
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield, fluoride-free and large-scale synthesis of MIL-101(Cr)†‡
Tian
Zhao
a,
Felix
Jeremias
ab,
Ishtvan
Boldog
a,
Binh
Nguyen
*c,
Stefan K.
Henninger
*b and
Christoph
Janiak
*a
aInstitut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, Universitätsstr. 1, D-40225 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
bDepartment of Thermally Active Materials and Solar Cooling, Fraunhofer Institute for Solar Energy Systems (ISE), Heidenhofstr. 2, D-79110 Freiburg, Germany. E-mail: stefan.henninger@ise.fraunhofer.de
cChemCon GmbH, Engesserstr. 4b, D-79108 Freiburg, Germany. E-mail: Binh.Nguyen@chemcon.com
First published on 19th August 2015
Abstract
MIL-101(Cr), one of the most important prototypical MOFs, is well investigated and widely used in many scientific fields. With regard to MOF synthesis in general, the addition of a modifier is commonly used to improve the properties of the products. The effect of inorganic (mineral) and organic acid modifiers was thoroughly investigated in the synthesis of MIL-101(Cr) and HNO3 could increase the yield to over 80% of a product with average SBET > 3200 m2 g−1 in repeated experiments (from an average of 50% in most published syntheses) in small-scale laboratory synthesis. The large-scale synthesis could use the finding of HNO3 addition and produce MIL-101(Cr) in >100 g quantities with yields near 70% and BET-surface areas near 4000 m2 g−1. The addition of acetic acid (CH3COOH) together with seeding could decrease the reaction temperature, the lowest being 160 °C (from typically 220 °C in published procedures), with still relatively good yield and BET surface area of the product. The use of other strong inorganic or weak carboxylic acids as modulators typically caused a decrease in yield and porosity.
Introduction
Metal–organic frameworks (MOFs) receive continuous attention1,2 due to their high porosity which promises applications in, e.g., gas storage,3,4 gas5–7 and liquid8 separation processes, drug delivery,9 heterogeneous catalysis,10 heat transformation11–13etc.14MIL-101(Cr)15 is a three-dimensional chromium terephthalate-based porous material with the empirical formula [Cr3(O)X(bdc)3(H2O)2] (bdc = benzene-1,4-dicarboxylate, X = OH or F). Its structure resembles the augmented MTN zeolite topology. MIL-101(Cr) has two types of inner cages with diameters of 29 Å and 34 Å, and pore aperture window diameters of up to 16 Å (Fig. 1 and Fig. S1 in the ESI‡) with a high surface area (BET surface area of 4000 m2 g−1).15 MIL-101(Cr) has terminal water molecules connected to the octahedral trinuclear Cr(III)3O building units, which can be removed under high vacuum, thus creating potential Lewis acid sites.16,17 MIL-101(Cr)12 and its ligand-modified derivatives18,19 show remarkable stability towards water, which makes it most suitable for applications in the presence of moisture/water.12,18,19
 | ||
| Fig. 1 (a) Zeolite-type framework presentation of the MIL-101 structure by showing the topological connectivity (in green) of the centers of the vertex-sharing supertetrahedra (b), with the {Cr3(μ3-O)X(H2O)2} (X = OH or F) secondary building unit at the vertices of a tetrahedron. Thereby, two types of mesoporous cages (a) with pentagonal and hexagonal windows are formed. The smaller cage with only pentagonal windows has a van-der-Waals pore diameter of 2.9 nm, and the larger cage with pentagonal and hexagonal windows has a pore diameter of 3.4 nm. For further presentations of the pore and window size see Fig. S1 in the ESI.‡ Building blocks for MIL-101, [Cr3(μ3-O)X(bdc)3(H2O)2], generated from the deposited X-ray data file at the Cambridge Structure Database (CSD-Refcode OCUNAK)15 using the program DIAMOND.20 | ||
MIL-101(Cr) has evolved into one of the most important prototypical MOFs. MIL-101(Cr) or its derivatives are used as catalysts, e.g., in the oxidation of aryl sulfide to corresponding sulfoxide,21 epoxidation of alkenes22,23 in the presence of H2O2, cyanosilylation of aldehydes,24 desulfurization of dibenzothiophene etc.25 Amine-grafted MIL-101(Cr) has been used as a catalyst for the Knoevenagel condensation reaction with high yield and high selectivity.16 Pd loaded on amine-grafted MIL-101(Cr) was used as a catalyst for the Heck reaction;26 it showed high activity and strong durability in visible-light induced photocatalytic H2 production.27 Similarly Cu nanoparticles embedded in MIL-101(Cr) were high performance catalysts for the reduction of aromatic nitro compounds.28 Fe3O4 nanoparticles embedded in MIL-101(Cr) behaved as magnetic nanocatalysts for the solvent free oxidation of benzyl alcohol in the presence of TBHP.29 CoAl2O4 nanoparticles embedded in MIL-101(Cr) have proved to be efficient catalysts for oxidative catalysis.30 Recently MIL-101(Cr) and its phospho-tungstic acid (PTA) composite material have been studied as heterogeneous acid catalysts in the acetalization of aldehydes with alcohols.31 Functionalized MIL-101(Cr) has been investigated for heterogeneous catalysis in the condensation reaction of aldehydes with alcohols.32
MIL-101 (Cr) with pyridine adsorbed has been used for the high-performance liquid chromatographic separation of tocopherols.33 The adsorption properties of MIL-101(Cr) towards 1,2-dichloroethane (DCE), ethyl acetate (EA), and benzene in the presence of water vapour were investigated. The order of affinity was found to be DCE > EA > benzene.34
MIL-101(Cr) in polysulfone (PSF) mixed-matrix membranes (MMMs) exhibited a remarkable four-fold increase in the permeability of O2 and CO2 compared to pure PSF for possible O2/N2, CO2/N2 or CO2/CH4 gas separations. MIL-MMMs for O2/N2 separation had a constant selectivity of 5–6,35 and those for CO2 over N2 or CH4 the selectivity increases from about 20 to 25 with increasing MIL wt%.36 MIL-101(Cr) loads of up to 24% were achieved in PSF, and the MIL-101(Cr) particles showed excellent adhesion with polysulfone in the mixed-matrix membranes and remarkable long term stability.
Post-synthetic modification (PSM) of the benzene-1,4-dicarboxylate ligand in MIL-101(Cr) using nitrating acid (HNO3/H2SO4) can influence, for example, the water uptake behaviour.37 PSM can create or modify suitable functionalities in the organic linkers in preformed MOFs.38,39 Nitro and amino functional groups were introduced in MIL-101(Cr) under extremely strong acidic conditions (nitrating acid for nitration and SnCl2/conc. HCl for reduction to –NH2).35 Moreover, partially functionalized materials with two different organic ligands in the same framework have been synthesized, which are difficult to obtain through direct synthesis.18 Mixed-linker MIL-101(Cr) was recently synthesized with bdc derivatives containing –NH2, –NO2, –H, –SO3H, –Br, –OH, –CH3, and –COOH.19
The unique combination of properties highlighted above, such as outstanding hydrolytic stability, large surface area and pore sizes, low price of synthetic precursors, presence of functional metal sites and various possibilities for postsynthetic modification makes MIL-101(Cr) an excellent candidate for industrial applications.
Syntheses of MIL-101(Cr)
The initial MIL-101(Cr) synthesis reports a fluorine-free route but also the addition of an equimolar amount of hydrofluoric acid (HF) to chromium and H2bdc. The obtained products are isostructural.15 Accordingly, MIL-101(Cr) is given by the empirical sum formula [Cr3(O)X(bdc)3(H2O)2] (X = OH or F) where either F− or OH− and two aqua ligands occupy the terminal positions in the three Cr octahedra which form the trinuclear {Cr3(μ3-O)X(H2O)2} secondary building unit (Fig. S1 in the ESI‡). Small scale syntheses involve the use of HF, as a so-called mineralizing agent, which ensure unsurpassed crystallinity and a BET surface area up to 4200 m2 g−1.15,17 Several small-scale synthesis procedures are reported in the literature (see Table 1). Most follow the original synthesis procedure by Férey et al.15 In this original procedure HF was used and a yield of only ∼50% was stated after separation of MIL-101 from the terephthalic acid. A yield of ∼50% for MIL-101 is not very satisfying in view of the lengthy synthesis and necessary separation from unused terephthalic acid. Also, the use of dangerous HF is not desirable for large-scale syntheses. HF is classified as a chemical toxicant; it is a highly corrosive liquid, and also a contact poison. Because of the ability of hydrofluoric acid to penetrate tissue, life-threatening poisoning can occur readily through exposure of skin or eyes, and more readily when inhaled or even swallowed. HF must therefore be handled with extreme care, using protective equipment and safety precautions beyond those used with other mineral acids.40 Apart from HF, researchers also tried to use other additives in the small scale syntheses of MIL-101(Cr). For instances, using NaOH instead of HF to obtain nano-sized (50 nm) MIL-101(Cr) has been reported, and the product possesses relatively good BET surface area (∼3200 m2 g−1) and fairly good yield (37%).41 The particle size of MIL-101(Cr) can be controlled from 19(4) nm to 84(12) nm, by using monocarboxylic acid as a mediator, with a BET surface area reaching 2900 m2 g−1.42 Weakly alkaline lithium/potassium acetate was employed to assist the synthesis of high-quality MIL-101(Cr) (BET surface area up to 3400 m2 g−1).43 Hydrofluoric acid and sodium acetate were used as mineralizing agents to obtain hierarchically mesostructured MIL-101(Cr).44 Evidently, the influence of pH is not clear as both acidic and basic conditions are possible.| Additive | Time (h) | Temperature (°C) | Yieldb (%) | S BET (m2 g−1) | V pore (cm3 g−1) | Ref. |
|---|---|---|---|---|---|---|
| a The preparation followed the original hydrothermal synthesis procedure by Férey et al.15 b The yield is based on Cr and refers to the isolated material after the washing procedures. When no yields were given (n.a.) and the original procedure by Férey et al.15 was followed a yield of ∼50% can be assumed. We note that a significant water uptake of ∼1 g(H2O)/g(MIL) can occur at 40–50% room humidity which will lead to higher weights than the truly empty material. c TMAOH = tetramethylammonium hydroxide. d This is a singular high-yield synthesis differing from all other reports. e This value is singular and perhaps doubtful; the repeated syntheses in our group (ref. 32, 61 and 62) only gave a yield of about 50%. | ||||||
| HF | 8 | 220 | ∼50 | ∼4100 | 2.02 | 15 |
| HF | 8 | 220 | n.a.b | 2231 | 1.08 | 48 |
| HF | 8 | 220 | n.a.b | 2233 | 1.20 | 49 |
| HF | 8 | 220 | n.a.b | 2651 | 1.29 | 50 |
| HF | 8 | 220 | n.a.b | 2846 | 1.30 | 51 |
| HF | 8 | 220 | ∼47 | 2931 | 1.45 | 52 |
| HF | 8 | 220 | n.a.b | 2995 | 1.31 | 53 |
| HF | 8 | 220 | ∼53 | 3007 | 1.51 | 54 |
| HF | 8 | 220 | ∼50 | 3200 | 2.10 | 55 |
| NaOH | 24 | 210 | ∼37 | ∼3200 | 1.57 | 41 |
| None | 18 | 218 | ∼64d | ∼3460 | Not given | 56 |
| HF | 8 | 220 | n.a.b | ∼3020 | 1.80 | 26 |
| HF | 8 | 220 | ∼57 | ∼2367 | 1.46 | 24 |
| HF | 8 | 220 | n.a.b | ∼2693 | 1.30 | 57 |
| HF | 8 | 220 | n.a.b | ∼2220 | 1.13 | 58 |
| HF | 8 | 220 | n.a.b | ∼3115 | 1.58 | 59 |
| HF | 8 | 220 | n.a.b | ∼2059 | 1.10 | 12 |
| TMAOHc | 24 | 180 | ∼88e | ∼3197 | 1.73 | 60 |
| TMAOHc | 24 | 180 | ∼50 | ∼3060 | 1.45 | 61,62 |
| TMAOHc | 24 | 180 | ∼47 | ∼3055 | 1.51 | 32 |
| HF | 8 | 220 | n.a.b | ∼3318 | 2.02 | 63 |
| HF | 8 | 220 | n.a.b | ∼2800 | 1.47 | 18 |
The concomitant drawbacks are the toxicity of hydrofluoric acid, relatively low reported yield of approximately 50%15 and questionable reproducibility, as there is a large spread of surface area values, with most of them in the interval of 2400–3500 m2 g−1 (Table 1). The reported work-up procedures are tedious and could include size-selective/double filtrations for separation of larger crystals of terephthalic acid and prolonged washings, including such environmentally non-benign agents as NH4F.16 In order to obtain pure MIL-101(Cr) materials, the as-synthesized MIL-101(Cr) needs to be purified by washing processes using hot water (70 °C, 5 h), hot ethanol (60 °C, 3 h), hot aqueous NH4F solutions (60 °C, 10 h) and rinsing with hot water at least 5 times to remove fluoride and other starting material residues.16,17 Thus, the use of fluoride and the necessary washing procedures are major obstacles for large-scale manufacturing. Indeed, MIL-101 type of compound is not represented among the Basolite® series of MOFs produced by BASF and commercialized by Aldrich, despite recognized importance45 and interest towards its production that was announced as early as in 2009.46
Stimulated by this background we screened a number of acidic modulators,47 in the synthesis of MIL-101 and compared them with fluoride-assisted syntheses under simplified work-up conditions. We report an optimization study which resulted in the development of a high-yield procedure with proven scalability.
Results and discussion
By trying to replace HF as an acid additive we tested various inorganic and organic acids under otherwise identical conditions. During these experiments, two acid additives became noteworthy, nitric acid and acetic acid. Thereby we found that HNO3 led to a reproducible increase in yield to over 80% with BET surface areas around and over 3200 m2 g−1. On the other hand the use of acetic acid in MIL-101(Cr) synthesis allowed to significantly decrease the reaction temperature. A significant amount of terephthalic acid can still be present inside the pores and is mixed with the MIL crystallites. The residual reactants and eventually the solvent needed to be removed from the pores in order to obtain a material as porous as possible. All synthesis products underwent the same purification, that is, washing and drying (activation) procedures. The products synthesized in this work are typically synthesized without HF, thus, the formulae of MIL-101(Cr) frameworks from this work do not contain F atoms and should be [Cr3(O)(OH)(bdc)3(H2O)2].High-yield, small-scale synthesis
According to the literature, a typical synthesis of MIL-101(Cr) lasts 8 h at ∼220 °C (Table 1). Additives are commonly used in the synthesis, especially HF which can act as a mineralizing agent to increase the crystallinity of microporous materials and favors the formation of highly crystalline phases in MOFs.64We tried to replace the modifier/additive HF by other mineral acids and also by organic acids, including both strong mineral and weak carboxylic acids with varying coordination capabilities of the corresponding anions. Acidic modulators received less attention compared to basic modulators, due to the perception that acidification should shift the equilibrium away from the formation of MIL-101(Cr) as nitric acid is released during the process. When the addition of HNO3 showed promising results, HNO3 synthesis experiments with varying amounts were conducted to reveal the optimal synthetic conditions. The reaction was carried out with addition of 0.25, 0.5, 0.75, 1.0, 1.25 and 1.5 equivalents of nitric acid (N) with respect to chromium nitrate. Products are designated as N-0.25, N-0.5, N-0.75, N-1.0, N-1.25 and N-1.5 respectively. The sample without HNO3 or other additives was named N-0. All these experiments were carried out with 1.0 mmol each of chromium(III) nitrate nonahydrate and terephthalic acid in a PTFE- (Teflon-) lined autoclave at 220 °C for 8 hours (see the Experimental section). After the hydrothermal reaction, the purification of products followed as described in the Experimental section. The analytical results showed that a considerable improvement in yield and product quality could be reached compared to experiments without addition of nitric acid (Table 2).
| HNO3 (N)-equivalentsa | Yielda (%) | S BET (m2 g−1) | S Langmuir (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
a HNO3 equivalents with respect to Cr and bdcH2. The Cr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bdc ratio is always 1:1, and the yield is based on Cr.
b Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ±50 m2 g−1.
c Calculated from N2 sorption isotherms at 77 K (p/p0 = 0.95) for pores ≤20 nm.
d Repeated experiments to demonstrate reproducibility. :bdc ratio is always 1:1, and the yield is based on Cr.
b Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ±50 m2 g−1.
c Calculated from N2 sorption isotherms at 77 K (p/p0 = 0.95) for pores ≤20 nm.
d Repeated experiments to demonstrate reproducibility.
|
||||
| N-0 | 56.6 | 2410 | 3270 | 1.30 |
| N-0.25 | 67.5 | 2740 | 3600 | 1.33 |
| N-0.5 | 71.7 | 2770 | 3690 | 1.40 |
| N-0.75 | 73.6 | 2890 | 3910 | 1.38 |
| N-1.0a | 82.3 | 3450 | 4610 | 1.66 |
| N-1.0bd | 81.6 | 3130 | 4330 | 1.58 |
| N-1.0cd | 80.8 | 3420 | 4640 | 1.69 |
| N-1.25 | 69.9 | 3060 | 4220 | 1.52 |
| N-1.5 | 66.2 | 2540 | 3320 | 1.26 |
Nitrogen sorption isotherms of HNO3-variable MIL-101(Cr) are shown in Fig. 2, which are typical type I sorption isotherms65 as reported in the literature for MIL-101(Cr).15 Obviously, MIL-101(Cr) with one equivalent of HNO3 (N-1.0, red curve) possesses the highest Brunauer Emmett Teller (BET) surface area (3450 m2 g−1). With less or more HNO3 equivalents the surface area decreases. Fig. 2 also shows the curve of HNO3 equivalents versus the corresponding SBET value. It is obvious that the addition of 1.0 equivalent of nitric acid in synthesis produced the best porosity of MIL-101(Cr). In addition, N-1.0 gave an excellent high yield of around 80% after product purification (Table 2). Additionally, N-0.25, N-0.5, N-0.75, N-1.25 and N-1.5 also showed a significant increase in yields compared to the original literature yield by Férey et al.15 Analysis of the literature (Table 1) shows that only the use of TMAOH as a basic modulator might give comparable yields and quality in porosity of the material, however, this claim is yet to be proven, as there are two reports indicating much lower yields under comparable conditions (Table 1).
 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms of HNO3-variable MIL-101(Cr); filled symbols are for adsorption, and empty symbols for desorption. (b) Plot of SBET values versus HNO3 equivalents. | ||
To confirm that the experiment N-1.0 can be reproduced, two repeated experiments were carried out. The results are also included in Table 2. Overall, the synthesis with 1.0 eq. of HNO3 is reproducible with yields above 80% and BET surface areas above 3100 m2 g−1. The nitrogen sorption isotherms of each repeated experiment can be found in Fig. S2 in the ESI.‡ Yields and surface areas are near the high end of syntheses reported in the literature (Table 1).
Furthermore, higher equivalents of nitric acid (2.0 eq. and 5.0 eq.) were also tested in MIL-101(Cr) synthesis. However, under too high nitric acid concentration conditions, there is no positive effect anymore. In the 2.0 eq. experiment, SBET decreased to 1990 m2 g−1, while 5.0 eq. gave a grey powder product without porosity. Thus, in a small-scale MIL-101(Cr) synthesis (5 mL scale), using 1.0 eq. HNO3 is the optimum quantity.
The purity of N-1.0 was analyzed by CHN and EDX elemental analyses. Before elemental analysis, N-1.0 was dried in a vacuum oven (120 °C, 12 mbar) for 2 h. Calcd. for [Cr3(O)(OH)(bdc)3(H2O)2]·2H2O: C 38.26, H 2.81, N 0, Cr 20.70; found C 38.43, H 2.91, N 0.00. From EDX analysis, the atom ratio C/Cr = 8.5 (calc. 8.0) is in good agreement with the formula.
The powder X-ray diffractograms of the MIL-101(Cr) samples with HNO3 as an additive can all be positively matched to the simulated XRD pattern which was generated from the deposited X-ray data file at the Cambridge Structure Database (CSD-Refcode OCUNAK) using the program Mercury (Fig. 3).
 | ||
| Fig. 3 Powder X-ray diffractograms of (a) N-1.0 samples and (b) samples with other HNO3 equivalents in comparison with the simulated pattern (Cu-Kα radiation). | ||
The pore size distribution curve and cumulative pore volume curve for N-1.0 were analyzed by the NL-DFT method (Fig. S3 in the ESI‡) and are similar to the reported pore size distribution for MIL-101(Cr).66–68
Upscaling the synthesis of MIL-101(Cr)
Another objective of this work was to transfer the synthesis of MIL-101(Cr) to a larger scale of about 100 g product in a single batch. Therefore, the scale-up to a reaction volume of 3 L (see 3 L autoclave in Fig. S4 in the ESI‡) was successively investigated and achieved. According to the resulting analytical data the procedure for the synthesis of MIL-101(Cr) was adapted and optimized as follows to produce the material in good quality and quantity.In the literature the MIL-101 synthesis at smaller scales was conducted without stirring.69–71 Since the reaction volume was substantially increased, it was interesting to see, whether stirring would be required in larger scale reactions to maintain homogeneity for the formation of the desired product. The results of the experiments indicate that stirring of the reaction mixture seems not to be necessary to form MIL-101(Cr) in good quality. In fact, stirring leads to a material that showed a lower BET surface area (Fig. 4).
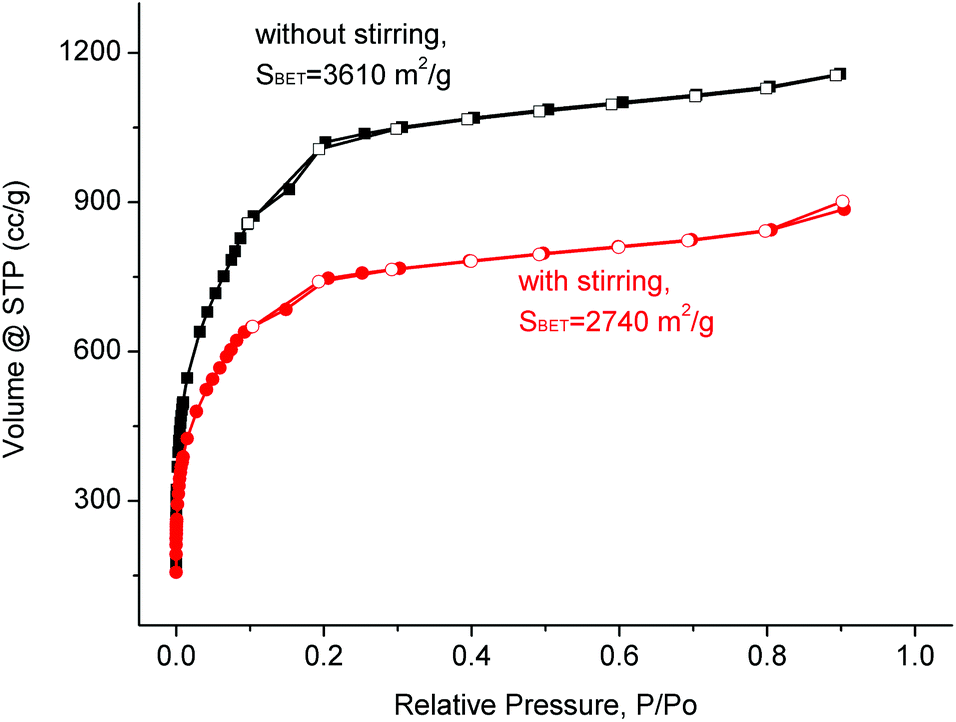 | ||
| Fig. 4 Nitrogen sorption isotherms and their corresponding BET surface areas for non/stirring-experiments of large-scale preparations of MIL-101(Cr), filled symbols are for adsorption, and empty symbols for desorption. The synthesis and treatment of the products both followed the general procedure. The BET surface areas were calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ±50 m2 g−1. | ||
Also, the influence of the reaction temperature on the resulting material was studied. Experiments at smaller scales are usually conducted at 220 °C (Table 1). A series of runs were carried out that covered a temperature range of 180–220 °C. The results suggested that a lower reaction temperature also forms the desired product in good quality. Unexpected was the observation, that a reaction temperature of 220 °C at a larger scale seemed to produce an unknown phase instead of the intended MIL-101(Cr) product (Fig. 5). This result also indicated that in the case of large scale synthesis, the optimized conditions can be different from small scale synthesis.
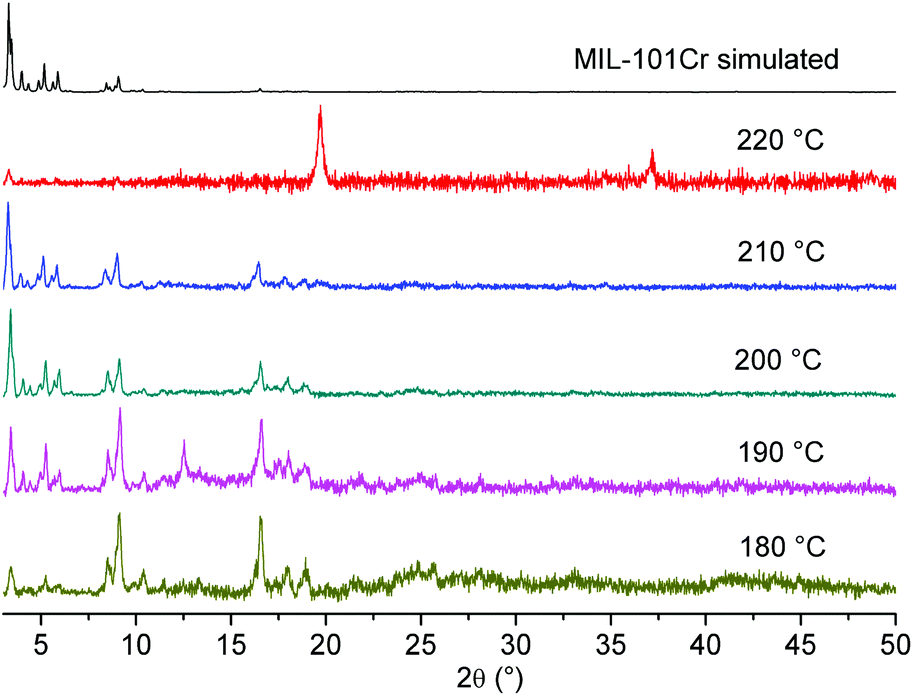 | ||
| Fig. 5 PXRD of large-scale preparations of MIL-101(Cr) at different temperatures (diffractograms were recorded on a Bruker D8 Advance with DaVinci™, Cu-Kα radiation). | ||
In addition the reaction time (6–16 h) and cooling time (4–24 h) were extended to study their impact on the resulting material and yield in the large-scale synthesis. The results indicated no significant effect of reaction and cooling time on quality and quantity of the resulting material. A possible explanation is that crystallization takes place very quickly. Once the material crystallizes, there is only a minor equilibrium process of dissolution and recrystallization.
Furthermore an optimized washing procedure was established for large-scale synthesis. In the initial procedure the raw product was suspended in DMF by using half of the reaction volume, that is, 1.5 L, stirred for 15 min and isolated by centrifugation. This washing procedure was repeated with 1.5 L ethanol and 1.5 L water. Using this procedure we obtained a material with considerable amounts of impurities still remaining in the pores of the product. Hence, we decided to prolong the washing step and to employ solvent volumes 1.5 times of the reaction volume (i.e. 4.5 L) to ensure the removal of impurities from the pores. The product obtained after washing according to the new procedure (for details see the Experimental section) gave materials with higher purity. Consequently, increased nitrogen sorption capacity was observed for all materials after using the new washing procedure (Fig. 6). The method proved to be very efficient to remove starting material or impurities that remained in the pores of the product. For a large-scale product the BET surface of 3870 m2 g−1 can be considered remarkable.
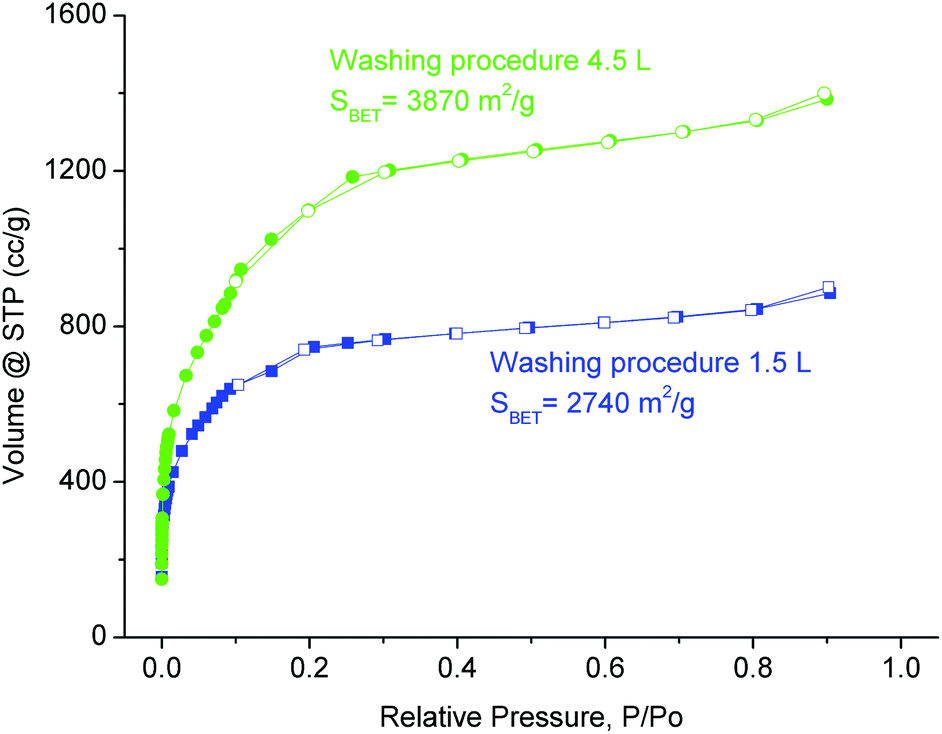 | ||
| Fig. 6 N2 sorption isotherms and BET surface areas of materials from large scale synthesis and different washing procedures. The noted procedure “1.5 L – blue” (“4.5 L – green”) refers to washing with 1.5 L (4.5 L) DMF – separation – 1.5 L (4.5 L) ethanol – separation – 1.5 L water; see the Experimental section for details of the 4.5 L procedure. | ||
Finally, the addition of 0.5, 1.0 and 1.5 equivalents of HNO3 with respect to chromium nitrate was investigated (Fig. 7). The experiments confirmed the results of the small scale reactions, in which the yield was considerably improved by the addition of nitric acid. The large scale preparations also gave about 20% more material for all conducted experiments with addition of nitric acid (0.5, 1.0 and 1.5 eq.) compared to experiments without addition of nitric acid. We assume that the addition of nitric acid affects the particle growth, yielding material with a larger particle size, thus, leading to material that is more efficiently isolated by centrifugation (centrifugation is a major issue in large-scale preparation). The yields were around 48% for experiments without the addition of nitric acid. Addition of nitric acid gave yields of around 66% (for 0.5 eq.), 68–72% (for 1.0 eq.) and 67% (for 1.5 eq.).
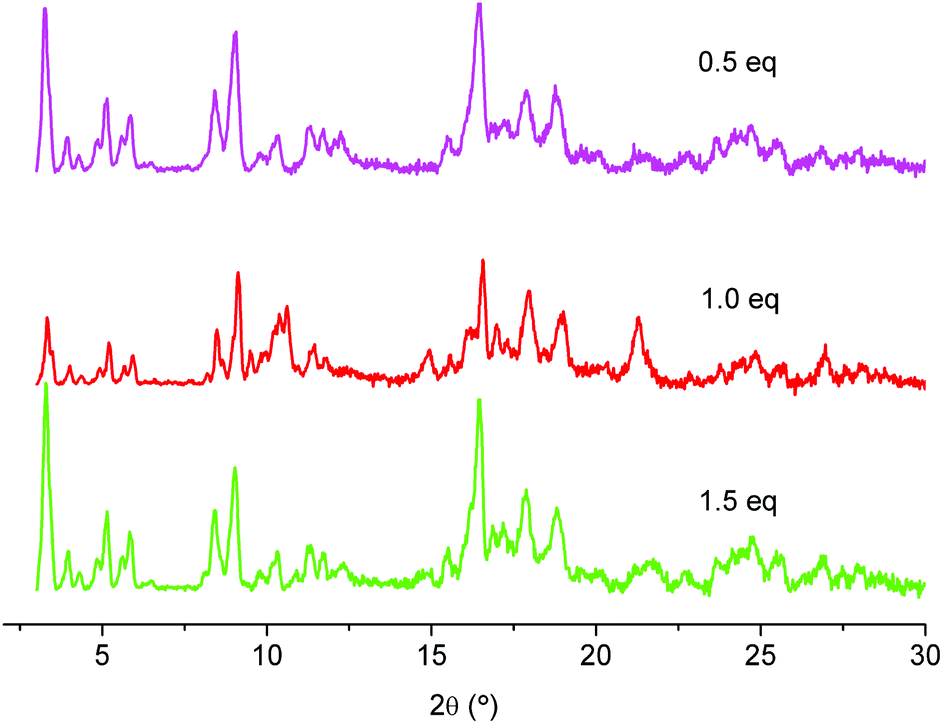 | ||
| Fig. 7 PXRD of large-scale preparations of MIL-101(Cr) with different equivalents of HNO3. (PXRD data were recorded on a Bruker D8 Advance with DaVinci™, Cu-Kα radiation). | ||
Low temperature synthesis of MIL-101(Cr) with acetic acid
Normally, MIL-101(Cr) syntheses are carried out at 220 °C in a Teflon liner within autoclaves. Using microwave irradiation as the heating source one can decrease the reaction temperature to 210 °C.72 Using TMAOH as a mineralizing agent and prolonging the reaction time to 24 h one can reduce the reaction temperature to 180 °C. But until now we are not aware of a report to obtain MIL-101(Cr) at a temperature below 180 °C. We have also tested the addition of several other inorganic and organic acids in MIL-101(Cr) synthesis (see Table 5 below). Among them, acetic acid shows a special effect in so far, that it can largely decrease the reaction temperature without significant influence on the porosity of the material. A lower synthesis temperature can be desired not only from a viewpoint of lower energy and setup costs, but also, e.g., when MIL-101(Cr) is intended as a host for sensitive guest molecules.48–51 In cases, where SBET of MIL-101(Cr) is only 2000 m2 g−1, there is still enough space for the storage of guest molecules.73 The porosity of the MIL-101 material is not the only consideration, but a lower synthesis temperature may allow for the inclusion of temperature-sensitive molecules during the in situ formation of MIL-101(Cr). For example, one could directly synthesize MIL-101(Cr) in the presence of guest molecules at lower temperature. With the addition of acetic acid, the MIL-101(Cr) synthesis temperature can be decreased to as low as 160 °C. In some experiments, the addition of several milligrams of pre-formed MIL-101(Cr) seeding powder was necessary.When 1 equivalent of acetic acid with respect to chromium nitrate is added in the MIL-101(Cr) synthesis at 220 °C, the product is of competitive purity, yield and BET surface relative to other MIL-101(Cr) products. If the quantity of acetic acid is increased up to 8.3 equivalents with respect to chromium, a green glassy-looking material is formed (Fig. 8, left). When the equivalent of addition of acetic acid with respect to chromium is lower than 8.3, the product is a green powder (similar to N-1.0 in Fig. 8, right). Upon addition of more than 8.3 equivalents of acetic acid with respect to chromium no product was isolated. The yield and porosity information of acetic acid-variable MIL-101(Cr) is listed in Table 3. Powder X-ray diffractograms of MIL-101(Cr) with acetic acid can be positively matched to the simulated pattern of MIL-101(Cr) (Fig. 9).
 | ||
| Fig. 8 Image of MIL-101(Cr) with acetic acid (8.3 equivalents, 220 °C) (left) and HNO3 (N-1.0) (right) as a modifier. | ||
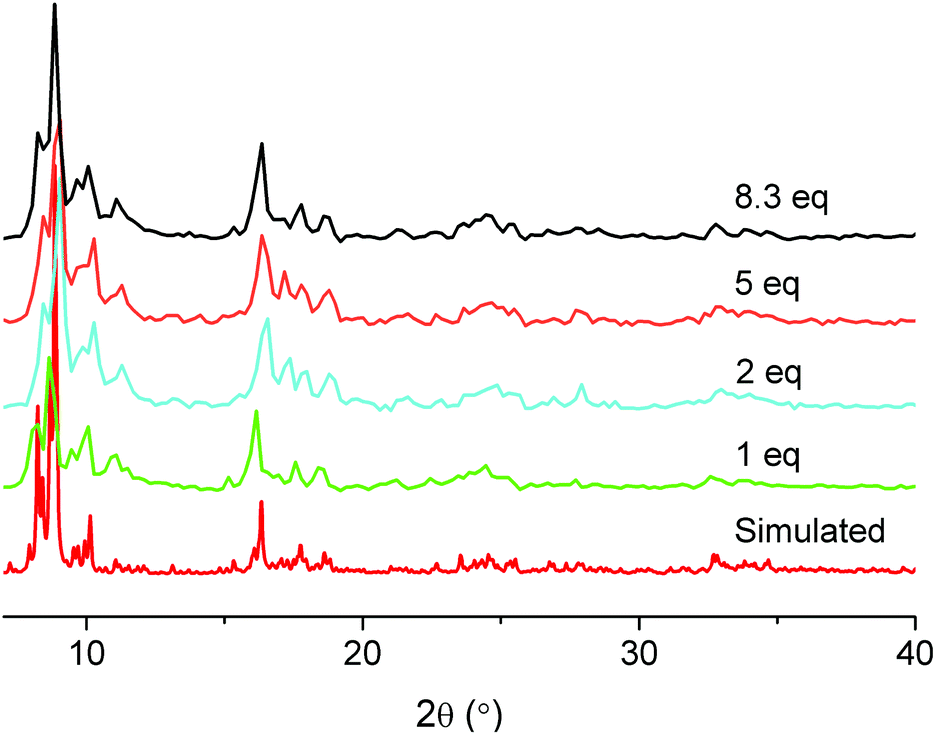 | ||
| Fig. 9 The PXRD patterns of acetic acid-variable MIL-101(Cr) compared with the simulated pattern (Cu-Kα radiation). | ||
| Acetic acid equivalentsa | Yielda (%) | S BET (m2 g−1) | S Langmuir (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
|
a Acetic acid equivalents with respect to Cr and bdcH2. The Cr:bdc ratio is always 1:1, and the yield is based on Cr. The reaction temperature was 220 °C. No seed crystals were added – different from reactions in Table 4.
b Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ±50 m2 g−1.
c Calculated from N2 sorption isotherms at 77 K (p/p0 = 0.95) for pores ≤20 nm.
|
||||
| 1 eq. | 65.3 | 2680 | 3490 | 1.17 |
| 2 eq. | 52.1 | 2450 | 3210 | 1.28 |
| 5 eq. | 36.4 | 2660 | 3480 | 1.27 |
| 8.3 eq. | 24.4 | 2750 | 3620 | 1.55 |
| 10 eq. | None | — | — | — |
MIL-101 formed with 1 eq. and 8.3 eq. acetic acid had BET surface areas of 2680 m2 g−1 and 2750 m2 g−1 respectively. Considering literature reports of MIL-101 with surface areas in the range of 2000–3000 m2 g−1 (Table 1), these values are acceptable. The synthesis of MIL-101Cr with 8.3 eq. of acetic acid was also carried out at temperatures of 220, 200, 180 and 160 °C (Table 4). Upon lowering the temperature, the yield decreased considerably. We found, however, that the addition of about 5 milligrams of pre-formed MIL-101(Cr) powder as seeds would significantly raise the yield again (Table 4). The PXRD patterns of the “seeded” samples are in good agreement with the simulated pattern of MIL-101(Cr) (Fig. 10). The BET results of these “seeded” samples are typically better than those of the “non-seeded” samples (Table 4).
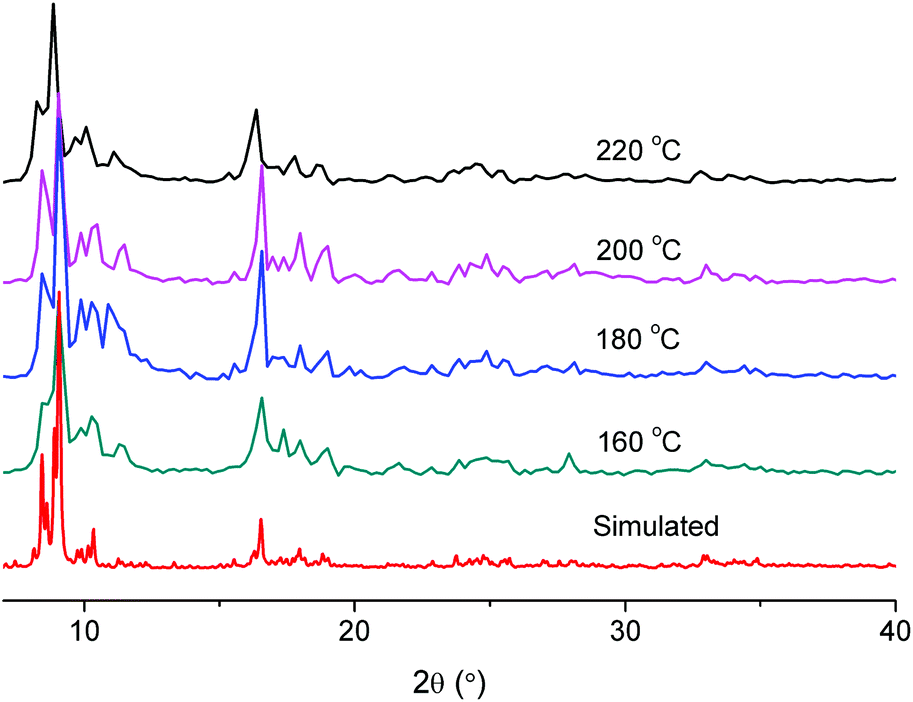 | ||
| Fig. 10 Powder X-ray diffractograms of “seeded” samples with 8.3 eq. of acetic acid at different temperatures compared with the simulated MIL-101(Cr) pattern. | ||
| Acetic acid equivalentsa | Seeded | Temp. (°C) | Yielda (%) | S BET (m2 g−1) | S Langmuir (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|---|---|
|
a Acetic acid equivalents with respect to Cr and bdcH2. The molar Cr:bdc ratio is always 1:1 with 1.0 mmol (400 mg) Cr(NO3)3·9H2O and 1.0 mmol (166 mg) of bdcH2, and the yield is based on Cr.
b Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ±50 m2 g−1. Nitrogen sorption isotherms of the seeded samples are presented in Fig. S5 in the ESI.
c Calculated from N2 sorption isotherms at 77 K (p/p0 = 0.95) for pores ≤20 nm.
|
||||||
| A-8.3-220 | No | 220 | 24.4 | 2750 | 3620 | 1.55 |
| A-8.3-200 | No | 200 | 24.4 | 2360 | 3060 | 1.31 |
| A-8.3-180 | No | 180 | 13.2 | 3240 | 4400 | 1.61 |
| A-8.3-160 | No | 160 | None | — | — | — |
| A-8.3-220s | Yes | 220 | 48.9 | 2810 | 3680 | 1.85 |
| A-8.3-200s | Yes | 200 | 44.5 | 2790 | 3600 | 1.30 |
| A-8.3-180s | Yes | 180 | 43.2 | 2750 | 3630 | 1.30 |
| A-8.3-160s | Yes | 160 | 33.5 | 2700 | 3500 | 1.38 |
Noteworthily, the BET and porosity results did not show a clear dependence on temperature. For example, a material obtained from a non-seeded synthesis at 180 °C with low-yield had the highest BET surface area of 3240 m2 g−1 (Table 4). For the nitrogen sorption isotherms of “seeded” samples at different synthesis temperature see Fig. S5 in the ESI.‡
It is worth to note that the presence of seeds in the synthesis of MIL-101(Cr) allowed a temperature as low as 160 °C at which no product was obtained without seeds. At temperatures below 160 °C no MIL-101(Cr) products could be obtained anymore even if seeding was applied.
The purity of A-8.3-160 was analyzed by CHN and EDX elemental analyses. Before analysis, A-8.3-160 was dried in a vacuum oven (120 °C, 12 mbar) for 2 h. Calcd for [Cr3(O)(OH)(bdc)3(H2O)2]·2H2O: C 38.26, H 2.81, N 0.00; found C 37.92, H 2.65, N 0.00. From EDX analysis, the atom ratio C/Cr = 7.7 (calc. 8.0) is in good agreement with the formula.
Other acids as modifiers in MIL-101(Cr) synthesis
We also compared the use of hydrofluoric acid, trifluoroacetic acid, sulfuric acid, hydrochloric acid, phenylphosphonic acid, benzoic acid, formic acid, fumaric acid, citric acid and succinic acid as modifiers in MIL-101(Cr) synthesis (Table 5).| Additive/modifiera | Yielda (%) | S BET (m2 g−1) | S Langmuir (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
|
a The quantities of all additives were 1.0 equivalent with respect to chromium. The Cr:bdc ratio is always 1:1, and the yield is based on Cr. All products were purified in the same way as described in the experimental section.
b Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherms at 77 K with an estimated standard deviation of ± 50 m2 g−1.
c Calculated from N2 sorption isotherms at 77 K (p/p0 = 0.95) for pores ≤20 nm.
|
||||
| Hydrofluoric acid | 47.4 | 3620 | 4990 | 1.82 |
| None | 56.6 | 2410 | 3270 | 1.30 |
| (Sorted by approximate acid strength) | ||||
| Hydrochloric acid | 36.3 | 1560 | 2030 | 0.79 |
| Sulfuric acid | 48.2 | 1750 | 2200 | 0.81 |
| Nitric acid (N-1.0a) | 82.3 | 3450 | 4610 | 1.66 |
| Trifluoroacetic acid | 73.8 | 2650 | 3620 | 1.34 |
| Phenylphosphonic acid | 51.9 | 2460 | 3350 | 1.49 |
| Fumaric acid | 28.7 | 760 | 1040 | 0.69 |
| Citric acid | 37.2 | 740 | 1050 | 0.58 |
| Formic acid | 27.1 | 590 | 720 | 0.56 |
| Succinic acid | 59.8 | 2510 | 3250 | 1.28 |
| Benzoic acid | 39.4 | 1760 | 2290 | 0.93 |
| Acetic acid (8.3 eq.) | 24.4 | 2750 | 3620 | 1.55 |
The addition of HF was used to repeat the original experiment.15 Indeed, using HF as an additive yielded a MIL-101(Cr) product with the highest BET surface area of all small-scale samples and a yield of around 50%, very near to the values reported in the literature (cf.Table 1).15 An additive-free method gave only 2410 m2 g−1 of BET surface area and the yield was similar to the HF-experiment. The PXRD patterns (see Fig. S6 in the ESI‡) of the products with the various additives showed more or less byproduct formation. Fumaric and citric acid failed to function as sensible additives.
Conclusions
In summary, only nitric acid (HNO3) and acetic acid (CH3COOH) proved advantageous in the synthesis of MIL-101(Cr) from a series of tested mineral and organic acid additives/modifiers as alternatives to HF. An advantage of HNO3 was a >30% increase in yield with the BET surface area (>3100 m2 g−1) lagging only ∼10% behind compared to the use of HF. Nitric acid also worked well in large-scale MIL-101(Cr) synthesis (3 L) where the toxic nature of HF would be of increased concern. The combined use of acetic acid and seeding of the reaction mixture with MIL-101(Cr) allowed to decrease the temperature of the synthesis to 160 °C, retaining a yield of ∼50% and an acceptable BET surface area of 2700–2800 m2 g−1. The demonstrated singularly low temperature synthesis broadens the range of synthetically tolerable conditions for potential functional guests, which could be embedded in MIL-101(Cr).Experimental section
Materials
Chromium(III) nitrate nonahydrate (98.5%, Alfa Aesar or 99%, Acros), benzene-1,4-dicarboxylic acid, terephthalic acid (bdcH2, 99+%, Acros), nitric acid (65 wt%, VWR or p. A. Applichem), acetic acid (99.7+%, Alfa Aesar), hydrofluoric acid (48 wt% in H2O, Sigma-Aldrich), sulfuric acid (95.0%–98.0%, Sigma-Aldrich), hydrochloric acid (36 wt% in H2O, Alfa Aesar), trifluoroacetic acid (99%, Sigma-Aldrich), phenylphosphonic acid (98%, Sigma-Aldrich), benzoic acid (99.5%, Sigma-Aldrich), formic acid (97%, Alfa Aesar), fumaric acid (99.0%, Fluka), citric acid (99.5%, Sigma-Aldrich), succinic acid (99.5%, Fluka), N,N-dimethylformamide (DMF, 99%, VWR or Alfa Aesar) and ethanol (99.8%, Carl Roth or p.a. Applichem). All chemicals were used as obtained from commercial sources without further purification.Instrumentation
Powder X-ray diffraction (PXRD) measurements were carried out on samples at ambient temperature with a Bruker D2 Phaser using a flat silicon, low background sample holder and Cu-Kα radiation (λ = 1.54184 Å) at 30 kV and 0.04° s−1 as shown in Fig. 3, 9 and S6.‡ Diffractograms were obtained on flat layer sample holders with a beam scattering protection blade installed, which led to the low relative intensities measured at 2θ < 7°. Simulated PXRD patterns were calculated from single crystal data using the Mercury 3.0.1 software suite from CCDC. A shown in Fig. 5 and 7 the PXRD data were recorded on a Bruker D8 Advance with DaVinci™, using a rotating sample holder, a Cu anode tube at 40 kV/40 mA, with a Ni filter and constant sample illumination spot size (broadness: 12 mm); step size 0.02°, 0.2 s per step, Cu-Kα radiation.Nitrogen physisorption isotherms at 77 K were obtained using a NOVA-4000e instrument within a partial pressure range of 10−6–1.0 as shown in Fig. 2, S2 and S5,‡ and Tables 2–5. Before measurements, the samples were degassed at 120 °C for 2 h. Alternatively, nitrogen sorption isotherms were measured at 77 K using a Quantachrome Autosorb iQ MP gas sorption analyzer as shown in Fig. 4 and 6. Ultra high purity (UHP, grade 5.0, 99.999%) nitrogen, and helium gases were used; the latter was used for performing cold and warm free space correction measurements. Samples were degassed for 2 h at 120 °C with the built-in oil-free vacuum system of the instrument (ultimate vacuum <10−8 mbar).
The samples were transferred to pre-weighed sample tubes capped with a septum. Then the sample tube was connected to the preparation port of the sorption analyzer and degassed under vacuum for the specified time and temperature. After weighing, the sample tube was then transferred to the analysis port of the sorption analyzer. Helium gas was used for the determination of the cold and warm free space of the sample tubes. DFT calculations for the pore size distribution curves were carried out using the native ASiQWin 1.2 software employing the ‘N2 at 77 K on carbon, slit pore, NLDFT equilibrium’ model.74–76
Elemental (C, H, N) analysis was done with a Perkin-Elmer Series 2 Elemental Analyser 2400.
Energy dispersive X-ray spectrometric (EDX) measurements were carried out on a Jeol scanning electron microscope JSM-6510 with a tungsten (W) cathode and an EDX unit. The samples were coated with Au for 20 s at 30 mA by using a Jeol JFC-1200 sputter coater (JSM-6510).
The contents of the autoclave were transferred to two centrifuge tubes and the supernatant solution was carefully removed after centrifugation. Water (5 mL) was added in each tube and the solid was evenly dispersed in the aqueous phase. After renewed centrifugation and removal of the supernatant solution, DMF (5 mL) was added to each tube which were placed in a hot (80 °C) ultrasonic bath and sonicated for 1 h. Centrifugation was again performed to separate MIL-101 and DMF. The precipitate was transferred to a 25 mL beaker where it was stirred with 10 mL of water at 70 °C for 5 h. After separation by centrifugation, the same washing procedure but using ethanol was repeated once more at the same temperature. The final product was obtained by centrifugation and dried in a vacuum oven (120 °C, 12 mbar) for 2 h.
Acknowledgements
The work was supported by the Federal German Ministry of Economics (BMWi) under grant 0327851A/B/C. T. Z. thanks the China Scholarship Council (CSC) for a doctoral fellowship and I. B. thanks the Alexander von Humboldt foundation for a postdoctoral fellowship.Notes and references
- Introductions to special MOF issues: J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC; H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC; S.-H. Jhung, N. A. Khan and Z. Hasan, CrystEngComm, 2012, 14, 7099–7109 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC; M. Paik Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed; L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC; J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; G. Férey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. De Weireld, A. Vimont, M. Daturif and J.-S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC; J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef PubMed.
- B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC; T.-Z. Zhang, Z.-M. Zhang, Y. Lu, F. Hai and E.-B. Wang, CrystEngComm, 2013, 15, 459–462 RSC; C. Hou, Q. Liu, T.-a. Okamura, P. Wang and W.-Y. Sun, CrystEngComm, 2012, 14, 8569–8576 RSC; W.-Y. Gao, Y. Niu, Y. Chen, L. Wojtas, J. Cai, Y.-S. Chen and S. Ma, CrystEngComm, 2012, 14, 6115–6117 RSC; C. Li, W. Qiu, W. Shi, H. Song, G. Bai, H. He, J. Li and M. J. Zaworotko, CrystEngComm, 2012, 14, 1929–1932 RSC.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Dalton Trans., 2012, 41, 14003–14027 RSC; B. Zornoza, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2013, 166, 67–78 CrossRef CAS PubMed; G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 Search PubMed.
- K. A. Cychosz, R. Ahmad and A. J. Matzger, Chem. Sci., 2010, 1, 293–302 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- J.-Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; J. Guo, J. Yang, Y.-Y. Liu and J.-F. Ma, CrystEngComm, 2012, 14, 6609–6617 RSC; B. J. Burnett, P. M. Barron and W. Choe, CrystEngComm, 2012, 14, 3839–3846 RSC; H.-j. Pang, H.-y. Ma, J. Peng, C.-j. Zhang, P.-p. Zhang and Z.-m. Su, CrystEngComm, 2011, 13, 7079–7085 RSC.
- F. Jeremias, D. Fröhlich, C. Janiak, S. Henninger, C. Janiak and S. K. Henninger, Chimia, 2013, 67, 419–424 CrossRef CAS PubMed; C. Janiak and S. K. Henninger, Nachr. Chemie, 2013, 61, 520–523 CrossRef PubMed; S. K. Henninger, F. Jeremias, H. Kummer and C. Janiak, Eur. J. Inorg. Chem., 2012, 2625–2634 CrossRef PubMed.
- J. Ehrenmann, S. K. Henninger and C. Janiak, Eur. J. Inorg. Chem., 2011, 471–474 CrossRef CAS PubMed.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC; F. Jeremias, S. K. Henninger and C. Janiak, Chem. Commun., 2012, 48, 9708–9710 RSC; F. Jeremias, V. Lozan, S. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC; F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, RSC Adv., 2014, 4, 24073–24082 RSC; D. Fröhlich, S. K. Henninger and C. Janiak, Dalton Trans., 2014, 43, 15300–15304 RSC.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC; G. Férey, Dalton Trans., 2009, 4400–4415 RSC; M. J. Prakash and M. S. Lah, Chem. Commun., 2009, 3326–3341 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS PubMed.
- A. Khutia, H. U. Rammelberg, T. Schmidt, S. Henninger and C. Janiak, Chem. Mater., 2013, 25, 790–798 CrossRef CAS.
- M. Lammert, S. Bernt, F. Vermoortele, D. E. De Vos and N. Stock, Inorg. Chem., 2013, 52, 8521–8528 CrossRef CAS PubMed.
- K. Brandenburg, Diamond (Version 3.2), crystal and molecular structure visualization, Crystal Impact, K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2007–2012 Search PubMed.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, H. Seo, M. Yoon, J. Kim, S. H. Jhung, C. Serre and G. Férey, Appl. Catal., A, 2009, 358, 249–253 CrossRef CAS PubMed.
- C. M. Granadeiro, A. D. S. Barbosa, P. Silva, F. A. Almeida Paz, V. K. Saini, J. Pires, B. de Castro, S. S. Balula and L. Cunha-Silva, Appl. Catal., A, 2013, 453, 316–326 CrossRef CAS PubMed.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS PubMed.
- A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC.
- D. Julião, A. C. Gomes, M. Pillinger, L. Cunha-Silva, B. de Castro, I. S. Gonçalves and S. S. Balula, Fuel Process. Technol., 2015, 131, 78–86 CrossRef PubMed.
- S.-N. Kim, S.-T. Yang, J. Kim, J.-E. Park and W.-S. Ahn, CrystEngComm, 2012, 14, 4142–4147 RSC.
- M. Wen, K. Mori, T. Kamegawa and H. Yamashita, Chem. Commun., 2014, 50, 11645–11648 RSC.
- F. Wu, L.-G. Qiu, F. Ke and X. Jiang, Inorg. Chem. Commun., 2013, 32, 5–8 CrossRef CAS PubMed.
- M. Saikia, D. Bhuyan and L. Saikia, New J. Chem., 2015, 39, 64–67 RSC.
- C. M. Granadeiro, M. Karmaoui, E. Correia, D. Julião, V. S. Amaral, N. J. O. Silva, L. Cunha-Silva and S. S. Balula, RSC Adv., 2015, 5, 4175–4183 RSC.
- L. Bromberg and T. A. Hatton, ACS Appl. Mater. Interfaces, 2011, 3, 4756–4764 CAS.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- F. Yang, C.-X. Yang and X.-P. Yan, Talanta, 2015, 137, 136–142 CrossRef CAS PubMed.
- S. Xian, Y. Yu, J. Xiao, Z. Zhang, Q. Xia, H. Wang and Z. Li, RSC Adv., 2015, 5, 1827–1834 RSC.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Chem. Commun., 2012, 48, 2140–2142 RSC.
- H. B. Tanh Jeazet, T. Koschine, C. Staudt, K. Raetzke and C. Janiak, Membranes, 2013, 3, 331–353 CrossRef PubMed.
- S. Bernt, V. Guillerm, C. Serreb and N. Stock, Chem. Commun., 2011, 47, 2838–2840 RSC.
- S. M. Cohen, Chem. Sci., 2010, 1, 32–36 RSC.
- M. Kim; and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC.
- M. Yamashita, M. Suzuki, H. Hirai and H. Kajigaya, Care Med., 2001, 29(8), 1575–1578 CrossRef CAS.
- N. A. Khan, I. J. Kang, H. Y. Seok and S. H. Jhung, Chem. Eng. J., 2011, 166, 1152–1157 CrossRef CAS PubMed.
- D. Jiang, A. D. Burrows and K. J. Edler, CrystEngComm, 2011, 13, 6916–6919 RSC.
- J. Zhou, K. Liu, C. Kong and L. Chen, Bull. Korean Chem. Soc., 2013, 34(6), 1625–1631 CrossRef CAS.
- T. Shen, J. Luo, S. Zhang and X. Luo, J. Environ. Chem. Eng., 2015, 3, 1372–1383 CrossRef CAS PubMed.
- M. Jacoby, Chem. Eng. News, 2008, 86, 13–16 Search PubMed.
- Chemistry World, 2009 (February), 49; http://www.rsc.org/images/MOFtcm18142948.pdf.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- P. Á. Szilágyi, E. Callini, A. Anastasopol, C. Kwakernaak, S. Sachdeva, R. van de Krol, H. Geerlings, A. Borgschulte, A. Züttelc and B. Damb, Phys. Chem. Chem. Phys., 2014, 16, 5803–5809 RSC.
- N. Cao, J. Su, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 9726–9734 CrossRef CAS PubMed.
- X. Zhou, W. Huang, J. Shi, Z. Zhao, Q. Xia, Y. Li, H. Wang and Z. Li, J. Mater. Chem. A, 2014, 2, 4722–4730 CAS.
- J. Pires, M. L. Pinto, C. M. Granadeiro, A. D. S. Barbosa, L. Cunha-Silva, S. S. Balula and V. K. Saini, Adsorption, 2014, 20, 533–543 CrossRef CAS.
- Y. Li and R. T. Yang, AIChE J., 2008, 54, 269–279 CrossRef CAS PubMed.
- L. H. Wee, F. Bonino, C. Lamberti, S. Bordigab and J. A. Martensa, Green Chem., 2014, 16, 1351–1357 RSC.
- W. Salomon, F.-J. Yazigi, C. Roch-Marchal, P. Mialane, P. Horcajada, C. Serre, M. Haouas, F. Taulelle and A. Dolbecq, Dalton Trans., 2014, 43, 12698–12705 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS.
- I. Senkovska and S. Kaskel, Microporous Mesoporous Mater., 2008, 112, 108–115 CrossRef CAS PubMed.
- N. V. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. Shmakov, Yu. A. Chesalov, D. N. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS PubMed.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- J. Yang, Q. Zhao, J. Li and J. Dong, Microporous Mesoporous Mater., 2010, 130, 174–179 CrossRef CAS PubMed.
- M. Wickenheisser and C. Janiak, Microporous Mesoporous Mater., 2015, 204, 242–250 CrossRef CAS PubMed.
- M. Wickenheisser, A. Herbst, R. Tannert, B. Milow and C. Janiak, Microporous Mesoporous Mater., 2015, 215, 143–153 CrossRef CAS PubMed.
- B. B. Saha, I. I. El-Sharkawy, T. Miyazaki, S. Koyama, S. K. Henninger, A. Herbst and C. Janiak, Energy, 2015, 79, 363–370 CrossRef CAS PubMed.
- T. Loiseau and G. Férey, J. Fluorine Chem., 2007, 128, 413–422 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquero and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- X. X. Huang, L. G. Qiu, W. Zhang, Y. P. Yuan, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, CrystEngComm, 2012, 14, 1613–1617 RSC.
- F. Yin, G. Li and H. Wang, Catal. Commun., 2014, 54, 17–21 CrossRef CAS PubMed.
- Z. Rui, Q. Li, Q. Cui, H. Wang, H. Chen and H. Yao, Chin. J. Chem. Eng., 2014, 22, 570–575 CrossRef CAS.
- S. Wang, L. Bromberg, H. Schreuder-Gibson and T. A. Hatton, ACS Appl. Mater. Interfaces, 2013, 5, 1269–1278 CAS.
- E. V. Ramos-Fernandez, M. Garcia-Domingos, J. Juan-Alcañiz, J. Gascon and F. Kapteijn, Appl. Catal., A, 2011, 391, 261–267 CrossRef CAS PubMed.
- C. X. Yang and X. P. Yan, Anal. Chem., 2011, 83, 7144–7150 CrossRef CAS PubMed.
- S. H. Jhung, J.-H. Lee, J. W. Yoon, C. Serre, G. Férey and J.-S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef CAS PubMed.
- H. Dai, J. Su, K. Hua, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 4947–4953 CrossRef CAS PubMed.
- L. D. Gelb, K. E. Gubbins, R. Radhakrsihan and M. Sliwinska-Bartowiak, Rep. Prog. Phys., 1999, 62, 1573–1659 CrossRef CAS.
- N. A. Sedron, J. P. R. B. Walton and N. Quirke, Carbon, 1989, 27, 853–861 CrossRef.
- A. Vishnyakov, P. Ravikovitch and A. V. Neimark, Langmuir, 2000, 16, 2311–2320 CrossRef.
Footnotes |
| † Dedicated to Prof. F. Ekkehardt Hahn on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Graphics of MIL-101(Cr) structure elements; additional N2 sorption isotherms, pore size distribution curves, powder X-ray diffractograms, and the picture of a 3 L autoclave. See DOI: 10.1039/c5dt02625c |
| This journal is © The Royal Society of Chemistry 2015 |