe, T. Suzuki, T. Nishioka and K. Isobe, J. Organomet. Chem., 1997, 549, 117–125 CrossRef CAS
- M. Nomura, E. Tsukano, C. Fujita-Takayama, T. Sugiyama and M. Kajitani, J. Organomet. Chem., 2009, 694, 3116–3124 CrossRef CAS
- J.-J. Liu, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2015, 44, 10281–10288 RSC
- F. Takagi, H. Seino, M. Hidai and Y. Mizobe, J. Chem. Soc., Dalton Trans., 2002, 3603–3610 RSC
- Z.-J. Yao, B. Xu, X.-K. Huo and G.-X. Jin, J. Organomet. Chem., 2013, 747, 85–89 CrossRef CAS
- M. R. Churchill, S. A. Julis and F. J. Rotella, Inorg. Chem., 1977, 16, 1137–1141 CrossRef CAS
- M. R. Churchill and S. A. Julis, Inorg. Chem., 1977, 16, 1488–1494 CrossRef CAS
-
W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 6th edn, 2009 Search PubMed
- K. Yui, Y. Aso, T. Otsubo and F. Ogura, Bull. Chem. Soc. Jpn., 1988, 61, 953–959 CrossRef CAS
- E. Block, V. Eswarakrishnan, M. Gernon, G. Ofori-Okai, C. Saha, K. Tang and J. Zubieta, J. Am. Chem. Soc., 1989, 111, 658–665 CrossRef CAS
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1410515–1410521. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02542g |
| This journal is © The Royal Society of Chemistry 2015 |