
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterobimetallic complexes with highly flexible 1,1′-bis(phospholanoalkyl)ferrocene ligands†‡
Andy
Schmied
,
Axel
Straube
,
Toni
Grell
,
Sascha
Jähnigen
and
Evamarie
Hey-Hawkins
*
Institute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
First published on 24th July 2015
Abstract
The two highly flexible ligands 1,1′-bis(4-phospholanobutyl)ferrocene (5a) and 1,1′-bis(5-phospholanopentyl)ferrocene (5b) reacted with [PtCl2(cod)] (cod = 1,5-cyclooctadiene), [AuCl(tht)] (tht = tetrahydrothiophene) and [{RhCl(CO)2}2] to give the respective chelate complexes cis-[PtCl2(5a,b-κ2P,P′)] (7a,b), [AuCl(5a,b-κ2P,P′)] (8a,b) and trans-[RhCl(CO)(5b-κ2P,P′)] (9b). Treatment of 5a,b with selenium gave the corresponding selenides 6a,b. All compounds have been fully characterised by NMR (1H, 13C, 31P) and IR spectroscopy and mass spectrometry. In addition, crystal structures of 6a, 7b, 8a,b and 9b were determined by X-ray crystallography. Furthermore, the rhodium-catalysed hydroformylation of 1-octene has been studied with ligand 5b.
Introduction
Bis(phosphino)ferrocenes are commonly used ligands in a variety of homogeneously catalysed reactions.1 Enormous progress has been made in the chemistry of 1,1′-bis(diphenylphosphino)ferrocene (dppf)2 and its achiral3 and chiral4,5 derivatives. However, less rigid ligands in which the ferrocene moiety and the phosphine are separated by a flexible carbon-based spacer are rare,6 and the preparation of the corresponding metal complexes often suffers from low selectivity. Usually, complex product mixtures resulting from intra- and intermolecular reactions are obtained due to insufficient differentiation caused by the high conformational flexibility of 1,1′-substituted ferrocene derivatives. Thus, no metal complexes of the type [Fe{C5H4(CH2)nPR2}2] (n = 2, R = Ph, tBu; n = 3, R = Ph) could be isolated.7 However, flexibility of a bidentate ligand may be important for the acceleration of specific transitions within a catalytic cycle and, therefore, may have a significant effect on the catalyst's efficiency.8Recently, we have shown that highly flexible bis-phospholane ligands based on solely aliphatic backbones react selectively with [AuCl(tht)] (tht = tetrahydrothiophene) to give macrocyclic complexes [Au2Cl2{μ-(C4H8P)(CH2)n(PC4H8)-κ2P,P′}2] (n = 5, 7, 9, 11),9 without using high-dilution techniques or templates to avoid polymerisation.10
As the phospholane moiety apparently increases the selectivity of metal complex formation and bisphospholanes proved to be privileged structures in catalysis,11 we have prepared the ferrocene-based bis-phospholanes [Fe{C5H4(CH2)nP(C4H8)}2] [n = 4 (5a), 5 (5b)] and studied their coordination behaviour in platinum(II), gold(I) and rhodium(I) complexes.
Results and discussion
Synthesis of bis-phospholane ligands and selenides
Two bis-phospholane ligands having highly flexible alkylene spacers with four or five methylene groups between the ferrocene moiety and the phospholane were synthesised by a modification of a strategy published by Haddow et al. in 2009 (Scheme 1).12 The starting materials, 1,1′-bis(ω-bromoalkyl)ferrocenes, were prepared by Friedel–Crafts acylation of ferrocene with ω-bromoacyl chlorides and subsequent reduction of the obtained 1,1′-acylferrocenes 1a,b.13 The crystal structure of 1,1′-bis(5-bromopentanoyl)ferrocene (1b) shows no unusual features and can be found in the ESI.‡ Treatment of two equivalents of 1-phenylphospholane with 1,1′-bis(ω-bromoalkyl)ferrocenes 2a,b gave the corresponding phosphonium salts 3a and 3b in quantitative yield as orange solids. Basic hydrolysis of the P−C(phenyl) bond by aqueous sodium hydroxide solution led to the formation of the phosphine oxides 4a and 4b. From these, the bis-phospholane ligands 5a and 5b were obtained as viscous, orange oils by reduction with a mixture of lithium aluminium hydride and trimethylsilyl chloride. | ||
| Scheme 1 Synthesis of bis-phospholane ligands 5a and 5b. (i) AlCl3, 2 equiv. Br(CH2)nCOCl, CHCl3 (n = 3, 4); (ii) AlCl3, LiAlH4, Et2O; (iii) 2 equiv. 1-phenylphospholane, CH3CN, 80 °C; (iv) NaOHaq, 95 °C; (v) LiAlH4, SiMe3Cl, THF. | ||
The corresponding selenides 6a and 6b were prepared by treating 5a and 5b with two equivalents of grey selenium. Crystals of 6a suitable for X-ray diffraction (crystal structure shown in the ESI‡) could be obtained from ethyl acetate/n-hexane. In contrast, 6b is a yellow oil at room temperature. In the 31P{1H} NMR spectra of both compounds, a sharp singlet at δ = 45.6 ppm is observed (1JP–Se = 693 Hz, determined from the 77Se satellites). Since 1JP–Se is directly related to the s character of the lone pair of electrons,146a and 6b appear to be stronger σ donors in comparison with FerroLANE (FerroLANE = 1,1′-bis(phospholano)ferrocene) ligands (cf. JP–Se = 738 Hz in (S)-1,1′-bis(2,5-dimethylphospholanoselenide)ferrocene).5
Metal complexes
Treatment of [PtCl2(cod)] (cod = 1,5-cyclooctadiene) with one equivalent of the bis-phospholanes 5a and 5b in dichloromethane afforded the corresponding complexes [PtCl2(5a-κ2P,P′)] (7a) and [PtCl2(5b-κ2P,P′)] (7b) (Scheme 2). While 7a is formed selectively, the reaction of 5b with [PtCl2(cod)] gave a mixture containing 7b and another platinum complex (two singlets, 1JP–Pt ≈ 3450 Hz, determined from the 195Pt satellites, consistent with cis-coordinated phosphines, are observed in the 1P{1H} NMR spectrum of the reaction mixture). In analogy to observations made by Guino-o et al. on the formation of platinum complexes with FerroLANE ligands, the minor side product may contain 5b in a bridging binding mode.15 Compounds 7a and 7b, the latter isolated from the product mixture by crystallisation, were obtained as slightly hygroscopic orange solids and were fully characterised by NMR and IR spectroscopy, mass spectrometry, cyclic voltammetry and elemental analysis. In the 31P{1H} NMR spectra, a downfield shift is observed on coordination, from δ ≈ −26.8 ppm (free ligand) to sharp signals at 17.4 (7a) and 15.7 ppm (7b). Crystal structures of both complexes were obtained (Fig. 1). 7a crystallises in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) from CH2Cl2/n-hexane, while 7b crystallises in space group P21/n. The platinum atoms in 7a and 7b are coordinated in a square-planar fashion by two phospholane moieties and two chlorido ligands in cis configuration, as expected from the 1JP–Pt values of 3426 and 3452 Hz, respectively. The Pt–P and Pt–Cl bond lengths as well as the P–Pt–P and Cl–Pt–Cl bond angles are in agreement with those of related bis-phospholane platinum(II) complexes (e.g., cis-[PtCl2{PPh(C4H8)}2]: Pt–P 223.9(1) pm, Pt–Cl 236.7(1) pm, P–Pt–P 92.60(6)° Cl–Pt–Cl 88.30(7)°).16
from CH2Cl2/n-hexane, while 7b crystallises in space group P21/n. The platinum atoms in 7a and 7b are coordinated in a square-planar fashion by two phospholane moieties and two chlorido ligands in cis configuration, as expected from the 1JP–Pt values of 3426 and 3452 Hz, respectively. The Pt–P and Pt–Cl bond lengths as well as the P–Pt–P and Cl–Pt–Cl bond angles are in agreement with those of related bis-phospholane platinum(II) complexes (e.g., cis-[PtCl2{PPh(C4H8)}2]: Pt–P 223.9(1) pm, Pt–Cl 236.7(1) pm, P–Pt–P 92.60(6)° Cl–Pt–Cl 88.30(7)°).16
 | ||
| Scheme 2 Synthesis of metal complexes. | ||
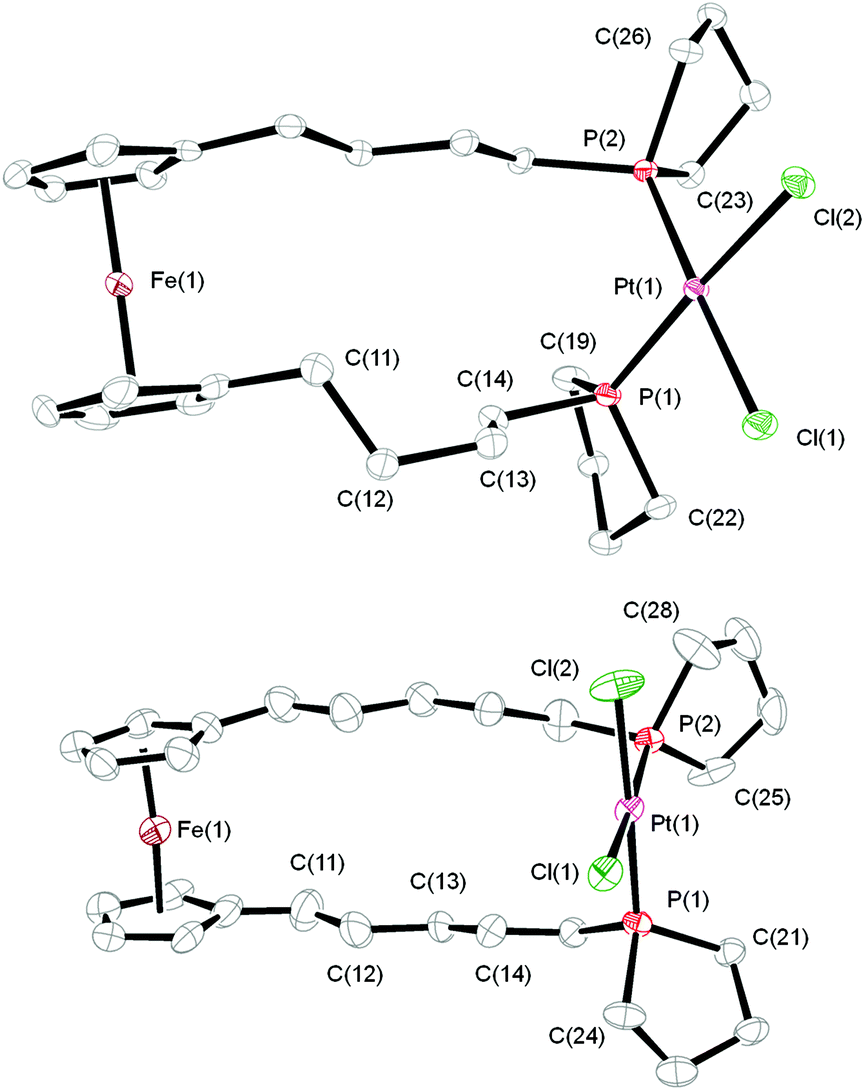 | ||
| Fig. 1 Molecular structures of 7a (top) and 7b (bottom). Ellipsoids are drawn at the 50 and 30% probability level, respectively. Hydrogen atoms are omitted for clarity. Selected bond lengths (in pm) and angles (in °): 7a: Pt1–Cl1 234.9(1), Pt1–Cl2 237.1(2), Pt1–P1 223.2(1), Pt1–P2 223.8(1), P1–C19 184.7(3), P1–C22 182.0(3), P2–C23 182.4(4), P2–C26 184.0(4); P1–Pt1–P2 94.27(4), Cl2–Pt1–Cl1 87.71(4), C22–P1–C19 95.1(2), C23–P2–C26 94.1(2); dihedral angle C11–C12–C13–C14 71.2(3). 7b: Pt1–Cl1 234.7(2), Pt1–P1 223.8(2), Pt1–P2 222.7(2), P1–C21 182.5(8), Pt1–Cl2 234.3(3), P1–C24 184.7(8), P2–C25 180(1), P2–C28 183(1); P1–Pt1–P2 94.52(8), Cl2–Pt1–Cl1 87.62(9), C21–P1–C24 93.9(4), C25–P2–C28 94.2(5); dihedral angle C11–C12–C13–C14 167.5(8). | ||
While the environment of the platinum atoms in 7a and 7b is very similar, differences are observed in the structural parameters of the ferrocene moieties and the aliphatic spacers (Table 1).
| Compound | Θ (°) | τ (°) | CpR ring conformation |
|---|---|---|---|
| a Dihedral angle between mean planes through cyclopentadienyl (CpR) rings. b Torsional twist about CpR(centroid)⋯Fe⋯CpR(centroid) axis. | |||
| 1b | 1.40 | 142.12 | Anticlinal |
| 6a | 1.55 | 146.83 | Anticlinal |
| 7a | 2.10 | 10.50 | Synperiplanar |
| 7b | 5.54 | 11.41 | Synperiplanar |
| 8b | 2.81 | 4.32 | Synperiplanar |
| 9b | 0.54 | 72.30 | Synclinal |
Although the cyclopentadienyl rings in both complexes adopt a synperiplanar conformation, the dihedral angle between the mean planes through the cyclopentadienyl rings is significantly larger in 7b (5.54 vs. 2.10°).
A gauche conformation (θ = 71.2(3)°) is observed in one of the two alkylene spacers of 7a. In contrast, all methylene groups in the aliphatic backbone of 7b have an almost perfect antiperiplanar conformation.
Addition of [AuCl(tht)] to a solution of one equivalent of 5a or 5b led to the selective formation of the corresponding complexes [AuCl(5a-κ2P,P′)] (8a) and [AuCl(5b-κ2P,P′)] (8b) (Scheme 2). The formation of polymeric products, known for the dppf ligand and several dppf derivatives, was not observed.17 The 31P{1H} NMR spectra of 8a and 8b exhibit sharp singlets at δ = 34.3 and 37.4 ppm, respectively. In the mass spectra (ESI+ mode), [M − Cl]+ peaks are observed. All attempts to obtain single crystals of 8a suitable for structure determination were unsuccessful. The molecular structure of 8b, which crystallises in the orthorhombic space group P212121, is shown in Fig. 2. In 8b, the gold atom is almost linearly coordinated by the two phospholane moieties (P1–Au1–P2 173.06(5)°). The distortion occurs due to an additional, weak coordination of a terminal chlorido ligand. The Au–Cl bond (287.1(1) pm vs. 250.0(4) pm in [AuCl(PPh3)2]18) is conspicuously long, but is in line with non-ferrocene-based analogues, as are the Au–P bond lengths (cf. [Au2Cl2{μ-(C4H8P)(CH2)7(PC4H8)}2]: Au–Cl 290.3(1) pm, Au–P 229.7(1) and 229.4(1) pm).9 The cyclopentadienyl rings adopt a synperiplanar arrangement, and a gauche conformation is observed in analogy to 7a in one of the two alkylene spacers.
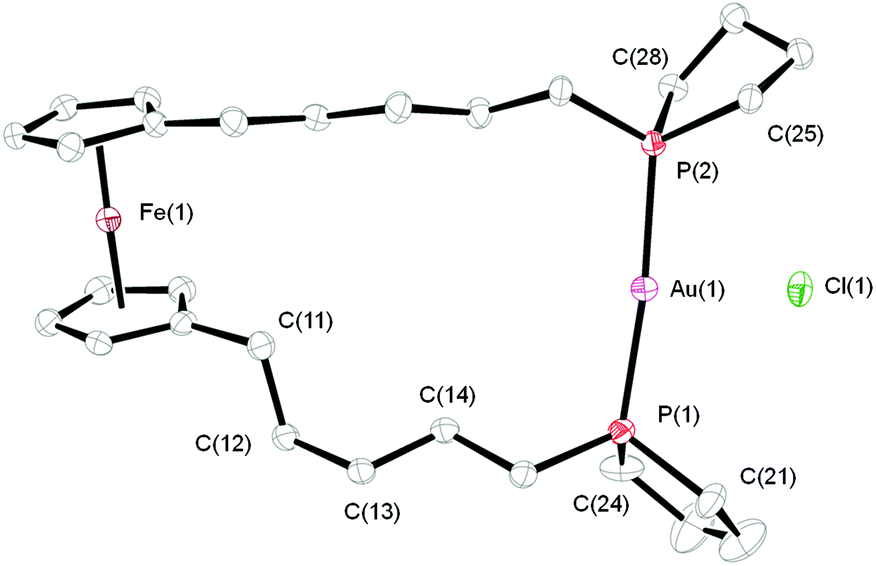 | ||
| Fig. 2 Molecular structure of 8b. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (in pm) and angles (in °): Au1–P1 229.1(1), Au1–P2 229.5(1), Au–Cl1 287.1(1), P1–C21 183.6(6), P1–C24 181.9(6), P2–C25 183.3(5), P2–C28 183.1(5); P1–Au1–P2 173.06(5), P1–Au1–Cl1 96.20(5), P2–Au1–Cl1 90.66(5), C21–P1–C24 95.5(3), C25–P2–C28 95.2(2); dihedral angle C11–C12–C13–C14 70.10(3). | ||
Treatment of [{RhCl(CO)2}2] with two equivalents of 5b in dichloromethane gave the corresponding complex [RhCl(CO)(5b-κ2P,P′)] (9b). All attempts to isolate pure [RhCl(CO)(5a-κ2P,P′)] (9a) from the obtained product mixture were unsuccessful. However, the appearance of an [M]+ peak in the mass spectrum (ESI+ mode) supports the formation of 9a.
A sharp doublet is observed at δ = 26.5 ppm in the 31P{1H} NMR spectrum of 9b. The coupling constant of 1JP–Rh = 115.4 Hz indicates a trans configuration. The IR spectrum shows one absorption for νCO at 1952 cm−1 (cf. 1968 cm−1 in [Rh2Cl2(CO)2{μ-(C4H8P)(CH2)3(PC4H8)}2]).12 Compound 9b crystallises from CH2Cl2/Et2O in the monoclinic space group P21/n (Fig. 3). The rhodium atom is coordinated in a square-planar fashion by the trans-coordinated bis-phospholane, a carbonyl and a chlorido ligand. Bond lengths and angles are in agreement with those of structurally related bis-phospholane rhodium complexes.12
 | ||
| Fig. 3 Molecular structure of 9b. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (in pm) and angles (in °): Rh1–P1 231.10(8), Rh1–P2 230.42(8), Rh1–Cl1 236.38(6), Rh1–C29 180.1(2), P1–C21 184.5(2), P1–C24 183.7(2), P2–C25 184.0(2), P2–C28 184.7(2), C29–O1 114.9(2); P1–Rh1–P1 176.66(1), C21–P1–C24 94.43(7), Cl1–Rh1–C29 175.27(5), C25–P2–C28 94.36(8), Rh1–C29–O1 177.9(2); dihedral angle C11–C12–C13–C14 70.58(1). | ||
The cyclopentadienyl rings in 9b adopt a synclinal conformation, in contrast to the obtained platinum and gold complexes. Similar to 7a and 8b, a gauche conformation is observed in one of the two alkylene spacers.
Cyclovoltammetry
In the cyclic voltammograms (see ESI‡), a single, reversible oxidation occurs in all isolated complexes (6a,b, 7a,b, 8a,b and 9b) at E1/2 = −0.06, 0.10 or −0.13 V versus Fc+/FcH, attributed to the FeII/FeIII redox couple.Theoretical considerations
DFT calculations were conducted to comprehend the gauche conformation observed in 7a, 8b and 9b.Three conformers of 9b in which the ferrocene moieties adopt a synclinal conformation were optimised (Fig. 4). One of the two alkylene chains exhibits a gauche conformation in C1 and C2, the latter similar to that in the crystal structure. In contrast, all methylene groups in C3 are antiperiplanar to each other. Furthermore, the ferrocene moiety in C3 exhibits a stronger tilt towards the plane that contains the chloride and the metal atoms (angle of slope 54° vs. 60° in C1). However, energy differences between the three conformers (Grel) are negligible (see Fig. 4.). Therefore, we suggest that the gauche conformation arises from effects of crystal packing. The appearance of C2 instead of the energetically favoured conformer C3 supports this suggestion.
 | ||
| Fig. 4 Optimised structures of the most stable conformers of 9b. | ||
Hydroformylation catalysis
Bis-phospholanes with a flexible aliphatic backbone are interesting ligands for the rhodium-catalysed hydroformylation of alkenes.12 Total conversion of 1-octene is observed with 5b/[{RhCl(CO)2}2] as catalyst over 18 h at 40 bar CO/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) pressure and 50 °C (Scheme 3). The regioselectivity (n:iso = 1.8) is similar to those of non-ferrocene-based bis-phospholane systems.12
:1) pressure and 50 °C (Scheme 3). The regioselectivity (n:iso = 1.8) is similar to those of non-ferrocene-based bis-phospholane systems.12
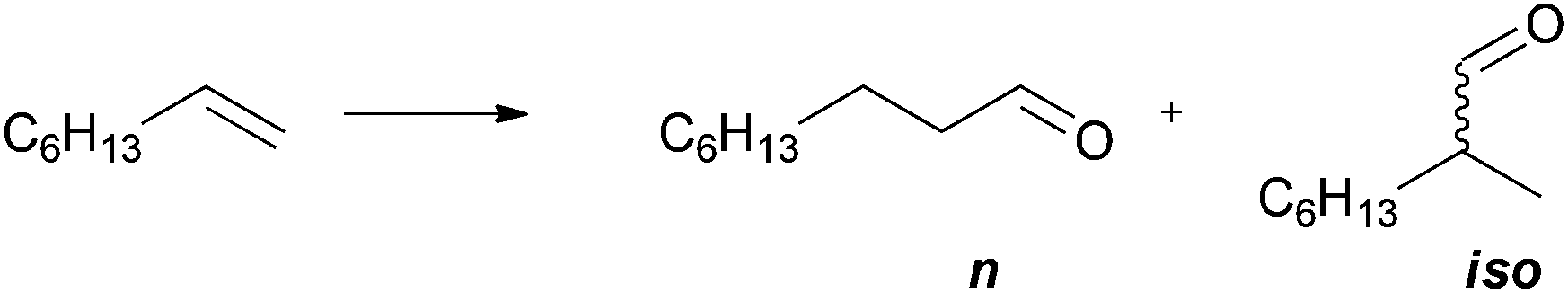 | ||
| Scheme 3 Hydroformylation of 1-octene. Reaction conditions: 50 °C, 40 bar CO/H2 (1:1) for 18 h, 0.05 mol% [{RhCl(CO)2}2], 5b:Rh = 5:2. See Experimental part for details. Conversion 100%, n:iso = 1.8. | ||
Conclusions
A new class of highly flexible 1,1′-bis(phospholano)ferrocene ligands was prepared. Their selective reaction with [PtCl2(cod)], [{RhCl(CO)2}2] and [AuCl(tht)], independent of the number of methylene groups in the spacer molecule, was shown. Chelate complexes from intramolecular reactions were obtained exclusively. Furthermore, all complexes show a reversible FeII/FeIII redox process. To the best of our knowledge, the reported metal complexes are the first 1,1′-bis(phosphino)ferrocene-based heterobimetallic complexes with highly flexible alkylene backbones that could be isolated and structurally characterised by X-ray diffraction.The reported results are essential for a deeper understanding of the influence of the substituent at the phosphorus atom on the metal complex formation of flexible bis(phosphino)ferrocenes.
Experimental
General methods
All reactions were carried out in a nitrogen atmosphere by using standard Schlenk techniques19 and anhydrous solvents, which were purified with an MB SPS-800 solvent purification system from MBRAUN or as mentioned in the literature.20 1-Phenylphospholane,21 [AuCl(tht)],22 [PtCl2(cod)]23 and 2a,b13 were prepared according to the literature. All other chemicals were used as purchased. NMR spectra were recorded at 298 K on a Bruker AVANCE DRX 400 spectrometer or a Bruker VARIAN 300 spectrometer. The chemical shifts δ of 1H, 13C, 31P are reported in parts per million (ppm) at 400.12 (300.23), 100.63 (75.49) and 162.02 MHz, respectively, with tetramethylsilane as an internal standard and referencing to the unified scale.24 Coupling constants J are given in Hz. FTIR spectra were recorded on a Perkin-Elmer Spectrum 2000 FTIR spectrometer, scanning between 400 and 4000 cm−1, using KBr pellets. Wavenumbers![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) are reported in cm−1. Mass spectra were recorded on an ESQUIRE 3000 plus spectrometer. Elemental analyses were carried out with a Heraeus VARIO EL oven. Melting and decomposition points were measured in sealed capillaries by using a variable heater from Gallenkamp. Cyclic voltammetry experiments were conducted at ambient temperature with an SP-50 potentiostat from BioLogic Science Instruments. All scans were performed with 0.1 M tetrabutylammonium tetrafluoridoborate as supporting electrolyte in CH2Cl2 and a standard three-electrode cell (glassy carbon working electrode, platinum counter electrode, silver wire reference electrode). Ferrocene was used as internal reference.
are reported in cm−1. Mass spectra were recorded on an ESQUIRE 3000 plus spectrometer. Elemental analyses were carried out with a Heraeus VARIO EL oven. Melting and decomposition points were measured in sealed capillaries by using a variable heater from Gallenkamp. Cyclic voltammetry experiments were conducted at ambient temperature with an SP-50 potentiostat from BioLogic Science Instruments. All scans were performed with 0.1 M tetrabutylammonium tetrafluoridoborate as supporting electrolyte in CH2Cl2 and a standard three-electrode cell (glassy carbon working electrode, platinum counter electrode, silver wire reference electrode). Ferrocene was used as internal reference.
Synthesis and characterisation
1,1′-Bis(4-(phenylphospholanium)butyl)ferrocene dibromide (3a). Yield 1.56 g (99%). Decomp. >156 °C. Found C, 58.4; H, 6.6. Calc. for C38H50Br2FeP2: C, 58.2; H, 6.4. IR
3054w, 2935s, 2359w, 1636m, 1438s, 1404m, 1264m, 1165w, 1120s, 1020m, 879m, 805m, 751m, 692m, 516m. 1H NMR (CD3OD, 400 MHz): δ 7.93 (m, 4H), 7.83 (m, 2H), 7.74 (m, 4H), 3.95 (m, 4H), 3.93 (m, 4H), 2.74–2.58 (m, 12H), 2.35 (t, J = 7.2, 4H), 2.30–2.08 (m, 8H), 1.66–1.47 (m, 8H). 13C{1H} NMR (CD3OD, 100 MHz): δ 135.5 (d, JC,P = 3.2), 133.2 (d, JC,P = 9.5), 131.2 (d, JC,P = 12.3), 121.0 (d, JC,P = 77.1), 89.3 (s), 69.8 (s), 69.0 (s), 33.1 (d, JC,P = 15.6), 29.6 (s), 27.2 (d, JC,P = 5.9), 23.5 (d, JC,P = 45.3), 23.3 (d, JC,P = 52.3), 22.9 (d, JC,P = 4.6). 31P{1H} NMR (CD3OD, 162 MHz): δ 50.1 (s). MS (ESI(+), CH3OH) m/z 703.2 [M − Br]+, 312.1 [M − 2Br]+.
1,1′-Bis(5-(phenylphospholanium)pentyl)ferrocene dibromide (3b). Yield 1.61 g (99%). Decomp. >220 °C. Found C, 59.1; H, 6.7. Calc. for C40H54Br2FeP2: C, 59.3; H 6.8. IR
2964m, 2932m, 2854w, 1456m, 1438m, 1404w, 1262m, 1097m, 1024m, 803m, 749w, 693w, 518w. 1H NMR (CD3OD, 400 MHz): δ 7.96 (m, 4H), 7.80 (m, 2H), 7.71 (m, 4H), 3.93 (m, 4H), 3.92 (m, 4H), 2.75–2.61 (m, 12H), 2.3 (t, J = 6.8, 4H), 2.27–2.08 (m, 8H), 1.60–1.40 ppm (m, 12H). 13C{1H} NMR (CD3OD, 100 MHz): δ 135.5 (d, JC,P = 2.9), 133.1 (d, JC,P = 9.5), 131.2 (d, JC,P = 12.1), 121.1 (d, JC,P = 77.1), 89.8 (s), 69.8 (s), 69.0 (s), 31.7 (s), 31.2 (d, JC,P = 15.5), 30.2 (s), 27.2 (d, JC,P = 5.8), 23.6 (d, JC,P = 45.3), 23.3 (d, JC,P = 52.3), 23.3 (d, JC,P = 4.5). 31P{1H} NMR (CD3OD, 162 MHz): δ 50.0 (s). MS (ESI(+), CH3OH) m/z 733.2 [M − Br]+, 326.1 [M − 2Br]+.
1,1′-Bis(4-(phospholano oxide)butyl)ferrocene (4a). Yield 1.28 g (96%). Found C, 62.3; H, 8.1. Calc. for C26H40FeO2P2: C, 62.2; H, 8.0. IR
3092w, 2935s, 2864m, 1635m, 1451m, 1407m, 1266s, 1162s, 1111m, 1056m, 1023m, 862m, 803m, 729m, 518m. 1H NMR (CDCl3, 300 MHz): δ 3.91 (d, J = 1.6, 4H), 3.88 (d, J = 1.6, 4H), 2.29 (t, J = 7.2, 4H), 1.94 (m, 4H), 1.81–1.46 (m, 24H). 13C{1H} NMR (CDCl3, 75 MHz): δ 88.3 (s), 68.7 (s), 67.9 (s), 32.7 (d, JC,P = 13.5), 30.8 (d, JC,P = 61.6), 29.1 (s), 26.9 (d, JC,P = 64.8), 24.5 (d, JC,P = 7.8), 22.0 (d, JC,P = 4.0). 31P{1H} NMR (CDCl3, 162 MHz): δ 71.2 (s). MS (ESI(+), CH2Cl2, CH3CN) m/z 503.5 [M + H]+.
1,1′-Bis(5-(phospholano oxide)pentyl)ferrocene (4b). Yield 1.38 g (99%). Found C, 63.4; H, 8.2. Calc. for C28H44FeO2P2: C, 63.4; H, 8.4. IR
3094w, 2925s, 2860m, 1464m, 1405m, 1264s, 1169s, 1110s, 1053s, 1042s, 1021s, 858m, 816s, 803s, 752w, 728m, 514m, 498m. 1H NMR (CDCl3, 300 MHz): δ 3.96 (m, 4H), 3.94 (m, 4H), 2.32 (t, J = 7.2, 4H), 2.01 (m, 4H), 1.88–1.58 (m, 20H), 1.58–1.34 (m, 8H). 13C{1H} NMR (CDCl3, 75 MHz): δ 88.6 (s), 68.4 (s), 67.6 (s), 30.8 (s), 30.7 (d, JC,P = 61.5), 30.7 (d, JC,P = 14.0), 29.0 (s), 26.8 (d, JC,P = 64.7), 24.3 (d, JC,P = 7.8), 21.9 (d, JC,P = 4.0). 31P{1H} NMR (CDCl3, 162 MHz): δ 70.9 (s). MS (ESI(+), CH2Cl2, CH3CN) m/z 530.2 [M]+.
1,1′-Bis(4-(phospholano)butyl)ferrocene (5a). Yield 0.97 g (69%). Found C, 66.1; H, 8.25. Calc. for C26H40FeP2: C, 66.4; H, 8.6. IR
3081w, 2930s, 2852m, 1458w, 1447w, 1262s, 1106s, 1040s, 1021s, 872m, 802s, 709w, 485m. 1H NMR (CDCl3, 400 MHz): δ 3.89 (m, 4H), 3.88 (m, 4H), 2.24 (t, J = 6.0, 4H), 1.77–1.56 (m, 12H), 1.55–1.23 (m, 16H). 13C{1H} NMR (CDCl3, 100 MHz): δ 89.0 (s), 68.6 (s), 67.7 (s), 33.9 (d, JC,P = 10.8), 29.2 (s), 28.8 (d, JC,P = 14.8), 27.8 (s), 26.8 (d, JC,P = 14.7), 25.9 (d, JC,P = 10.4). 31P{1H} NMR (CDCl3, 162 MHz): δ −26.7 (s). MS (ESI(+), CHCl3, CH3CN) m/z 471.2 [M + H]+.
1,1′-Bis(5-(phospholano)pentyl)ferrocene (5b). Yield 0.97 g (65%). Found C, 67.45; H, 8.5. Calc. for C28H44FeP2: C, 67.5; H, 8.9. IR
3087m, 2925s, 2855s, 1447m, 1262s, 1095s, 1022s, 803s, 743w, 705m, 668m, 488m. 1H NMR (CDCl3, 400 MHz): δ 3.88 (m, 4H), 3.87 (m, 4H), 2.22 (t, J = 7.4, 4H), 1.77–1.53 (m, 12H), 1.45–1.27 (m, 16H), 1.21 (m, 4H). 13C{1H} NMR (CDCl3, 75 MHz): δ 89.2 (s), 68.2 (s), 67.7 (s), 31.3 (d, JC,P = 9.1), 31.2 (s), 29.4 (s), 29.0 (d, JC,P = 13.0), 27.9 (s), 26.8 (d, JC,P = 13.0), 26.0 (d, JC,P = 8.8). 31P{1H} NMR (CDCl3, 162 MHz): δ −26.8 (s). MS (ESI(+), CHCl3, CH3CN) m/z 499.4 [M + H]+.
1,1′-Bis(4-(phospholanoselenide)butyl)ferrocene (6a). Yield 94.2 mg (75%). mp 107 °C (n-hexane/ethyl acetate) Found C, 49.6; H, 6.6. Calc. for C26H40FeP2Se2: C, 49.7; H, 6.4. IR
3084w, 2928s, 2861m, 1656w, 1649w, 1460m, 1401m, 1336w, 1301w, 1256w, 1178w, 1114m, 1101m, 1067m, 1040w, 1017m, 881m, 860m, 820m, 766w, 750w, 728m, 704m, 675m, 528m. 1H NMR (CDCl3, 400 MHz): δ 3.92 (m, 4H), 3.89 (m, 4H), 2.30 (t, J = 7.6, 4H), 2.25–1.89 (m, 16H), 1.83–1.61 (m, 8H), 1.55 (m, 4H). 13C{1H} NMR (CDCl3, 400 MHz): δ 88.3 (s), 68.1 (s), 67.8 (s), 33.8 (d, JC,P = 45.3), 33.3 (d, JC,P = 39.2), 32.1 (d, JC,P = 14.8), 29.1 (s), 26.5 (d, JC,P = 5.1), 23.7 (d, JC,P = 3.2). 31P{1H} NMR (CDCl3, 162 MHz): δ 45.6 (s, 77Se satellites d, 1JP,Se = 693). MS (ESI(+), CH2Cl2, CH3CN) 630.0 [M]+.
1,1′-Bis(5-(phospholanoselenide)pentyl)ferrocene (6b). Yield 99.8 mg (76%). Found C, 51.1; H, 6.65. Calc. for C28H44FeP2Se2: C, 51.2; H, 6.8. IR
3085w, 2929s, 2856m, 1656w, 1631w, 1446m, 1409m, 1262s, 1108s, 1068s, 1023s, 919w, 860m, 802s, 699w, 661w, 527m. 1H NMR (CDCl3, 400 MHz): δ 3.91 (m, 4H), 3.89 (m, 4H), 2.26 (t, J = 7.4), 2.22–1.88 (m, 16H), 1.83–1.57 (m, 8H), 1.51–1.31 (m, 8H). 13C{1H} NMR (CDCl3, 100 MHz): δ 88.9 (s), 68.8 (s), 67.9 (s), 33.9 (d, JC,P = 45.4), 33.3 (d, JC,P = 39.2), 30.8 (s), 30.2 (d, JC,P = 15.0), 29.2 (s), 26.5 (d, JC,P = 5.1), 23.7 (d, JC,P = 3.4). 31P{1H} NMR (CDCl3, 162 MHz): δ 45.6 (s, 77Se satellites d, 1JP,Se = 693). MS (ESI(+), CH2Cl2, CH3CN) 658.1 [M]+.
cis-Dichlorido-[1,1′-bis(4-(phospholano)butyl)ferrocene-κ2P,P′]-platinum(II) (7a). Yield 123.7 mg (84%). Decomp. >180 °C. Found C, 42.55; H, 5.4. Calc. for C26H40Cl2FeP2Pt: C, 42.4; H, 5.5. IR
3078w, 2932s, 2858m, 1697w, 1636m, 1448m, 1262m, 1111s, 1074s, 1024s, 882m, 808s, 714w, 685w, 514m, 498m, 418m. 1H NMR (CD2Cl2, 400 MHz): δ 4.08 (m, 8H), 2.40 (m, 4H), 2.33–2.08 (m, 8H), 2.05–1.77 (m, 12H), 1.78–1.55 (m, 8H). 13C{1H} NMR (CD2Cl2, 75 MHz): 88.9 (s), 69.1 (s), 68.2 (s), 32.4 (m), 29.5–28.5 (m), 27.9–26.0 (m), 25.4 (s). 31P{1H} NMR (CD2Cl2, 162 MHz): δ 17.4 (s, 195Pt satellites d, 1JP,Pt = 3426). MS (ESI(−), CH2Cl2, CH3OH) m/z 771.0 [M + Cl]−.
cis-Dichlorido-[1,1′-bis(5-(phospholano)pentyl)ferrocene-κ2P,P′]-platinum(II) (7b). Yield 85.6 mg (56%). Decomp. >160 °C. Found C, 43.6; H, 6.1. Calc. for C28H44Cl2FeP2Pt: C, 44.0; H, 5.8. IR
3084w, 2964m, 2828m, 2854m, 1456m, 1419w, 1263s, 1096s, 1024s, 804s, 689w, 661w, 498m. 1H NMR (CD2Cl2, 400 MHz): δ 4.04 (m, 4H), 3.96 (m, 4H), 2.31 (m, 4H), 2.12–1.96 (m, 8H), 1.93–1.76 (m, 12H), 1.68 (m, 4H), 1.47–1.58 (m, 8H). 13C{1H} NMR (CD2Cl2, 75 MHz): δ 90.4 (s), 69.8 (s), 68.8 (s), 31.4–30.8 (m), 29.8 (s), 29.7–29.0 (m), 28.1–27.5 (m), 27.2–26.5 (m), 26.3–25.6 (m). 31P{1H} NMR (CD2Cl2, 162 MHz): δ 15.7 (s, 195Pt satellites d, 1JP,Pt = 3452). MS (ESI(−), CH2Cl2, CH3CN) m/z 800.1 [M + Cl]−. MS (ESI(+), CH2Cl2, CH3CN) m/z 764.2 [M − Cl]+.
Crystals suitable for X-ray diffraction could be obtained through vapour diffusion of diethyl ether into a solution of 8b in dichloromethane over two weeks at 4 °C.
[1,1′-Bis(4-(phospholano)butyl)ferrocene-κ2P,P′]gold(I) chloride (8a). Yield 136.3 mg (97%). Decomp. >170 °C. Found C, 44.2; H, 5.4. Calc. for C28H44AuClFeP2: C, 44.4; H, 5.7. IR
2928m, 2865m, 1653m, 1631s, 1456m, 1262s, 803m, 694w, 516m, 490m, 417w. 1H NMR (CD2Cl2, 400 MHz): δ 3.99 (m, 8H), 2.44–2.22 (m, 8H), 2.10–1.75 (m, 16H), 1.75–1.56 (m, 8H). 13C{1H} NMR (CD2Cl2, 75 MHz): δ 88.9 (s), 68.9 (s), 68.0 (s), 32.2 (bs), 29.1 (s), 28.5 (m), 27.1 (s), 27.0–26.2 (m). 31P{1H} NMR (CD2Cl2, 162 MHz): δ 34.3 (s). MS (ESI(+), CH2Cl2, CH3CN) m/z 667.1 [M − Cl]+.
[1,1′-Bis(5-(phospholano)pentyl)ferrocene-κ2P,P′]gold(I) chloride (8b). Yield 143.3 mg (98%). Decomp. >175 °C. Found C, 45.7; H, 6.1. Calc. for C28H44AuClFeP2: C, 46.0; H, 6.1. IR
3084w, 2963m, 2927m, 2854m, 1437w, 1419w, 1263s, 1099s, 1023s, 803s, 701w, 668m, 488m. 1H NMR (CD2Cl2, 400 MHz): δ 4.02 (m, 4H), 3.92 (m, 4H), 2.42 (m, 4H), 2.31 (m, 4H), 2.05 (m, 4H), 1.96–1.77 (m, 12H), 1.70 (m, 4H), 1.60 (m, 4H), 1.50 (m, 4H). 13C{1H} NMR (CD2Cl2, 75 MHz): δ 90.0 (s), 69.1 (s), 67.5 (s), 31.7–31.4 (m), 31.4 (s). 29.5–28.9 (m), 28.8 (s), 27.4 (bs), 27.2 (m). 31P{1H} NMR (CD2Cl2, 162 MHz): δ 37.4 (s). MS (ESI(+), CH2Cl2, CH3CN) m/z 695.2 [M − Cl]+. MS (ESI(−), CH2Cl2, CH3CN) m/z 765.4 [M + Cl]−.
trans-Carbonylchlorido-[1,1′-bis(5-(phospholano)butyl)ferrocene-κ2P,P′]rhodium(I) (9a). 9a could not be isolated in pure form and was therefore only characterised by 31P{1H} NMR and ESI-MS. 31P{1H} NMR (CD2Cl2, 162 MHz): 26.9 (d, 1JP,Rh = 115.6). MS (ESI(+), CH2Cl2, CH3CN) m/z 636.2 [M]+.
trans-Carbonylchlorido-[1,1′-bis(5-(phospholano)pentyl)ferrocene-κ2P,P′]rhodium(I) (9b). Yield 109.2 mg (82%). Decomp. >125 °C. Found C, 52.6; H, 6.4. Calc. for C29H44ClFeOP2Rh: C, 52.4; H, 6.7. IR
3079w, 2928s, 2854m, 1952w (CO), 1656m, 1627m, 1558w, 1506m, 1498w, 1405m, 1263m, 1111m, 1053m, 1022m, 853w, 802m, 658w, 517w. 1H NMR (CD2Cl2, 400 MHz): δ 3.91(m, 4H), 3.87 (m, 4H), 2.30 (m, 4H), 2.20 (m, 4H), 1.96–1.66 (m, 20H), 1.62–1.41 (m, 8H). 13C{1H} NMR (CD2Cl2, 75 MHz): δ 188.9–187.3 (m, CO), 89.7 (s), 68.6 (s), 66.8 (s), 31.5–30.8 (m), 29.5–29.0 (m), 28.0 (s), 27.0 (bs), 26.3–25.5 (m). 31P{1H} NMR (CD2Cl2, 162 MHz): 26.5 (d, 1JP,Rh = 115.4). MS (ESI(+), CH2Cl2, CH3CN) m/z 629.2 [M − Cl]+.
Crystal structure determinations
Data for compounds 1b, 6a, 7a, 7b, 8b and 9b were collected on an Oxford Diffraction CCD Xcalibur-S diffractometer (data reduction with CrysAlis Pro,25 including the program SCALE 3 ABSPACK26 for empirical absorption correction) by using MoKα irradiation (λ = 71.073 pm) and ω-scan rotation. Structures were solved with the SIR tool.27 Refinement was performed with SHELXL97.28 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined by constrained methods using the riding model. The refinement was carried out with the least-squares method on F2. Final R indices were calculated as follows: R1 = ∑||Fo|−|Fc||/∑|Fo| and wR2 = {∑[w(Fo2 − Fc2)2]/∑w(Fo2)2}1/2. Figures were drawn with ORTEP.29 CCDC 1410621 (1b), 1410623 (6a), 1410618 (7a), 1410619 (7b), 1410620 (8b), 1410622 (9b) contain the supplementary crystallographic data for this paper. Table 2 summarises the details of the data collection, structure solution and refinement for 7ab, 8b, and 9b.| Compound | 7a | 7b | 8b | 9b |
|---|---|---|---|---|
| Formula | C26H40Cl2FeP2Pt | C28H44Cl2FeP2Pt | C28H44AuClFeP2 | C29H44ClFeOP2Rh |
| Formula weight | 736.36 | 764.41 | 730.84 | 664.79 |
| Temperature [K] | 130(2) | 130(2) | 130(2) | 130(2) |
| Crystal system | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group |
P |
P21/n | P212121 | P21/n |
| Unit cell | ||||
| a [pm] | 885.8(5) | 1986.9(5) | 1229.6(5) | 1077.3(5) |
| b [pm] | 1049.0(5) | 777.4(5) | 1352.6(5) | 1218.3(5) |
| c [pm] | 1540.0(5) | 2021.6(5) | 1697.4(5) | 2216.6(5) |
| α [°] | 72.024(5) | 90 | 90 | 90 |
| β [°] | 85.158(5) | 112.348(5) | 90 | 97.477(5) |
| γ [°] | 76.207(5) | 90 | 90 | 90 |
| V [Å3] | 1322(1) | 2888(2) | 2823(2) | 2885(2) |
| Z | 2 | 4 | 4 | 4 |
| ρ calcd. [g cm−3] | 1.850 | 1.758 | 1.720 | 1.531 |
| μ [mm−1] | 6.172 | 5.653 | 5.928 | 1.300 |
| F(000) | 728 | 1520 | 1456 | 1376 |
| Crystal size [mm] | 0.15 × 0.10 × 0.10 | 0.10 × 0.10 × 0.02 | 0.25 × 0.15 × 0.04 | 0.30 × 0.30 × 0.30 |
| Θ Min–ΘMax [°] | 2.09–26.37 | 2.45–25.35 | 2.54–26.37 | 2.54–36.32 |
| Collected reflections | 10398 |
13600 |
24262 |
64818 |
| Indep. reflections (Rint) | 5383 (0.0287) | 5292 (0.0510) | 5759 (0.0386) | 13973 (0.0359) |
| Completeness to ΘMax | 100.0% | 99.9% | 99.9% | 100.0% |
| Restraints/parameters | 0/289 | 0/307 | 0/298 | 0/492 |
| GooF (F2) | 0.917 | 1.045 | 1.050 | 1.041 |
| R 1, wR2 (I > 2σ(I)) | 0.0229, 0.0452 | 0.0520, 0.0931 | 0.0268, 0.0603 | 0.0315, 0.0668 |
| R 1, wR2 (all data) | 0.0319, 0.0478 | 0.0795, 0.1027 | 0.0301, 0.0615 | 0.0427, 0.0714 |
| Residual electron density [e Å−3] | 0.768/−1.375 | 1.063 /−0.679 | 0.866/−0.366 | 1.632/−0.725 |
| Absolute structure parameter | — | — | −0.010(5) | — |
Hydroformylation catalysis
Catalytic reactions were performed with an AMTEC SPR16 generation 2 slurry-phase reactor system and stock solutions in toluene. Typical procedure: 15 mL steel autoclaves were purged with argon. 0.25 μmol [{RhCl(CO)2}2] and 1.25 μmol 5b were transferred into the reactor and stirred for 30 min at rt. Toluene (to achieve a bulk volume of 3 mL) and 0.5 mmol 1-octene were added. The autoclave was then pressurised with 40 bar of CO/H2 (1:1) and heated to 50 °C. The mixture was stirred vigorously for 18 h. Analysis of the reaction products was performed by GC-MS with a GCMS-QP2010 spectrometer from Shimadzu.
Computational studies
For all quantum chemical calculations, geometry optimisations as well as vibrational analysis, the programme suite ORCA30 version 3.0.2 was used. DFT calculations were performed using the B3LYP functional31 with the basis set Def2-TZVP.32 The convergency criterion was set to 10−8 Hartree in all calculations. The vibrational analyses were carried out numerically. All structures were verified as local energy minima by only positive eigenvalues of the Hessian matrix. The simulation of the solvent environment was performed using the COSMO model33 for which the dielectric constant ε was set to 8.93 (CH2Cl2). The picture was generated with PYMOL.34Acknowledgements
We gratefully acknowledge financial support from the Fonds der Chemischen Industrie (FCI, doctoral grant to A. Schmied), the Studienstiftung des deutschen Volkes (doctoral grant for T.G.) and the Graduate School Leipzig School of Natural Sciences – Building with Molecules and Nano-objects (BuildMoNa). We thank S. Märcker, M. Röckl, B. Fritzsche, and R. Zäbe for measurements of IR and NMR spectra and M. Schmied-Tobies for help with GC-MS analysis.Notes and references
- (a) P. Štěpnička, Ferrocenes. Ligands, Materials and Biomolecules, John Wiley & Sons Ltd., West Sussex, 2008 Search PubMed; (b) A. Togni and T. Hayashi, Ferrocenes. Homogeneous Catalysis. Organic Synthesis. Material Science, VCH, New York, Weinheim, 1995 Search PubMed.
- (a) G. Bandoli and A. Dolmella, Coord. Chem. Rev., 2000, 209, 161–196 CrossRef CAS; (b) I. R. Butler, W. R. Cullen, T.-J. Kim, S. J. Rettig and J. Trotter, Organometallics, 1985, 4, 972–980 CrossRef CAS; (c) C. Nataro, A. N. Campbell, M. A. Ferguson, C. D. Incarvito and A. L. Rheingold, J. Organomet. Chem., 2003, 673, 47–55 CrossRef CAS; (d) G. Pilloni, B. Longato and B. Corain, J. Organomet. Chem., 1991, 420, 57–65 CrossRef CAS; (e) P. J. Stang, B. Olenyuk, J. Fan and A. M. Arif, Organometallics, 1996, 15, 904–908 CrossRef CAS; (f) D. A. Clemente, G. Pilloni, B. Corain, B. Longato and M. Tiripicchio-Camellini, Inorg. Chim. Acta, 1986, 115, L9 CrossRef CAS; (g) O. J. Curnow, T. Jones, T.-J. Kim and F. W. B. Einstein, Organometallics, 1985, 4, 346–351 CrossRef.
- (a) A. Acosta-Ramírez, M. Muñoz-Hernández, W. D. Jones and J. J. García, Organometallics, 2007, 26, 5766–5769 CrossRef; (b) F. N. Blanco, L. E. Hagopian, W. R. McNamara, J. A. Golen, A. L. Rheingold and C. Nataro, Organometallics, 2006, 25, 4292–4300 CrossRef CAS; (c) A. Fihri, P. Meunier and J.-C. Hierso, Coord. Chem. Rev., 2007, 251, 2017–2055 CrossRef CAS; (d) L. E. Hagopian, A. N. Campbell, J. A. Golen, A. L. Rheingold and C. Nataro, J. Organomet. Chem., 2006, 691, 4890–4900 CrossRef CAS; (e) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158–163 CrossRef CAS; (f) S. L. Kahn, M. K. Breheney, S. L. Martinak, S. M. Fosbenner, A. R. Seibert, W. S. Kassel, W. G. Dougherty and C. Nataro, Organometallics, 2009, 28, 2119–2126 CrossRef CAS; (g) J.-F. Ma and Y. Yamamoto, J. Organomet. Chem., 1999, 574, 148–154 CrossRef CAS; (h) J. H. L. Ong, C. Nataro, J. A. Golen and A. L. Rheingold, Organometallics, 2003, 22, 5027–5032 CrossRef CAS; (i) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313–328 RSC; (j) P. Štěpnička, I. Císařová and J. Schulz, Organometallics, 2011, 30, 4393–4403 CrossRef; (k) M. Trivedi, S. K. Ujjain, G. Singh, A. Kumar, S. K. Dubey and N. P. Rath, J. Organomet. Chem., 2014, 772–773, 202–209 CrossRef CAS; (l) M. A. Zuideveld, B. H. G. Swennenhuis, M. D. K. Boele, Y. Guari, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2002, 2308 RSC; (m) M. J. Burk, T. P. Harper, J. R. Lee and C. Kalberg, Tetrahedron Lett., 1994, 35, 4963–4966 CrossRef CAS.
- (a) U. Berens, M. J. Burk, A. Gerlach and W. Hems, Angew. Chem., Int. Ed., 2000, 39, 1981–1983 CrossRef CAS; (b) R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 CrossRef PubMed; (c) D. Heller, H.-J. Drexler, J. You, W. Baumann, K. Drauz, H.-P. Krimmer and A. Börner, Chem. – Eur. J., 2002, 8, 5196–5203 CrossRef CAS PubMed; (d) J. You, H.-J. Drexler, S. Zhang, C. Fischer and D. Heller, Angew. Chem., Int. Ed., 2003, 42, 913–916 CrossRef CAS PubMed; (e) M. J. Burk and M. F. Gross, Tetrahedron Lett., 1994, 35, 9363–9366 CrossRef CAS.
- C. L. Mandell, S. S. Kleinbach, W. G. Dougherty, W. S. Kassel and C. Nataro, Inorg. Chem., 2010, 49, 9718–9727 CrossRef CAS PubMed.
- (a) N. J. Goodwin, W. Henderson and B. K. Nicholson, Inorg. Chim. Acta, 1999, 295, 18–24 CrossRef CAS; (b) J. J. Adams, O. J. Curnow, G. Huttner, S. J. Smail and M. M. Turnbull, J. Organomet. Chem., 1999, 577, 44–57 CrossRef CAS; (c) T. Höcher, S. Blaurock and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2002, 1174–1180 CrossRef; (d) Y. Yamamoto, T. Tanase, I. Mori and Y. Nakamura, J. Chem. Soc., Dalton Trans., 1994, 3191–3192 RSC; (e) D. M. Bensley and E. Mintz, J. Organomet. Chem., 1988, 353, 93–102 CrossRef CAS; (f) O. J. Curnow, G. Huttner, S. J. Smail and M. M. Turnbull, J. Organomet. Chem., 1996, 524, 267–270 CrossRef CAS; (g) T. Kauffmann, J. Ennen, H. Lhotak, A. Rensing, S. Fritz and A. Woltermann, Angew. Chem., Int. Ed. Engl., 1980, 19, 328–329 CrossRef; (h) N. E. Schore and B. E. LaBelle, J. Organomet. Chem., 1981, 46, 2306–2310 CrossRef CAS.
- R. T. Kettenbach, W. Bonrath and H. Butenschön, Chem. Ber., 1993, 126, 1657–1669 CrossRef CAS.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed.
- M. Streitberger, A. Schmied and E. Hey-Hawkins, Inorg. Chem., 2014, 53, 6794–6804 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, UK, 2nd edn, 2009 Search PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle and R. Papp, Dalton Trans., 2009, 202–209 RSC.
- J. Bhatt, B. M. Fung and K. M. Nicholas, Liq. Cryst., 1992, 12, 263–272 CrossRef CAS.
- (a) D. W. Allen, I. W. Nowell and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1985, 2505–2508 RSC; (b) D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51–54 RSC.
- M. A. Guino-o, A. H. Zureick, N. F. Blank, B. J. Anderson, T. W. Chapp, Y. Kim, D. S. Glueck and A. L. Rheingold, Organometallics, 2012, 31, 6900–6910 CrossRef CAS.
- R. A. Baber, M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle, A. Haynes, G. L. Williams and R. Papp, Organometallics, 2007, 26, 713–725 CrossRef CAS.
- (a) M. C. Gimeno, A. Laguna, S. Cristina and P. G. Jones, Inorg. Chem., 1993, 32, 5926–5932 CrossRef CAS; (b) A. Houlton, M. D. P. Mingos, D. M. Murphy, D. J. Williams, L.-T. Phang and A. T. S. Hor, J. Chem. Soc., Dalton Trans., 1993, 3629–3630 RSC; (c) P. Štěpnička and I. Císařová, J. Organomet. Chem., 2012, 716, 110–119 CrossRef.
- G. A. Bowmaker, J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai and A. H. White, J. Chem. Soc., Dalton Trans., 1987, 1089–1097 RSC.
- S. Herzog and J. Dehnert, Z. Chem., 1964, 4, 1–11 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, U.K., 3rd edn, 1988 Search PubMed.
- (a) G. Grüttner and E. Krause, Chem. Ber., 1916, 49, 437–444 CrossRef; (b) G. Grüttner and M. Wiernik, Chem. Ber., 1915, 48, 1473–1486 CrossRef.
- G. R. Bourret, P. J. G. Goulet and R. B. Lennox, Chem. Mater., 2011, 23, 4954–4959 CrossRef CAS.
- J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521–6528 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- CrysAlis Pro, Oxford Diffraction Ltd., Oxfordshire, U.K., 2010 Search PubMed.
- SCALE3 ABSPACK, Oxford Diffraction Ltd., Oxfordshire, U.K., 2010 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, SHELX97: Programs for Crystal Structure Analysis (Release 97–2), Universität Göttingen, Göttingen, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 73–78 CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 785–789 CrossRef CAS.
- (a) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 2571–2577 CrossRef CAS; (b) F. Weigand and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 3297–3305 RSC.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 2235–2245 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC Search PubMed.
Footnotes |
| † Dedicated to Professor Christian Robl on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystal structures of 1b and 6a, crystallographic data, and cyclic voltammograms of 5ab, 7ab, 8ab and 9b. CCDC 1410618–1410623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02567b |
| This journal is © The Royal Society of Chemistry 2015 |