
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration and ion pair formation in aqueous Y3+–salt solutions†
Wolfram W.
Rudolph
*a and
Gert
Irmer
b
aMedizinische Fakultät der TU Dresden, Institut für Virologie im MTZ, Fiedlerstr. 42, 01307 Dresden, Germany. E-mail: Wolfram.Rudolph@tu-dresden.de
bTechnische Universität Bergakademie Freiberg, Institut für Theoretische Physik, Leipziger Str. 23, 09596 Freiberg, Germany
First published on 29th September 2015
Abstract
Raman spectra of aqueous yttrium perchlorate, triflate (trifluoromethanesulfonate), chloride and nitrate solutions were measured over a broad concentration range (0.198–3.252 mol L−1). The spectra range from low wavenumbers to 4200 cm−1. A very weak mode at 384 cm−1 with a full width at half height at 50 cm−1 in the isotropic spectrum suggests that the Y3+ – octa-aqua ion is thermodynamically stable in dilute perchlorate solutions (∼0.5 mol L−1) while in concentrated perchlorate solutions outer-sphere ion pairs and contact ion pairs are formed. The octa-hydrate, [Y(OH2)8]3+ was also detected in a 1.10 mol L−1 aqueous Y(CF3SO3)3 solution. Furthermore, very weak and broad depolarized modes could be detected which are assigned to [Y(OH2)8]3+(aq) at 100, 166, 234 and 320 cm−1 confirming that a hexa-hydrate is not compatible with the hydrated species in solution. In yttrium chloride solutions contact ion pair formation was detected over the measured concentration range from 0.479–3.212 mol L−1. The contact ion pairs in YCl3(aq) are fairly weak and disappear with dilution. At a concentration <0.2 mol L−1 almost all complexes have disappeared. In YCl3 solutions, with additional HCl, chloro-complexes of the type [Y(OH2)8−nCln]+3−n (n = 1,2) are formed. The Y(NO3)3(aq) spectra were compared with a spectrum of a dilute NaNO3 solution and it was concluded that in Y(NO3)3(aq) over the concentration range from 2.035–0.198 mol L−1 nitrato-complexes [Y(OH2)8−n(NO3)ln]+3−n (n = 1,2) are formed. The nitrato-complexes are weak and disappear with dilution <0.1 mol L−1. DFT geometry optimizations and frequency calculations are reported for both the yttrium–water cluster in the gas phase and the cluster within a polarizable continuum model in order to implicitly describe the presence of the bulk solvent. The bond distance and angle for the square antiprismatic cluster geometry of [Y(OH2)8]3+ with the polarizable dielectric continuum is in good agreement with data from recent structural experimental measurements. The DFT frequency of the Y–O stretching mode of the [Y(OH2)8]3+ cluster, in a polarizable continuum, is at 372 cm−1 in satisfactory agreement with the experimental value.
1. Introduction
Yttrium is a silvery-metallic transition metal in group 3 and chemically similar to the lanthanides. Yttrium is almost always found combined with the lanthanides in rare earth minerals but is never found in nature as a free element. The most important use of yttrium is in making phosphors and also in the production of electrolytes, electronic filters, lasers and superconductors.1–3 Its radioactive isotope, 90Y, has a wide use in radiation therapy to treat cancer.4 Yttrium has no known biological role though it is found in most, if not all, organisms and tends to concentrate in the liver, kidney, spleen, lungs, and bones of humans.5 The coordination numbers in crystalline Y3+-compounds vary widely from 3 to 126 but eight and nine fold coordination are the most frequent.7–11In aqueous solution, yttrium exists exclusively in the trivalent state, Y3+, and the ion is strongly hydrated judging by its high hydration enthalpy,3 due to its high charge to radius ratio. The hydration number for Y3+ in aqueous solution was determined based on structural techniques such as X-ray diffraction (XRD), anomalous X-ray diffraction (AXD) and large angle X-ray scattering (LAXS).12–16 The local structure of aqueous Y3+ salt solutions was also studied applying extended X-ray absorption fine structure (EXAFS), and X-ray absorption fine structure (XAFS) methods.17–20 A combined study of neutron scattering and EXAFS measurements applying reversed modeling on YCl3(aq) must be mentioned.21 A summary of experimental structural results is presented in Table 1. From the Table, it is clear that Y3+ is hydrated by 8 water molecules in the first-shell forming a square-antiprismatic geometry. The Y3+–O bond distance was found at 2.366 Å. In addition to the experimental work, a computer simulation significantly contributed to clarifying the details of the structure and dynamics of the waters in the first hydration shell of Y3+.22 This simulation study confirmed the Y3+ octa-hydrate and its square antiprismatic geometry. Furthermore, the structural results show that X-ray measurements and EXAFS investigations were carried out at high and low solute concentrations using different anions as counter ions. This, however, poses a problem because contact ion pairs may form in concentrated solutions and this fact has been reported in an earlier XRD study on yttrium selenate solutions.12 A recent notable structural study on an aqueous 1 mol kg−1 YCl3 solution applying a local structure refinement model of disordered material21 revealed contact ion pairs between Y3+ and Cl−.
| Method | Solution composition | La3+/mol L−1 | CN | Y–O bond distance/Å | Remarks | Ref. |
|---|---|---|---|---|---|---|
| XRD | Y(ClO4)3 | 1.094 | 8.0 ± 0.3 | 2.37 ± 0.005 | Second sphere: 4.52 Å | 12 |
| Y2(SeO4)3 | 0.77 | 8.0 ± 0.3 | 2.33 ± 0.005 | Second sphere: 4.47 Å | ||
| 0.89 | 2.325 ± 0.005 | Second sphere: 4.47 Å | ||||
| AXD | YCl3 | 0.5 | 8.1 ± 0.3 | 2.46 ± 0.02 | 13 | |
| 1.0 | 8.2 ± 0.5 | 2.51 ± 0.02 | ||||
| EXAFS | YCl3 | 0.6 | 7.5 ± 0.3 | 2.34 ± 0.02 | Y K-edge additionally Raman spectra of YCl3 in H2O and D2O are given | 17 |
| 1.2 | 7.6 ± 0.3 | 2.34 ± 0.02 | ||||
| 2.3 | 7.9 ± 0.3 | 2.34 ± 0.02 | ||||
| YBr3 | 0.5 | 7.5 ± 0.3 | 2.33 ± 0.02 | |||
| 1.0 | 8.5 ± 0.3 | 2.33 ± 0.02 | ||||
| 2.1 | 7.5 ± 0.3 | 2.34 ± 0.02 | ||||
| XRD | YCl3 | 2.73 | 7.7 ± 0.3 | 2.36 ± 0.02 | 14 | |
| YBr3 | 2.67 | 7.8 ± 0.3 | 2.36 ± 0.02 | |||
| EXAFS | YCl3 | 0.1 | 9.1 ± 0.2 | 2.36 ± 0.02 | Temperature dependent measurements from 25–340 °C | 18 |
| Y(NO3)3 | 0.05 | 9.4 ± 0.2 | 2.367 ± 0.006 | |||
| EXAFS/LAXS | Y(ClO4)3 acidified with HClO4 | 1.71 | 8.0 ± 0.1 | 2.368 ± 0.005 | Mean value of second hydration sphere consisting of ∼16 oxygen atoms at a mean Y–O distance of 4.40(4) Å | 19 |
| 1.14 | ||||||
| 0.43 | ||||||
| EXAFS/XANES | YBr3 | 0.005 mol kg−1 | 8.6 ± 0.1 | 2.352 ± 0.003 | Concentration profile from 0.005–2.00 mol kg−1 | 20 |
| 2.00 mol kg−1 | 8.8 ± 0.1 | 2.353 ± 0.001 | ||||
| AXD | YCl3 | 3.5 mol kg−1 | 8 | 2.33 | Second sphere at 4.77 YCl3; conctact ion pairs between Y3+ and Cl− | 15 |
| XRD | Y(NO3)3 | 2.89 | 8.0 ± 0.1 | 2.36 ± 0.01 | Additionally Raman spectrum of Y(NO3)3 in H2O was given | 16 |
| ND/EXAFS | YCl3 | 1 mol kg−1 | ∼7.4 ± 0.5 Y–Ow | 2.33 | Contact ion pairs between Y3+ and Cl− were established | 21 |
| ∼0.8 ± 0.2 Y-Cl |
Raman spectroscopy which probes the immediate environment of metal ions in solution was frequently used to study hydrated cations by measuring the vibrational modes of a [M(H2O)n]x+ species.23–28 A highly polarized band in the 250–550 cm−1 region due to the symmetrical metal–oxygen mode of the hydrated cation is the most characteristic peak. This peak is sensitive to possible ion pair formation. Occasionally, all the bands of the metal–oxygen modes may be observed and used to support the assignment of the point group symmetry and coordination number in aqueous solution. Raman scattering measurements on Y3+(aq) should allow, in principle, the characterization of the solution structure in greater detail. However, aqueous Y3+ solutions were measured by Raman spectroscopy only on a few occasions.16,17,29 Due to the limitations of Raman spectroscopic technique at the time, the quality of the spectra in the low frequency range17,29 was poor. Furthermore, no reduced spectral treatment of the data in the low frequency range was applied. It has been shown on a variety of aqueous metal salt solutions that for meaningful Raman spectroscopic analysis, reduced spectra (spectra in R-format) are necessary in the terahertz frequency range23–28 in order to account for the Bose-Einstein correction and scattering factor.23,30
The present study was undertaken to characterize the hydration and speciation in aqueous Y3+ solutions and to this end Y3+ – salt solutions with common anions (ClO4−, CF3SO3−, Cl−, NO3−) were studied over a broader concentration range and down to the terahertz frequency range. Triflate and perchlorate are considered non-complex-forming anions and were therefore chosen to measure the Y–O modes in aqueous solution so as to identify and assign bands unique to the first hydration sphere of Y3+(aq). A Y(ClO4)3 solution in heavy water was also measured in order to characterize the vibrational isotope effect by changing from [Y(H2O)8]3+ to [Y(D2O)8]3+. In a variety of di- and trivalent metal ion solutions with chloride and nitrate as counterions, however, it has been shown that these anions readily form complexes24–28 and the question arises as to whether these complexes also occur with Y3+(aq). Specifically, the following aqueous systems were measured by Raman spectroscopy at 23 °C: Y(ClO4)3 and Y(ClO4)3 plus HClO4, Y(CF3SO3)3, YCl3 and YCl3 plus additional HCl, and Y(NO3)3. Y(ClO4)3 solutions in heavy water were measured not only to determine the isotope effect on the Y–O modes (Y–OH2/D2O) but also to measure the influence of the salts on the water and heavy water stretching and deformation bands as a function of the Y3+ salt concentration.
Calculations on yttrium–water clusters with six and eight waters in the first sphere were considered applying density functional theory (DFT) in the gas phase. However, gas phase clusters do not reflect the aqua-cluster geometry nor the vibrational modes of metal ions in solution23–28 and therefore, Y3+– clusters with a polarizable dielectric continuum were simulated in order to take into account the effects of the bulk solvent. Frequency calculations were carried out via the calculation of the second derivative of the energy with respect to the nuclear positions of Y3+–water clusters.
2. Experimental details; data analysis and DFT calculations on Y3+–water clusters
A. Preparation of solutions
The yttrium ion concentrations of all solutions were analysed by complexometric titration.31 The solution densities were determined with a pycnometer at 23 °C and the molar ratios water per salt were calculated (Rw-values). For Raman spectroscopic measurements the solutions were filtered through a fine sintered glass frit (1–1.6 μm pore size). The solutions showed no Tyndall effect and were “optically empty”.32Yttrium perchlorate solutions were prepared from Y2O3 (Sigma-Aldrich, 99.9%) and HClO4 in a beaker until all oxide dissolved. A Y(ClO4)3 stock solution was prepared at 2.559 mol L−1 (Rw = 16.25). This solution was acidified with a slight amount of HClO4 and a pH value at ∼3.5 for the concentrated solutions was measured. From this stock solution, the following dilution series was prepared: 2.378 mol L−1 (Rw = 21.46), 1.785 mol L−1 (Rw = 25.67), 1.280 mol L−1 (Rw = 37.81), 0.853 mol L−1 (Rw = 59.45), 0.640 mol L−1 (Rw = 81.10) and 0.256 mol L−1 (Rw = 210.9). The solutions were analysed for dissolved chloride with a 5% AgNO3 solution. The absence of a white AgCl precipitate was proof that the stock solution was free of Cl−.
Two Y(ClO4)3 solutions in heavy water were prepared from a deuterated Y(ClO4)3 stock solution and with 99.9 atom % D (Sigma-Aldrich) at 2.30 mol L−1 and 1.73 mol L−1.
A 1.10 mol L−1 Y(CF3SO3)3 solution was prepared from anhydrous Y(CF3SO3)3 (Sigma-Aldrich, 99.9%) and triply distilled water.
Seven YCl3 solutions were prepared from YCl3·6H2O (Sigma, 99.9%) and triply distilled water. The solution concentrations were found at 3.212 mol L−1 (Rw = 15.40), 2.378 mol L−1 (Rw = 21.46), 2.141 mol L−1 (Rw = 24.00), 1.175 (Rw = 45.30), 1.010 mol L−1 (Rw = 53.53), 0.803 mol L−1 (Rw = 67.19) and 0.479 mol L−1 (Rw = 113.82). Furthermore, a solution series with an excess of HCl was prepared (YCl3 plus HCl): (A) 2.13 mol L−1 YCl3 + 4.03 mol L−1 HCl and (B) 2.17 mol L−1 YCl3 + 1.31 mol L−1 HCl. Two HCl solutions were also prepared at 3.97 mol L−1 and at 1.325 mol L−1.
Three Y(NO3)3 solutions were prepared from Y(NO3)3·6H2O and triply distilled water: 2.035 mol L−1 (RW = 23.04), 1.036 mol L−1 (RW = 49.20) and 0.198 mol L−1 (RW = 275.88).
B. Spectroscopic measurements
Raman spectra were measured in the macro chamber of the T 64000 Raman spectrometer from Jobin Yvon in a 90° scattering geometry at 23 °C. These measurements have been described elsewhere.30,33 A quartz cuvette from Hellma Analytics (Müllheim, Germany) with 10 mm path length and a volume 1000 μL was used. Briefly, the spectra were excited with the 514.532 nm line of an Ar+ laser at a power level of 1100 mW at the sample. After passing the spectrometer in subtractive mode, with gratings of 1800 grooves per mm, the scattered light was detected with a cooled CCD detector. IVV and IVH spectra were obtained with fixed polarization of the laser beam by rotating the polarizator at 90° between the sample and the entrance slit to give the scattering geometries:| IVV = I(Y[ZZ]X) = 45α′2 + 4γ′2 | (1) |
| IVH = I(Y[ZY]X) = 3γ′2. | (2) |
The isotropic spectrum, Iiso is then constructed:
| Iiso = IVV − 4/3·IVH. | (3) |
The polarization degree of the Raman bands, ρ (ρ = IVH/IVV) was determined using a polarizer and if necessary adjusted before each measuring cycle using CCl4. A detailed account on this procedure may be found in ref. 30 and 33.
In order to obtain spectra defined as R(![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ) which are independent of the excitation wavenumber νL, the measured Stokes intensity should be corrected for the scattering factor (νL − ())3. In the case of counting methods used, the measured count rates were corrected with the factor (νL − ())3. The spectra were further corrected for the Bose-Einstein temperature factor, B = [1 − exp(−hc/kT)] and the frequency factor, , to give the so called reduced spectrum, R(). It is also possible to calculate the isotropic spectrum in R-format from the corrected RVV and RVH spectra according to eqn (4):
) which are independent of the excitation wavenumber νL, the measured Stokes intensity should be corrected for the scattering factor (νL − ())3. In the case of counting methods used, the measured count rates were corrected with the factor (νL − ())3. The spectra were further corrected for the Bose-Einstein temperature factor, B = [1 − exp(−hc/kT)] and the frequency factor, , to give the so called reduced spectrum, R(). It is also possible to calculate the isotropic spectrum in R-format from the corrected RVV and RVH spectra according to eqn (4):
| R()iso = R()VV − 4/3R()VH. | (4) |
In the low wavenumber region, the I() and RQ() spectra are significantly different and only the spectra in R-format are presented. It should be noted that one of the advantages of using isotropic R-spectra is that the baseline is almost flat in the 50–700 cm−1 wavenumber region allowing relatively unperturbed observation of the presence of any weak modes.23,30,33
C. DFT calculations
The calculations were carried out using the Gaussian03 package34 employing the unrestricted B3LYP functional.35 The LANL2DZ (Los Alamos National Laboratory 2 Double Zeta) basis set was used which adequately reproduces the geometrical parameters, in particular the experimentally observed Y–O distance. LANL2DZ uses a relativistic effective core potential (ECP) for the inner electrons of Y3+, and its valence electrons. All electrons for the other atoms are described by D95 V (Dunning/ Huzinaga valence double-zeta) functions. The 6-31G (double-zeta Pople type) basis set and application of diffuse and polarization functions for the atoms O and H resulted in small changes. Placing the cluster in a solvent continuum employing the Polarized Continuum Model (PCM) which takes into account the solvation effect of bulk water gave significantly better results compared with the experimental frequencies. The PCM used was the version described in ref. 36 where the solvent is modelled as an isotropic and homogeneous continuum, characterized by its dielectric properties. The cavity is defined as a set of interlocking spheres attached to the solute atoms. The electrostatic solute–solution interaction is calculated introducing an apparent charge distribution spread on the cavity surface.Geometries of gas phase clusters with 8 water molecules surrounding the Y3+ ion, were optimized applying different basis sets and several cluster geometries for [Y(H2O)8]3+ with different symmetries namely D4, C2 and S8 were considered. A [Y(H2O)8]3+ cluster with symmetry S8 including a polarizable dielectric continuum (PC) was simulated in order to take into account the solvent effect. The cluster with S8 symmetry was the only cluster which led to an energy minimum without imaginary frequencies. The frequencies of the [Y(H2O)8]3+ cluster including a polarizable dielectric continuum were calculated (B3LYP/LANL2DZ). The DFT simulations on [Y(H2O)6]3+ gave a stable geometry with symmetry Th. The [Y(H2O)6]3+ cluster imbedded in a PC was optimized and frequencies were calculated. The geometric data and the frequency of the breathing mode of the [Y(H2O)8]3+ cluster in comparison with the [Y(H2O)6]3+ will be discussed below.
3. Results and discussion
3.1. The [Y(OH2)8]3+(aq) ion
Y3+ is strongly hydrated in aqueous solution as indicated by its large standard molar enthalpy of hydration (ΔHohyd) at ∼ −3640 kJ mol−1 but it should be stressed that ΔHohyd – values reported in the literature scatter from −3583 to −3733 kJ mol−1.37–42 Our DFT value for ΔHohyd is −3951 kJ mol−1 which is smaller than the thermodynamic value (calculation procedure in Appendix A). The Y3+ ion, hydrated by eight water molecules in the first hydration shell with a Y–O bond distance at 2.366 Å, and a second hydration shell, much more diffuse, at 4.40 Å consisting of ∼16 water molecules was observed.19 The optimized [Y(H2O)8]3+ geometry in the gas phase and also with a polarizable continuum gave a square antiprismatic YO8 coordination polyhedron with symmetry S8 (Fig. 1). The cluster, [Y(H2O)8]3+, was also optimized applying higher basis sets which led to geometries with symmetry D4 and C2 (Table S1†). However, only the [Y(H2O)8]3+ cluster with S8 symmetry imbedded in a PC could be successfully simulated. The calculated Y–O bond distance of the [Y(H2O)8]3+ cluster with a solvation sphere, taking into account the influence of the bulk water, is in good agreement with the experimental value (Table 1). The [Y(H2O)8]3+ cluster in the gas phase, however, shows a larger value compared to the experimental Y–O bond distance (lack of bulk water). These DFT results are given in Table 2 together with the experimental data and a Car–Parrinello molecular dynamics (CPMD) simulation result.22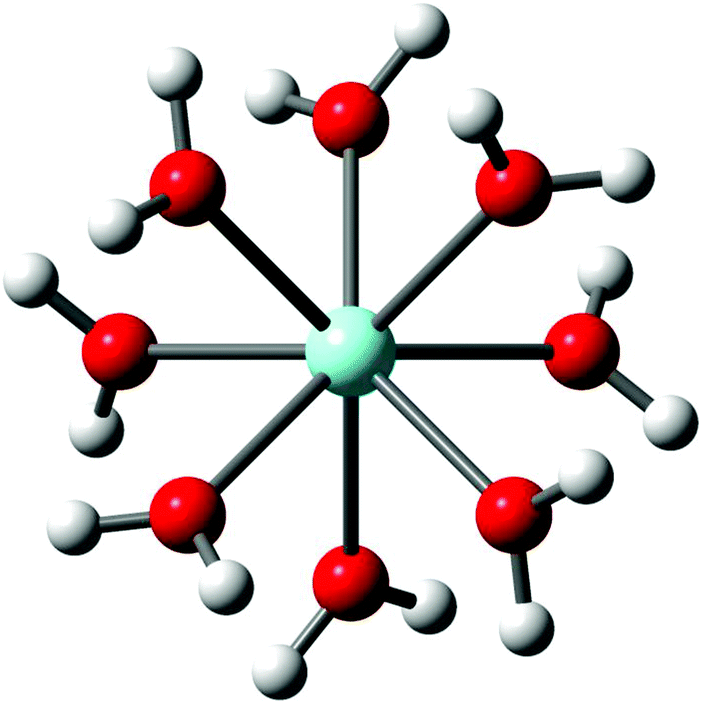 | ||
| Fig. 1 Structure of the octa-aqua Y3+-ion (S8 – symmetry). | ||
| Bond distances (Å) and angels (°) | DFT data/gas phase cluster | DFT data/cluster + PC model |
Ref. 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Ref. 12b |
Ref. 19c |
Ref. 18d |
|---|---|---|---|---|---|---|
| a CPMD; O–H bond distance and H–O–H angle from Fig. 5.22 b XRD: results for Y(ClO4)3(aq). c EXAFS: results for Y(ClO4)3(aq). d EXAFS: results on diluted YBr3(aq). | ||||||
| Y–O | 2.399 | 2.354 | 2.38 | 2.37 | 2.368 | 2.618 |
| O–H | 0.981 | 0.989 | 0.96 | |||
| H–O–H | 109.6 | 110.9 | 106 | |||
A Y3+-hexa-hydrate was also successfully optimized but a noticeably smaller Y–O bond distance (Table S2†) was found compared to the measured one at 2.366 Å (ref. 19) which shows that the hexa-hydrate may be ruled out as a valid species in aqueous solution.‡ The results for the [Y(H2O)6]3+ in the gas phase and modelled with a solvation sphere are also given in Table S2.† (In contrast to solution state, in crystalline compounds CNs vary from 3–12.3,6)
The vibrational analysis of the [Y(OH2)8]3+ (S8 symmetry) with its 69 normal modes (n.m.'s) leads to the following irreducible representation: Γv(S8) = 8a(Ra) + 9b(i.r.) + 18e1(Ra, i.r.) + 18e2 (n.a.) + 16e3 (n.a.). The modes with character a and e1 are Raman active and those with character b and e1 are infrared active. The vibrations can be divided into 48 internal and external vibrations of the eight coordinated water molecules plus 21 n. m.'s of the YO8 skeleton.
The vibrations of the water modes are decoupled from those of the YO8 skeleton and the water molecules of the first shell are considered point masses. The YO8 skeleton (D4d symmetry) with its 9 atoms leads to 21 n.m.'s. The irreducible representation of these skeleton modes is as follows: Γv(D4d) = 2a1 (Ra) + b1(i.a.) + 2b2(i.r.) + 3e1(i.r.) + 3e2(Ra) + 2e3(Ra). Although the YO8 skeleton possesses no symmetry centre, the mutual exclusion rule is nevertheless effective. The 7 modes with the character a1, e2 and e3 are Raman active while the six modes with the character b1, b2 and e1 are infrared allowed. The symmetric Y–O stretch, the breathing mode, is only Raman active and appears strongly polarized in the Raman spectrum as the strongest band of the skeleton. Two additional depolarized Raman active stretching modes are expected (character e2 and e3) as well as four other Raman deformation modes (character a1, e2 and e3). In I.R., two stretching modes (character b2 and e1) are expected and the remaining are deformations. In reality, however, the skeleton modes may not always be easily detected because they appear quite broad, weak and even obscured.
Considering the coordinated water molecules of the [Y(OH2)8]3+ cluster, the internal and external n.m.'s of the water molecules may be divided into 24 internal and 24 external vibrations. These vibrations are derived from the rotational- and translational degrees of freedom of the isolated water molecule. The n.m.'s of these waters, the external modes, are librations such as wag, twist, and rock.43 Generally, the water molecules in the metal-aqua complexes possess weak, very broad modes below 1200 cm−1. In addition to these librations, internal water modes are observed: the deformation mode, ν2(H2O) and two stretching OH modes, ν1 and ν3. The deformation mode in liquid water is found at 1640 cm−1 and the stretching modes at ∼3400 cm−1 appear as a very broad structured band of H-bonded water molecules. The water modes are modified when coordinated to metal ions such as Y3+ but are difficult to separate from the contributions of the librational and internal water modes of the bulk phase. In neat liquid water, the H-bonded water molecules show broad and weak librational modes and internal water modes, the deformation band, δ H–O–H, and the stretching O–H bands.43 Spectra of liquid water and heavy water, bands and band assignments are given elsewhere.44–46
The hydration sphere of Y3+(aq) is somewhat labile and a water-exchange rate constant kex at 25 °C was given at 2 × 107 s−1 (from H2O–SO42− interchange rates) with a water residence time τ = 50 ns. The hydration sphere of Y3+(aq), however, is less labile compared to [La(OH2)9]3+(aq).47,48§ The mechanistic path for the water exchange for [Ho(OH2)8]3+ follows an associative water exchange process.51–53 It is safe to assume a similar water exchange mechanism for Y3+(aq) because of its similar ionic radius compared to Ho3+(aq).54
Chemically, yttrium resembles the heavy rare earth elements more closely than scandium,3,6 its neighbor in the periodic table, and especially holmium in its chemical behavior.3 Y3+(aq) is less basic in aqueous solution (logKh,1 = −7.8) compared to La3+(aq) (logKh,1 = −8.81).55 A first principle MD simulation on the stepwise hydrolysis reaction of Y3+ in aqueous solution has been presented recently.56
3.2. Y(ClO4)3 and Y(CF3SO3)3 solutions in water and heavy water
 | ||
| Fig. 2 Overview Raman spectrum (polarized and depolarized) of a 1.28 mol L−1 Y(ClO4)3 solution (Rw = 37.7). Upper panel: bands of ClO4−(aq) and the water deformation mode at 1626 cm−1. The inset at the LHS shows spectra in the terahertz frequency rage in greater detail. The inset at the RHS shows the ν1(a1) ClO4− band at full detail. Lower panel: OH-stretching band of water and the O–H⋯O* band of the weakly H-bonded water, ClO4−⋯H2O at 3538 cm−1. The peak positions of the O–H water bands are given at 3240 and 3428 cm−1. | ||
The ClO4− ion (Td symmetry) has nine vibrational degrees of freedom and its internal vibrations span the representation Γvib(Td) = a1(Ra) + e(Ra) + 2f2(Ra, i.r.). All four n.m.'s are Raman active, but in i.r. only the f2 modes are active. The Raman and infrared spectra of ClO4−(aq) are well characterized24–26 and therefore shall be only, briefly, discussed. In dilute solution, the symmetric Cl–O stretch, ν1(a1) ClO4− appears at 931.5 cm−1, is totally polarized (ρ = 0.005) and has a band width equal to 7.1 cm−1. The antisymmetric stretch, ν3(f2) ClO4− centred at 1105 cm−1 and the deformation modes ν4(f2) ClO4− at 629 cm−1 and ν2(e) ClO4− at 458 cm−1 are depolarized. In a Y(ClO4)3 solution at 0.256 mol L−1 , the ν1(a1) ClO4− band, the strongest Raman band, appears at 931.8 cm−1 (fwhh at 7.4 cm−1), shifts to 934.5 cm−1 and broadens considerably (fwhh = 13.1 cm−1) in a solution of 2.559 mol L−1. At the same time, the antisymmetric stretch, ν3(f2) ClO4− shifts to slightly higher wavenumbers and broadens. The concentration behavior of the ClO4− modes in a similar system, La(ClO4)3(aq), is given in ref. 25.
The Raman spectra of Y(ClO4)3(aq) reveal, in addition to the perchlorate-bands, weak bands in the terahertz frequency region in the RVV orientation which will be discussed further below (Fig. 2, upper panel). In the isotropic scattering profile, a band appears at 384 cm−1 but does not exist in NaClO4(aq) or HClO4(aq). The band must therefore stem from vibrations connected to the YO8 skeleton. This strongly polarized band is assigned to the symmetric stretching mode, the breathing mode of the [Y(OH2)8]3+ cluster. In a fairly dilute Y(ClO4)3 solution at 0.256 mol L−1 the band appears symmetrical with a fwhh = 50 cm−1 (Fig. S1†). With increasing Y(ClO4)3 concentration, it widens and shifts to slightly lower wavenumbers. In a concentrated solution at 2.559 mol L−1, the breathing mode, ν1YO8 of the [Y(OH2)8]3+ cluster appears at 382 cm−1. The concentration profiles of the isotropic breathing mode, YO8 is shown in Fig. 3. The integrated band intensity of this ν1YO8 mode, depending on the Y(ClO4)3 solute concentration, plots as a straight line and the linear function: A384 = 1331.2·C0 (coefficient of determination, R2 = 0.997) with A384 the integrated band intensity of ν1YO8 and C0 the stoichiometric Y(ClO4)3 concentration. Replacing water with heavy water leads to an isotope shift to lower wavenumbers by a factor of ∼0.948 in Y(ClO4)3(D2O). The effect of deuteration on the YO8 breathing mode was measured in Y(ClO4)3–D2O solutions and a band at 365 cm−1 observed. The theoretical shift of ν1 on deuteration (H2O/D2O considered as point masses) is given according to:
| ν′1 = ν1[m(H2O)/m(D2O)]1/2 = (384 cm−1)·1.054 = 364.2 cm−1. | (5) |
 | ||
| Fig. 3 Concentration plot of isotropic Raman spectra (R-format) of five Y(ClO4)3 solutions. From bottom to top: (A) 0.256 mol L−1 (Rw = 211.0), (B) 0.640 mol L−1 (Rw = 81.4), (C) 0.853 mol L−1 (Rw = 59.3), (D) 1.280 mol L−1 (Rw = 37.7) and (E) 2.559 mol L−1 (Rw = 15.7). Note, that the peak position of the YO8 breathing mode at 384 cm−1 for a 0.256 mol L−1 solution shifts only slightly to lower wavenumbers at 382 cm−1 for a 2.559 mol L−1 Y(ClO4)3 solution. At the same time, the band broadens and a shoulder at 336 cm−1 appears for the two most concentrated solutions. | ||
Relative intensity measurements confirm that the scattering intensity of the ν1Y–O mode is very weak with the scattering coefficient, Sh = 0.0380. The Sh values, defined as the R-corrected relative scattering efficiency of the M–O bands, were published for a variety of stretching modes of hexa-aqua metal ions in solution.24,25,27,28 The ν1 breathing mode for [La(H2O)9]3+ at 343 cm−1 (ref. 26) also possesses a small relative intensity of 0.025. Both ions, Y3+ and La3+, have a low polarizability and are classified as hard cations according to the HSAB concept of Pearson.57
In addition to the breathing mode of the [Y(OH2)8]3+ cluster, four other modes appear at 100.5, 166, 233.5 and 319.5 cm−1 in the anisotropic scattering (Fig. 4). These weak, broad modes stem from the YO8 skeleton fundamentals of the [Y(OH2)8]3+ and break the symmetry of the YO8 skeleton and therefore appear only in the anisotropic scattering but not in the isotropic profile. These depolarized modes were also observed slightly down-shifted in Y(ClO4)3(D2O) which is caused by the isotope effect going from [Y(OH2)8]3+ to [Y(OD2)8]3+. In [Y(OD2)8]3+(D2O) these bands appear at 90, 143, 221, and 302.5 cm−1. The isotope effect on the coordinated heavy water modes was calculated according to eqn (5), however, ν1 was substituted with νi (where index i denotes the frequency of the depolarized YO8 modes). Table 3 shows the summarized band parameters of the five observed skeleton modes of YO8 in water and heavy water. From these Raman spectroscopic results, it follows directly that the Y3+ hydration shell cannot constitute a hexa-hydrate with Th symmetry which has been, for instance, characterized for [Al(OH2)6]3+(aq).43,58 Group theoretical considerations expect only three skeleton modes in Raman; one of which should be totally polarized (breathing mode for the AlO6 skeleton) and the remaining two depolarized. All of these bands were detected in the Raman spectrum of an Al(ClO4)3 solution with the symmetric stretching mode of [Al(OH2)6]3+ at 525 cm−1 strongly polarized and the other two at 438 cm−1 and 332 cm−1 depolarized.43,58
 | ||
| Fig. 4 Raman scattering profiles (RVV and RVH) of a 2.559 mol L−1 Y(ClO4)3 solution (Rw = 15.7). Upper panel: polarized and depolarized scattering profiles dominated by the ClO4−(aq) deformation bands at 459 cm−1 and 628 cm−1. The inset shows the very weak, depolarized scattering contributions at 100, 166, 235, 319 cm−1 and the strongly polarized band 382 cm−1 in the polarized scattering (RVV = 45α′2 + 4γ′2). Lower panel: isotropic scattering profile (Riso) showing the breathing mode, ν1 YO8 and the weak and broad restricted translational band at ∼170 cm−1. | ||
| [Y(H2O)8]3+ | [Y(D2O)8]3+ | ||||
|---|---|---|---|---|---|
| Peak position/cm−1 | Band area | fwhh/cm−1 | Peak position/cm−1 | Band area | fwhh/cm−1 |
| dp = depolarized mode; pp strongly polarized. | |||||
| 100.5 (dp) | 78 | 50 | 90 (dp) | 33.5 | 76 |
| 166 (dp) | 696 | 87.5 | 143 (dp) | 138 | 69.8 |
| 233.5 (dp) | 570 | 63 | 221 (dp) | 189 | 63 |
| 319.5 (dp) | 331 | 67 | 302.5 (dp) | 131 | 53 |
| 382 (pp) | 3410 | 58 | 365 (pp) | 2050 | 53 |
Although the perchlorate ion was chosen as the counter ion because it is known as a non-complexing anion, penetration into the first hydration sphere of the Y3+ seems plausible in the most concentrated Y(ClO4)3(aq). In the Y(ClO4)3(aq) at 2.559 mol L−1 the mole ratio solute to water is 16.3. This water content is enough to completely hydrate the Y3+ ion with 8.3 water molecules remaining. It is clear that in these concentrated solutions solvent – separated ion pair formation is simply forced on Y3+ because of the lack of water. At such a concentration state the outer-sphere ion pairs, [Y(OH2)8]3+·ClO4−, may exist in equilibrium with contact ions pairs (Y3+·ClO4−). These ion pairs may explain the slight concentration dependence of the peak position, the shoulder at 336 cm−1 mentioned previously and the broadening of the ν1YO8 band. In a 0.256 mol L−1 Y(ClO4)3 solution, however, the ν1YO8 band occurs as a symmetrical band at 384 cm−1 and a fwhh at ∼50 cm−1. Furthermore, to rule out a hydrolysis effect, ternary solutions Y(ClO4)3/HClO4/H2O were studied. Y(ClO4)3 solution at 2.160 mol L−1 plus 1.518 mol L−1 HClO4 and a more dilute one at 1.080 mol L−1 Y(ClO4)3 plus 0.759 mol L−1 HClO4 were measured in the terahertz frequency range in order to check the influence of the hydrolysis of Y3+(aq). Isotropic bands at 382.5 and 381.5 cm−1 appeared with a 15% larger band width than those in Y(ClO4)3(aq) without additional HClO4. This result shows that the hydrolysis of Y3+ cannot be the reason for the variation of the band parameters such as peak position and fwhh of the isotropic Y–O mode and reinforces the assumption of contact-ion-pair formation in the most concentrated solutions.
The DFT frequency of the ν1YO8 for [Y(H2O)8]3+ imbedded in a polarizable dielectric continuum simulating the bulk water phase was calculated at 372 cm−1 in satisfactory agreement with the measured value (Table 3) and in contrast to the breathing mode for the cluster in the gas phase at 336.5 cm−1. Imbedding the [Y(H2O)8]3+ species in a PC to simulate the bulk water is necessary to achieve acceptable agreement with the experimental frequencies. The DFT frequency of the ν1YO8 mode for [Y(D2O)8]3+ imbedded in a PC was calculated at 351 cm−1 and in satisfactory agreement with the measured value at 365 cm−1 (Table 3). DFT frequencies for the whole [Y(H2O)8]3+ cluster which includes YO8 skeletal, librational and internal water modes are given in Table S3 ESI.† The Y–O bond distance of the [Y(H2O)8]3+ is 2.354 Å in good agreement with the experimental structural data (Table 1).
Similar cluster calculations on Y3+ with 6 water molecules surrounded by a polarizable dielectric continuum lead to a stable cluster geometry [Y(H2O)6]3+ which possesses Th symmetry. The Y3+-hexa-hydrate has a much shorter Y–O bond distance at 2.28 Å. The DFT frequency for the breathing mode, ν1YO6 at 408 cm−1 is 7% higher than the experimental one. These results on the [Y(H2O)6]3+ cluster show that the hexa-hydrate can be ruled out as a valid cluster geometry for Y3+ in solution. The ν1YO6 frequency, geometrical parameters and the electronic energy of the cluster [Y(H2O)6]3+ with a solvation sphere, modelled with a PC and for comparison with a gas phase cluster (no bulk-sphere taken into account) are given in Table S3.† To reiterate, the DFT results for [Y(H2O)8]3+ are in better agreement with the experimental data than for [Y(H2O)6]3+.
The Raman spectroscopic analysis clearly shows that in dilute Y(ClO4)3(aq) a mode appears at 384 cm−1 (fwhh = 50 cm−1) which represents the breathing mode of the YO8 skeleton. However, four additional weak and broad bands, depolarized in character, could be characterized in concentrated Y(ClO4)3(aq). The five bands observed in Y(ClO4)3(aq) show that the Y3+ cannot form a hexa-hydrate in aqueous solution. In concentrated Y(ClO4)3(aq) > 1.5 mol L−1, outer-sphere ion pair formation is observed indicated by the slight shift of the ν1YO8 mode, an asymmetry at 336 cm−1. DFT simulations support the existence of the [Y(H2O)8]3+ cluster and the calculated breathing mode of the cluster is in satisfactory agreement with the measured one.
3.3. YCl3(aq)
An overview Raman spectrum of a 3.212 mol L−1 YCl3 solution (Rw = 15.37) is presented in Fig. 5 and 6 from 100–4200 cm−1. Additionally, concentration plots of four YCl3(aq) solution spectra from 2.378 to 0.479 mol L−1 are presented in the low frequency region of the spectrum (50–1100 cm−1) in Fig. S6† and the O–H stretching region of the solvent, water, in Fig. S7† (2500–4200 cm−1). The broad bands in Fig. 5 in the polarized scattering at 189 cm−1, which is overlapped with a component at 248 cm−1 and at 660 cm−1 are caused by the solute, water. The broad band at 189 cm−1 represents the restricted translation and the band at 660 cm−1 librations and a concentration plot in this wavenumber region for four YCl3 solutions from 2.378 to 0.479 mol L−1 is given in Fig. S6.† The deformation band of water appears at 1648 cm−1 in the 3.212 mol L−1 YCl3 solution and the O–H profile shows peaks at 3228 cm−1 and 3430 cm−1 (Fig. 6). Scattering profiles for the same four YCl3 solutions in the water O–H stretching region at the high frequency range from 2500–4200 cm−1 showing the O–H stretching profiles are given Fig. S7.† The influence of the solute YCl3 on the restricted translation, librations, the H2O deformation mode and O–H stretching band (Fig. S6 and S7†) with increasing YCl3 concentration is discussed for a similar system, CaCl2(aq) in greater detail.24 The similarity of both spectral profiles of CaCl2(aq) and YCl3(aq) in the O–H stretching region reinforces two facts: one that the O–H stretching profile is mostly influenced by anions, in our case Cl− and second that Cl− forms H-bonds of the type O–H⋯Cl− almost as strong as the ones between water molecules (O–H⋯O) in neat water.24 This stands in contrast to the influence of ClO4− on the O–H stretching band of water which forms very weak H-bonds with the solvent as mentioned in section 3.1. | ||
| Fig. 5 Raman scattering spectra (R-format) of a 3.212 mol L−1 YCl3 solution. The two top curves are the polarized and depolarized scattering profiles and the lower curve represents the isotropic scattering. The Y–O breathing mode to 379 cm−1 is slightly downshifted compared to the one in Y(ClO4)3(aq) at 384 cm−1 which is caused by the penetration of Cl− into the first hydration sphere, forming a species. In the polarized scattering profile, the mode at 248 cm−1 is due to an anisotropic [Y(OH2)8−nCln]+3−n skeleton vibration and the broad overlapped band at 189 cm−1 to the restricted translation of water/Cl−(aq). The extremely broad mode at 660 cm−1 is due to the librational water band. The band at 1648 cm−1 is due to the deformation mode of the solvent, water. The inset shows the isotropic Raman spectrum in the terahertz region in greater detail. The mode at 204 cm−1 is due to the restricted translational band, O–H⋯O/Cl− of water/Cl−(aq) and the broad feature 263 cm−1 is assigned to a ν Y3+–Cl− mode. | ||
 | ||
| Fig. 6 Raman scattering spectrum (polarized, isotropic (darker line) and depolarized scattering profiles) of a 3.212 mol L−1 YCl3 solution from 1400–4200 cm−1. The inset (only polarized and depolarized scattering) gives the deformation mode at 1648 cm−1 and its combination band, which is split into two components at 2075 and 2280 cm−1, of the solvent, water, in greater detail. The O–H stretching band shows two band components at 3226 cm−1 and 3430 cm−1. | ||
Focusing further on the terahertz frequency region, the Y–O stretching mode in this solution appears at 379 cm−1 and is down shifted compared to the unassociated cluster which occurs at 384 cm−1. In addition to the very broad isotropic component at 202 cm−1, mentioned above, a broad shoulder appears at ∼263 cm−1 and this new isotropic component must stem from a Y3+–Cl− stretching mode. This finding is clear evidence that Cl− has penetrated into the first hydration shell of Y3+ forming a [Y(OH2)8−nCln]+3−n (n = 1, possibly 2). With dilution, the Y–O stretching mode shifts to higher wavenumbers from 379 cm−1 for a 3.212 mol L−1 solution (Rw = 15.4) (Fig. 5) to 384 cm−1 obtained in a 0.479 mol L−1 solution (Rw = 113.82) (see dilution plot of four YCl3 spectra from 2.378 to 0.479 mol L−1 in Fig. S6†). This fact shows that with dilution of the concentrated solution, the chloro–complex formation disappears rapidly and extrapolation of our Raman data shows that in YCl3(aq) < 0.2 mol L−1 the Y3+ cation is fully hydrated. Additionally, the intensity of the band of ν1YO8 as a function of concentration does not increase linearly, which would be expected if the octa-hydrated Y3+ species remained stable, but curves toward smaller intensities at concentrated solutions. This shows that chloro – complexes must have formed in higher concentrated solutions at the expense of the fully hydrated Y3+, [Y(H2O)8]3+ (Fig. S8†).
To further verify chloro-complex formation in YCl3(aq), addition of excess HCl was investigated. Isotropic spectra of YCl3–HCl solutions at a fixed concentration of YCl3 at 2.03 mol L−1 with 0, 1.0 and 4.0 mol L−1 HCl added are shown in Fig. 7. The isotropic band for YCl3(aq) without added HCl is at 383 cm−1, shifts to 380 cm−1 with 1.0 mol L−1 HCl added and to 376 cm−1 with 4.0 mol L−1 additional HCl. The decreasing peak position of the ν1YO8 band with increasing HCl concentration reflects the rise in the concentration of the Y3+-chloro-complex, [Y(OH2)8−nCln]+3−n (n = 1, 2). Fig. 8 presents a difference spectrum of YCl3(aq) at 2.03 mol L−1 plus 4.0 mol L−1 HCl(aq) from which the isotropic scattering profile of a 4 mol L−1 HCl(aq) was subtracted and reveals broad bands at 326 cm−1, at 228 cm−1 and a small scattering contribution at 102 cm−1. Clearly chloro-complexes are formed in these solutions. The formation of the chloro-complex increases with an increase in Cl− concentration and the first equilibrium steps according to the eqn (6) may be formulated:
| [Y(OH2)8]3+ + Cl− ↔ [Y(OH2)7Cl]2+ + H2O. | (6) |
 | ||
| Fig. 7 Isotropic Raman spectra of a 2.13 mol L−1 YCl3 solution and two additional solutions where HCl is added: (A) 2.03 mol L−1 YCl3 without HCl; (B) 2.13 mol L−1 YCl3 + 1.13 mol L−1 HCl and (C) 2.13 mol L−1 YCl3 + 3.97 mol L−1 HCl. The broad shoulder at 263 cm−1 in (A) increases in intensity in solutions B and C and may be assigned to Y3+–Cl− stretch. | ||
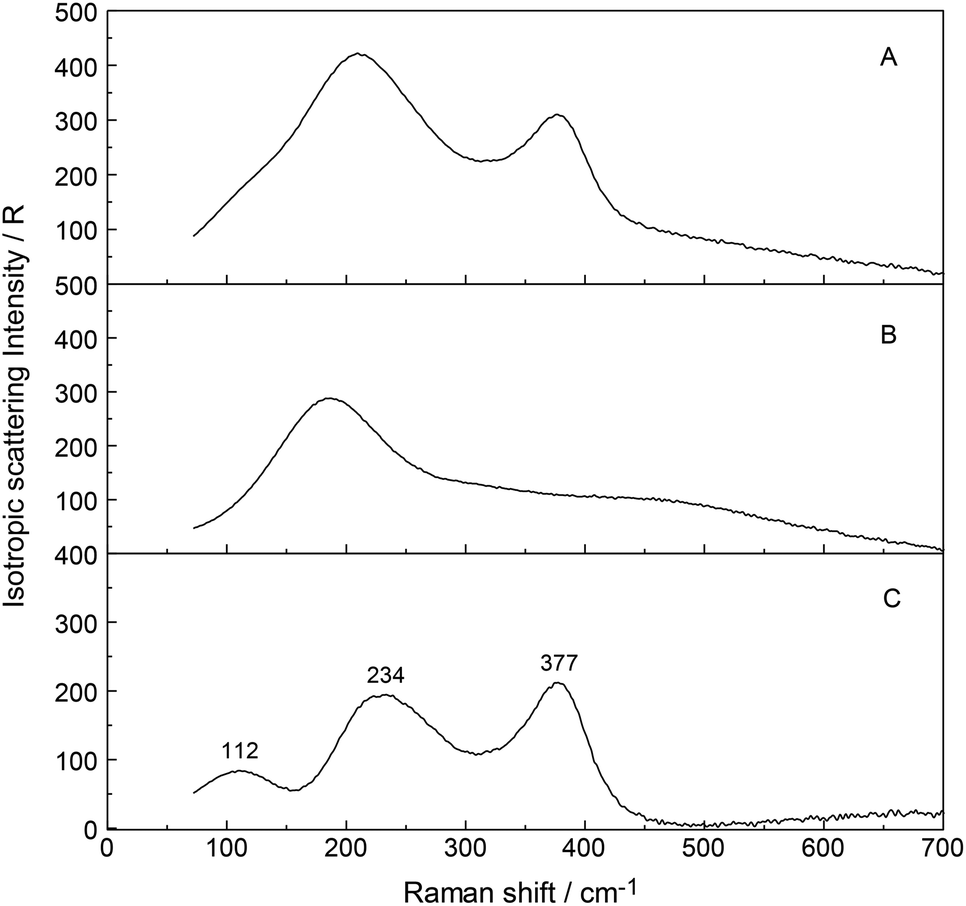 | ||
| Fig. 8 Difference Raman spectrum (trace in panel C) of the isotropic scattering profile of a 2.13 mol L−1 YCl3(aq) plus 3.97 mol L−1 HCl (trace in panel A) from which an isotropic profile of HCl(aq) at 3.98 mol L−1 (trace in panel B) was subtracted. Panel C: the band at 377 cm−1 stems from the Y–O mode of the complex, [Y(OH2)8−nCln]+3−n (n = 1,2) and the broad mode at 234 with a shoulder at 263 cm−1 and a band at 112 cm−1 are due to Y3+–Cl− vibrations of the chloro-complex species. | ||
Results of anion exchange experiments of metal ions in aqueous HCl(aq) including Y3+ showed that yttrium has a negligible adsorption.59 Therefore, higher Y3+-chloro-complexes (complex anions) with n > 3 may be ruled out. The formation of a second complex cation, the di-chloro-complex according to eqn (7) may be possible:
| [Y(OH2)7Cl]2+ + Cl− ↔ [Y(OH2)6Cl2]+ + H2O. | (7) |
The estimated value of the logβ1 for eqn (5) equal to ∼0.1 reveals the weak nature of the chloro-complex in YCl3(aq) at 23 °C. Thermodynamic data on YCl3(aq) confirm the weak nature of the complex/complexes and equilibrium constants for reaction 6 may be found in ref. 60. The existence of a weak chloro-complex species in LaCl3(aq) using Raman spectroscopy in the terahertz region was presented recently.26 In the most concentrated YCl3 solution, the mole ratio solute to water is 1 to 15.4 which means that there are not enough water molecules to form a complete second hydration sphere around all ions forcing outer-shell ion pairing. The chloride ions penetrate the flexible hydration shell of Y3+ into the first hydration shell, while in LaCl3(aq) weak chloro-complex formations occur,26 in AlCl3 solutions, however, even in very concentrated ones, Cl− does not penetrate into the first hydration shell of Al3+.43,58 The hydration shell of [Al(OH2)6]3+ is quite inert. Further experimental support for chloride penetrating into the first hydration shell of Y3+ and formation of a chloro-complex stems from a recent combined neutron scattering and X-ray scattering study in which information from EXAFS were incorporated20 into their applied analytical data treatment. This high quality structural study21 on a 1.0 mol kg−1 YCl3(aq) showed that Y3+ is hydrated by seven water molecules and one chloride ion, forming an inner-sphere ion complex in which the water molecules maintain angular configurations consistent with a square antiprismatic configuration. In an earlier EXAFS study on YCl3(aq) and YCl3(aq) plus an excess of LiCl, it was also demonstrated that Cl− leads to chloro-complex formation in concentrated YCl3 solutions.13
To summarize, the [Y(OH2)8−nCln]+3−n modes in chloride solutions could be detected and formation of weak chloro-complexes with Y3+ were detected. In dilute solutions (c < 0.2 mol L−1) the chloro-complex species disappeared upon dilution and [Y(OH2)8]3+(aq) and Cl−(aq) are formed. The chloro-complex formation in concentrated solutions may be one reason for the data scatter for Y–O bond distance and coordination numbers presented for Y3+(aq) and this fact was highlighted recently.21
3.4. Y(NO3)3(aq)
When nitrate replaces water in the first coordination sphere of a cation, marked changes occur in the spectrum of the ligated nitrate, so that it is possible to differentiate between the modes of the ligated and the unligated nitrate. Nitrato-complex formation has been observed for a variety of divalent and trivalent metals in solutions.24–28 The modes of the unligated nitrate, the fully hydrated nitrate, NO3−(aq), were measured recently24–26 and for a 0.409 mol L−1 NaNO3 solution (RW = 134.1) the Raman spectrum is given in ref. 26 together with the vibrational spectroscopic data of the bands and their assignments. Nitrato-complex formation is not only characterized by the ligated NO3− but also by the fact that the M–O symmetric stretch of the metal ions (M = metal ion) show two bands, a band of the fully hydrated cation and a band of the partially hydrated metal ion. A ligand mode M–L (L = NO3−) should appear at low frequencies as well.A brief summary for the vibrational data on NO3−(aq) shall be presented before discussing the spectroscopic features in Y(NO3)3(aq). The “free” NO3− anion possesses D3h symmetry and four modes should be observed (two doubly degenerate). The n.m.'s span the representation Γvib(D3h) = a′1(Ra) + a′′2(i.r.) + 2e′(Ra,i.r.). The ν1(a′1) symmetric stretching mode N–O is Raman active but forbidden in infrared, while the e′ modes, ν3 (N–O antisymmetric stretch) and ν4 (bending mode) are Raman and infrared active. The out-of plane deformation mode with the character a′′2 is only infrared active. In NO3−(aq), the polarized Raman mode at 1047.4 cm−1 is the strongest band in Raman scattering and appears quite narrow with a fwhh = 6.55 cm−1. This band is assigned to the symmetric N–O stretch vibration, ν1(a′1). Its depolarization degree at 0.034 is quite low and confirms that it represents a symmetrical normal mode. The mode ν3(e′), the asymmetric N–O stretching mode which is depolarized and relatively weak appears not as a single band but as two band components at 1347 and 1408 cm−1 and appears as a doublet even in very dilute solutions of NaNO3(aq). Asymmetric hydration of the nitrate anion in aqueous solution explains the double band (cf. for instance24,60). The depolarized mode ν4(e′) at 716 cm−1 is active in Raman and infrared and is much weaker than the symmetric stretching mode ν1(a′1). The infrared active mode, ν2(a′′2) occurs at 830 cm−1 and its overtone 2 × ν2 at 1660 cm−1 is Raman active (a′1) and appears polarized.
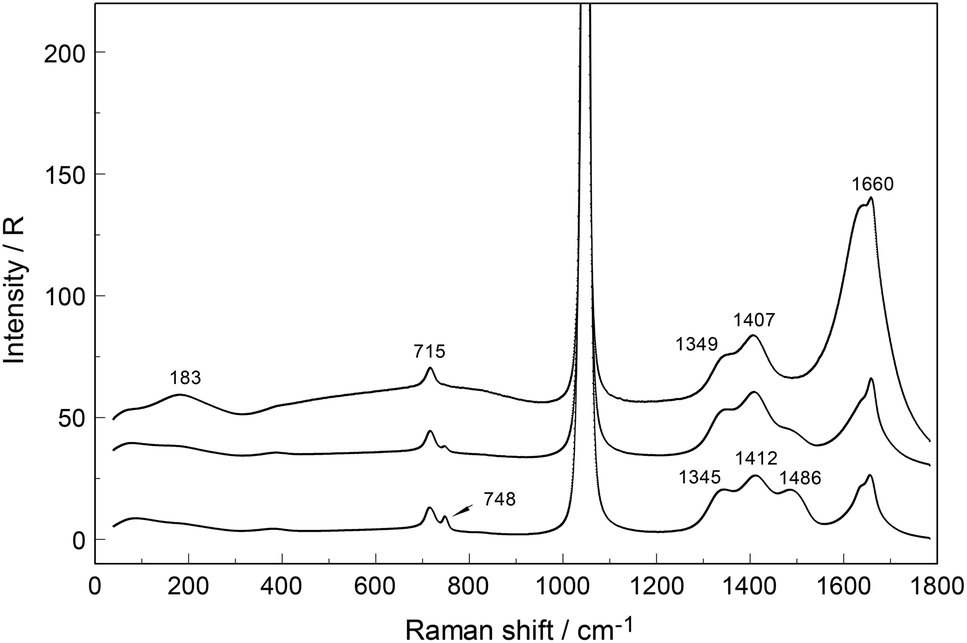
An overview Raman spectrum (R-format) of a 2.035 mol L−1 Y(NO3)3 solution ranging from 40–1800 cm−1 is presented in Fig. 9. A concentration series of polarized Raman spectra of three Y(NO3)3 solutions at 2.035 mol L−1 (RW = 23.04), 1.036 mol L−1 (RW = 49.20) and 0.198 mol L−1 (RW = 257.9) from 100 to 1800 cm−1 are presented in Fig. 10. A comparison of Raman spectra of a solution with 2.035 mol L−1 Y(NO3)3 with a solution at 0.198 mol L−1 in the low frequency range from 45–900 cm−1 is detailed in Fig. S9.† Additionally, in Fig. S10† the same concentrations are compared in the high frequency region from 800–1800 cm−1 including the ν3(e′) antisymmetric stretching region and 2 ν2 overtone. In these spectra, the nitrate split into bands of free, hydrated nitrate,  and nitrate bound,
and nitrate bound, 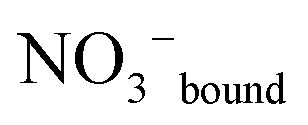 to Y3+. This is a result of the fact that nitrate penetrates into the first hydration sphere forming inner-sphere complexes. The Raman spectra of Y(NO3)3(aq) are, therefore, more complex than the ones for NaNO3(aq) resulting in bands of “free” nitrate and bound nitrate. The Raman spectrum of the concentrated Y(NO3)3 solution, at 2.035 mol L−1, may illustrate this (Fig. 9). The first evidence of the ligated nitrate is the split of the ν4(e′) NO3− bending mode into bands at 715 cm−1 (fwhh = 21 cm−1), at 724.5 cm−1 (fwhh = 18.3 cm−1) and 748 cm−1 (fwhh = 13.5 cm−1). The band at 715 cm−1 represents the NO3− bending mode of “free” NO3−(aq) and the bands at 724.5 cm−1 and 748 cm−1 represent the bending modes of the bound nitrate. Furthermore, a very weak and broad band at ∼826 cm−1, normally Raman forbidden ν2(a′′2), appears in the concentrated Y(NO3)3 solution (Fig. 9). The ν1 N–O symmetric stretching band appears broadened (fwhh = 12.2 cm−1) and slightly shifted to higher wavenumbers at 1049.3 cm−1 compared with the N–O stretching band in dilute NaNO3(aq) solutions. A moderately weak shoulder containing ∼11% of the whole band contour at lower wavenumbers is also visible at 1037 cm−1 (fwhh = 21 cm−1). The antisymmetric stretching mode, ν3(e′) broadens and the split of the two band components mentioned earlier widens considerably. Additional isotropic band contributions of the N–O antisymmetric stretch appear at 1328 and 1470 cm−1 and this is shown in Fig. 9 and S10.† Finally, the Raman active overtone 2ν2, which is broadened, appears at 1658 cm−1. All these features appear in concentrated Y(NO3)3(aq) solutions and are clear indications that NO3− penetrated into the first hydration sphere of Y3+. With dilution the bands of the bound nitrate disappear rapidly and are vanished in dilute solutions (Fig. 10). It is noteworthy to mention that Cabaco et al.16 could not detect inner-sphere nitrato-complexes in a concentrated Y(NO3)3 at 2.89 mol L−1 using XRD analysis and Raman spectroscopy (no R-normalized Raman spectra have been used).
to Y3+. This is a result of the fact that nitrate penetrates into the first hydration sphere forming inner-sphere complexes. The Raman spectra of Y(NO3)3(aq) are, therefore, more complex than the ones for NaNO3(aq) resulting in bands of “free” nitrate and bound nitrate. The Raman spectrum of the concentrated Y(NO3)3 solution, at 2.035 mol L−1, may illustrate this (Fig. 9). The first evidence of the ligated nitrate is the split of the ν4(e′) NO3− bending mode into bands at 715 cm−1 (fwhh = 21 cm−1), at 724.5 cm−1 (fwhh = 18.3 cm−1) and 748 cm−1 (fwhh = 13.5 cm−1). The band at 715 cm−1 represents the NO3− bending mode of “free” NO3−(aq) and the bands at 724.5 cm−1 and 748 cm−1 represent the bending modes of the bound nitrate. Furthermore, a very weak and broad band at ∼826 cm−1, normally Raman forbidden ν2(a′′2), appears in the concentrated Y(NO3)3 solution (Fig. 9). The ν1 N–O symmetric stretching band appears broadened (fwhh = 12.2 cm−1) and slightly shifted to higher wavenumbers at 1049.3 cm−1 compared with the N–O stretching band in dilute NaNO3(aq) solutions. A moderately weak shoulder containing ∼11% of the whole band contour at lower wavenumbers is also visible at 1037 cm−1 (fwhh = 21 cm−1). The antisymmetric stretching mode, ν3(e′) broadens and the split of the two band components mentioned earlier widens considerably. Additional isotropic band contributions of the N–O antisymmetric stretch appear at 1328 and 1470 cm−1 and this is shown in Fig. 9 and S10.† Finally, the Raman active overtone 2ν2, which is broadened, appears at 1658 cm−1. All these features appear in concentrated Y(NO3)3(aq) solutions and are clear indications that NO3− penetrated into the first hydration sphere of Y3+. With dilution the bands of the bound nitrate disappear rapidly and are vanished in dilute solutions (Fig. 10). It is noteworthy to mention that Cabaco et al.16 could not detect inner-sphere nitrato-complexes in a concentrated Y(NO3)3 at 2.89 mol L−1 using XRD analysis and Raman spectroscopy (no R-normalized Raman spectra have been used).
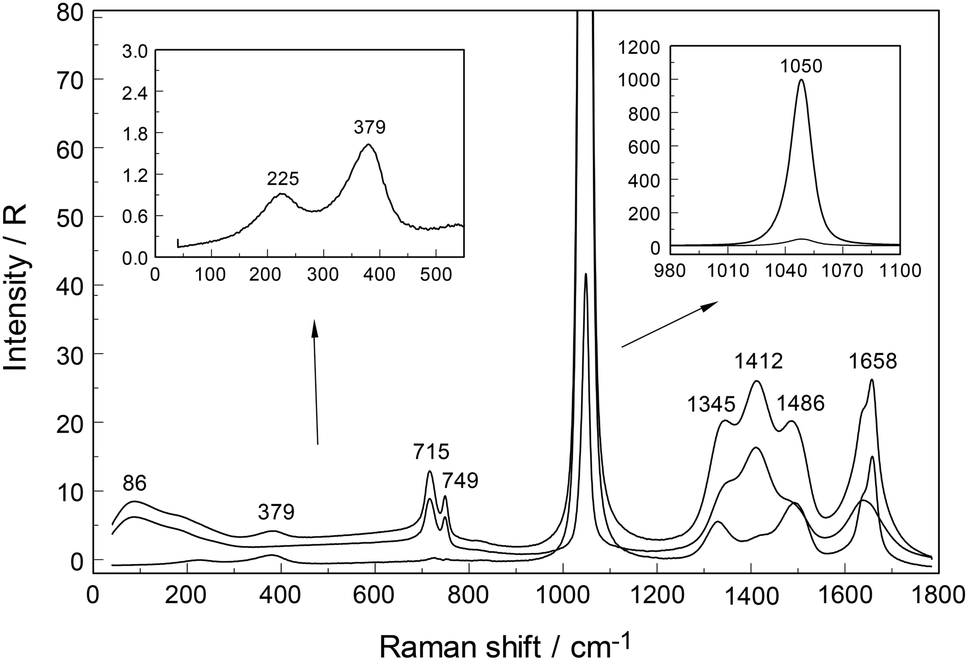 | ||
| Fig. 9 Raman spectrum of a 2.035 mol L−1 (RW = 23.04) Y(NO3)3 solution from 45–1800 cm−1. The curves for Rpol and Rdepol scattering are shown on top of Riso (darker, dotted line). The inset at the LHS shows the isotropic scattering at the low wavenumber range while the inset at the RHS depicts the NO3−(aq) mode at 1050 cm−1 at its full intensity scale. | ||
 | ||
| Fig. 10 Raman spectra (R-polarized) of three Y(NO3)3 solutions from bottom to top: at 2.035, 1.036 and 0.198 mol L−1 solute concentrations. The Raman bands in the low frequency range from 40–900 cm−1 at the highest and lowest solution concentration (2.035 and 0.198 mol L−1) are given in greater detail in Fig. S9† (panel (A): 2.035 and (B): 0.198 mol L−1). Note, that the ν1 N–O stretching mode at ∼1047 cm−1 is presented at its full scale in insets of Fig. S10† (panel (A): 2.035 and (B): 0.198 mol L−1) for both concentrations including the bands between 1200 and 1800 cm−1. | ||
In the aqueous Y(NO3)3(aq), the weak symmetric stretching mode Y–O at 384 cm−1 shows a dependence on Y(NO3)3 concentration. With an increase in Y(NO3)3 concentration the band shifts to lower wavenumbers, namely from 384 cm−1 in dilute solutions to 379 cm−1 in a 2.035 mol L−1 solution and the band broadens as well with increasing concentration. For quantitative purposes, the broad ν1Y–O band was fitted with two Gauss–Lorentz bands resulting in a component at 384 cm−1 (fwhh = 51.6 cm−1) indicative of the octa-aqua yttrium3+ species while the second band component at 347 cm−1 (fwhh = 84 cm−1) is due to the hydrated yttrium–nitrato complex, [Y(OH2)8−n(NO3)n]+3−n. The results of the curve fit are given in Fig. 11. The ligand mode of the nitrato-complex appears as a polarized band at 223 cm−1. In the concentrated solution, 45% of Y3+ exists in the form of a nitrato-complex. With dilution the nitrato-complex formation diminishes quickly and in the dilute solution at 0.198 mol L−1 only ∼10% of Y3+ is coordinated by nitrate. Upon further dilution the nitrato-complex disappears and from extrapolations of α-values (the degree of nitrato-complex formation) as a function of solution concentration, it becomes obvious that in solutions <0.1 mol L−1 99% of the Y3+ is fully hydrated and the nitrato-complex with only ∼1% becomes insignificant. The nitrate is tentatively assumed to be monodentate, although in solid state compounds of hydrated Y(NO3)3 such as Y(OH2)4(NO3)3·2H2O nitrate occurs in bidentate fashion.61 It may be deduced from the work by Dobler et al.62 investigating Ln(III) cations interacting with NO3−, that in aqueous solution with enough water to fully hydrate the trivalent cation Ln(III) (Ln = Lu3+) nitrate acts as a monodentate ligand. A second shell of water molecules stabilizes the first water sphere via hydrogen bond formation including the nitrate. Therefore, the formation of the nitrato-complex with Y3+ may be expressed by eqn (8):
| [Y(OH2)8]3+ + NO3− ↔ [Y(OH2)7ONO2]2+ + H2O. | (8) |
| [Y(OH2)7ONO2]2+ + NO3− ↔ [Y(OH2)6(ONO2)2]+ + H2O. | (9) |
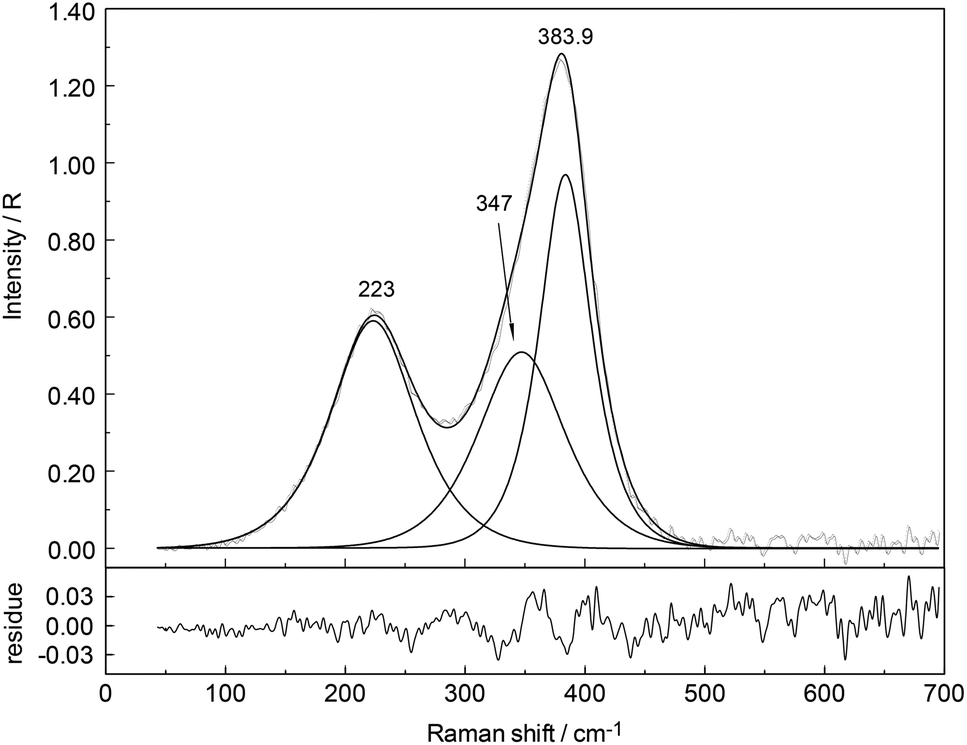 | ||
| Fig. 11 Isotropic Raman spectrum (R-format) of 2.035 mol L−1 Y(NO3)3 solution in the terahertz frequency range (40–700 cm−1). Given are the measured spectrum (darker line), the sum curve and the component bands of the fit at 223 cm−1 (fwhh = 165 cm−1; A1 = 58.02), 347 cm−1 (fwhh = 165 cm−1; A2 = 49.6) and 384 cm−1 (fwhh = 51.6 cm−1; A3 = 59.3). The index i denotes the integrated band intensities of the band intensities Ai (i = 1–3). Note the residue curve as the difference between measured curve and sum curve is given underneath. | ||
An estimate of the logβ1 value at ∼0.1 reveals the weak nature of the nitrato-complex in Y(NO3)3(aq) at 23 °C. Thermodynamic data on the weak nitrato complex formation may be found in ref. 63. Ultrasound absorption measurements were carried out on yttrium nitrate solutions applying a one-step mechanism and revealed the formation of a mono-nitrato complex.64 The behaviour of the Y3+–nitrato-complex solution is comparable to the nitrato-complex formation in La(NO3)3(aq)26 where the complex formation is apparent albeit weak. In contrast to the weak nitrato-complex formation in aqueous Y(NO3)3, in Al(NO3)3 solutions from dilute to high concentrations, the aluminium(III) hexahydrate, [Al(OH2)6]3+ exists as a stable cluster. The Raman scattering bands, especially the breathing mode at 525 cm−1, indicate the existence of the hexahydrate.58 In concentrated solutions outer-sphere ion pairs, [Al(OH2)6]3+·NO3− are formed. However, nitrate does not penetrate the first hydration shell of Al3+ indicated by the unchanged Raman signature of the AlO6 of [Al(OH2)6]3+.58
4. Conclusions
Raman spectra of aqueous Y3+ perchlorate, triflate, chloride and nitrate solutions were measured over a broad concentration range (0.198 mol L−1–3.252 mol L−1). The terahertz frequency range has been measured as well as spectra in the O–H stretching region. The weak, polarized mode at 384 cm−1 (fwhh = 50 cm−1) was assigned to ν1Y–O of the YO8 skeleton. In deuterated Y(ClO4)3 solution, a mode at 365 cm−1 was assigned to the breathing mode, ν1Y–O of the [Y(OD2)8]3+. Raman spectroscopic data suggest that the [Y(OH2)8]3+ ion is thermodynamically stable in dilute perchlorate and triflate solutions. But inner-sphere complexes could not be detected, spectroscopically. Outer-sphere ion pairs of the type [Y(OH2)8]3+·ClO4− and possibly inner-sphere ion pairs are formed in concentrated Y(ClO4)3(aq). DFT frequency calculations of a [Y(OH2)8]3+ imbedded in a polarizable dielectric continuum gave a ν1 Y–O equal to 372 cm−1 in fair agreement with the experiment. Bond distances and angles of the [Y(OH2)8]3+ imbedded in a polarizable dielectric continuum were also presented. The symmetry of the octa-aqua complex is S8 while the YO8 skeleton has D4d symmetry.In YCl3 solutions, Cl− penetrates the first hydration sphere of Y3+(aq) and chloro-complexes are formed. However, the chloro-complexes disappear rapidly upon dilution and at a concentration <0.2 mol L−1 the chloro-complexes have almost vanished. This Raman spectroscopic finding is substantiated from recent results applying neutron – and X-ray scattering as well as EXAFS.12,16
In concentrated Y(NO3)3 solutions, NO3− penetrates the first hydration sphere of Y3+ and a nitrato-complex was characterized; the nitrato-complex disappears fairly rapidly upon dilution and in solutions <0.1 mol L−1 no nitrato-complexes could be observed.
Appendix A
The binding energy for the cluster in the gas phase consisting of Y3+ ion with 8 water molecules in the first sphere was calculated according to eqn (10):| ΔEe = Ee[Y(OH2)8(g)]3+ − Ee[Y3+(g)] − 8·Ee[OH2(g)]. | (10) |
This equation corresponds to a gas-phase reaction at T = 0 K of isolated molecules with stationary nuclei, but thermochemical measurements are carried out with vibrating molecules usually at 298 K. Comparison of the theoretical with experimental data, therefore, requires correction for the translational, rotational and vibrational energy. For one mole molecules of an ideal gas, one obtains the enthalpy:
| H(T) = Ee + Et(T) + Er(T) + Ev(T) + RT. | (11) |
Here, Ee stands for the electronic energy, Et(T) the translational energy, Er(T) the rotational energy, and 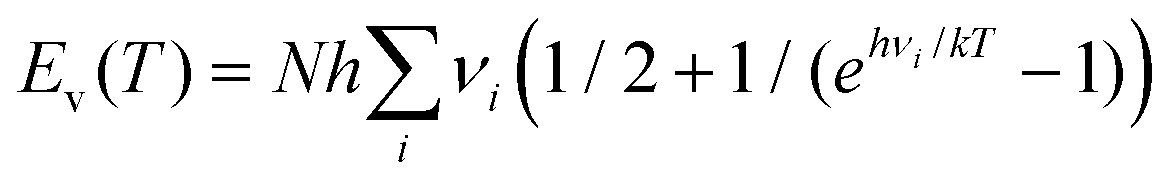 is the thermal vibrational energy. The gas-phase hydration enthalpy ΔHhyd(g)(T) is then obtained with an equation similar to eqn (10). Using results of our DFT calculations and harmonic frequencies, we obtain for the cluster [Y(OH2)8]3+ the standard hydration enthalpy ΔHhyd(g) (298) = −2541 kJ mol−1 in the gas phase. The estimation of the hydration enthalpy in the liquid state, ΔHhyd(l) for [Y(OH2)6]2+ requires consideration of additional contributions. According to a Born-Haber cycle it follows:
is the thermal vibrational energy. The gas-phase hydration enthalpy ΔHhyd(g)(T) is then obtained with an equation similar to eqn (10). Using results of our DFT calculations and harmonic frequencies, we obtain for the cluster [Y(OH2)8]3+ the standard hydration enthalpy ΔHhyd(g) (298) = −2541 kJ mol−1 in the gas phase. The estimation of the hydration enthalpy in the liquid state, ΔHhyd(l) for [Y(OH2)6]2+ requires consideration of additional contributions. According to a Born-Haber cycle it follows:
| ΔHhyd(l) ≈ ΔHhyd(g) + ΔHvap + ΔHsolv. | (12) |
Here, ΔHvap is the heat of water vaporization65 and ΔHsolv means the solvation energy resulting from the transfer of the gas phase cluster to the liquid state. The solvation enthalpy of the cluster can be estimated with the Polarized Continuum Model by placing the [Y(OH2)6]3+ gas phase cluster in a polarizable dielectricum and so taking implicitly into account the bulk water and comparing with the naked cluster. From the difference of the calculated enthalpies, the solvation energy ΔHsolv (298) results in −1762 kJ mol−1. Taking into account the vaporization heat of the 8 water molecules, we obtain for the hydration enthalpy ΔHhyd(l) (298) = −3951 kJ mol−1.
Experimental values for the standard molar hydration enthalpy ΔHhyd(l) (298) of Y3+ in the liquid state are: −3710 kJ mol−1,37 −3599 kJ mol−1,38 −3733 kJ mol−1,39 −3583 kJ mol−1,40 −3590 kJ mol−1,41 −3620 kJ mol−1.42 The correspondence with the theoretical values is satisfactory in consideration of the simple set of basis functions that has been used.
Acknowledgements
WWR and GI wish to thank Frau B. Ostermay for her skilful technical assistance.References
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons, Chichester, West Sussex, 2006 Search PubMed.
- J. Emsley, Nature's Building Blocks: An A-Z Guide to the Elements, Yttrium, Oxford University Press, Oxford, 2011, pp. 617–620 Search PubMed.
- V. R. Sastri, J. R. Perumareddi, V. Ramachandra Rao, G. V. S. Rayudu and J.-C. Bünzli, Modern Aspects of Rare Earths and their Complexes, Elsevier, Amsterdam, 2003 Search PubMed.
- A. Kheyfits, Radiol. Today, 2010, 11, 20 Search PubMed.
- N. S. MacDonald, R. E. Nusbaum, G. V. Alexander, F. Ezmirlian, P. Spain and D. E. Rounds, J. Biol. Chem., 1952, 195, 837–841 CAS.
- C.-H. Huang and Z. Bian, Introduction in Rare Earth Coordination Chemistry: Fundamentals and Applications, ed. C.-H. Huang, Wiley, Singapore, 2010, pp. 1–39 Search PubMed.
- J. McB. Harrowfield, D. L. Kepert, J. M. Patrick and A. H. White, Aust. J. Chem., 1983, 36, 483–492 CrossRef CAS.
- R. D. Rogers and L. K. Kurihara, Inorg. Chim. Acta, 1986, 116, 171–177 CrossRef CAS.
- R. W. Broach, J. M. Williams, G. P. Felcher and D. G. Hinks, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 2317–2321 CrossRef.
- F. A. Cotton, A. Davison, V. W. Day, M. F. Fredrich, C. Orvig and R. Swanson, Inorg. Chem., 1982, 21, 1211–1214 CrossRef CAS.
- Y. Ohki, Y. Suzuki, T. Takeuchi and A. Ouchi, Bull. Chem. Soc. Jpn., 1988, 61, 393–405 CrossRef CAS.
- G. Johansson and H. Wakita, Inorg. Chem., 1985, 24, 3047–3052 CrossRef CAS.
- E. Matsubara, K. Okuda and Y. Waseda, J. Phys.: Condens. Matter, 1990, 2, 9133–9143 CrossRef CAS.
- M. I. Cabaco, M. A. Marques, M. I. de Barros Marques, G. Bushell-Wye, M. M. Costa, M. J. de Almeida and L. C. Andrade, J. Phys.: Condens. Matter, 1995, 7, 7409–7418 CrossRef CAS.
- S. Ramos, G. W. Neilson, A. C. Barnes and A. Mazuelas, J. Phys. Chem. B, 2001, 105, 2694–2698 CrossRef CAS.
- M. I. Cabaco, M. I. de Barros Marques, M. A. Marques, A. M. Gaspar and M. L. de Almeida, J. Mol. Liq., 2005, 117, 69–76 CrossRef CAS PubMed.
- M. I. de Barros Marques, M. A. Marques and J. R. Rodrigues, J. Phys.: Condens. Matter, 1992, 4, 7679–7690 CrossRef.
- K. V. Ragnarsdottir, E. H. Oelkers, D. M. Sherman and C. R. Collins, Chem. Geol., 1998, 151, 29–39 CrossRef.
- P. Lindqvist-Reis, K. Lamble, S. Pattanaik, I. Persson and M. Sandström, J. Phys. Chem. B, 2000, 104, 402–408 CrossRef CAS.
- S. Diaz-Moreno, A. Munoz-Paez and J. Chaboy, J. Phys. Chem. A, 2000, 104, 1278–1286 CrossRef CAS.
- D. T. Bowron and S. Diaz-Moreno, J. Phys. Chem. B, 2007, 111, 11393–11399 CrossRef CAS PubMed.
- T. Ikeda, M. Hirata and T. Kimura, J. Chem. Phys., 2005, 122, 024510–024515 CrossRef PubMed.
- W. W. Rudolph, M. H. Brooker and C. C. Pye, J. Phys. Chem., 1995, 99, 3793–3797 CrossRef CAS.
- W. W. Rudolph and G. Irmer, Dalton Trans., 2013, 3919–3935 RSC.
- W. W. Rudolph and G. Irmer, Dalton Trans., 2013, 14460–14472 RSC.
- W. W. Rudolph and G. Irmer, Dalton Trans., 2015, 44, 295–305 RSC.
- W. W. Rudolph and C. C. Pye, Phys. Chem. Chem. Phys., 2002, 4, 4319–4327 RSC.
- W. W. Rudolph, D. Fischer, M. R. Tomney and C. C. Pye, Phys. Chem. Chem. Phys., 2004, 6, 5145–5155 RSC.
- V. A. Sipachev and A. I. Grigor'ev, Zh. Strukt. Khim., 1969, 10, 710–714 Search PubMed.
- W. W. Rudolph and G. Irmer, Appl. Spectrosc., 2007, 61, 1312–1327 CrossRef CAS PubMed.
- A. I. Vogel, A Text-Book of Quantitative Inorganic Analysis, Longman, London, 3rd edn, 1961 Search PubMed.
- F. H. Spedding, M. J. Pikal and B. O. Ayers, J. Phys. Chem., 1966, 70, 2440–2449 CrossRef CAS ; see p. 2441.
- W. W. Rudolph, D. Fischer and G. Irmer, Appl. Spectrosc., 2006, 60, 130–144 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- C. Lee, W. Yang and R. C. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS PubMed.
- R. Noyes, J. Am.. Chem. Soc., 1962, 84, 513–522 CrossRef CAS.
- D. R. Rosseinsky, Chem. Rev., 1965, 65, 467–490 CrossRef CAS.
- J. E. Desnoyers and C. Jolicoeur, in Modern Aspects of Electrochemistry, ed. J. O'M. Bockris and B. E. Conway, Plenum Press, New York, N. Y., 1969, ch. 1, vol. 5 Search PubMed.
- D. W. Smith, J. Chem. Educ., 1977, 54, 540–542 CrossRef CAS; D. W. Smith, J. Chem. Educ., 1977, 54, 540–542 CrossRef.
- Y. Marcus, Biophys. Chem., 1994, 51, 111–127 CrossRef CAS.
- J. Burgess Ions in Solution: Basic Principles of Chemical Interactions, Ellis Horwood, Chichester/Halsted Press, New York, 1988 Search PubMed.
- W. Rudolph and S. Schönherr, Z. Phys. Chem., 1989, 270, 1121–1134 CAS.
- B. Auer, R. Kumar, J. R. Schmidt and J. L. Skinner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14215–14220 CrossRef CAS PubMed.
- B. M. Auer and J. L. Skinner, J. Chem. Phys., 2008, 128, 224511 CrossRef CAS PubMed.
- M. H. Brooker, G. Hancock, B. C. Rice and J. Shapter, J. Raman Spectrosc., 1989, 20, 683–694 CrossRef CAS PubMed.
- N. Purdie and C. A. Vincent, Trans. Faraday Soc., 1967, 63, 2745–2757 RSC.
- D. P. Fay, D. Litchinsky and N. Purdie, J. Phys. Chem., 1969, 73, 544–552 CrossRef CAS.
- G. E. Walrafen, J. Chem. Phys., 1962, 36, 1035–1042 CrossRef CAS PubMed.
- I. A. Heisler, K. Mazur and S. R. Meech, J. Phys. Chem. B, 2011, 115, 1863–1873 CrossRef CAS PubMed.
- C. Cossy, L. Helm and A. E. Merbach, Inorg. Chem., 1989, 28, 2699–2703 CrossRef CAS.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923–1959 CrossRef CAS PubMed.
- J. Zhang, N. Heinz and M. Dolg, Inorg. Chem., 2014, 53, 7700–7708 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- G. D. Klungness and R. H. Byrne, Polyhedron, 2000, 19, 99–107 CrossRef CAS.
- X. Liu, X. Lu, R. Wang and H. Zhou, Chem. Geol., 2012, 334, 37–43 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- W. W. Rudolph, R. Mason and C. C. Pye, Phys. Chem. Chem. Phys., 2000, 2, 5030–5040 RSC.
- K. A. Kraus, F. Nelson and G. W. Smith, J. Phys. Chem., 1954, 58, 11–17 CrossRef CAS.
- J. Thøgersen, J. Réhault, M. Odelius, T. Ogden, N. K. Jena, S. J. Jensen, S. R. Keiding and J. Helbing, J. Phys. Chem. B, 2013, 117, 3376–3388 CrossRef PubMed.
- B. Ribar, N. Milinski and Z. Budovalcev, Cryst. Struct. Commun., 1980, 9, 203–206 CAS.
- M. Dobler, P. Guilbaud, A. Dedieu and G. Wipff, New J. Chem., 2001, 25, 1458–1465 RSC.
- J. Schijf and R. H. Byrne, Geochim. Cosmochim. Acta, 2004, 68, 2825–2837 CrossRef CAS PubMed.
- H. Wang and P. Hemmes, J. Phys. Chem., 1974, 78, 261–265 CrossRef CAS.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA Key Values for Thermodynamics, Hemisphere Publishing Corp., New York, 1989 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt02586a |
| ‡ In early literature29 a Y3+-hexahydrate was assumed in the solution state. |
| § The vibrational duration for the Y–O breathing mode is 0.09 ps. Approximately ∼5.6 × 105 vibrations occur before the cluster experiences a water exchange. |
| ¶ In addition to the stretching mode ν(O–H⋯ClO4−) at 3538 cm−1 a very weak, broad mode appears in the terahertz region at ∼170 cm−1 in Y(ClO4)3 solutions (Riso) as very broad and very weak. This mode has an equivalent in pure water at ∼175 cm−1 where it is moderately intense and slightly polarized (restricted translational mode of the H-bonded water molecules (O⋯O–H)). In concentrated Y(ClO4)3 solutions other H-bonds are important, namely OH⋯ClO4− and the intensity of the band due to HOH⋯ClO4− is extremely weak in the isotropic Raman scattering.24,49,50 |
| This journal is © The Royal Society of Chemistry 2015 |