
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MeL2Zn2(μ-1,6-Ph2-N6) – a building block for new hexazene complexes†‡
S.
Gondzik
,
C.
Wölper
,
R.
Haack
,
G.
Jansen
and
S.
Schulz
*
University of Duisburg-Essen, Universitätsstr. 5-7, S07 S03 C30, 45117 Essen, Germany. Fax: Int (+)201 1833830; Tel: Int (+)201 1834635E-mail: stephan.schulz@uni-due.de
First published on 5th August 2015
Abstract
The zinc hexazene complex MeL2Zn2(μ-1,6-Ph2-N6) 1 (MeL = HC[C(Me)N(2,4,6-Me3C6H2)]2) is a suitable hexazene transfer reagent in reactions with main group metal and transition metal complexes containing M–Me units. The reactions proceed with elimination of MeLZnMe and the resulting complexes were characterized by NMR and IR spectroscopy and single crystal X-ray diffraction (5, 8). Quantum chemical calculations were performed to investigate the electronic structure of 5′ and 8′ in more detail and to identify the absorption bands of the hexazene unit.
Introduction
Metal complexes containing a metal-bonded hexazene dianion, [RNNNNNNR]2−, which have recently received growing interest, are still rare and only eight complexes have been reported to date (Scheme 1). They are typically synthesized by one-electron reductive coupling reactions of two organic azide molecules RN3, in particular the sterically demanding 1-azidoadamantane (AdN3), with iron(I)1,2 and magnesium(I)3,4 complexes. These strongly reducing metal complexes are typically kinetically stabilized by monoanionic, N,N′-chelating β-diketiminato or guanidinato groups. In addition, an iron(II) hexazene complex was recently prepared by reaction of AdN3 with a less reducing bis(alkoxide) iron(II) complex.5 | ||
| Scheme 1 Structurally characterized metal hexazene complexes. | ||
According to density functional theory studies, the reaction proceeds via initial formation of a dimeric iron azide reaction intermediate, in which the azide group is monoreduced to a bridging azide radical and the iron centers are oxidized to the formal +3 oxidation state. The iron azide then reacts to the hexazene complex.5 In contrast, the reaction of the Fe(I) complex (MeL′Fe)2N2 (MeL′ = HC[C(Me)N(2,6-i-Pr2C6H3)]2) with AdN3 proceeds through a different reaction mechanism as can be already derived from the observation, that this reaction is strongly solvent-dependant. The hexazene-bridged diiron(II) complex [(MeL′Fe)2(μ–η2:η2-PhN6Ad)] was obtained in high yields in non-coordinating solvents,1 while the tetrazene complex MeLFe(AdNNNNAd) containing a monoanionic radical tetrazene ligand (AdNNNNAd˙−) and a Fe(II) metal center were obtained in the presence of coordinating solvents such as t-Bu-pyridine.6 QM/MM computations showed that the formation of [(MeL′Fe)2(μ–η2:η2-PhN6Ad)] could not result from simple radical dimerization of two molecules of LMeFe(N3Ad˙) since there is not a significant thermodynamic driving force for this dimerization.7 Instead, the diiron organoazide intermediate (MeLFe)2(μ-N3Ad) is initially formed, which gives the hexazene complex upon addition of a second AdN3 molecule. In contrast, displacement of N2 in LMeFeNNFeLMe by a coordinating solvent yields a monometallic complex, which reacts with AdN3 with formation of non-isolable LMeFeII(N3Ad˙−) containing a radical monoanion, which eliminates N2 with subsequent formation of the Fe(III) imido complex LMeFe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NAd. The imido complex then reacts in a dipolar 1,3-cycloaddition reaction to the tetrazene complex as was previously observed in reactions of other low-valent metal complexes with organic azides.8–11 The reactions most likely involve the initial formation of a transient metal imide (MNR) intermediate, which subsequently undergoes a dipolar 1,3-cycloaddition reaction with a second equivalent of the azide RN3 as was shown for isolated metal-imido complexes.12–15
NAd. The imido complex then reacts in a dipolar 1,3-cycloaddition reaction to the tetrazene complex as was previously observed in reactions of other low-valent metal complexes with organic azides.8–11 The reactions most likely involve the initial formation of a transient metal imide (MNR) intermediate, which subsequently undergoes a dipolar 1,3-cycloaddition reaction with a second equivalent of the azide RN3 as was shown for isolated metal-imido complexes.12–15
We recently reported on reactions of the one-electron reducing Zn(I) agent (MeL)2Zn2 (MeL = HC[C(Me)N(2,4,6-Me3C6H2)]2) with organic azides including the sterically less demanding PhN3, yielding the first Zn(II) hexazene complex [(MeLZn)2(μ–η2:η2-PhN6Ph)] 1.16 The solid state structure of 1 is almost identical to those of other hexazene complexes and the bonding situation in the dianionic hexazene unit is best described with a central N–N single bond and two allyl-like, monoanionic N3 moieties with delocalized π-electrons. In remarkable contrast to neutral hexaazadienes,17–21 metal hexazene complexes are thermally very stable due to the sterically demanding organic substituents. To the best of our knowledge, the chemical reactivity of hexazene complexes has not been investigated in detail, to date. Herein we show that 1 with the sterically less demanding Ph-substituted hexazene moiety is a powerful hexazene transfer reagent in reactions with transition metal and main group metal complexes. Moreover, quantum chemical calculations were performed to identify the N6 vibrations in the resulting complexes.
Results and discussion
Reactions of 1 with equimolar amounts of Me2Zn, Me3Al and MeLi (Scheme 2) in C6D6 occurred with elimination of MeLZnMe and subsequent formation of the heteroleptic hexazene complex 2 and the heterobimetallic hexazene complexes 3 and 4. Organozinc complexes are long known transfer reagents and their capability to serve as starting reagents for the synthesis of heterometallic complexes has recently been shown.22 | ||
| Scheme 2 Synthesis of the heteroleptic hexazene complex 2 and heterobimetallic hexazene complexes 3 and 4 by metal exchange reactions of [(MeLZn)2(μ–η2:η2-N6Ph2)] 1 with equimolar amounts of different metal alkyl complexes (MeLi, Me2Zn, Me3Al). | ||
The formation of the homobimetallic hexazene complex [(MeLZn)(μ–η2:η2-N6Ph2)(ZnMe)] 2 and the heterobimetallic hexazene complexes [(MeLZn)(μ–η2:η2-N6Ph2)(AlMe2)] 3 and [(MeLZn)(μ–η2:η2-N6Ph2)(Li)] 4 was confirmed by in situ1H NMR spectroscopy. Since 2 and 3 show a comparable solubility as the by-product MeLZnMe, numerous attempts to separate 2 and 3 from MeLZnMe by fractional crystallisation from different organic solvents failed. Therefore, pure samples of the heteroleptic complexes 2 and 3 could not be isolated. In contrast, pure 4 was isolated by filtration since it is almost insoluble in organic solvents.
The in situ1H NMR spectra of 2 and 3 (Fig. 1) showed the expected resonances of the by-product MeLZnMe as well as a set of resonances of the MeL substituent, the Ph groups of the hexazene moiety and the metal-Me substituent in a relative intensity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (2) and 1:2:2 (3), respectively, clearly proving the substitution of only one MeLZn unit of 1 by a ZnMe (2) and AlMe2 (3) unit. Since 4 is completely insoluble in organic solvents, an in situ1H NMR spectrum of the reaction of 1 and one equivalent of MeLi only showed the resonances of resulting MeLZnMe. Unfortunately, crystals of 2–4 suitable for single crystal X-ray diffraction studies were not obtained.
:2:1 (2) and 1:2:2 (3), respectively, clearly proving the substitution of only one MeLZn unit of 1 by a ZnMe (2) and AlMe2 (3) unit. Since 4 is completely insoluble in organic solvents, an in situ1H NMR spectrum of the reaction of 1 and one equivalent of MeLi only showed the resonances of resulting MeLZnMe. Unfortunately, crystals of 2–4 suitable for single crystal X-ray diffraction studies were not obtained.
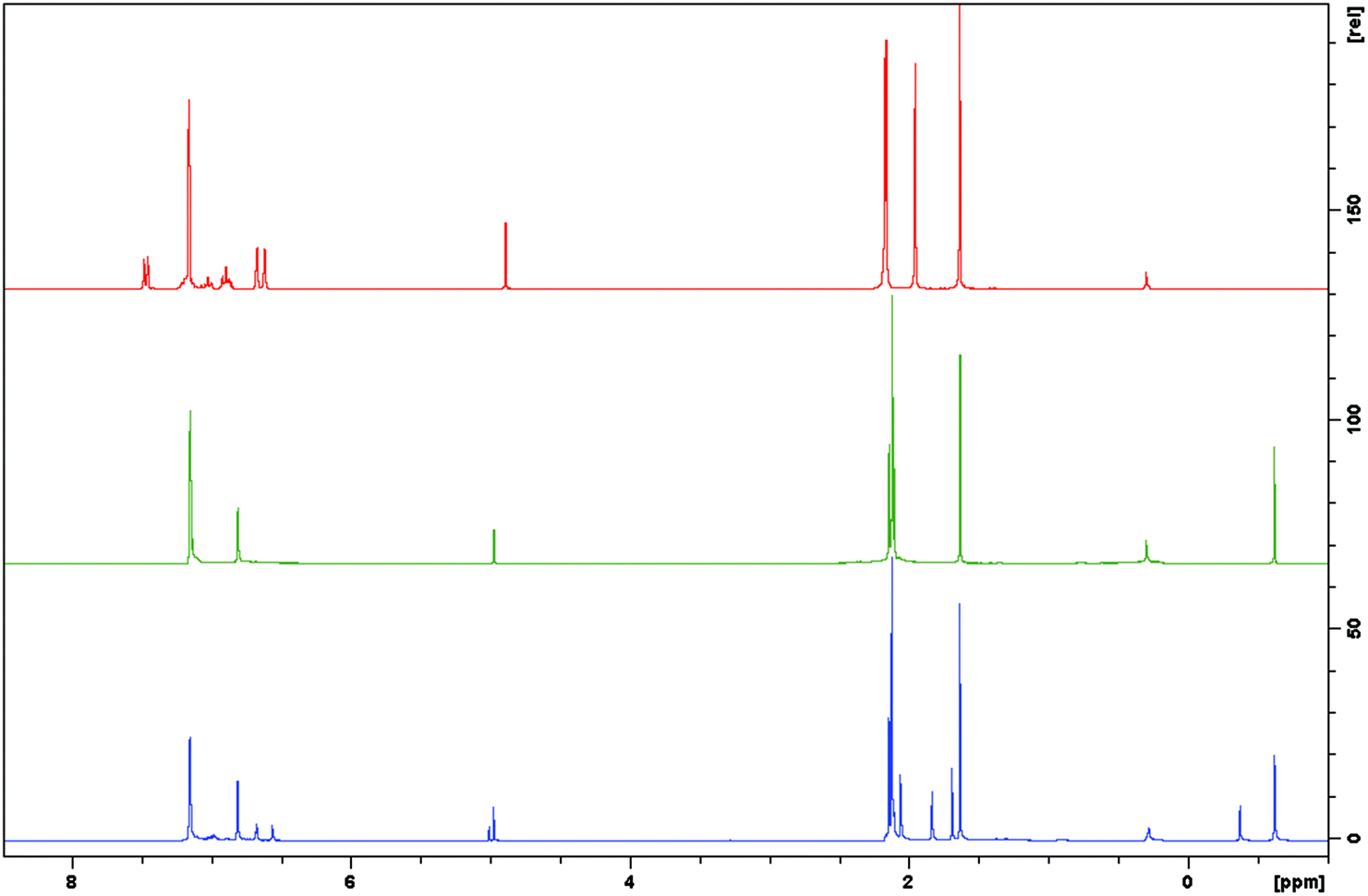 | ||
| Fig. 1 1H NMR spectra of 1 (top), MeLZnMe (middle) and the reaction of 1 and Me2Zn (bottom), demonstrating the substitution of only one MeLZn moiety by a MeZn unit. | ||
Detailed time-dependant in situ1H NMR spectroscopic studies showed, that 2 slowly rearranged in solution into the corresponding homoleptic complexes [(MeLZn)2(μ–η2:η2-PhN6Ph)] 1 and [MeZn)2(μ–η2:η2-PhN6Ph)] 5 and after five days, the formation of 1 and 5, which crystallized in the NMR tube as deep red crystals due to its limited solubility, was complete. The resulting 1H NMR spectrum only showed resonances of 1 and MeLZnMe. As a consequence, we investigated the substitution of both MeLZn moieties in 1 by reaction with two equivalents of Me2Zn and Me3Al (Scheme 3), respectively.
 | ||
| Scheme 3 Synthesis of homobimetallic hexazene complexes 5 and 6 by metal exchange reactions of 1 with two equivalents of Me-substituted metal complexes and the donor-stabilized complex 8. | ||
The formation of two equivalents of MeLZnMe in the resulting reaction mixtures was clearly confirmed by in situ1H NMR spectroscopy. In addition, one set of resonances due to the AlMe2 (6) groups and the Ph groups of the hexazene unit in a relative intensity of 1:1 are visible, pointing to the formation of the homobimetallic hexazene complex [Me2Al)2(μ–η2:η2-PhN6Ph)] 6. Since 5 is insoluble in non-coordinating deuterated solvents (benzene, toluene, fluorobenzene) even at elevated temperatures, it immediately formed a red crystalline powder in the NMR tube and as a consequence, NMR data could not be obtained in these solvents. In contrast, 5 dissolved easily in coordinating solvents such as thf-d8 with a color change to yellow. The 1H NMR spectrum of the resulting, most likely thf-coordinated, complex show the expected resonances of the Zn–Me and Ph groups in a relative intensity of 1:1. In the 13C NMR spectrum the Zn–Me3 signal was not visible as is often observed for Zn–Me compounds.
The transmetalation reactions occurred stepwise as was proven by in situ1H NMR spectroscopy for the reaction of 1 with Me2Zn (Fig. 2). Addition of one equivalent of Me2Zn to 1 yielded 2, which then reacted with a second equivalent of Me2Zn with formation of 5 and MeLZnMe, respectively. 5 was also obtained from the reaction of 2 with one equivalent of Me2Zn.
 | ||
| Fig. 2 1H NMR spectra of 1 +one equivalent of Me2Zn (top), +two equivalents of Me2Zn (middle), 2 with one equivalent of Me2Zn (bottom), clearly demonstrating the stepwise substitution reaction of the MeLZn moieties. | ||
In remarkable contrast to the reactions of 1 with two equivalents of ZnMe2 and AlMe3, its reaction with two equivalents of MeLi was completely different. 1 reacts with the first equivalent MeLi with formation of MeLZnMe and 4, which then reacts with a second equivalent of MeLi to MeLLi and 7 (Scheme 3). No further reaction was observed with a third equivalent of MeLi.
Single crystals of 5 were obtained from a solution of 1 in C6D6, which was carefully stratified with a solution of ZnMe2, upon standing (without stirring) at ambient temperature for 6 hours. An X-ray diffraction study revealed the formation of the homobimetallic hexazene complex [(MeZn)2(μ–η2:η2-PhN6Ph)] 5 (Fig. 3), which crystallizes in the monoclinic space group C2/c with the molecule placed on a centre of inversion.
 | ||
| Fig. 3 Solid state structure of 5 (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. | ||
The N4Zn metallacycle is almost perfectly planar (r.m.s. deviation from the least-squares plane 0.056(6) Å). The central N6Ph2 hexazene unit adopts a bridging position and is connected to two three-coordinated Zn atoms. The Zn–C bond lengths (Zn–C 1.953(3) Å) are in the range of known zinc methyl complexes.23–25 The different N–N bond lengths within the hexazene unit give insight into the electron localization within the complex. The central N3–N3′ bond (1.378(5) Å) is significantly elongated compared to the almost identical N1–N2 (1.305(3) Å) and N2–N3 (1.299(3) Å) bonds, hence the hexazene unit is best described as a dianionic ligand with a central N3–N3′ single bond and two allyl-like, monoanionic N3 moieties containing delocalized π-electrons between N1, N2 and N3. The N1–N2–N3 bond angle of 118.4(2)° and the N–N bond lengths within the hexazene unit are comparable to those previously observed in hexazene complexes (Table 1).1–5,16 In contrast to as-described hexazene complexes, the π-electrons with the five-membered ring of dianionic tetrazene complexes MN4 (Ni, Fe, Al, Cr) are not delocalized. According to singly crystal X-ray diffraction studies and computational studies, the tetrazene complexes have a central N–N double bond and two N–N single bonds.6,26–28
| N1–N2 | N2–N3 | N3–N3′ | N1–N2–N3 | |
|---|---|---|---|---|
| a This work; L = CH3CN; Pipiso = [(DipN)2C(cis-NC5H8Me2-2,6)]–. | ||||
| (MeL′Fe)2N6Ada21 |
1.304 | 1.305 | 1.411 | 117.107 |
| (t-BuL′Fe)2N6Ad21 |
1.307 | 1.318 | 1.408 | 116.485 |
| [(Pipiso)Fe]2N6Ada22 |
1.300 | 1.309 | 1.415 | 117.487 |
| [(RO)2Fe]2N6Ada25 |
1.295 | 1.308 | 1.399 | 117.592 |
| (MeL′Mg)2N6Ada23 |
1.301 | 1.314 | 1.410 | 117.827 |
| (t-BuL′Mg)2N6Ada24 |
1.304 | 1.312 | 1.419 | 117.984 |
| (MeLMg)2N6Ada24 |
1.303 | 1.310 | 1.424 | 117.853 |
| (MeLZn)2N6Ph2116 |
1.304 | 1.302 | 1.403 | 117.350 |
| (MeZn)2N6Ph25a |
1.305 | 1.299 | 1.377 | 118.374 |
| [Me(L)Zn]2N6Ph28a |
1.310 | 1.297 | 1.402 | 117.352 |
Intermolecular Zn⋯N interactions (2.275(2) Å) between adjacent hexazene complexes in 5, which result from the electron deficiency of the threefold-coordinated Zn atom, result in the formation of ladder-like one-dimensional polymeric structure (Fig. 4).
 | ||
| Fig. 4 Intermolecular interactions in 5 leading to ladder-type 1-dimensional chains in the solid state (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. | ||
The intermolecular Zn⋯N interactions can be disrupted in reactions with stronger Lewis bases as was exemplarily shown in the reaction of 5 with two equivalents of CH3CN, resulting in the formation of the Lewis base-coordinated hexazene complex (MeZn)2(μ–η2:η2-PhN6Ph)(CH3CN)28. Crystals of 5 were suspended in C6D6, yielding a deep red suspension, which immediately turned to a yellow solution after addition of CH3CN. Since the solubility of the resulting CH3CN-stabilized complex [(MeZn)2(μ–η2:η2-N6Ph2)(CH3CN)] 8 is significantly better than that of base-free 5, 1H and 13C NMR spectra could be recorded. 8 shows the expected resonances due to the metal–Me substituent, the Ph groups of the hexazene moiety and the coordinating acetonitrile in a relative intensity of 1:1:1. The NMR signal of the acetonitrile molecule (2.12 ppm) is significantly shifted to lower field compared to free CH3CN (0.58 ppm).
Single crystals of 8 were obtained from a concentrated solution in C6D6 after storage at +4 °C for 12 hours (Fig. 5). 8 crystallizes in the monoclinic space group P21/n with two molecules in the unit cell. The zinc atoms in 8 are slightly out of the plane of the N4Zn rings (r.m.s. deviation from the least-squares plane 0.142(3) Å). The N–N bond lengths (N1–N2 1.310(16) Å, N2–N3 1.2968(16) Å and N3–N3′ 1.402(2) Å) and the N1–N2–N3 bond angle of 117.35(11)° within the hexazene unit are comparable to those observed in 1 and 5.
 | ||
| Fig. 5 Solid state structure of 8 (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. | ||
The individual molecules of 8 form chains via CH⋯π interactions. It is a trifurcated interaction involving all three hydrogen atoms of the acetonitrile. The individual bond angles are comparably small (Table 2), however the whole group is nearly perpendicular to the plane of the phenyl ring (angle between best plane of the ring and C8–C9 vector: 83.4°). The neighboring molecules within the chain are related by inversion (Fig. 6). The chains are oriented along [−110] and in turn interconnected by CH⋯π interactions via screw-axis and glide-plane symmetry. Again a hydrogen of the acetonitrile is involved. Its interaction with the phenyl ring's π-systems shows the typical orientation of a strong interaction. The second non-classical hydrogen bond is formed between the phenyl ring (H5) and the π-electrons of the hexazene unit (represented by the N1–N2–N3–N3′ centroid). The values of its geometric parameters suggest a similar strength. The remaining voids in the packing are filled with benzene molecules.
 | ||
| Fig. 6 CH⋯π interactions in 8. | ||
| DH⋯Aa | Symmetry | H⋯A/Å | DH⋯A/° |
χb/° |
|---|---|---|---|---|
| a az: centroid of the hexazene unit as defined below. Ph: centroid of the phenyl ring. b χ is the angle between the best plane of the ring and the H/centroid vector. | ||||
| C9–H9a⋯Ph | −x + 1, −y, −z + 1 | 2.9976 | 101.27 | 72.7 |
| C9–H9b⋯Ph | −x + 1, −y, −z + 1 | 3.4053 | 77.34 | 63.4 |
| C9–H9c⋯Ph | −x + 1, −y, −z + 1 | 3.0481 | 98.11 | 76.1 |
| C9–H9b⋯Ph | −x + 3/2, y − 1/2, −z + 3/2 | 2.7031 | 152.87 | 88.2 |
| C5–H5⋯az | x − 1/2, −y + 1/2, z − 1/2 | 3.1145 | 133.98 | 68.9 |
Quantum chemical calculations
In order to identify the spectroscopic signatures of the hexazene unit, we have carried out density functional theory calculations with the BP86 functional29 including a third generation dispersion correction (DFT + D3)30,31 using a quadruple-zeta basis set32 for the free monomers of 5′ and 8′. In case of 5 this represents a drastic approximation since the bridging function of the zinc atoms connecting different monomers within the crystal thus is ignored. Geometry optimization leads to a C2h symmetrical structure for 5′ (Zn–N1 2.035, Zn–N3 2.001, N1–N2 1.306, N2–N3 1.308, N3–N3′ 1.394 Å, N1–N2–N3 116.6°) and to a structure symmetry Ci for 8′ (Zn–N1 2.062, Zn–N3 2.031, N1–N2 1.311, N2–N3 1.301, N3–N3′ 1.400 Å, N1–N2–N3 116.4°). The most important difference between the quantum chemical gas phase structure of 8′ and the corresponding crystal structure is a bending of the acetonitrile molecules towards the phenyl rings to form intramolecular CH–π bonds. According to a natural population analysis33 the total charge on the N6 unit is −1.80e for 5′ (−0.50e for N1, −0.38e for N3) and −1.73e for 8′ (−0.50e for N1, −0.34e for N3) while the charge on Zn amounts to 1.37e for 5′ and 1.42e for 8′, respectively.The most intense line above 1000 cm−1 in the experimental IR spectrum of 8 at 1262 cm−1 according to the DFT + D3 calculations has to be ascribed to an antisymmetric linear combination of N1–C (phenyl) and N1′–C′(phenyl′) stretching vibrations (with contributions from phenyl ring deformations and a slight N1–N2 stretch contribution), computed at 1269 cm−1. The second most intense peak at 1203 cm−1 is ascribed to the antisymmetric N1–N2/N1′–N2′ stretch vibration (with a contribution from C–C–Hortho bend of the phenyl rings), computed at 1201 cm−1. We assign the experimental line at 1341 cm−1 to the antisymmetric N2–N3/N2′–N3′ stretch vibration (with phenyl ring deformation contributions), which in the theoretical spectrum is located at 1352 cm−1. Note that the line at 1301 cm−1 corresponds to an in plane deformation of the phenyl rings, calculated at 1299 cm−1. Finally the first intense line below 1000 cm−1 at 943 cm−1 is assigned to an antisymmetric linear combination of the N1–N2–N3 and N1′–N2′–N3′ bending modes (again with contributions of in plane phenyl ring deformations), computed at the same frequency. These findings essentially agree with those for the Zn–hexazene complex 1.16
The assignment of the experimental IR spectrum of 5 is less unambiguous due to the fairly different structures of 5 in the crystal and 5′ in the gas phase: the intense line at 1258 cm−1 is probably the antisymmetric C–N1/C′–N1′ stretch vibration, computed at 1264 cm−1. However in the computed spectrum this line has a much lower intensity than that of the N2–N3/N2′–N3′ stretch vibration computed at 1294 cm−1, which seems to be absent in the experimental spectrum, possibly due to the involvement of the N2 atom in a long bond to the zinc atom of a neighbouring molecule. The two small peaks at 1198 cm−1 and 1343 cm−1 on the other hand are likely to be ascribed to the antisymmetric N1–N2/N1′–N2′ and N2–N3/N2′–N3′ stretch vibrations, computed at 1204 and 1347 cm−1, respectively. Again we find the antisymmetric N1–N2–N3/N1′–N2′–N3′ bending mode in the computed spectrum at 947 cm−1 as the most likely explanation for the small experimental peak at 954 cm−1. One may speculate that the intense absorption bands at 1079 and 1011 cm−1 are due to Zn–N stretch vibrations along the corresponding weak Zn–N bonds between two neighbouring molecules in the crystal of 5.
The most intense line at 1065 cm−1 in the experimental Raman spectrum of 5, which is also found in the spectrum of 8, according to our calculations corresponds to the symmetrical linear combination of the N1–N2–N3 and N1′–N2′–N3′ bending vibrations with some admixture of N3–N3′ stretch, computed at 1031 and 1028 cm−1, respectively. The neighbouring line at 1084 cm−1, the most prominent feature in the experimental spectrum of 8, however is missing in the spectrum of 5. Thus it is likely due to an acetonitrile ligand vibration, the best candidate of which is CH3-wagging mode. The latter is computed at 1021 cm−1 but might be strongly shifted due to the different environment of the methyl group in the crystal of 8. The intense Raman lines in the experimental spectra at 1295 (5) and 1289 cm−1 (8) were identified in our computations as the symmetric C–N1/C′–N1′ stretch vibrations, calculated at 1279 (5′) and 1290 cm−1 (8′), respectively. The symmetrical linear combination of the N2–N3 and N2′–N3′ stretch vibrations is computed to have frequencies of 1361 (5′) and 1370 cm−1 (8′), respectively, i.e., in a frequency range where in the experimental spectrum it is difficult to distinguish lines from noise. However, based on the computed frequencies of 1194 (5′) and 1192 cm−1 (8′), we assign the experimental Raman bands at 1211 (5) and 1209 cm−1 (8) to the symmetrical N1–N2/N1′–N2′ stretch vibration. The band at 938 cm−1 can be assigned with the help of the computed line position of 922 cm−1 to the symmetrical linear combination of acetonitrile C–C stretch vibrations, and the experimental bands at 895 (5) and 897 cm−1 (8) correspond to the N3–N3′ stretch vibration, found at 882 (5′) and 860 cm−1 (8′) in our calculations.
Conclusions
We demonstrated the promising potential of [(MeLZn)2(μ–η2:η2-PhN6Ph)] 1 to serve as building block for the synthesis of both hetero- and homobimetallic main group metal and transition metal hexazene complexes. Our particular reaction pathway offers a general access to metal hexazene complexes and doesn't requires any reduction potential of the metal fragment that should be incorporated into the hexazene complex, which is in remarkable contrast to the previously reported route using low-valent reducing reagents such as Fe(I), Mg(I) and Zn(I) complexes. Quantum chemical calculations at the DFT + D3 level yielded structures (5′, 8′), which were in good agreement with experimental ones for the central N6 unit and which allowed an assignment of the most prominent features related to the N6 unit in the IR and Raman spectra.Experimental
Synthetic procedures
All manipulations were performed in a glovebox under an Ar atmosphere or using standard Schlenk techniques. Solvents were carefully dried over Na/K and degassed prior to use. 1 was prepared according to literature method.161H (300 MHz) and 13C{1H} (75.5 MHz) NMR spectra (δ in ppm) were recorded using a Bruker Avance DPX-300 spectrometer and are referenced to the trace of the respective protonated solvent impurities present in the deuterobenzene (C6D5H, 1H: δ = 7.154; 13C: δ = 128.0) and deuterotetrahydrofuran (C4D8O, 1H: δ = 3.580; 13C: δ = 67.4). IR spectra were measured in an ALPHA-T FT-IR spectrometer equipped with a single reflection ATR sampling module. The spectrometer was placed in a glovebox so as to perform the FT-IR measurements in inert gas atmosphere. Raman spectra were measured 100 s with a Bruker Senterra Microscope at 785 nm and 20 mW. The microanalyses were performed at the elemental analysis laboratory of University of Duisburg-Essen.Yield: 43 mg (74.3%). – M.P.: 273 °C. – C35H39N8ZnLi (642.94): calcd C 65.32, H 6.07, N 17.42; found C 65.1, H 6.11, N 17.49. – IR: ν = 2962, 2913, 1605, 1549, 1516, 1452, 1395, 1351, 1323, 1258, 1199, 1085, 1012, 973, 858, 793, 758, 723, 693, 633, 491, 452, 390 cm−1. – 1H-NMR (300 MHz, C6D6, 25 °C): MeLZnMe: δ = −0.61 (s, 3H, Zn–CH3), 1.63 (s, 6H, CCH3), 2.12 (s, 12H, o-CCH3), 2.14 (s, 6H, p-CCH3), 4.97 (s, 1H, γ-CH), 6.81 (s, 4H, m-H).
Yield: 18 mg (86%). – M.P.: 217 °C (decomp.). – C14H16N6Zn2 (398): calcd C 42.21, H 4.02, N 21.10; found C 42.3, H 4.03, N 21.14. – IR: ν = 2962, 2906, 2819, 1595, 1485, 1398, 1343, 1257, 1197, 1079, 1010, 954, 861, 790, 724, 688, 661, 496, 398 cm−1. – Raman: ν = 359, 391, 946, 1150, 1286, 1578, 1746, 1810, 2130, 2349 cm−1. – 1H-NMR (300 MHz, C6D6, 25 °C): MeLZnMe: δ = −0.62 (s, 6H, Zn–CH3), 6.93 (m, 1H, Ph-p-H), 7.11 (m, 2H, Ph-m-H), 7.41 (m, 2H, Ph-o-H). The solubility of 5 in C6D6 and toluene-d8 was too low to obtain suitable NMR spectra. However, 5 is soluble in coordinating solvents such as thf-d8. 1H-NMR (300 MHz, thf-d8, 25 °C): δ = −0.55 (s, 3H, Zn–CH3), 7.01 (m, 2H, Ph-p-H), 7.29 (m, 4H, Ph-m-H), 7.51 (m, 4H, Ph-o-H). – 13C-NMR (75 MHz, thf-d8, 25 °C): δ = 119.38 (p-Ph), 124.60 (m-Ph), 129.89 (o-Ph), 150.31 (i-Ph).
Yield: 16 mg (84%). – M.P.: 224 °C. – C16H22N6Al2 (351.96): calcd C 54.55, H 6.25, N 23.87; found C 55.1, H 6.27, N 24.01. – IR: ν = 2962, 2914, 1594, 1554, 1485, 1453, 1397, 1344, 1257, 1197, 1087, 1012, 946, 859, 793, 755, 719, 690, 634, 495, 455, 392 cm−1. – 1H-NMR (300 MHz, C6D6, 25 °C): δ = −0.62 (s, 12H, Al-CH3), 6.88 (m, 4H, Ph-o-H), 7.26 (m, 4H, Ph-m-H), 7.46 (m, 2H, Ph-p-H). – 13C-NMR (75 MHz, C6D6, 25 °C): δ = −18.05 (Al-CH3), 124.46 (p-Ph), 132.19 (m-Ph), 133.31 (o-Ph), 152.85 (i-Ph).
Yield: 15 mg (53%). – M.P.: 326 °C. – C13H13N6ZnLi (324.94): calcd C 48.01, H 4.00, N 25.85; found C 48.1, H 4.03, N 25.91. – IR: ν = 2960, 1625, 1481, 1453, 1259, 1200, 1084, 1013, 981, 857, 792, 695, 582, 440 cm−1. – 1H-NMR (300 MHz, C6D6, 25 °C): MeLLi: δ = 1.69 (s, 6H, C–CH3), 2.04 (s, 12H, o-CCH3), 2.30 (s, 6H, p-CCH3), 4.79 (s, 1H, C–H), 6.93 (s, 4 H, Mes-H). MeLZnMe: δ = −0.61 (s, 3H, Zn–CH3), 1.63 (s, 6H, CCH3), 2.12 (s, 12H, o-CCH3), 2.14 (s, 6H, p-CCH3), 4.97 (s, 1H, γ-CH), 6.81 (s, 4H, m-H).
Yield: 13.3 mg (92.4%). – M.P.: 237 °C (decomp.). – C18H22N8Zn2 (481.17): calcd C 44.93, H 4.61, N 23.29; found C 45.02, H 4.60, N 23.42. – IR: ν = 3026, 2906, 2276, 1586, 1453, 1340, 1301, 1262, 1203, 1072, 999, 943, 896, 810, 755, 721, 667, 629, 547, 510, 493, 471, 442 cm−1. – Raman: ν = 222, 244, 356, 389, 946, 1032, 1152, 1286, 1325, 1570, 1734, 1807, 2134, 2349 cm−1. – 1H-NMR (300 MHz, C6D6, 25 °C): δ = −0.36 (s, 6H, Zn–CH3), 2.12 (s, 6 H, NCCH3), 7.00 (m, 4H, Ph-o-H), 7.28 (m, 4H, Ph-m-H), 7.76 (m, 2H, Ph-p-H). – 13C-NMR (75 MHz, C6D6, 25 °C): δ = −15.04 (Zn–CH3), 1.03 (NCCH3), 119.60 (NCCH3), 124.58 (p-Ph), 129.23 (m-Ph), 130.08 (o-Ph), 150.07 (i-Ph).
Crystallography
Crystallographic data of 5 and 8 were collected on a Bruker AXS SMART diffractometer (MoKα radiation, λ = 0.71073 Å) at 100(1) K. The structures were solved by Direct Methods (SHELXS-97)34 and refined anisotropically by full-matrix least-squares on F2 (SHELXL-2014).35,365 was refined as non-merohedral twin based on HKL5 data. Absorption corrections were performed semi-empirically from equivalent reflections on basis of multi-scans (Bruker AXS APEX2, TWINABS). Hydrogen atoms were refined using a riding model or rigid methyl groups.Acknowledgements
S.S. and G.J. gratefully acknowledge financial support by the University of Duisburg-Essen.Notes and references
- R. E. Cowley, J. Elhaïk, N. A. Eckert, W. W. Brennessel, E. Bill and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6074 CrossRef CAS PubMed.
- L. Fohlmeister and C. Jones, Aust. J. Chem., 2014, 67, 1011 CrossRef CAS.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., 2009, 121, 3017 ( Angew. Chem. Int. Ed. , 2009 , 48 , 2973 ) CrossRef PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938 CrossRef CAS PubMed.
- J. A. Bellow, P. D. Martin, R. L. Lord and S. Groysman, Inorg. Chem., 2013, 52, 12335 CrossRef CAS PubMed.
- R. E. Cowley, E. Bill, F. Neese, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2009, 48, 4828 CrossRef CAS PubMed.
- R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172 CrossRef CAS PubMed.
- C. Cui, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., 2000, 112, 4705 ( Angew. Chem., Int. Ed. , 2000 , 39 , 4531 ) CrossRef.
- N. J. Hardman and P. P. Power, Chem. Commun., 2001, 1184 RSC.
- H. Zhu, Z. Yang, J. Magull, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2005, 24, 6420 CrossRef CAS.
- M. T. Mock, C. V. Popescu, G. P. A. Yap, W. G. Dougherty and C. G. Riordan, Inorg. Chem., 2008, 47, 1889 CrossRef CAS PubMed.
- A. H. Obenhuber, T. L. Ganietti, X. Berrebi, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 2994 CrossRef CAS PubMed.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974 CrossRef CAS.
- A. M. Geer, C. Tejel, J. A. López and M. A. Ciriano, Angew. Chem., 2014, 126, 5720 ( Angew. Chem., Int. Ed. , 2014 , 53 , 5614 ) CrossRef PubMed.
- R. I. Michelman, R. G. Bergman and R. A. Andersen, Organometallics, 1993, 12, 2741 CrossRef CAS.
- S. Gondzik, S. Schulz, D. Bläser, C. Wölper, R. Haack and G. Jansen, Chem. Commun., 2014, 50, 927 RSC.
- F. R. Berson, The High Nitrogen Compounds, Wiley, New York, 1984 Search PubMed.
- O. Nuyken, C. Scherer, A. Baindl, A. R. Brenner, U. Dahn, R. Gärtner, S. Kaiser-Röhrich, R. Kollefrath, P. Matusche and B. Voit, Prog. Polym. Sci., 1997, 22, 93 CrossRef CAS.
- K. A. Hofmann and H. Hock, Chem. Ber., 1911, 44, 2946 CrossRef PubMed.
- D. Mackay, D. D. McIntyre and N. J. Taylor, J. Org. Chem., 1982, 47, 532 CrossRef CAS.
- C. M. Fitchett, C. Richardson and P. Steel, J. Org. Biomol. Chem., 2005, 3, 498 RSC.
- T. Kaczorowski, I. Justyniak, D. Prochowicz, K. Zelga, A. Kornowicz and J. Lewiński, Chem. – Eur. J., 2012, 18, 13460 CrossRef CAS PubMed; W. Clegg, E. Crosbie, S. H. Dale-Black, E. Hevia, G. W. Honeyman, A. R. Kennedy, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Organometallics, 2015, 34, 2580 CrossRef.
- S. Marks, T. K. Panda and P. W. Roesky, Dalton Trans., 2010, 39, 7230 RSC.
- M. S. Hill and P. B. Hitchcock, Dalton Trans., 2002, 4694 RSC.
- A. Kasani, R. McDonald and R. G. Cavell, Organometallics, 1999, 18, 3775 CrossRef CAS.
- S. W. Lee, G. A. Miller, C. F. Campana and W. C. Trogler, Inorg. Chem., 1988, 27, 1215 CrossRef CAS.
- H. P. Zhu, Y. Zhi, J. Magull, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2005, 24, 6420 CrossRef CAS.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Inorg. Chim. Acta, 2011, 369, 103 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- S. Grimme, WIREs Comput. Mol. Sci., 2011, 1, 211 CrossRef CAS PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, WIREs Comput. Mol. Sci., 2014, 4, 91 CrossRef CAS PubMed.
- (a) F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753 CrossRef CAS PubMed; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed; (b) A. E. Reed, L. A. Curtis and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-2014, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen (Germany), 2014. (see also CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed ).
- shelXle, A Qt GUI for SHELXL, C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof R. Boese on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data of 5 and 8; 1H (2–8), 13C (5, 6, 8), IR (2–8) and Raman spectra (5, 8); quantum chemical calculations of 5′ and 8′. CCDC 1408085 (5) and 1408086 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02423d |
| This journal is © The Royal Society of Chemistry 2015 |