
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral thioether and selenoether macrocyclic coordination to Group 1 cations (Li–Cs) – synthesis, spectroscopic and structural properties†
Martin J. D.
Champion
,
William
Levason
,
David
Pugh
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 24th August 2015
Abstract
The complexes [M(L)][BArF] (BArF = tetrakis{3,5-bis(trifluoromethyl)-phenyl}borate), L = [18]aneO4S2 (1,4,10,13-tetraoxa-7,16-dithiacyclooctadecane): M = Li–Cs; L = [18]aneO2S4 (1,10-dioxa-4,7,13,16-tetrathiacyclooctadecane): M = Li, Na, K; L = [18]aneO4Se2 (1,4,10,13-tetraoxa-7,16-diselenacyclooctadecane): M = Na, K, as well as [Na(18-crown-6)][BArF], are obtained in good yield as crystalline solids by reaction of M[BArF] with the appropriate macrocycle in dry CH2Cl2. X-ray crystallographic analyses of [Li([18]aneO4S2)][BArF] and [Li([18]aneO2S4)][BArF] show discrete distorted octahedral cations with hexadentate coordination to the macrocycle. The heavier alkali metal complexes all contain hexadentate coordination of the heterocrown, supplemented by M⋯F interactions via the anions, producing extended structures with higher coordination numbers; Na: CN = 7 or 8; K: CN = 8; Rb: CN = 9; Cs: CN = 8 or 10. Notably, all of the structures exhibit significant M–S/Se coordination. The crystal structures of the potassium and rubidium complexes show two distinct [M(heterocrown)]+ cations, one with M⋯F interactions to two mutually cis [BArF]− anions, and the other with mutually trans [BArF]− anions, giving 1D chain polymers. Solution multinuclear (1H, 13C, 7Li, 23Na, 133Cs) NMR data show that the macrocyclic coordination is retained in CH2Cl2 solution.
Introduction
Thioethers and selenoethers are soft, moderate σ-donor ligands with a high affinity for medium and low oxidation state transition metal ions in particular, whilst a significant body of work concerning high oxidation state early transition metals, f- and p-block acceptors has emerged over the last decade or so.1 In contrast, examples showing thioether or selenoether coordination towards the oxophilic s-block cations are much rarer.2–4 Within Group 2, while there are no complexes with neutral acyclic thio- or seleno-ether coordination, a small number of examples containing Ca–S/Se and Sr–S/Se coordination, based upon 15- and 18-membered oxa-thia and oxa-selena macrocycles, have been reported recently.2,3 These include [CaI2([18]aneO2S4)] and [MI2([18]aneO4E2)] (M = Ca, Sr; E = S, Se; [18]aneO2S4 = 1,10-dioxa-4,7,13,16-tetrathiacyclooctadecane, [18]aneO4S2 = 1,4,10,13-tetraoxa-7,16-dithiacyclooctadecane; [18]aneO4Se2 = 1,4,10,13-tetraoxa-7,16-diselenacyclooctadecane), all of which are eight-coordinate with the macrocycle hexadentate (d(Ca–S/Se) ∼ 3.0 Å) and the iodides mutually cis. The lower lattice energies of the alkaline earth diiodides (MI2) compared to MCl2 or M(CF3SO3)2, etc., leads to MI2 being suitable metal sources for this chemistry due to their higher solubilities in non-competitive solvents.3The lower charge![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :radius ratio of the Group 1 cations is expected to lead to considerably lower affinity of these cations for soft thio- or seleno-ether coordination. This is supported by early data on binding constants for alkali metal cations towards the oxa-thia analogues of 18-crown-6. Thus, the stability constant for K+/[18]aneO5S (1,4,7,10,13-pentaoxa-16-thiacyclooctadecane) and K+/[18]aneO4S2 are more than 102 and 106 times lower than for K+/18-crown-6, respectively, in methanol.5 Unsurprisingly therefore, well-characterised complexes of the Group 1 metals with similar soft donor ligands are extremely rare. There are no complexes with simple neutral acyclic thioethers, and only two prior examples with oxa-thia macrocycles, the K+ and Na+ cations with [18]aneO4S2 are structurally characterised. These are based upon lamellar halo-cuprate species, in which the K⋯S distances are 3.322(5)–3.496(4) Å and Na⋯S = 3.11(1) Å, although the presence of Cu–S coordination in these structures clearly has a significant influence.4 We have exploited the high solubility of alkali metal salts with diffuse, weakly coordinating anions in weakly coordinating (non-competitive) solvents, as a source of ‘naked’ alkali metal cations, as a synthetic entry to promote their coordination towards soft, neutral donor ligands. Even so, it was surprising to us to find that this can lead to homoleptic octathia coordination to Na+ in the macrocyclic complex [Na([24]aneS8)][BArF] ([24]aneS8 = 1,4,7,10,13,16,19,22-octathiacyclotetracosane; BArF = tetrakis{3,5-bis(trifluoromethyl)-phenyl}borate), containing distorted dodecahedral S8-coordination at the sodium centre, with d(Na–S) = 2.9561(15)–3.0524(15) Å.6 DFT calculations suggested that the pre-organisation of the [24]aneS8 macrocycle contributed to the successful isolation of this unique cation. Subsequently, using a similar approach we have also found that using even softer, neutral diphosphines leads to homoleptic Li+ and Na+ hexaphosphine cations in [Li(L–L)3][Al{OC(CF3)3}4] and [Na(L–L)3][BArF] (L–L = Me2PCH2CH2PMe2 or o-C6H4(PMe2)2).7 Further interest in caesium–sulfur coordination comes from the important role of Cs2CO3 in the formation of thioether macrocycles via high dilution cyclisation reactions in dmf solution.8 The key role in the cyclisation reaction is the formation of a Cs+–thiolate ion pair, however, despite the widespread application of this procedure for thioether macrocycle formation, evidence of caesium–thioether coordination, i.e. following ring-closure, has remained elusive.
:radius ratio of the Group 1 cations is expected to lead to considerably lower affinity of these cations for soft thio- or seleno-ether coordination. This is supported by early data on binding constants for alkali metal cations towards the oxa-thia analogues of 18-crown-6. Thus, the stability constant for K+/[18]aneO5S (1,4,7,10,13-pentaoxa-16-thiacyclooctadecane) and K+/[18]aneO4S2 are more than 102 and 106 times lower than for K+/18-crown-6, respectively, in methanol.5 Unsurprisingly therefore, well-characterised complexes of the Group 1 metals with similar soft donor ligands are extremely rare. There are no complexes with simple neutral acyclic thioethers, and only two prior examples with oxa-thia macrocycles, the K+ and Na+ cations with [18]aneO4S2 are structurally characterised. These are based upon lamellar halo-cuprate species, in which the K⋯S distances are 3.322(5)–3.496(4) Å and Na⋯S = 3.11(1) Å, although the presence of Cu–S coordination in these structures clearly has a significant influence.4 We have exploited the high solubility of alkali metal salts with diffuse, weakly coordinating anions in weakly coordinating (non-competitive) solvents, as a source of ‘naked’ alkali metal cations, as a synthetic entry to promote their coordination towards soft, neutral donor ligands. Even so, it was surprising to us to find that this can lead to homoleptic octathia coordination to Na+ in the macrocyclic complex [Na([24]aneS8)][BArF] ([24]aneS8 = 1,4,7,10,13,16,19,22-octathiacyclotetracosane; BArF = tetrakis{3,5-bis(trifluoromethyl)-phenyl}borate), containing distorted dodecahedral S8-coordination at the sodium centre, with d(Na–S) = 2.9561(15)–3.0524(15) Å.6 DFT calculations suggested that the pre-organisation of the [24]aneS8 macrocycle contributed to the successful isolation of this unique cation. Subsequently, using a similar approach we have also found that using even softer, neutral diphosphines leads to homoleptic Li+ and Na+ hexaphosphine cations in [Li(L–L)3][Al{OC(CF3)3}4] and [Na(L–L)3][BArF] (L–L = Me2PCH2CH2PMe2 or o-C6H4(PMe2)2).7 Further interest in caesium–sulfur coordination comes from the important role of Cs2CO3 in the formation of thioether macrocycles via high dilution cyclisation reactions in dmf solution.8 The key role in the cyclisation reaction is the formation of a Cs+–thiolate ion pair, however, despite the widespread application of this procedure for thioether macrocycle formation, evidence of caesium–thioether coordination, i.e. following ring-closure, has remained elusive.
We report here the results of a systematic study of the preparation, spectroscopic and structural features of a series of complexes of the Group 1 cations with hexadentate, 18-membered ring macrocycles containing both hard (O) and soft (S, Se) donor functions, which demonstrate that coordination of neutral thioether functions to alkali metal cations is not limited to just sodium and potassium, but extends to examples with all five cations from Li+ to Cs+. We also report the first examples of neutral selenoether coordination to a Group 1 cation, including structural authentication for [M([18]aneO4Se2)][BArF], M = Na and K.
Experimental
All preparations were carried out under a dry dinitrogen atmosphere using standard Schlenk and glove box techniques. [Li(thf)4][BArF] and Na[BArF]·2thf were synthesised using a slight modification of the literature procedure.9 The lithium salt was isolated as [Li(OH2)4][BArF], which was converted to the thf adduct by stirring in thf for 16 h over 4 Å molecular sieves. Filtration and removal of solvents afforded the thf adduct. Crude Na[BArF] was recrystallised from thf/n-hexane to remove the last traces of coloured impurity, resulting in the isolation of Na[BArF]·2thf. K[BArF], Rb[BArF] and Cs[BArF] were synthesised via cation exchange of Na[BArF]·2thf in water at 95 °C with excess (5 mol equiv.) KNO3, RbNO3 or CsNO3, respectively.10 The macrocycles [18]aneO4S2, [18]aneO2S4 and [18]aneO4Se2 were prepared using literature procedures.11 18-Crown-6 was purchased from Sigma and dried using SOCl2. CH2Cl2 was dried by distillation from CaH2 and n-hexane distilled from Na/K alloy. 1H and 13C{1H} NMR spectra were recorded in CD2Cl2 solution at 295 K using a Bruker AV II-400 spectrometer and are referenced to the residual CH2Cl2 resonance. 7Li, 23Na and 133Cs NMR spectra were obtained in CH2Cl2 solution on a Bruker AV II-400 spectrometer at 298 K (unless otherwise stated) and referenced to a 0.1 mol dm−3 solution of LiCl, NaCl or CsNO3 in D2O, respectively. NMR properties: 7Li: I = 3/2, N = 92.6%, Rc = 1.54 × 103, Q = −3.7 × 10−30 m2, Ξ = 38.87 MHz; 23Na: I = 3/2, N = 100%, Rc = 5.24 × 102, Q = 0.10 × 10−28, Ξ = 26.43; 39K: I = 3/2, N = 93.3%, Rc = 2.69, Q = 5.5 × 10−30, Ξ = 4.67; 85Rb: I = 5/2, N = 72.1%, Rc = 43.4, Q = 0.25 × 10−28, Ξ = 9.69; 133Cs: I = 7/2, N = 100%, Rc = 2.75 × 102, Q = −0.3 × 10−30, Ξ = 13.21.12 Microanalyses were undertaken at London Metropolitan University.X-ray crystallography
Crystals were obtained as described below. Details of the crystallographic data collection and refinement are in Table 1. Diffractometer: Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator (λ1 = 0.71073 Å) with VHF Varimax optics (70 or 110 μm focus). Cell determination and data collection: CrystalClear-SM Expert 3.1 b27, data reduction, cell refinement, and absorption correction: CrystalClear-SM Expert 2.1 b29.13 Structure solution and refinement were carried out using Olex2 or WinGX and software packages within.14 Disorder in the CF3 groups of the [BArF]− anions was present in all of the structures, which is often observed in compounds containing [BArF]−,15 and this was satisfactorily modelled using DFIX, DANG, DELU and SIMU restraints. Positional disorder was also present in the macrocycle ligands in one of the crystallographically independent cations in each of [Na(L)][BArF] complexes (L = [18]aneO4S2, [18]aneO4Se2 and [18]aneO2S4) and was modelled similarly. H-atoms were placed in geometrically-assigned positions with C–H distances of 0.95 Å (CH) or 0.98 Å (CH2) and refined using a riding model with Uiso(H) = 1.2Ueq(C). enCIFer was used to prepare material for publication.16| Compound | [Li([18]aneO4S2)][BArF] | [Na([18]aneO4S2)][BArF] | [K([18]aneO4S2)][BArF] | [Rb([18]aneO4S2)][BArF] |
| Formula | C44H36O4BF24LiS2 | C44H36O4BF24NaS2 | C88H72O8B2F48K2S4 | C44H36O4BF24RbS2 |
| M/g mol−1 | 1166.60 | 1182.65 | 2397.51 | 1245.13 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group (no.) | Pc (7) | Pc (7) | P21/c (14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
| a/Å | 12.753(3) | 12.682(3) | 16.9189(12) | 13.923(3) |
| b/Å | 22.349(5) | 22.573(5) | 13.6483(10) | 16.765(3) |
| c/Å | 17.450(4) | 17.452(4) | 44.153(3) | 22.402(5) |
| α/° | 90 | 90 | 90 | 97.029(4) |
| β/° | 95.618(4) | 97.460(3) | 97.6620(10) | 105.185(5) |
| γ/° | 90 | 90 | 90 | 90.376(3) |
| U/Å3 | 4950(2) | 4954(2) | 10104.4(13) |
5005(2) |
| Z | 4 | 4 | 4 | 4 |
| μ(Mo-Kα)/mm−1 | 0.240 | 0.249 | 0.318 | 1.204 |
| F(000) | 2352 | 2384 | 4832 | 2488 |
| Total reflections | 65730 |
37772 |
79011 |
58188 |
| Unique reflections | 21600 |
16023 |
17773 |
17632 |
| R int | 0.064 | 0.038 | 0.102 | 0.071 |
| Goodness-of-fit on F2 | 1.022 | 1.015 | 0.961 | 1.023 |
| R 1 [Io > 2σ(Io)] | 0.069 | 0.042 | 0.077 | 0.055 |
| R 1 (all data) | 0.103 | 0.051 | 0.126 | 0.083 |
| wR2b [Io > 2σ(Io)] | 0.165 | 0.096 | 0.194 | 0.132 |
| wR2 (all data) | 0.188 | 0.102 | 0.229 | 0.146 |
| Compound | [Cs([18]aneO4S2)][BArF] | [Li([18]aneO2S4)][BArF] | [Na([18]aneO4Se2)][BArF] | [Na([18]aneO2S4)][BArF] |
| Formula | C44H36O4BCsF24S2 | C44H36BF24LiO2S4 | C44H36BF24O4NaSe2 | C44H36BF24NaO2S4 |
| M/g mol−1 | 1292.57 | 1198.72 | 1276.45 | 1214.77 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group (no.) | Pbca (61) | P21 (4) | Cc (9) | C2/c (15) |
| a/Å | 17.974(5) | 16.596(4) | 12.7830(12) | 22.415(5) |
| b/Å | 19.582(5) | 19.366(5) | 67.748(7) | 8.9368(17) |
| c/Å | 28.567(7) | 23.794(6) | 17.4535(18) | 25.072(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 93.056(5) | 96.603(3) | 99.731(4) |
| γ/° | 90 | 90 | 90 | 90 |
| U/Å3 | 10055(5) |
7637(3) | 15015(3) |
4950.2(17) |
| Z | 8 | 6 | 12 | 4 |
| μ(Mo-Kα)/mm−1 | 0.950 | 0.312 | 1.617 | 0.330 |
| F(000) | 5120 | 3624 | 7584 | 2448 |
| Total reflections | 68140 |
42404 |
70507 |
13830 |
| Unique reflections | 11507 |
28887 |
26208 |
5638 |
| R int | 0.097 | 0.063 | 0.065 | 0.047 |
| Goodness-of-fit on F2 | 1.016 | 1.024 | 0.984 | 0.988 |
| R 1 [Io > 2σ(Io)] | 0.060 | 0.086 | 0.065 | 0.038 |
| R 1 (all data) | 0.118 | 0.145 | 0.079 | 0.056 |
| wR2b [Io > 2σ(Io)] | 0.127 | 0.191 | 0.155 | 0.090 |
| wR2 (all data) | 0.155 | 0.234 | 0.167 | 0.096 |
| a Common items: T = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo2]1/2. | |||
|---|---|---|---|
| Compound | [Na(18-crown-6)][BArF] | [K([18]aneO2S4)][BArF] | [K([18]aneO4Se2)][BArF] |
| Formula | C44H36BF24NaO6 | C44H36BF24KO2S4 | C88H72B2F48K2O8Se4 |
| M/g mol−1 | 1150.53 | 1230.88 | 2585.11 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group (No.) |
P (2) |
Cc (15) | P21/c (14) |
| a/Å | 10.495(2) | 22.558(4) | 16.622(3) |
| b/Å | 11.085(2) | 8.9230(14) | 13.876(2) |
| c/Å | 21.361(4) | 25.072(4) | 44.516(8) |
| α/° | 83.314(10) | 90 | 90 |
| β/° | 77.451(8) | 99.950(2) | 98.567(2) |
| γ/° | 79.135(10) | 90 | 90 |
| U/Å3 | 2374.7(8) | 4970.8(14) | 10153(3) |
| Z | 2 | 4 | 4 |
| μ(Mo-Kα)/mm−1 | 0.175 | 0.403 | 1.668 |
| F(000) | 1160 | 2480 | 5120 |
| Total reflections | 27127 |
15982 |
58673 |
| Unique reflections | 8375 | 5069 | 17383 |
| R int | 0.068 | 0.039 | 0.043 |
| Goodness-of-fit on F2 | 1.064 | 1.043 | 1.023 |
| R 1 [Io > 2σ(Io)] | 0.099 | 0.050 | 0.065 |
| R 1 (all data) | 0.149 | 0.061 | 0.104 |
| wR2b [Io > 2σ(Io)] | 0.260 | 0.123 | 0.163 |
| wR2 (all data) | 0.303 | 0.131 | 0.193 |
Preparations
Results and discussion
Coordination of the Group 1 cations to a range of 18-membered oxa-thia and oxa-selena macrocyclic ligands was achieved by the direct reaction of M[BArF] with the appropriate macrocycle in anhydrous CH2Cl2 solution (Scheme 1). The isolated complexes are highly soluble in CH2Cl2 and were obtained in good yield as white powders or colourless crystals. Their formulations were confirmed by microanalysis, solution 1H, 13C{1H}, 7Li, 23Na and 133Cs NMR spectroscopic data as appropriate, and by single crystal X-ray structure analysis. No solvent incorporation was evident in any of the products. The 1H and 13C{1H} NMR spectra confirm a 1:1 ratio of the macrocycle and the [BArF]− anion in each case, but were otherwise not very informative.
 | ||
| Scheme 1 Synthesis of the complexes reported in this work. | ||
Lithium-7 NMR spectra recorded for [Li([18]aneO4S2)]+ and [Li([18]aneO2S4)]+ each contain a singlet, at +0.07 and +1.85 ppm respectively, i.e. very small differences from LiCl in water (δ = 0). This is not unexpected given the small chemical shift range observed for 7Li (typically ca. 15 ppm).12 For the sodium complexes, 23Na NMR spectra (CH2Cl2) show a shift of the resonance to higher frequency as the S/Se donor atoms are introduced to the macrocycle (18-crown-6: −14.4 ppm; [18]aneO4S2: −1.9 ppm; [18]aneO4Se2: +2.3 ppm; [18]aneO2S4: +2.4 ppm). These chemical shifts compare with those for [Na([24]aneS8)][BArF] (S8 donor set, δ23Na = +4.0),6 [Na(thf)x][BArF] (δ23Na = −4.8),16 [Na(Me3tacn)2][BArF] (δ23Na = +6.2; Me3tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane) and [Na(Me4cyclam)(thf)][BArF] (δ23Na = +11.4; Me4cyclem = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane).17 Small positive chemical shifts were also seen for [Na{Me2P(CH2)2PMe2}3]+ and [Na{o-C6H4(PMe2)2}3]+,7 whereas δ23Na for solutions of NaBPh4 with 15-crown-5 in a variety of O-donor solvents are reported to be to low frequency of the reference irrespective of the concentration of the crown ether.18 The quadrupole moment (Q) for the 133Cs nucleus is small, hence NMR spectra are readily observed. The 133Cs NMR spectrum recorded for [Cs([18]aneO4S2)][BArF] (CH2Cl2) is a singlet at +45.2 ppm, significantly to high frequency of the chemical shifts observed for [Cs(crown)]+ (crown = 12-crown-4, 15-crown-5, 18-crown-6) in various solvents,19 whilst [Cs(Me6[18]aneN6)]+ shows a singlet +54 ppm at 298 K.20 The large Q values for 39K and 85Rb result in fast relaxation in these low-symmetry environments and no resonances were seen. Overall, the spectroscopic data for the complexes reported herein are consistent with coordination of the macrocycles to the alkali metal cations, the complexes being dynamic in solution, most likely through ‘ring-whizzing’ within the complex cations (it is unlikely that the [BArF]− coordination is retained in solution). However, these measurements do not provide unequivocal evidence for coordination of the soft S or Se donor atoms.
X-ray structures and comparisons
The paucity of complexes with thio- or seleno-ether coordination to a Group 1 cation in the literature means that structural authentication of the new complexes was essential to establish their identities. Furthermore, it is difficult to draw comparisons with other structures, and determining the significance of particular metal–ligand interactions is somewhat uncertain. Table 2 provides the sums of the relevant radii, on one hand the sum of the ionic radii of the cations and the covalent radii of the donor atoms (O, S, Se and F) and on the other the sum of the van der Waals radii for the relevant metal-donor atom combinations. Distances close to the former are considered to be ‘normal’ coordinate bonds, while in identifying weak interactions, for example, between the metal cation and the CF3 groups from [BArF]− anions, we have chosen to include any M⋯X distances at least 0.5 Å below the sum of the van der Waals radii. Although the precise cut-off is somewhat arbitrary, it seems to be a reasonable basis on which to describe the overall coordination in these complexes. It is also pertinent to note that due to the disorder present in the CF3 groups of the [BArF]− anions in the structures, while the gross coordination at the metal and the ligand conformations are secure, care should be exercised in any detailed analyses of bond lengths and angles.| M–X | X = O | X = S | X = Se | X = F | ||||
|---|---|---|---|---|---|---|---|---|
| ∑(ionic + cov radii)a | ∑(vdW radii)b | ∑(ionic + cov radii)a | ∑(vdW radii)b | ∑(ionic + cov radii)a | ∑(vdW radii)b | ∑(ionic + cov radii)a | ∑(vdW radii)b | |
| a Ionic radii of Group 1 cations taken from ref. 21 and covalent radii for ligand donor atoms taken from ref. 22. b van der Waals radii taken from ref. 23. c Based upon CN = 6. d Based upon CN = 8. | ||||||||
| M = Lic | 1.56 | 3.62 | 1.95 | 4.01 | 2.10 | 3.94 | 1.47 | 2.58 |
| M = Nad | 1.98 | 4.00 | 2.37 | 4.39 | 2.52 | 4.32 | 1.89 | 3.96 |
| M = Kd | 2.31 | 4.23 | 2.70 | 4.54 | 2.85 | 4.55 | 2.22 | 4.19 |
| M = Rbd | 2.41 | 4.71 | 2.80 | 5.10 | 2.95 | 5.03 | 2.32 | 4.67 |
| M = Csd | 2.54 | 4.98 | 2.93 | 5.37 | 3.08 | 5.30 | 2.45 | 4.94 |
The crystal structure of [Li([18]aneO4S2)][BArF] comprises discrete cations (Fig. 1) and anions, with two of each in the asymmetric unit. Each Li cation is six-coordinate with the hexadentate macrocycle, leading to a severely distorted octahedral geometry. The Li–O and Li–S bond distances show a range of values, with d(Li–O) = 2.055(12) to 2.349(13) Å and d(Li–S) = 2.724(11) to 2.788(11) Å. The angles, S1–Li1–S2 = 106.2(4)° and S3–Li2–S4 = 111.3(4)°, whilst the angles involved in the five-membered chelate rings all vary between ∼70–80°, indicating a significant degree of distortion from a regular octahedron. The irregularity of the coordination may in part reflect the poor size match of the 18-membered ring for the small Li+ centre, coupled with the packing of the large [BArF]− anions around the cations in the crystal lattice.
 | ||
| Fig. 1 The structure of the Li1-centred cation in [Li([18]aneO4S2)][BArF] with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. The Li2 cation adopts a similar geometry, with some variation in the bond distances and angles. Selected bond lengths (Å) and angles (°): Li1–O4 = 2.055(12), Li1–O2 = 2.094(12), Li1–O3 = 2.150(13), Li1–O1 = 2.349(13), Li1–S1 = 2.745(12), Li1–S2 = 2.761(11), O4–Li1–O3 = 78.5(5), O2–Li1–O1 = 75.4(4), O4–Li1–S1 = 81.2(4), O1–Li1–S1 = 71.7(3), O2–Li1–S2 = 77.7(4), O3–Li1–S2 = 75.2(4), S1–Li1–S2 = 106.2(4). | ||
Substituting two further S donor atoms into the macrocyclic ring, as in [Li([18]aneO2S4)][BArF], also leads to a structure comprising of discrete cations and anions, with three crystallographically independent, but structurally similar, variants of each in the asymmetric unit. Each Li is six-coordinate, encapsulated by the macrocycle which is folded to accommodate a distorted octahedral geometry at lithium (Fig. 2). The distortion from ideal octahedral is significantly less than in the [18]aneO4S2 analogue above, and the conformation of the coordinated macrocycle has the –S(CH2)2O(CH2)2S– linkages occupying meridional coordination sites, placing the CCOC torsion angles anti. The Li–O and Li–S distances vary between the different cations, and are ∼2.1 Å and ∼2.6 Å, respectively. The latter appear to be slightly shorter than those in [Li([18]aneO4S2)]+ above.
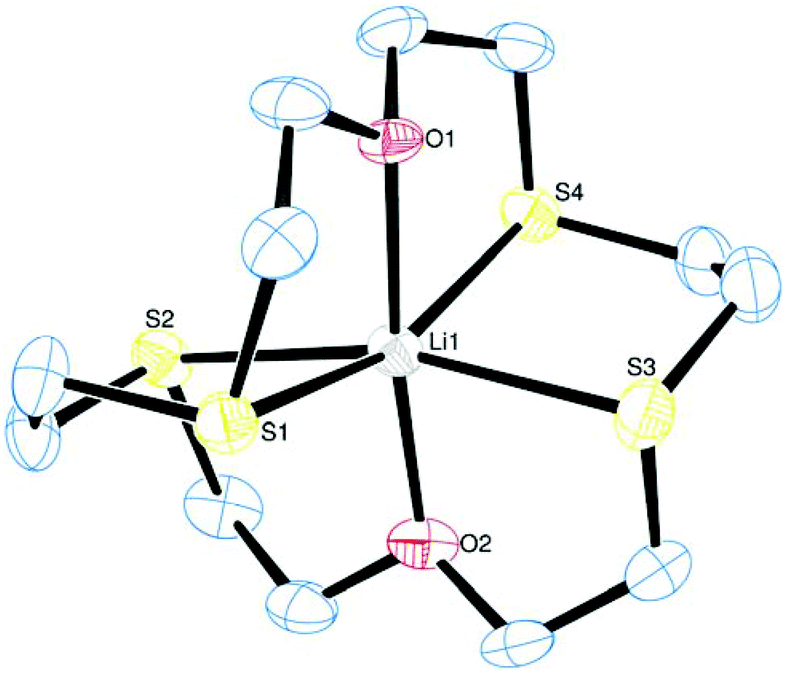 | ||
| Fig. 2 The structure of the Li1-centred cation in [Li([18]aneO2S4)][BArF] with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–O2 = 2.082(18), Li1–O1 = 2.118(18), Li1–S3 = 2.539(16), Li1–S1 = 2.573(16), Li1–S2 = 2.593(16), Li1–S4 = 2.604(15), O2–Li1–S3 = 80.9(6), O1–Li1–S1 = 82.1(6), O2–Li1–S2 = 84.2(6), S1–Li1–S2 = 81.5(4), O1–Li1–S4 = 79.9(5), S3–Li1–S4 = 85.6(5). | ||
The crystals of [Na([18]aneO4S2)][BArF] are isomorphous, but not isostructural, with their direct Li analogue, again with two crystallographically independent cations (the Na1-centred one of which is partly disordered, hence the structural description below is concerned with the Na2-centred cation) and two anions in the asymmetric unit. In this case, the Na-macrocycle coordination is quite similar to that in the Li analogue, with d(Na–O) in the range 2.355(4)–2.395(4) Å and d(Na2–S) (2.849(2) and 2.890(2) Å) all somewhat elongated. This is a consequence of the larger ionic radius of the Na+ centre, hence the ligand is less puckered, and the angles involved in the five-membered chelate rings span a narrower range (∼71–74°), with <S3–Na2–S4 = 129.77(8)°. Notably in this species there is an additional weak interaction to one F atom derived from a CF3 group in the anion, Na2⋯F26i = 3.306(3) Å (Fig. 3), leading to overall seven-coordination at the sodium centre through weakly associated ion pairs.
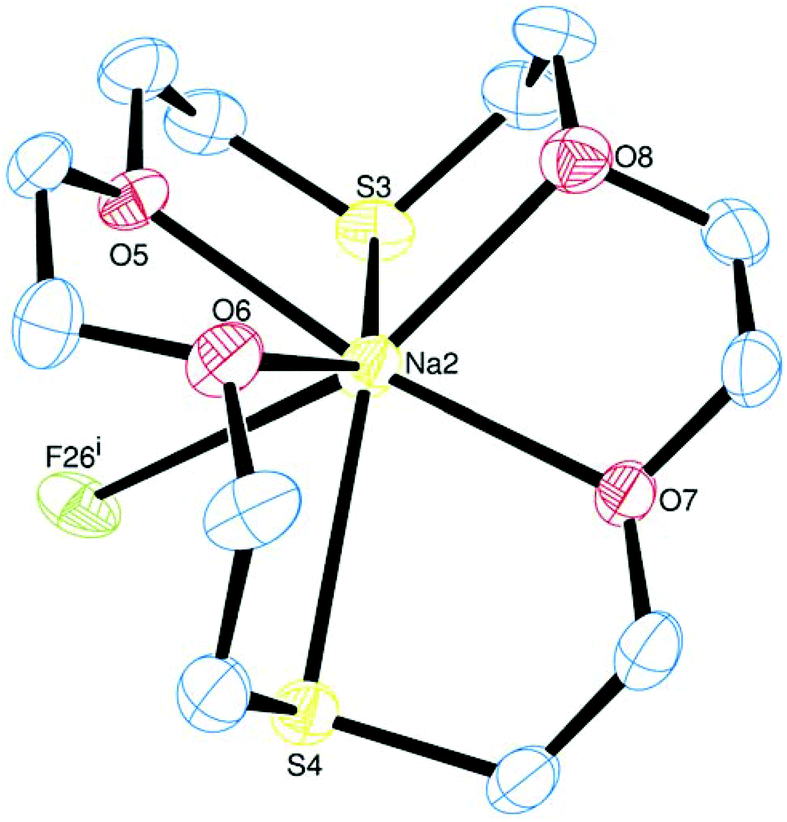 | ||
| Fig. 3 The structure of the Na2-centred cation in [Na([18]aneO4S2)][BArF] showing the interaction to a fluoride from the anion, with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Na2–O5 = 2.389(4), Na2–O6 = 2.355(4), Na2–O7 = 2.394(4), Na2–O8 = 2.395(4), Na2⋯F26i = 3.306(3), Na2–S3 = 2.849(2), Na2–S4 = 2.890(2), O6–Na2–O5 = 71.64(12), O7–Na2–O8 = 71.55(12), O5–Na2–S3 = 72.14(10), O8–Na2–S3 = 73.73(10), O6–Na2–S4 = 73.64(10), O7–Na2–S4 = 70.74(9), S3–Na2–S4 129.77(8). Symmetry operation: (i) −1 + x, −1 + y, z. | ||
[Na([18]aneO4Se2)][BArF] crystallises with three crystallographically distinct cations (the Na3-centred cation shows some disorder and is therefore excluded from the structural description) and three [BArF]− anions in the asymmetric unit. The Na1 centre is seven-coordinate through a puckered hexadentate macrocycle and one long, weak Na⋯F interaction (this is of borderline significance based on our criteria stated above; however the corresponding distances in the Na2 and Na3-centred cations are slightly shorter) (Fig. 4), with a near planar O3Se donor set and the remaining donor atoms above and below that plane. Similar coordination environments are present in the other cations, although there is a spread of bond distances, d(Na–O) = 2.315(7)–2.479(7), d(Na–Se) = 2.942(2)–2.986(4) Å. The latter are in line with the increased covalent radius of Se over S; otherwise the Na coordination environments are very similar to [Na([18]aneO4S2)][BArF] above.
 | ||
| Fig. 4 The structure of the Na1-centred cation in [Na([18]aneO4Se2)][BArF] showing the interaction to fluoride from the anion, with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Na1–O3 = 2.332(7), Na1–O1 = 2.380(7), Na1–O4 = 2.393(6), Na1–O2 = 2.396(7), Na1–Se1 = 2.942(3), Na1–Se2 = 2.967(3), Na1⋯F5 = 3.501(6), O3–Na1–O4 = 72.3(2), O1–Na1–O2 = 71.9(2), O1–Na1–Se1 = 74.90(18), O4–Na1–Se1 = 73.16(17), O3–Na1–Se2 = 74.54(17), O2–Na1–Se2 = 71.74(16), Se1–Na1–Se(2) = 121.13(13). | ||
The structure of [Na([18]aneO2S4)][BArF] is markedly different from its Li analogue above. It crystallises as a 1D chain polymer, with the Na in an eight-coordinate environment (Fig. 5a), and only one centrosymmetric cation and one anion (with crystallographic 2-fold symmetry) in the asymmetric unit. The macrocycle adopts a chair conformation with all four S atoms coordinated and the S4 donor set planar, d(Na–S) = 2.8823(6), 3.0718(7) Å, with the two oxygen atoms above and below that plane, d(Na–O) = 2.5526(13) Å. Two weak Na⋯F interactions (2.8766(13) Å) from bridging [BArF]− anions on opposite sides of the macrocycle give a distorted dodecahedral geometry overall at sodium, and give rise to the polymeric chain structure (Fig. 5b).
 | ||
| Fig. 5 (a) The structure of the centrosymmetric Na1-centred cation in [Na([18]aneO2S4)][BArF] showing the interactions to fluorides from the anions, with ellipsoids drawn at the 50% probability level. The other crystallographically independent cation adopts a similar structure. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Na1⋯F1 = 2.8766(13), Na1–O1 = 2.5526(13), Na1–S1 = 2.8823(6), Na1–S2 = 3.0718(7), O1–Na1–S1 = 67.26(3), O1–Na1–S2 = 66.22(3), S1i–Na1–S2 = 69.427(14), S1–Na1–S2i = 69.426(14). Symmetry operation: (i) 0.5 − x, 0.5 − y, 1 − z. (b) View of a portion of the polymeric chain with the [BArF]− anions greyed out for clarity. Colour key: gold = Na, yellow = S, red = O, green = F, black = C. | ||
To provide a benchmark comparison for the mixed donor macrocyclic complexes in this work, we also prepared and determined the structure of [Na(18-crown-6)][BArF]. The structure also shows a 1D chain polymer with eight-coordinate Na. There are two 50% occupancy (centrosymmetric) Na environments in the asymmetric unit, one of which (Na2-centred cation) shows severe rotational disorder of the macrocycle, while the other refines much better and is therefore the focus of the structural description and illustrated in Fig. 6a and b. The crown ether provides hexagonal planar coordination at Na, with irregular Na–O bond distances spanning >0.3 Å, and with a slightly puckered conformation as expected (<O–Na–O ∼ 62–65°), with two axial Na⋯F interactions (2.435(4) Å), similar motifs are evident in other salts containing the sodium-18-crown-6 cation.1 The Na⋯F distances are very much shorter than in the mixed donor macrocyclic cations described above.
 | ||
| Fig. 6 (a) View of the structure around Na1 of the centrosymmetric [Na(18-crown-6)][BArF], with ellipsoids drawn at the 50% probability level. The second crystallographically independent cation in the asymmetric unit contains a disordered 18-crown-6, but is otherwise similar. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Na1–O1 = 2.804(4), Na1–O2 = 2.489(4), Na1–O3 = 2.605(4), Na1⋯F1 = 2.435(4). Symmetry operation: (i) 2 − x, 2 − y, −z. (b) View of a portion of the polymeric chain formed through the bridging [BArF]− anions with the [BArF]− anions greyed out for clarity. Colour key: gold = Na, red = O, green = F, black = C. | ||
[K([18]aneO2S4)][BArF] is isostructural and isomorphous with [Na([18]aneO2S4)][BArF] described above, hence also forming a chain polymer with the K+ in an eight-coordinate environment via the O2S4 donor set from the hexadentate macrocycle (endocyclic) and weak axial interactions to CF3 groups from the bridging [BArF]− anions (Fig. 7). All of the bond distances at K1 are elongated by ca. 0.1 Å compared to the Na analogue. This is less than expected based purely on the difference in the ionic radii of the metal ions (0.33 Å for CN = 8), possibly reflecting a better size match between K+ and the [18]aneO2S4 binding cavity (although caution is required to avoid over-interpretation of such differences given that there is some disorder in the anion).
 | ||
| Fig. 7 View of the structure of the K1-centred cation in [K([18]aneO2S4)][BArF] showing the interactions to fluoride from the anions, with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): K1–O1 = 2.6253(18), K1⋯F7 = 2.9589(19), K1–S1 = 3.0445(8), K1–S2 = 3.1399(8), O1–K1–S1 = 65.74(4), O1–K1–S2 = 65.98(4), S1i–K1–S2 = 68.880(19). Symmetry operation: (i) −0.5 − x, 0.5 − y, −z. | ||
The structure of [K([18]aneO4S2)][BArF] is formed of a 1D chain polymer with three crystallographically independent cations in the asymmetric unit (as well as three [BArF]− anions). The K centres are eight-coordinate, through hexadentate endocyclic coordination of the macrocycle, with d(K–O) ∼ 2.7 Å and d(K–S) ∼ 3.2 Å, with two quite short K–F interactions (each ∼2.7 Å) completing the coordination environment (Fig. 8). The centrosymmetric K1- and K3-centred cations have significant K⋯F interactions on opposite sides of the macrocycle, while in the K2-centred cation the K⋯F interactions lie mutually cis, and <S3–K2–S2 = 154.90(6)°. Some puckering of the rings is evident from the angles subtended at K, which are all ∼64°.
 | ||
| Fig. 8 The structures of (a) the K1-centred cation and (b) the K2-centred cation in the asymmetric unit of [K([18]aneO4S2)][BArF], showing the interaction to fluoride from the anions, with ellipsoids drawn at the 50% probability level. The K3-centred cation is very similar to the K1-centred cation. Hydrogen atoms are omitted for clarity. The macrocycle around K1 is disordered positionally (up/down) around O2 with an occupancy split of roughly 3:2; the major component is shown. Selected bond lengths (Å) and angles (°): K1–F1 = 2.744(3), K1–S1 = 3.2161(12), K1–O1 = 2.703(3), K1–O2A = 2.706(7), K2–F7 = 2.690(6), K2–F25 = 2.733(3), K2–S2 = 3.2119(15), K2–S3 = 3.2075(16), K2–O5 = 2.709(3), K2–O3 = 2.731(3), K2–O4 = 2.744(3), K2–O6 = 2.750(3), S3–K2–S2 = 154.90(6). Symmetry operation: (i) 1 − x, 1 − y, −z. (c) View of a portion of the polymer chain with the [BArF]− anions greyed out for clarity. Colour key: blue = K, yellow = S, red = O, green = F, black = C. | ||
[K([18]aneO4Se2)][BArF] (Fig. 9) is isomorphous and isostructural with the tetraoxa-dithia analogue, and presents the first known complex containing potassium–selenoether coordination, d(K–Se) ca. 3.3 Å.
 | ||
| Fig. 9 The structures of the K1- and K2-centred cations in the asymmetric unit for [K([18]aneO4Se2)][BArF] showing the interactions to fluoride from the anions, with ellipsoids drawn at the 50% probability level. The K3-centred cation is very similar to the former. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): K1–O1 = 2.663(4), K1–O2 = 2.686(4), K1–F1 = 2.714(4), K1–Se1 = 3.3123(7), K2–O3 = 2.712(5), K2–O4 = 2.734(4), K2–O5 = 2.642(17), K2–O6 = 2.757(5), K2⋯F7 = 3.040(9), K2–F25 = 2.674(8), K2–Se2 = 3.3065(13), K2–Se3 = 3.307(4). Symmetry code: (i) −x, −y, 1 − z. | ||
The structure of [Rb([18]aneO4S2)][BArF] is also a 1D chain polymer, very similar to the K analogue, with the macrocycle hexadentate in each form of the cation, d(Rb–S) ∼ 3.3 Å (Fig. 10), although the crystal is not isomorphous. All the Rb–F distances are comparable to the Rb–O distances, indicating significant interactions that lie well within the sum of vdW radii. The geometry at Rb1 is higher than in the K analogue, as there are now three CF3 groups interacting with the Rb centre which appears to be ten-coordinate. The Rb2 (and Rb3: two half-occupancy centrosymmetric Rb centres in the asymmetric unit) are both 8-coordinate. The macrocycle donor set is closer to planar than in the K analogue.
 | ||
| Fig. 10 The structures of the two independent cations in [Rb([18]aneO4S2)][BArF] showing the interactions to fluoride from the anions, with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Rb1–O1 = 2.944(3), Rb1–O2 = 3.026(3), Rb1–O3 = 2.981(3), Rb1–O4 = 3.030(7), Rb1–S1 = 3.35564(12), Rb1–S2 = 3.2897(13), Rb1⋯F7 = 2.965(3), Rb1⋯F8 = 3.269(2), Rb1⋯F37 = 3.249(2), Rb1⋯F44 = 3.269(2), Rb2–O5 = 2.901(3), Rb2–O6 = 2.907(2), Rb2–S3 = 3.2690(12), Rb2⋯F23 = 2.818(7); O1–Rb1–O2 = 56.42(7), S2–Rb1–S1 = 146.62(3), O2–Rb1–S2 = 58.98(6), O3–Rb1–O4 = 57.34(14), O4–Rb1–S1 = 58.25(14), O1–Rb1–S1 = 57.99(6), O3–Rb1–S2 = 58.10(6), O5–Rb2–O6 = 59.10(8), O5–Rb2–S3 = 62.33(6), O6–Rb2–S3i = 61.72(6). Symmetry operation: (i) 1 − x, 1 − y, −z. | ||
Finally, the structure of [Cs([18]aneO4S2)][BArF] (Fig. 11) shows one cation and one anion in the asymmetric unit, and forms a 2D sheet polymer. The hexadentate macrocycle occupies one face of the Cs+ cation (Cs–S ∼ 3.5 Å), with four CF3 groups (each from a different [BArF]− anion) also coordinated through the other face, although the precise coordination at Cs cannot be confirmed due to disorder of the CF3 groups. There is also one longer Cs⋯F interaction through the centre of the macrocycle of ∼4.2 Å.
 | ||
| Fig. 11 The structure of the cation in [Cs([18]aneO4S2)][BArF] showing the interactions to fluoride from the anions, with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cs1–O1 = 3.374(7), Cs1–O2 = 3.068(9), Cs1–O3 = 3.083(9), Cs1–O4 = 3.092(9), Cs1–S1 = 3.568(4), Cs1–S2 = 3.505(3), Cs1⋯F5 = 3.235(3), Cs1⋯F8ii = 3.525(3), Cs1⋯F10iii = 3.448(7), Cs1⋯F12iii = 3.319(4), Cs1⋯F20i = 3.345(4), Cs1⋯F14iv = 4.27(2). Symmetry operations: (i) 1 − x, −0.5 + y, 1.5 − z; (ii) −0.5 + x, y, 1.5 − z; (iii) 1.5 − x, −0.5 + y, z; (iv) x, 0.5 − y, −0.5 + z. | ||
Conclusions
We have isolated and fully characterised a series of alkali metal complexes (Li+ to Cs+) with soft, neutral thioether and selenoether coordination via saturated 18-membered oxa-thia and oxa-selena macrocycles, and containing the large, weakly coordinating [BArF]− anion. There are no previously reported examples of lithium-, rubidium- or caesium-thioether coordination, and no Group 1 cation-selenoether complexes.The solution lability of the Group 1 complex cations limits the information that can be obtained from spectroscopic studies on these complexes, hence we have determined their crystal structures to establish unequivocally the metal coordination environments present. The M–S and M–Se distances determined, together with the endocyclic conformations of the macrocycles in all of the new complexes (which contrast with the exocyclic conformations adopted by the metal-free macrocycles3,24) are strongly indicative of significant, directional M–S/Se bonding interactions. Thus, in all the complexes reported, the macrocycle is hexadentate, while the flexibility of the saturated ring allows for very different degrees of ring puckering as the metal ion radius varies. Also notable, is the presence of M⋯F–CF2– interactions of variable significance across all examples, except with the smallest Li+ cation. These contacts serve to fill available spaces in the metal coordination spheres. Hence, while discrete distorted octahedral cations with O4S2 and O2S4 donor sets are present in the Li+ salts, higher coordination numbers are observed in all of the other cations; Na: CN = 7 or 8; K: CN = 8; Rb: CN = 9; Cs: CN = 8 or 10.
Although unexpected, these M–S/Se interactions clearly make a favourable contribution to the stability of the cations since other conformations of the macrocycles with O-only coordination are possible.1
Acknowledgements
We thank the EPSRC for supporting the SCFED project through a Programme Grant (EP/I033394/1). The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids.References
- (a) W. Levason, S. D. Orchard and G. Reid, Coord. Chem. Rev., 2002, 225, 159 CrossRef CAS; (b) W. Levason and G. Reid, J. Chem. Soc., Dalton Trans., 2001, 2953 RSC; (c) W. Levason, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 8491 RSC; (d) S. Kim, L. F. Lindoy and S. S. Lee, Coord. Chem. Rev., 2014, 280, 176 CrossRef CAS; (e) W. Levason and G. Reid, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley, New York, 2012, vol. 3, p. 785 Search PubMed; (f) L. Karmazin, M. Mazzanti and J. Pecaut, Chem. Commun., 2002, 654 RSC; (g) M. J. D. Champion, P. Farina, W. Levason and G. Reid, Dalton Trans., 2013, 42, 13179 RSC; (h) P. N. Bartlett, M. J. D. Champion, W. Levason, G. Reid and P. W. Richardson, Dalton Trans., 2015, 44, 2953 RSC; (i) J. W. Steed, Coord. Chem. Rev., 2001, 215, 171 CrossRef CAS.
- H. Park, K.-M. Park and S. S. Lee, Dalton Trans., 2010, 39, 9696 RSC.
- P. Farina, W. Levason and G. Reid, Dalton Trans., 2013, 42, 89 RSC.
- (a) T. Röttgers and W. S. Sheldrick, Z. Anorg. Allg. Chem., 2001, 627, 1976 CrossRef; (b) T. Röttgers and W. S. Sheldrick, J. Solid State Chem., 2000, 152, 271 CrossRef.
- (a) J. D. Lamb, R. M. Izatt, C. S. Swain and J. J. Christensen, J. Am. Chem. Soc., 1980, 102, 475 CrossRef CAS; (b) H. K. Frensdorff, J. Am. Chem. Soc., 1971, 93, 600 CrossRef CAS.
- M. J. D. Champion, J. M. Dyke, W. Levason, M. E. Light, D. Pugh, H. Bhakhoa, L. Rhyman, P. Ramasami and G. Reid, Inorg. Chem., 2015, 54, 2497 CrossRef CAS PubMed.
- M. Carravetta, M. Concistre, W. Levason, G. Reid and W. Zhang, Chem. Commun., 2015, 51, 9555 RSC.
- J. Buter and R. M. Kellogg, J. Org. Chem., 1981, 46, 4481 CrossRef CAS.
- M. Brookhart, B. Grant and A. F. Volpe Jr., Organometallics, 1992, 11, 3920 CrossRef CAS.
- I. Mon, D. A. Jose and A. Vidal-Ferran, Chem. – Eur. J., 2013, 19, 2720 CrossRef CAS PubMed.
- (a) J. S. Bradshaw, J. Y. Hui, B. L. Haymore, J. J. Christensen and R. M. Izatt, J. Heterocycl. Chem., 1973, 10, 1 CrossRef CAS; (b) J. S. Bradshaw, J. Y. Hui, Y. Chan, B. L. Haymore, R. M. Izatt and J. J. Christensen, J. Heterocycl. Chem., 1974, 11, 45 CrossRef CAS; (c) M. J. Hesford, W. Levason, M. L. Matthews and G. Reid, Dalton Trans., 2003, 2852 RSC; (d) P. Farina, T. Latter, W. Levason and G. Reid, Dalton Trans., 2013, 42, 4714 RSC.
- Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987 Search PubMed.
- (a) CrystalClear-SM Expert 3.1 b27, Rigaku Corporation, Tokyo, Japan, 2012 Search PubMed; (b) CrystalClear-SM Expert 2.1 b29, Rigaku Corporation, Tokyo, Japan, 2013 Search PubMed.
- (a) O. V. Dolomanov, L. H. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS; (b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. B. Chaplin and A. S. Weller, Eur. J. Inorg. Chem., 2010, 5124 CrossRef CAS.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335 CrossRef CAS.
- M. Everett, A. Jolleys, W. Levason, D. Pugh and G. Reid, Chem. Commun., 2014, 50, 5843 RSC.
- J. D. Lin and A. I. Popov, J. Am. Chem. Soc., 1981, 103, 3773 CrossRef CAS.
- A. Vendilo, K. Popov, M. Lajunen, V. Chistov, D. Djigailo, H. Rönkkömäki, V. Privalov and I. Pletnev, Polyhedron, 2014, 81, 341 CrossRef CAS.
- J. Dyke, W. Levason, M. E. Light, D. Pugh, G. Reid, H. Bhakhoa, P. Ramasami and L. Rhyman, Dalton Trans., 2015, 44, 13853 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverrıa, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- (a) N. K. Dalley, S. B. Larson, J. S. Smith, K. L. Matheson, R. M. Izatt and J. J. Christensen, J. Heterocycl. Chem., 1981, 18, 463 CrossRef CAS; (b) G. J. Grant, D. F. Galas, M. W. Jones, K. D. Loveday, W. T. Pennington, G. L. Schimek, C. R. Eagle and D. G. VanDerveer, Inorg. Chem., 1998, 37, 5299 CrossRef CAS.
Footnote |
| † Additional Data Available: Original NMR spectra for all complexes are available (DOI: 10.5258/SOTON/380541) and can be downloaded via <http://dx.doi.org/10.5258/SOTON/380541>. CCDC 1410981 ([Na(18-crown-6-][BArF]), 1410982 ([Li([18]aneO4S2)][BArF]), 1410983 ([Na([18]aneO4S2)][BArF]), 1410984 ([K([18]aneO4S2)][BArF]), 1410985 ([Rb([18]aneO4S2)][BArF]), 1410986 ([Cs([18]aneO4S2)][BArF]), 1410987 ([Li([18]aneO2S4)][BArF]), 1410988 ([Na([18]aneO2S4)][BArF]), 1410989 ([K([18]aneO2S4)][BArF]), 1410990 ([Na([18]aneO4Se2)][BArF]) and 1410991 ([K([18]aneO4Se2)][BArF]). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02583d |
| This journal is © The Royal Society of Chemistry 2015 |