
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antimalarial activity of ruthenium(II) and osmium(II) arene complexes with mono- and bidentate chloroquine analogue ligands†
Erik
Ekengard
a,
Lotta
Glans
a,
Irwin
Cassells
b,
Thibault
Fogeron
a,
Preshendren
Govender
b,
Tameryn
Stringer
b,
Prinessa
Chellan
b,
George C.
Lisensky
c,
William H.
Hersh
d,
Isa
Doverbratt
e,
Sven
Lidin
e,
Carmen
de Kock
f,
Peter J.
Smith
f,
Gregory S.
Smith
*b and
Ebbe
Nordlander
*a
aInorganic Chemistry Research Group, Chemical Physics, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: ebbe.nordlander@chemphys.lu.se
bDepartment of Chemistry, University of Cape Town, Private Bag, Rondebosch 7701, South Africa. E-mail: gregory.smith@uct.ac.za
cDepartment of Chemistry, Beloit College, 700 College St., Beloit, WI 53511, USA
dDepartment of Chemistry and Biochemistry, Queens College and the Graduate Center of the City University of New York, Queens, NY 11367-1597, USA
eCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Getingevägen 60, Box 124, SE-22100 Lund, Sweden
fDivision of Pharmacology, Department of Medicine, University of Cape Town Medical School, Observatory 7925, South Africa
First published on 16th September 2015
Abstract
Eight new ruthenium and five new osmium p-cymene half-sandwich complexes have been synthesized, characterized and evaluated for antimalarial activity. All complexes contain ligands that are based on a 4-chloroquinoline framework related to the antimalarial drug chloroquine. Ligands HL1–8 are salicylaldimine derivatives, where HL1 = N-(2-((2-hydroxyphenyl)methylimino)ethyl)-7-chloroquinolin-4-amine, and HL2–8 contain non-hydrogen substituents in the 3-position of the salicylaldimine ring, viz. F, Cl, Br, I, NO2, OMe and tBu for HL2–8, respectively. Ligand HL9 is also a salicylaldimine-containing ligand with substitutions in both 3- and 5-positions of the salicylaldimine moiety, i.e. N-(2-((2-hydroxy-3,5-di-tert-butylphenyl)methyl-imino)ethyl)-7-chloroquinolin-4-amine, while HL10 is N-(2-((1-methyl-1H-imidazol-2-yl)methylamino)ethyl)-7-chloroquinolin-4-amine) The half sandwich metal complexes that have been investigated are [Ru(η6-cym)(L1–8)Cl] (Ru-1–Ru-8, cym = p-cymene), [Os(η6-cym)(L1–3,5,7)Cl] (Os-1–Os-3, Os-5, and Os-7), [M(η6-cym)(HL9)Cl2] (M = Ru, Ru-HL9; M = Os, Os-HL9) and [M(η6-cym)(L10)Cl]Cl (M = Ru, Ru-10; M = Os, Os-10). In complexes Ru-1–Ru-8 and Ru-10, Os-1–Os-3, Os-5 and Os-7 and Os-10, the ligands were found to coordinate as bidentate N,O- and N,N-chelates, while in complexes Ru-HL9 and Os-HL9, monodentate coordination of the ligands through the quinoline nitrogen was established. The antimalarial activity of the new ligands and complexes was evaluated against chloroquine sensitive (NF54 and D10) and chloroquine resistant (Dd2) Plasmodium falciparum malaria parasite strains. Coordination of ruthenium and osmium arene moieties to the ligands resulted in lower antiplasmodial activities relative to the free ligands, but the resistance index is better for the ruthenium complexes compared to chloroquine. Overall, osmium complexes appeared to be less active than the corresponding ruthenium complexes.
Introduction
The development of organometallic complexes for medicinal purposes has progressed rapidly during the last decade.1–3 Although the development of anticancer agents is one of the largest research areas within the field, other applications, e.g. organometallics as antimalarial agents, have also generated scientific interest.4–6 Approximately half the world's population is at risk of contracting malaria and the disease kills around 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people per year. An African child dies from malaria every 45 seconds and widespread resistance to many of the available antimalarial drugs is undermining malaria control efforts.7 The recent reports of developing resistance against artemisinins have added renewed urgency to the development of new antimalarials.8 Chloroquine (CQ, Fig. 1) was the mainstay of malaria chemotherapy in large parts of the world for decades, but resistance has now prompted its replacement in most malaria-endemic countries.9 Ferroquine (FQ, Fig. 1), a ferrocene analogue of chloroquine, is a particularly successful organometallic antimalarial agent that has completed phase IIb clinical trials.10,11
000 people per year. An African child dies from malaria every 45 seconds and widespread resistance to many of the available antimalarial drugs is undermining malaria control efforts.7 The recent reports of developing resistance against artemisinins have added renewed urgency to the development of new antimalarials.8 Chloroquine (CQ, Fig. 1) was the mainstay of malaria chemotherapy in large parts of the world for decades, but resistance has now prompted its replacement in most malaria-endemic countries.9 Ferroquine (FQ, Fig. 1), a ferrocene analogue of chloroquine, is a particularly successful organometallic antimalarial agent that has completed phase IIb clinical trials.10,11
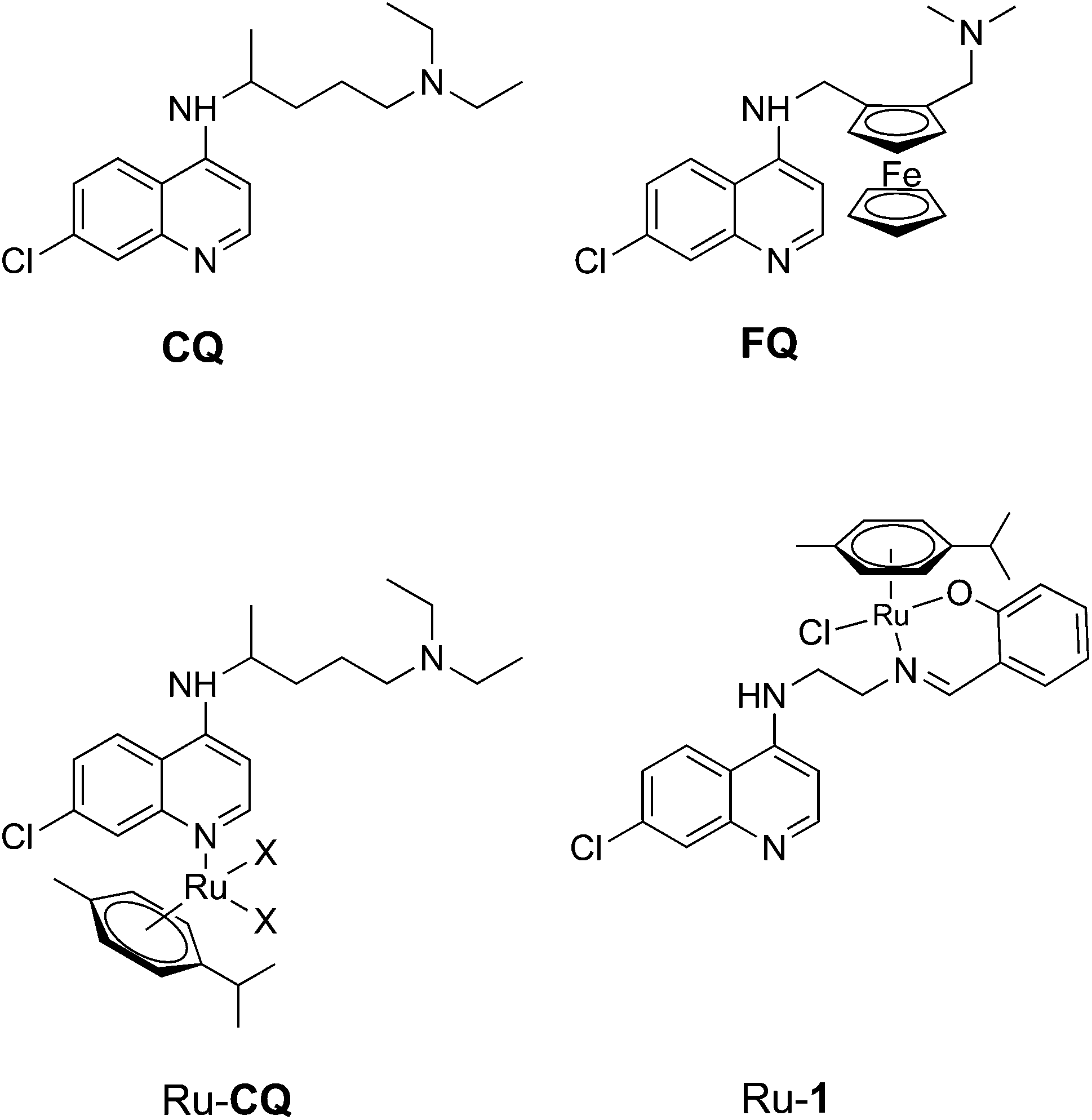 | ||
| Fig. 1 Chloroquine (CQ), Ferroquine (FQ) and reported Ru arene complexes with monodentate chloroquine or a bidentante chloroquine analogue as ligands; the chloroquine analogue ligand in Ru-1 is HL1 (cf. Scheme 1). | ||
Organometallic ruthenium arene complexes are a class of organometallic half-sandwich compounds that show both catalytic activity and promising anti-tumor activity, and such compounds have consequently been the subject of much research effort.12,13 It has been shown that ruthenium coordination complexes and organometallic ruthenium arene complexes of chloroquine (Fig. 1) exhibit increased activity towards chloroquine resistant malaria strains compared to chloroquine itself.14,15 In addition, the arene ruthenium chloroquine complexes showed anticancer activity against several tumor cell lines.16 In an earlier study, we reported half-sandwich ruthenium arene complexes with bidentate chloroquine analogue ligands.17 Relative to corresponding compounds with N,N-chelating ligands, a complex (Ru-1, Fig. 1) containing a chloroquine analogue with an N,O-chelating salicylaldiminate unit (HL1, Fig. 1) was shown to have the highest antimalarial activity.17
In comparison to the above-mentioned ruthenium half-sandwich complexes, arene complexes of the heavier congener osmium are much less studied, but recently osmium arene complexes with biological activity have been reported.18–25 Sadler and co-workers have synthesized and evaluated the anticancer activity of a large number of osmium arene complexes with bidentate ligands, including N,N-, N,O- and O,O-chelates.18–20 In addition, Keppler, Arion and co-workers have reported antiproliferative osmium arene complexes with ligands based on biologically active so-called paullones, containing an indolo-benzazepine that can be modified to create N,N-chelating ligands.23,24,26,27 Recently Pfeffer et al. have reported cyclometallated osmium arene complexes with C,N-chelating ligands that show activity against glioblastoma cells.25 In all three groups of compounds, the nature of the ligand and its mode of coordination have great impact on the anticancer activity of the complex.
Here, we wish to present the synthesis and characterization of a group of new ruthenium and osmium arene compounds with chloroquine analogue ligands similar to that used for Ru-1 (Fig. 1), which coordinate to the metal in bidentate or monodentate modes. This new set of complexes enables the study of (i) the effect of changing the metal from Ru to Os, (ii) the effect of the coordination mode of the ligand and (iii) the effect of the electronic properties of the substituent in para-position to the coordinating phenoxy group on antimalarial activity. The activity of the complexes was tested in vitro on Plasmodium falciparum malaria parasites; to our knowledge, these are the first osmium compounds tested for antimalarial activity.
Results and discussion
Synthesis
Considering the relatively good antimalarial activity exhibited by the ligand HL1 and its Ru p-cymene complex Ru-1 (vide supra and Fig. 1), we chose to explore this ligand framework for possible electronic influence exerted by substituent groups on the antimalarial activity of the ligands and their metal complexes. Thus, reaction of N1-(7-chloroquinolin-4-yl)ethane-1,2-diamine with the substituted salicylaldehydes S1–9 in refluxing ethanol yielded the Schiff bases HL1–9 in fair to excellent yields (51% to 99%) (Scheme 1), and these ligands were characterized by 1H-NMR and 13C NMR spectroscopy, IR spectroscopy and mass spectrometry (cf. Experimental section). Deprotonation of the (pro)ligands HL1–8 with Et3N, t-BuOK or NaH in dichloromethane followed by addition of [Ru(p-cymene)Cl2]2 gave the complexes [Ru(L)(p-cymene)Cl] (L = L1–8; Ru-1–Ru-8, respectively) in fair to excellent yields (53% to 93%), and in the corresponding reactions of HL1–3, HL5 and HL7 with [Os(p-cymene)Cl2]2, the analogous [Os(L)(p-cymene)Cl] complexes Os-1–Os-3, Os-5 and Os-7 were obtained in fair yields (36% to 72%, Scheme 2, cf. Experimental section).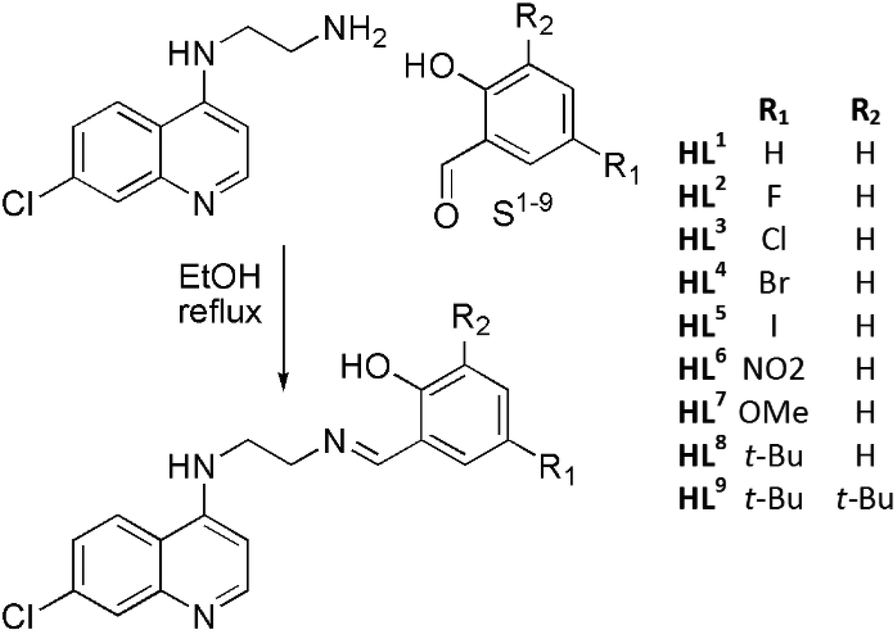 | ||
| Scheme 1 Synthesis of ligands HL1–9. | ||
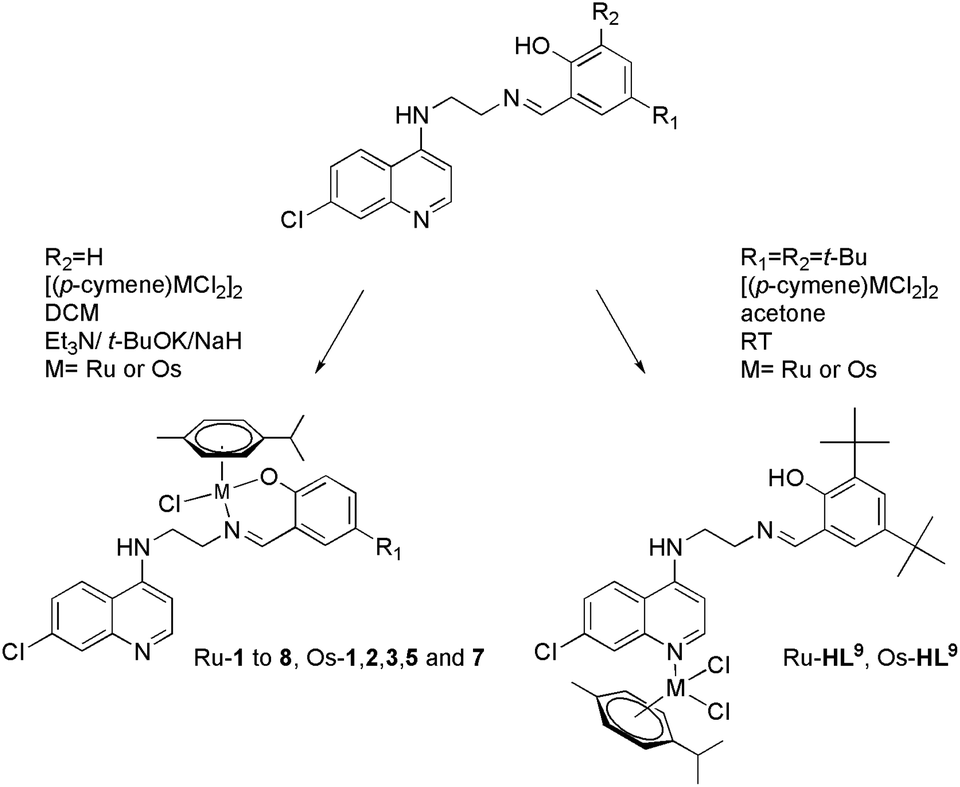 | ||
| Scheme 2 Synthesis of complexes Ru-1 to 8, Os-1, 2, 3, 5 and 7 and Ru-HL9 and Os-HL9, M = Ru or Os. | ||
The choices of base, workup and purification procedures were found to have only limited effect on the yields and purities of the complexes. Synthesis of the imidazolyl-containing ligand N-(2-((1-methyl-1H-imidazol-2-yl)methylamino)ethyl)-7-chloroquinolin-4-amine (L10) and its ruthenium p-cymene derivative, Ru-10, have been reported earlier.17 The complex [Os(p-cymene)(L10)Cl]Cl (Os-10) was synthesized by stirring [Os(p-cymene)Cl2]2 with ligand L10 in 1:2 molar ratio in acetone at room temperature (Scheme 3).
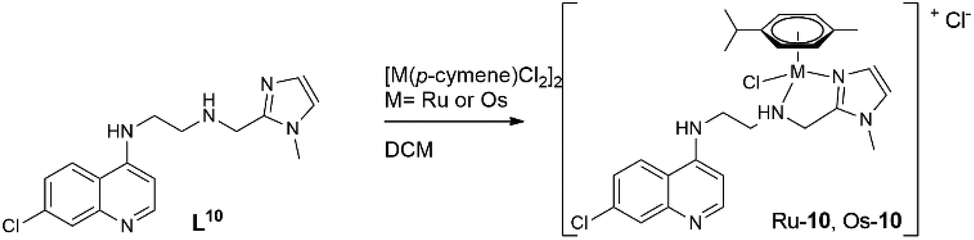 | ||
| Scheme 3 Synthesis of complex Ru-10 and Os-10. | ||
Under the conditions used to synthesize the above-mentioned half-sandwich ruthenium and osmium complexes, the 2,5-di-tert-butyl substituted ligand HL9 failed to give analogous ruthenium/osmium complexes; instead, inseparable mixtures of products were obtained. However, reaction of HL9 with half an equivalent of metal dimer in the absence of base gave products whose 1H-NMR spectra and mass spectrometry data (vide infra) were consistent with complexes of formulation [Ru(p-cymene)(HL9)Cl2], Ru-HL9, and [Os(p-cymene)(HL9)Cl2], Os-HL9. An NMR spectroscopy scale reaction showed that reaction of HL1 with the osmium dimer under the same conditions did not yield a monodentate complex analogous to Os-HL9, but rather resulted in a mixture of species that could not be properly identified. This is most likely due to the relative availability of the hydroxyl group, which is sterically protected by the tert-butyl groups in HL9 but available for coordination in HL1, which in the absence of base leads to competing coordination reactions that result in a complex mixture of products, including coordination through the salicylaldimine moiety and the quinoline nitrogen and coordination to both coordination sites. There are no examples in the literature of osmium-arene complexes with salicylaldiminate ligands containing a tert-butyl substituent in the 2-position, ortho to the phenolic group, and the few examples of ruthenium complexes are reported to be unstable in solution.28–30
Characterization
Coordination of unsymmetrical bidentate ligands to the Ru(η6-p-cymene)Cl and Os(η6-p-cymene)Cl moieties induces chirality at the metal atom, which results in loss of two-fold symmetry of the p-cymene ring. Hence, the observation of the aromatic protons of the p-cymene as four separate doublets and the p-cymene isopropyl group as two doublets in the 1H-NMR spectra of Ru-1–Ru-8 is consistent with a bidentate coordination mode of the salicylaldimine ligand. An upfield shift of the imine proton of approximately 1.1 ppm and the disappearance of the phenolic proton resonance between 13.5 and 12.5 ppm upon coordination of the ruthenium arene moiety strongly indicates that the structures of Ru-1–Ru-8 are as shown in Scheme 2. The IR-spectra of complexes Ru-1–Ru-8 show the imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch as a shoulder at 1617–1632 cm−1 and the phenolic C–O stretch at 1315–1330 cm−1, compared to 1640–1650 cm−1 and 1250 cm−1 in the free ligands. The distinct new vibration frequencies of the coordinated imine and phenoxylate moieties, as well as the disappearance of an O–H band around 3200 cm−1, further support the proposed structures of complexes Ru-1–Ru-8. The ESI mass spectra of these complexes all show molecular peaks ([M + H]+) (cf. Experimental section) as well as peaks corresponding to ([M − Cl]+) i.e. loss of a chloride ligand.
N stretch as a shoulder at 1617–1632 cm−1 and the phenolic C–O stretch at 1315–1330 cm−1, compared to 1640–1650 cm−1 and 1250 cm−1 in the free ligands. The distinct new vibration frequencies of the coordinated imine and phenoxylate moieties, as well as the disappearance of an O–H band around 3200 cm−1, further support the proposed structures of complexes Ru-1–Ru-8. The ESI mass spectra of these complexes all show molecular peaks ([M + H]+) (cf. Experimental section) as well as peaks corresponding to ([M − Cl]+) i.e. loss of a chloride ligand.
The NMR and IR spectra for the osmium complexes Os-1–Os-3, Os-5 and Os-7 closely resemble those for the corresponding ruthenium congeners. Their 1H-NMR spectra show the same features as the spectra for the ruthenium complexes: diastereotopic splitting of the aromatic and isopropyl protons of the p-cymene, an upfield shift of the imine proton resonance and the absence of the phenolic proton resonance. The one noteworthy difference is that the aromatic protons on the p-cymene ring appear slightly downfield from their positions in the ruthenium complexes; between 5.8 to 5.4 ppm for the osmium complexes, as compared to 5.5 to 5.0 ppm in the ruthenium complexes. This difference is similar to that observed in the parent dimers, where the osmium dimer has two doublets at 5.74 and 5.55 ppm while the lighter congener displays the same pattern at 5.46 and 5.33 ppm. Similarly to the ruthenium analogues, a shift to lower frequency is noted for the CN vibration; from ∼1645 cm−1 in the free ligand to ∼1610 cm−1 in Os-1–Os-3, Os-5 and Os-7, indicative of coordination of the imine nitrogen to osmium. Mass spectral analysis of Os-1–Os-3, Os-5 and Os-7 revealed that for each complex the base peak corresponds to the metal complex after loss of a chlorido ligand ([M − Cl]+). A peak for the protonated complex ([M + H]+) was also observed.
For complexes Ru-HL9 and Os-HL9, the 1H-NMR signals corresponding to the p-cymene show that the complex retains its two-fold symmetry, indicating monodentate coordination of the ligands. Sánchez-Delgado and co-workers have shown that coordination of chloroquine to ruthenium in [Ru(arene)(CQ)Cl2], Ru-CQ (Fig. 1), occurs through the quinoline nitrogen.14 These authors argued that the largest variations in the 1H chemical shifts (Δδ) of a bound ligand with respect to the values for the free form are observed for the nuclei in close vicinity of the coordination site.15,31 In Ru-HL9 and Os-HL9, the proton signals of ligand HL9 shift to a varying degree upon coordination, and Δδ values and signal assignments are reported in the ESI.† The largest 1H Δδ value for Ru-HL9 and Os-HL9 is observed for H14, which is adjacent to the nitrogen atom in the heterocyclic ring. This corresponds well with the 1H Δδ values observed for the dichloro species [Ru(p-cymene)(CQ)Cl2].14 In addition, 1H Δδ values for all protons on quinoline are large, while none of the protons of the di-tert-butyl phenyl moiety are shifted. The 1H Δδ values strongly suggest that the coordination mode of the ligand in Ru-HL9 and Os-HL9 is monodentate through the quinoline nitrogen, as illustrated in Scheme 2.
The 1H-NMR and IR spectra of Os-10 correspond closely to those of the reported ruthenium analogue.17 The bidentate bonding of L10 in the ruthenium complex Ru-10 was confirmed by X-ray diffraction17 and all the data strongly support the proposed structure for Os-10.
The spectroscopic and mass spectrometric data for complexes Ru-1–Ru-8, Ru-HL9 and Ru-10 and Os-1–Os-3, Os-5, Os-7, Os-HL9 and Os-10 are in full agreement with the proposed structures as depicted in Schemes 1 and 2. The exact bonding mode of ligand HL9 in complexes Ru-HL9 and Os-HL9 is difficult to assign with certainty on the basis of available data, but the 1H Δδ values clearly indicate that coordination occurs through the quinoline nitrogen.
Single crystal X-ray diffraction
The proposed coordination mode of the ligands was confirmed for complexes Ru-1, Ru-3, Ru-5, Os-5 and Os-7 by single crystal X-ray diffraction studies. Single crystals of Ru-1, Ru-3 and Ru-5 were grown by slow evaporation of a dichloromethane–petroleum ether solution of the respective complexes while for Os-5 and Os-7, crystals were grown by vapor diffusion of diethyl ether into a solution of the appropriate complex in dichloromethane. Fig. 2–6 show the molecular structures of Ru-1, Ru-3, Ru-5, Os-5 and Os-7, and Tables 1 and 2 contain a selection of relevant bond lengths and angles. Relevant crystal data and structure refinement parameters are listed in Table 5 in the Experimental section.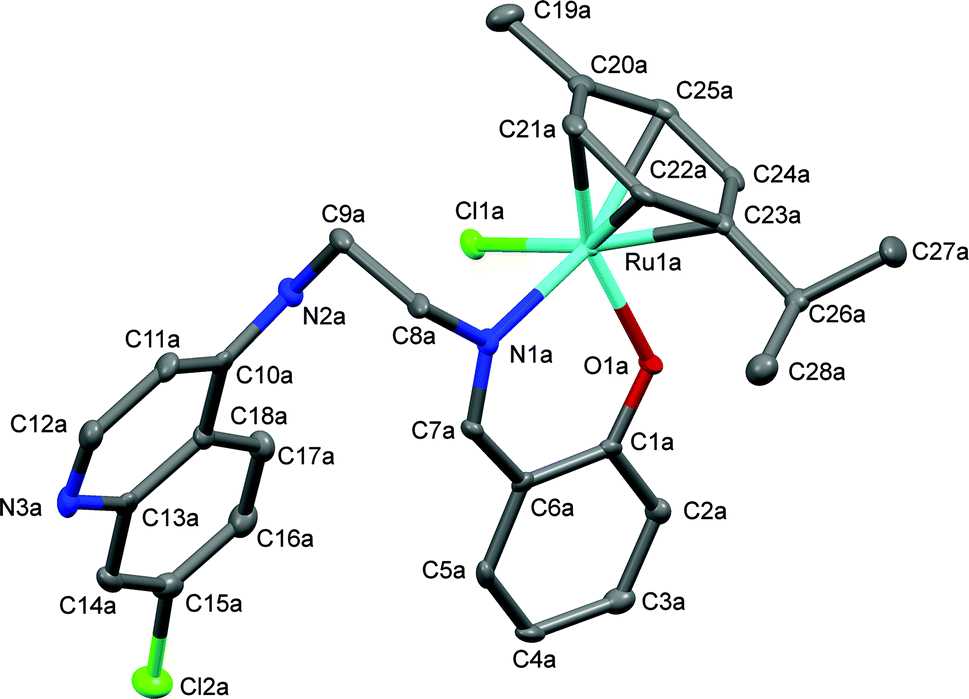 | ||
| Fig. 2 A Mercury plot of the molecular structure of complex [Ru(η6-cym)(L1)Cl], Ru-1. Hydrogen atoms have been omitted for the sake of clarity. Only one of the molecules in the asymmetric unit of the major polymorph is shown, see Fig. S-6 in the ESI† for the structure at the stacking fault. Thermal ellipsoids at the 50% level. | ||
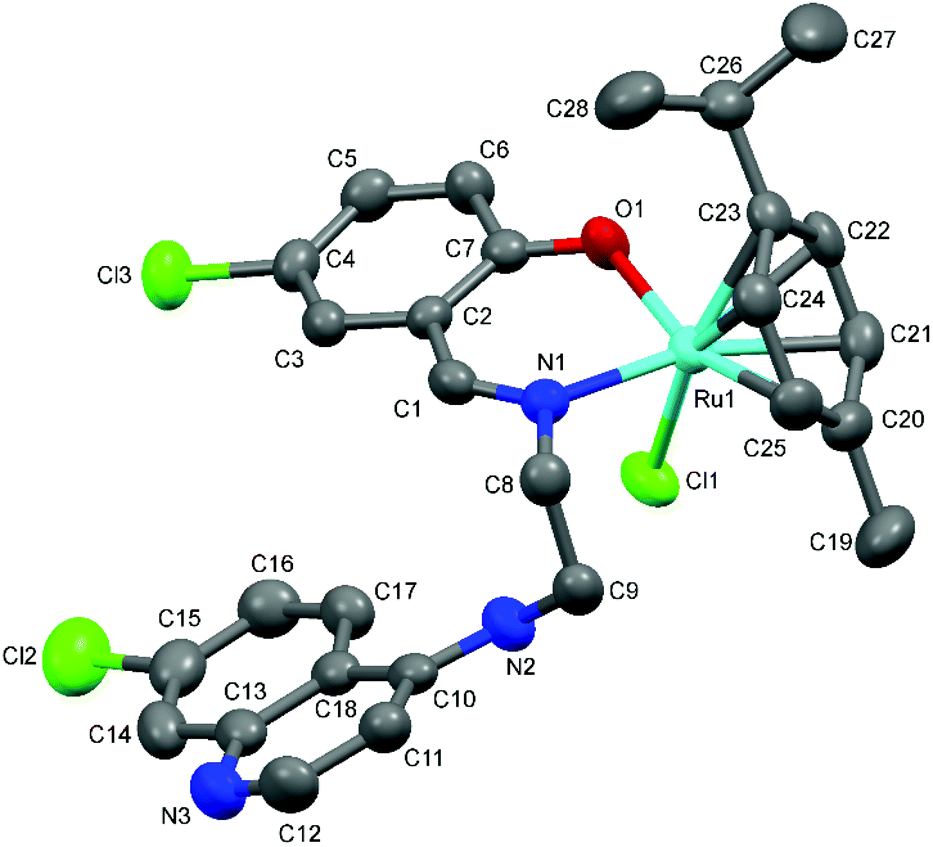 | ||
| Fig. 3 A Mercury plot of the molecular structure of complex [Ru(η6-cym)(L3)Cl] Ru-3. Hydrogen atoms have been omitted for the sake of clarity. Thermal ellipsoids at the 50% level. | ||
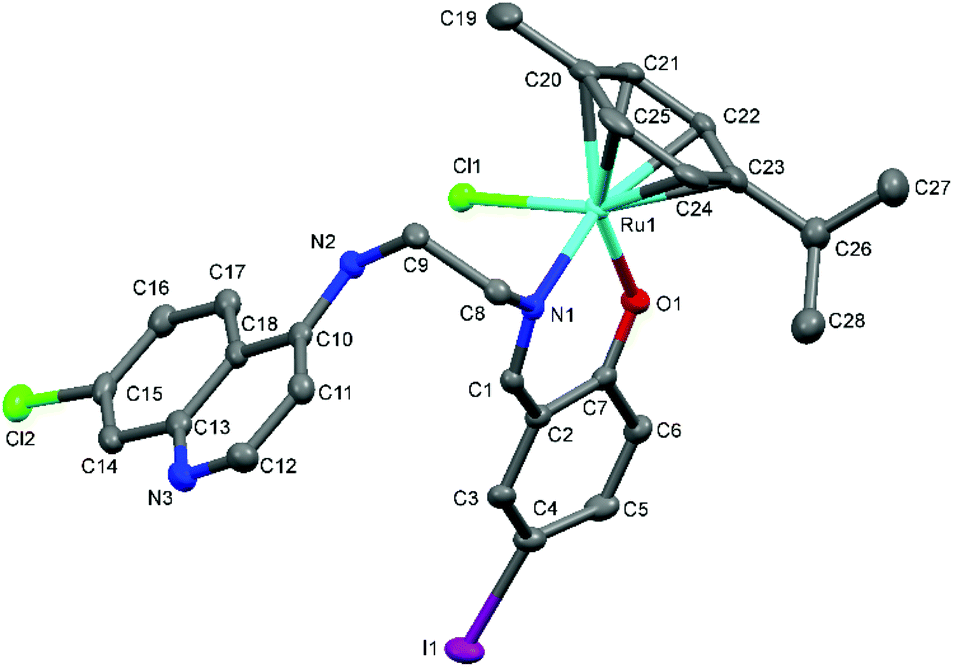 | ||
| Fig. 4 A Mercury plot of the molecular structure of complex [Ru(η6-cym)(L5)Cl] Ru-5. Hydrogen atoms have been omitted for the sake of clarity. Thermal ellipsoids at the 50% level. | ||
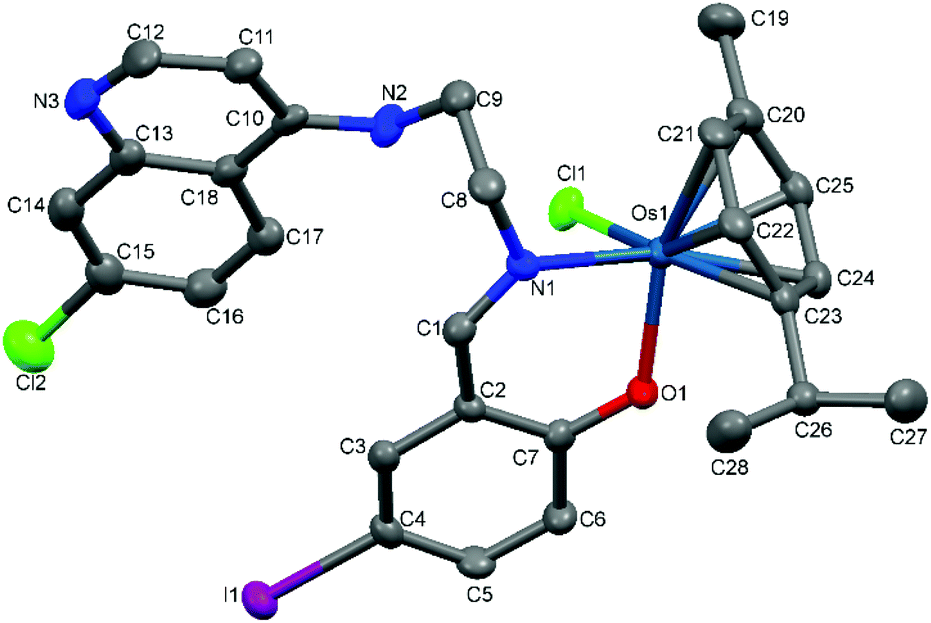 | ||
| Fig. 5 A Mercury plot of the molecular structure of complex [Os(η6-cym)(L5)Cl] Os-5. Hydrogen atoms have been omitted for the sake of clarity. Thermal ellipsoids at the 50% level. | ||
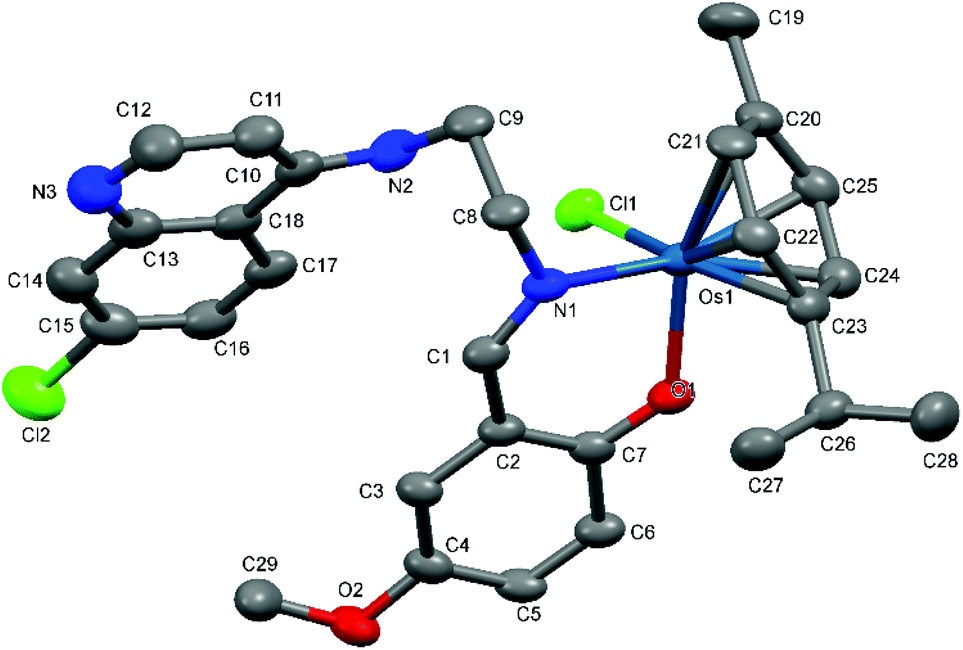 | ||
| Fig. 6 A Mercury plot of the molecular structure of complex [Os(η6-cym)(L7)Cl] Os-7. Hydrogen atoms have been omitted for the sake of clarity. Thermal ellipsoids at the 50% level. | ||
| Ru-1 | Ru-3 | Ru-5 | Ru-1 | Ru-3 | Ru-5 | ||
|---|---|---|---|---|---|---|---|
| Bond lengths (Å) | |||||||
| Ru1–Cycentroid | 1.672 | 1.668 | 1.680 | Ru1–O1 | 2.052(5) | 2.064(3) | 2.077(2) |
| Ru1–N1 | 2.105(5) | 2.098(4) | 2.100(2) | Ru1–Cl3 | 2.457(2) | 2.4502(12) | 2.4561(8) |
| Bond angles (°) | |||||||
| Cl1–Ru1–Cycentroid | 128.74 | 128.78 | 126.32 | O1–Ru1–Cl1 | 85.6(1) | 84.00(10) | 86.46(6) |
| O1–Ru1–Cycentroid | 123.54 | 123.60 | 124.17 | O1–Ru1–N1 | 88.1(2) | 87.33(15) | 87.43(8) |
| N1–Ru1–Cycentroid | 132.11 | 132.22 | 133.29 | Cl1–Ru1–N1 | 83.7(2) | 82.30(11) | 84.36(6) |
| Torsion angles (°) | |||||||
| C8–C9–N2–C10 | 73.9(7) | −84.2(4) | −73.9(3) | C1–N1–C8–C9 | −99.6(6) | 98.7(4) | 98.8(3) |
| Os-5 | Os-7 | Os-5 | Os-7 | ||
|---|---|---|---|---|---|
| Bond lengths (Å) | |||||
| Os1–Cycentroid | 1.656 | 1.659 | Os1–O1 | 2.059(2) | 2.064(3) |
| Os1–N1 | 2.103(3) | 2.098(4) | Os1–Cl1 | 2.4310(9) | 2.4502(12) |
| Bond angles (°) | |||||
| Cl1–Os1–Cycentroid | 129.37 | 130.37 | O1–Os1–Cl1 | 83.76(8) | 84.00(10) |
| O1–Os1–Cycentroid | 124.48 | 124.13 | O1–Os1–N1 | 87.45(10) | 87.33(15) |
| N1–Os1–Cycentroid | 132.73 | 132.50 | Cl1–Os1–N1 | 82.91(8) | 82.30(11) |
| Torsion angles (°) | |||||
| C8–C9–N2–C10 | 93.5(4) | 77.3(6) | C1–N1–C8–C9 | −96.2(4) | −96.5(5) |
When solving the structure of Ru-1 it was noticed that the refinement converges nicely for a solution that agrees well with expectations, but despite the good agreement between model and data (R1 = 4.37% Rw2 = 10.39%, GOOF = 1.84) there are some conspicuous residual electron densities that indicate that the solution is incomplete. Interestingly, these residuals appear in what seems to be random locations, neither close to the heavy atoms, nor in volumes where partially occupied solvent molecules may be expected. This seems to rule out local disorder and rather points to twinning or extended defects. It was noted that the five most prominent residual peaks fall into two categories, two having electron densities close to 4e− Å−3 and the remaining three having values less than half of this. Moreover, the two more pronounced residuals are both located at the same distance and direction (−0.785, 0.171) from the two independent Ru positions in the structure. In the same way, the three smaller peaks correspond to the translation of Cl positions. Translating the entire molecules along this vector in fact generates a packing without any short interatomic distances and with a packing efficiency comparable to that of the original structure. It thus seems probable that the residuals are generated by an alternative molecular packing in the ac-plane.
To test this hypothesis, a split model was introduced where copies of the two independent molecules in the unit cell appear at positions given by the vector between the main electron densities and the Ru atom positions. To avoid introducing a large number of new refinable parameters, the original model was fixed and the only refinable parameters (14) in the split model were the occupancies of the split positions and the rotations and translations of the major components to the new positions. These were allowed to refine independently and both occupancies came out very close to 5%, the translation vectors were essentially identical, and the rotations as expected were close to 0.
The refinement converged at R1 = 2.79%, Rw2 = 6.70%, GOOF = 1.20 with an essentially featureless electron difference density map. Despite the small number of refined parameters, this constitutes a major improvement on the original model and there is little doubt that this model correctly describes the local structure of the compound. Moreover, the same residual electron density in the same locations, corresponding to the same stacking fault, were observed in three other crystals of Ru-1, from two different batches of crystals. Fig. 2 shows the structure of one of the two non-equivalent molecules in the main morphology, see the ESI, Fig. S-6† for a representation of the two polymorphs in the crystal. The space group P21 would normally be associated with a single-enantiomer structure, but as discussed above, Ru-1 is stereogenic at the metal center but is present as the racemate, so the space group was surprising.32 Rather than finding a single enantiomer of Ru-1 in the asymmetric unit, both enantiomers were present, but with non-crystallographic inversion symmetry. That is, the two enantiomers were present in the asymmetric unit but with many minor conformational differences. Crystallization of a racemic mixture in a Sohnke space group was until recently thought to be extremely rare, with perhaps fewer than 50 examples.32,33 However, improved search methods have shown the number of these examples, referred to as kryptoracemates, to be a bit higher, and two recent publications describe 181 organic and 26 organometallic kryptoracemates.34,35 We now add Ru-1 to this list, although there is no particular reason that it crystallizes in this non-centrosymmetric space group. Bernal and Watkins have speculated that the crystallographic enantiomers in a non-Sohnke space group at room temperature could be converted to a non-crystallographic inversion pair at low temperature, but Ru-1 exhibits the same structure at room temperature as at 100 K.35 It is noteworthy that even though the structure in general is acentric the local structure at the stacking-fault is centrosymmetric.
Complexes Ru-3, Ru-5, Os-5 and Os-7 crystallize in centric monoclinic point groups, Ru-3, Ru-5 and Os-7 in P21/c and Os-5 in P21/n. They all refine well with standard procedures.
As suggested by the spectroscopic characterization, in all complexes the salicylaldiminato ligand (L1, L3, L5 and L7) coordinates to the metal in a bidentate manner via the phenolate oxygen and the imine nitrogen. The metal center adopts a typical ‘piano-stool’ conformation with the chlorido ligand and nitrogen and oxygen atoms of the salicylaldimine ring acting as the legs.
The metal is bound to the p-cymene ring in the expected η6 coordination mode. The bond lengths observed between the metal and the centroid of the p-cymene ring (M–Cycentroid) as well as the chlorido (M–Cl), oxygen (M–O) and nitrogen (M–N) atoms within each molecular structure agree with similar Ru(II) and Os(II)–arene complexes.17,36–40 For the osmium complexes Os-5 and Os-7, the bond angles formed between the centroid of the arene ring and the other atoms coordinated to the metal are in the range of 124–133° and bond angles close to 90° are noted for O1–Os1–Cl1, O1–Os1–N1 and Cl1–Os1–N1, in agreement with previously reported osmium arene structures.36–39 Torsion angles close to 90° are formed by the bonds of the imine and the aminoethyl chain (C8–C9–N2–C10 and C1–N1–C8–C9) as a consequence of the salicylaldehyde and quinoline rings sitting in planes that are almost parallel to each other.
The bond lengths and angles as well as the observed geometries around the metals in Os-5 and Os-7 are comparable Ru-1, Ru-3 and Ru-5 as well as other analogous [N,O]- and [N,N]-ruthenium arene complexes.17,40
Antimalarial activity
The antimalarial activity of ligands HL1–8 and complexes Ru-1–Ru-8, Os-1–Os-3, Os-5 and Os-7 was evaluated in vitro against chloroquine sensitive (CQS) NF54 and chloroquine resistant (CQR) Dd2 strains of Plasmodium falciparum (Table 3). The antimalarial activity of ligands L10 and HL1 as well as their ruthenium complexes Ru-10 and Ru-1 against the chloroquine sensitive (CQS) D10 and Dd2 strains has been reported previously17 and is included in Table 4 for comparison, together with the osmium analogues, Os-1 and Os-10, as well as HL9, Ru-HL9and Os-HL9. The relative biological activities are illustrated in Fig. 7.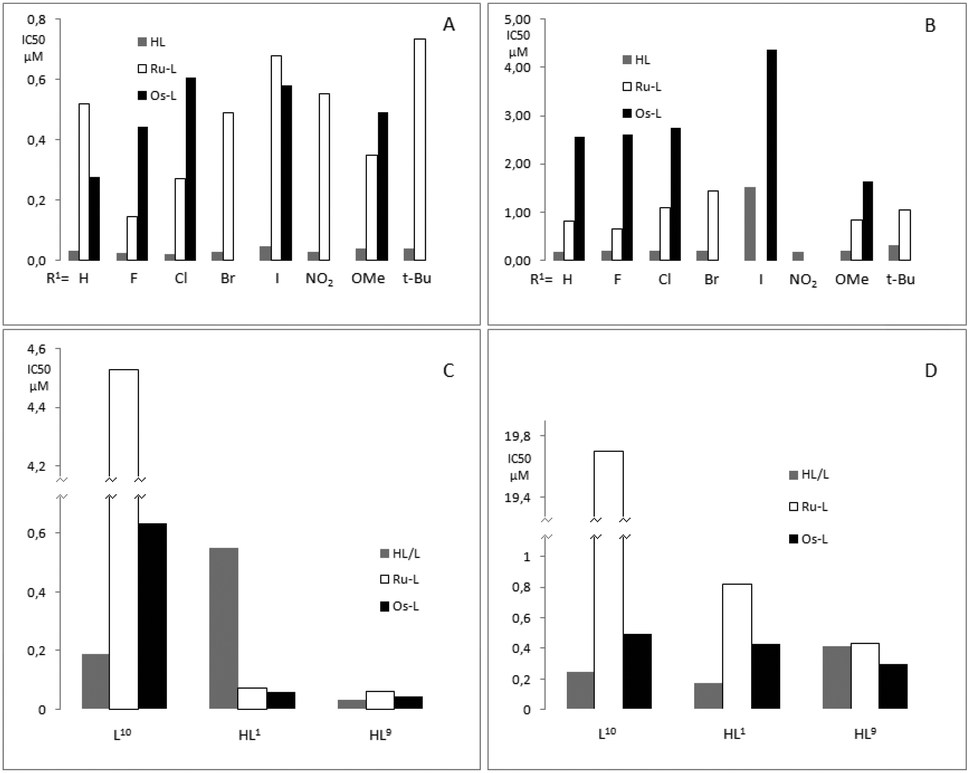 | ||
| Fig. 7 IC50 values for: (A) CQS, NF54 strain, HL1–8, Ru-1 to 8, and, Os-1,2,3,5 and 7, (B) CQR, Dd2 strain, HL1–8, Ru-1 to 8, and,Os-1,2,3,5 and 7, (C) CQS, D2 strain, L10, Ru-10 and Os-10, HL1, Ru-1 and Os-1 and HL9, Ru-HL9 and Os-HL9, (D) CQR, Dd2 strain, L10, Ru-10 and Os-10, HL1, Ru-1 and Os-1 and HL9, Ru-HL9 and Os-HL9. | ||
| Compound | R1 | NF54: IC50 (μM) | Dd2: IC50 (μM) | RI |
|---|---|---|---|---|
| a On another occasion no activity could be detected against Dd2 for HL1. b ND = no activity detected/IC50 > 30 μM, N/A = not applicable. | ||||
| HL1 | H | 0.032 ± 0.004 | 0.18 ± 0.02a | 5.5 |
| HL2 | F | 0.025 ± 0.007 | 0.2 ± 0.02 | 8 |
| HL3 | Cl | 0.021 ± 0.003 | 0.2 ± 0.04 | 9.6 |
| HL4 | Br | 0.027 ± 0.009 | 0.19 ± 0.03 | 7.2 |
| HL5 | I | 0.046 ± 0.006 | 1.52 ± 0.3 | 32.9 |
| HL6 | NO2 | 0.028 ± 0.008 | 0.18 ± 0.009 | 6.5 |
| HL7 | OMe | 0.04 ± 0.002 | 0.2 ± 0.03 | 5.2 |
| HL8 | t-Bu | 0.041 ± 0.002 | 0.31 ± 0.03 | 7.8 |
| Ru-1 | H | 0.52 ± 0.09 | 0.82 ± 0.05 | 1.6 |
| Ru-2 | F | 0.14 ± 0.04 | 0.66 ± 0.1 | 4.5 |
| Ru-3 | Cl | 0.27 ± 0.1 | 1.08 ± 0.3 | 4 |
| Ru-4 | Br | 0.49 ± 0.12 | 1.44 ± 0.07 | 2.9 |
| Ru-5 | I | 0.68 ± 0.2 | NDb | N/A |
| Ru-6 | NO2 | 0.55 ± 0.15 | NDb | N/A |
| Ru-7 | OMe | 0.35 ± 0.07 | 0.84 ± 0.2 | 2.4 |
| Ru-8 | t-Bu | 0.73 ± 0.2 | 1.04 ± 0.1 | 1.4 |
| Os-1 | H | 0.28 ± 0.1 | 2.56 ± 0.3 | 9.3 |
| Os-2 | F | 0.44 ± 0.07 | 2.6 ± 0.5 | 5.9 |
| Os-3 | Cl | 0.61 ± 0.09 | 2.75 ± 0.6 | 4.5 |
| Os-5 | I | 0.58 ± 0.09 | 4.37 ± 0.3 | 7.5 |
| Os-7 | OMe | 0.49 ± 0.05 | 1.64 ± 0.04 | 3.3 |
| CQ | N/A | 0.027 ± 0.01 | 0.22 ± 0.05 | 8.1 |
| R1 & R2 | D10: IC50 (μM) | Dd2: IC50 (μM) | RI | |
|---|---|---|---|---|
| a On another occasion no activity could be detected against Dd2 for HL1, N/A = not applicable. | ||||
| L10 | N/A | 0.19 ± 0.03 | 0.25 ± 0.06 | 1.3 |
| Ru-10 | N/A | 4.5 ± 0.7 | 19.7 ± 0.4 | 4.3 |
| Os-10 | N/A | 0.64 ± 0.06 | 0.5 ± 0.1 | 0.8 |
| HL1 | H | 0.55 ± 0.3 | 0.18 ± 0.02a | 0.32 |
| Ru-1 | H | 0.07 ± 0.007 | 0.82 ± 0.05 | 1.6 |
| Os-1 | H | 0.059 ± 0.01 | 0.43 ± 0.04 | 7.3 |
| HL9 | t-Bu | 0.032 ± 0.009 | 0.42 ± 0.09 | 13 |
| Ru-HL9 | t-Bu | 0.059 ± 0.01 | 0.43 ± 0.04 | 7.3 |
| Os-HL9 | t-Bu | 0.042 ± 0.01 | 0.3 ± 0.04 | 7.2 |
| CQ | N/A | 0.025 ± 0.009 | 0.14 ± 0.01 | 5.7 |
Ligands HL1–8 exhibit in vitro antiplasmodial activities similar to chloroquine, i.e. IC(NF54)50 ≈ 30 nM and IC(Dd2)50 ≈ 200 nM. The sole exception is HL5 (X = I), which shows very poor activity against the chloroquine resistant strain of P. falciparum (Dd2) (IC(Dd2)50 = 1520 nM). The ruthenium arene complexes Ru-1 to Ru-8 exhibit activities approximately an order of magnitude worse than the free ligands against the chloroquine sensitive strain, IC(NF54)50 = 140–730 nM. Against the chloroquine resistant strain two of the complexes, Ru-5 (X = I) and Ru-6 (X = NO2), showed no activity up to the highest concentration tested. The complexes that were active, displayed antiplasmodial behavior at concentrations 4 to 6 times higher than chloroquine and 3 to 7 times higher than the free ligands. However, the resistance index, i.e. the quotient of the activity against chloroquine resistant and chloroquine sensitive P. falciparum, is better compared to chloroquine for the complexes that were active against the resistant strain. While this is a promising result, it should not be overinterpreted, considering the relatively poor activities exhibited against the chloroquine sensitive strain.
The osmium complexes Os-1–Os-3 and Os-7 showed activities that were similar or slightly lower than their ruthenium analogues, but against the chloroquine resistant Dd2 strain they are consistently less active. The iodo-substituted complex Os-5 stands out, with slightly better activity than the ruthenium analogue Ru-5 against the CQS strain (IC(NF54)50 = 580 nM and IC(NF54)50 = 680 nM respectively) and some, although poor, activity against the CQR strain (IC(Dd2)50 = 4370 nM), against which the ruthenium analogue shows no activity.
Ligand HL9 was found to be significantly more active than the unsubstituted HL1, IC(D10)50 = 32 nM and IC(Dd2)50 = 550 nM respectively, and is almost as active as chloroquine against the CQS D10 strain. However, the dramatic increase in activity against the D10 strain that is observed upon cordination of Ru or Os to HL1 is not seen for HL9; metal coordination of HL9 results in a slight decrease in activity against the CQS strain for both Os-HL9 and Ru-HL9 relative to the free ligand and a slight increase in activity against the CQR strain in the case of Os-HL9. The small difference in activity for the complexes compared to the free ligand may indicate partial decomposition of the complexes under physiological conditions, and that the resultant antiplasmodial effect observed is primarily caused by the dissociated ligand, HL9. Coordination of L10 to osmium results in a complex (Os-10) which is less active than its free ligand against both parasite strains but is much more active than its ruthenium analogue, Ru-10.
Comparing osmium and ruthenium compounds, all the ruthenium salicylaldimine complexes performed better against the Dd2 strain (CQR) than their osmium congeners, while against NF54 (CQS) two complexes, Ru-1 and Ru-5, have lower activities than the osmium equivalents. For the compounds tested against D10 and Dd2, the imidazole-containing ruthenium complex Ru-10 is 7 and 40 times less active than Os-10 against the D10 and Dd2 strains, respectively. Both Ru-1 and Ru-HL9 have activities similar to Os-1 and Os-HL9 against the chloroquine sensitive D10 strain and are slightly less active against the Dd2 chloroquine resistant strain than Os-1 and Os-HL9. While several osmium complexes exhibited better activity than their ruthenium congeners, replacement of ruthenium by osmium in these complexes does not appear to offer a significant advantage for antiplasmodial activity.
If the antiplasmodial activities of ligands HL1–8 are plotted against the Hammett σ-constants of the substituents in R1 (cf. Scheme 1), a weak negative correlation can be seen (see ESI†) with the electron-withdrawing F, Cl, Br and NO2-substituted ligands, HL2–4,6, having the highest activities and HL7,8, bearing the electron-donating methoxy and tert-butyl groups having some of the lowest activities. However, no such trend can be seen for the corresponding ruthenium or osmium complexes. Similarly HL9 shows higher activity against the D10 strain than HL1, while the opposite is true for the CQR Dd2 strain. It is worth noting that the activity of HL1 and its complexes Ru-1 and Os-1 vary considerably between the two different chloroquine sensitive strains. The free ligand is almost 20 times more active against NF54 than D10, while the ruthenium (Ru-1) and osmium (Os-1) complexes are 7 and 5 times less active against NF54 than D10, respectively.
Since the overall trend is that the uncoordinated ligands are significantly more active than the metal complexes, it is possible that the complexes in themselves are inactive, while the active species are the decoordinated ligands. If this is the case, the differences in antiplasmodial activity between the ruthenium and osmium complexes could be the result of the greater lability of the coordinated ligands in the former, resulting in a higher concentration of free ligand at equal concentrations of the complexes. For the ligand HL9 and its complexes Ru-9 and Os-9, the IC50 data for the Dd2 P. Falciparum strain suggest a reverse trend, but the activities are actually not statistically different (cf.Table 4). The fact that there is no significant difference between the activities of this ligand and its complexes may indicate that the ligand is fully decoordinated from the metal under physiological conditions, and that the free ligand determines the activity.
Electrochemistry
The ruthenium complexes Ru-2,-3,-5,-6 and -7 were studied by cyclic and differential pulse voltammetry, see Fig. 8. All 5 complexes showed a single irreversible oxidation not present in the corresponding free ligand, presumed to be the oxidation of RuII to RuIII. The oxidation potential correlates well (ρ = 0.98) with the Hammett σ-constants of the substituents in the R1 position (cf. Scheme 1). However, as might be anticipated, no correlation between the oxidation potentials of the complexes and their anti-malarial activities could be observed.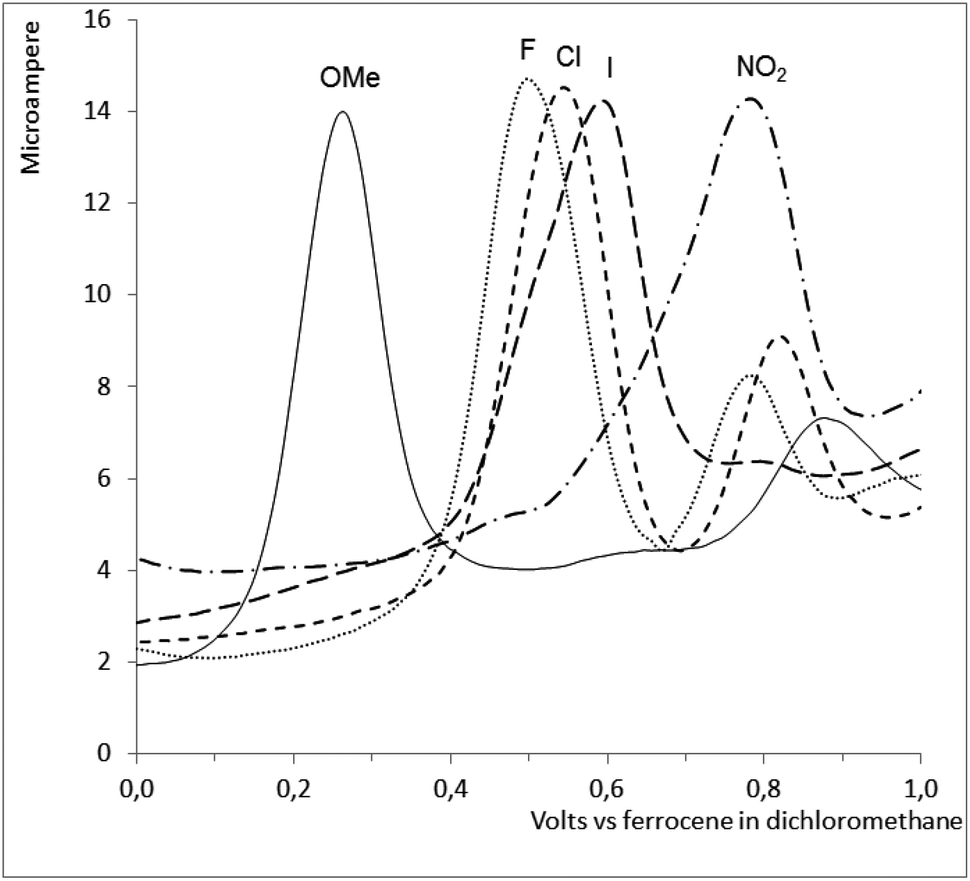 | ||
| Fig. 8 A differential pulse voltammetry (DPV) trace for complexes Ru-2,-3,-5,-6 and -7, showing the increase in oxidation potential when more electron withdrawing groups are introduced on the salicyl aldimne moiety. | ||
Experimental
General information
Ruthenium and osmium complexes were synthesized under dry argon or nitrogen using standard Schlenk and vacuum-line techniques. Solvents used were dried by distillation over appropriate drying reagents and stored over molecular sieves under nitrogen. Acetone was dried over molecular sieves overnight, followed by storage under argon. All chemicals were purchased from Sigma Aldrich, except OsCl3·xH2O which was purchased from Strem and used as received. [Ru(p-cymene)Cl2]2,41 [Os(p-cymene)Cl2]2,42,43N1-(7-chloroquinolin-4-yl)ethane-1,2-diamine,44N-(2-((1-methyl-1H-imidazol-2-yl)methylamino)ethyl)-7-chloroquinolin-4-amine (L10) and N-(2-((2-hydroxyphenyl)methyl-imino)ethyl)-7-chloroquinolin-4-amine (HL1)17 were prepared according to literature methods. NMR spectra were recorded on a Varian Inova 500 MHz (1H at 499.76 MHz, 13C at 125.68 MHz), a Varian Unity XR400 MHz (1H at 399.95 MHz, 13C at 100.58 MHz), Varian Mercury XR300 (1H at 300.08 MHz, 13C at 75.46 MHz) or Bruker Biospin GmbH (1H at 400.22 MHz, 13C at 100.65 MHz) spectrometer at ambient temperature using the residual solvent peak as internal standard for 1H NMR and 13C NMR. IR spectra were recorded on a Nicolet Avatar 360 FT-IR instrument or Perkin-Elmer Spectrum 100 FT-IR Spectrometer as KBR pellets. Electrospray ionization (ESI) mass spectra were recorded using a Waters Micromass Q-TOF micro mass spectrometer or a Waters API Quattro Micro instrument in either the positive or negative mode.Syntheses
C), 1614w (7-chloroquinoline), 1580s (7-chloroquinoline), 1547w (7-chloroquinoline), 1582m, 1449w, 1431w, 1380w, 1273m, 1257m, 1143m, 811m, 782w; MS (ESI+) m/z 344 ([M + H]+, 100%).
C), 1616w (7-chloroquinoline), 1584s (7-chloroquinoline), 1477m, 1369w, 1279m, 1146w, 804m; MS (ESI+) m/z 360 ([M + H]+, 100%).
C), 1615w (7-chloroquinoline), 1583s (7-chloroquinoline), 1474w, 1453w, 1432w, 1369m, 1280m, 1211w, 1145m, 1083w, 1049w, 1010w, 902w, 865w, 802m, 779w; MS (ESI+) m/z 406 ([M + H]+, 100%).
C), 1618w (7-chloroquinoline), 1582s (7-chloroquinoline), 1560w, 1542w (7-chloroquinoline), 1473w, 1366w, 1282m, 1145m, 802m; MS (ESI+) m/z 452 ([M + H]+, 100%).
C), 1607m (7-chloroquinoline), 1583s (7-chloroquinoline), 1541m (7-chloroquinoline), 1515w, 1481w, 1451w, 1327s, 1281w, 1231m, 1173w, 1094m, 892w, 841w, 800w; MS (ESI−) m/z 369 ([M − H]−, 100%).
C), 1614w (7-chloroquinoline), 1582s (7-chloroquinoline), 1548w (7-chloroquinoline), 1489w, 1451w, 1430w, 1371w, 1271s, 1209w, 1153w, 1143w, 1053w, 852w, 766m; MS (ESI+) m/z 356 ([M + H]+, 100%).
:heptane:EtOAc 5:25:70) to yield the desired product as a grey powder (0.523 g, 51%). 1H NMR (500 MHz, CDCl3) δ 12.75 (s, 1H), 8.55 (d, J = 5.2 Hz, 1H), 8.37 (s, 1H), 7.97 (s, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.39 (dd, J = 8.7, 1.9 Hz, 1H), 7.34 (dd, J = 8.8, 1.2 Hz, 1H), 7.18 (s, 1H), 6.92 (d, J = 8.6 Hz, 1H), 6.51 (d, J = 5.2 Hz, 1H), 5.40 (s, 1H), 3.95 (m), 3.74 (m, 2H); 13C{1H} NMR (125.68 MHz, DMSO-d6) δ 167.30, 158.24, 151.97, 149.95, 149.00, 140.69, 133.35H, 129.36, 127.88, 127.49, 124.10, 123.97, 117.96, 117.47, 115.93, 99.06, 56.79, 43.02, 33.66, 31.27; IR (KBr) vmax/cm−1 3229s (O–H), 2963m, 1639s (NC), 1609w (7-chloroquinoline), 1590s (7-chloroquinoline), 1543w (7-chloroquinoline), 1494w, 1455w, 1428w, 1367w, 1327w, 1254m, 1214w, 1139m, 1064w, 911w, 878w, 832m, 803w; MS (ES+) m/z 382 ([M + H]+, 100%).
:MeOH, 9:1 v/v) and the product (Ru-2) was dried under vacuum and obtained as a brown solid (24 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 8.48 (s, 1H), 7.93 (s, 1H), 7.72 (d, 1H, J = 8.9 Hz), 7.21 (s, 1H), 7.20 (s, 1H), 6.92 (t, 1H, J = 9.7 Hz), 6.84 (dd, 1H, J = 4.6 Hz, J = 9.3 Hz), 6.82 (br,1H), 6.44 (s, 1H), 6.09 (d, 1H, J = 6.1 Hz), 5.55 (d, 1H, J = 6.2 Hz), 5.50 (d, 1H, J = 6.1 Hz), 5.43 (d, 1H, J = 4.6 Hz), 5.02 (d, 1H, J = 4.8 Hz), 4.52 (m,1H), 4.23 (m,2H), 3.88 (m, 1H), 2.75 (td, 1H, J = 6.9 Hz, J = 14.0 Hz), 2.30 (d, 3H), 1.25 (d, 3H, J = 6.9 Hz), 1.15 (d, 3H, J = 6.8 Hz); 13C{1H} NMR (125.68 MHz, CDCl3) δ 164.78, 161.60, 152.46 (d, J = 232.4 Hz), 150.26, 147.59, 135.94, 126.82, 126.00, 123.77 (d, J = 23.7 Hz), 123.09, 122.93 (d, J = 7.4 Hz), 117.37 (d, J = 232.4 Hz), 117.04, 116.61 (d, J = 7.8 Hz), 100.68, 99.08, 98.22, 88.45, 83.15, 80.84, 80.47, 66.87, 42.30, 30.63, 22.92, 21.61, 18.87; Found 53.01% C, 4.83% H and 5.68% N. C28H28Cl2FN3ORu requires 54.82% C, 4.60% H and 06.85% N. IR (KBr) vmax/cm−1 3452 (br), 2962m, 1623sh (NO), 1613s (7-chloroquinoline), 1583s (7-chloroquinoline), 1541m (7-chloroquinoline), 1466s, 1427w, 1389w, 1316w, 1243w, 1210w, 1027w, 870w, 817m; MS (ESI+) m/z 614 ([M + H]+, 100%).
O), 1614s (7-chloroquinoline), 1581s (7-chloroquinoline), 1527w (7-chloroquinoline), 1460m, 1389w, 1322w, 1262w, 1090w, 1026m, 872w, 805m; MS (ESI−) m/z 630 ([M − H]−, 100%).
O), 1611s (7-chloroquinoline), 1581s (7-chloroquinoline), 1523w (7-chloroquinoline), 1459m, 1388w, 1320m, 1172w, 1137w, 1078w, 1028m, 872w, 819m; MS (ESI+) m/z 676 ([M + H]+, 100%).
O), 1612s (7-chloroquinoline), 1581s (7-chloroquinoline), 1541w, 1522w (7-chloroquinoline), 1459m, 1386w, 1320m, 1173w, 1139w, 1081m, 874w, 819m; MS (ESI+) m/z 722 ([M + H]+, 100%).
O), 1623s (7-chloroquinoline), 1603s (7-chloroquinoline), 1548w (7-chloroquinoline), 1474w, 1355m, 1195w, 1102m, 1034w; MS (ESI+) m/z 641 ([M + H]+, 100%).
O), 1610s (7-chloroquinoline), 1582s (7-chloroquinoline), 1560w, 1540m (7-chloroquinoline), 1466m, 1314w, 1261w, 1091w, 1027m, 869w, 804m; MS (ESI+) m/z 626 ([M + H]+, 100%).
O), 1618s (7-chloroquinoline), 1578s (7-chloroquinoline), 1541w (7-chloroquinoline), 1474m, 1457w, 1421w, 1389w, 1322m, 1255w, 1180w, 1142w, 1023w, 834w; MS (ESI+) m/z 651 ([M + H]+, 100%).
:1 (v/v). The solvent was evaporated to give the product (Os-1) as a red solid (50 mg, 72%). 1H NMR (500 MHz, CDCl3) δ8.53 (d, 1H, J = 5.1 Hz), 7.91 (s, 1H), 7.66 (d, 1H, J = 8.9 Hz), 7.22 (m, 2H), 7.18 (d, 1H, J = 8.9 Hz), 6.85 (d, 1H, J = 8.5 Hz), 6.53 (br s, 1H), 6.43 (d, 1H, J = 5.1 Hz), 6.32 (d, 1H, J = 7.4 Hz), 6.27 (t, 1H, J = 7.4 Hz), 5.81 (m, 2H), 5.79 (d, 1H, J = 5.0 Hz), 5.39 (d, 1H, J = 5.0 Hz), 4.38 (m, 1H), 4.30 (m, 1H), 4.11 (m, 1H), 3.77 (m, 1H), 2.64 (sept, 1H, J = 6.8 Hz), 2.38 (s, 3H), 1.26 (d, 3H, J = 6.8 Hz), 1.13 (d, 3H, J = 6.8 Hz); 13C-NMR; IR (KBr) vmax/cm−1 3300m (br), 2960m, 2923w, 2858w, 1633sh (NC) 1613s (7-chloroquinoline), 1588s (7-chloroquinoline), 1539m (7-chloroquinoline), 1469m, 1447m, 1322m, 1200w, 1142w, 1084w, 1034w; MS (ESI+) m/z 686 ([M + H]+, 3%), 650 ([M − Cl]+, 30%), 325.6 ([M − Cl + H]2+, 100%).
N), 1589 (s, CN). MS (ESI+) m/z 668.15 ([M − Cl]+, 100%), 704.12 ([M + H]+, 40%).
N), 1580 (s, CN). MS (ESI+) m/z 684.1 ([M − Cl]+, 100%), 720.09 ([M + H]+, 50%).
N), 1583 (s, CN). MS (ESI+) m/z.
N), 1587 (s, CN). MS (ESI+) m/z 680.2 ([M − Cl]+, 100%), 716.14 ([M + H]+, 20%).
X-Ray structure analysis
An orange-red crystal of Ru-1 was cut from a long spar and mounted on a 200 micron MiTeGen MicroMount in Paratone-N oil, and then cooled with an Oxford Cryosystems Series 700 low-temperature device; a single crystal of Ru-5 was found and treated in the identical manner. The X-ray intensity data were measured on a Bruker Smart Breeze CCD system equipped with a graphite monochromator (λ = 0.71073 Å) at 100(2) K. A total of 1464 frames were collected with a total exposure time of 4.07 hours for Ru-1 and 3.25 hours for Ru-5. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm, and data were corrected for absorption effects using the multi-scan method (SADABS).The structure of Ru-1 was first solved using the Bruker SHELXTL Software Package with anisotropic full-matrix least-squares refinement on F2. Hydrogen atom positions were calculated for all CH hydrogen atoms but thermal parameters were allowed to refine isotropically. The NH hydrogen atoms were located in the difference maps and then refined with isotropic thermal parameters and distance restraints (0.91 Å at 100 K).
Copies of the two independent molecules in the unit cell were then created and placed in positions given by the vector between the main residual electron densities and the Ru atom positions. This model was refined with respect to only the occupancies of the split positions and the rotations and translations of the major components to the new positions to yield the final structure. The final refinements were performed using the JANA2006 software.45
The red crystals of Ru-3 were fixed to glass fiber using Epoxy glue and the single crystal data was collected at room temperature on an Oxford Diffraction Xcalibur EOS CCD diffractometer with graphite monochromatised Mo-Kα radiation (λ = 0.71073 Å) operated at 50 kV and 40 mA, with a detector distance of 50 mm and 2θ range 3.0–28.8°. The Oxford CrysAlisPro RED software was used for data processing, including numerical absorption correction based on Gaussian integration over a multifaceted crystal model.46
Structural solution was accomplished using charge flipping as implemented in Superflip.47 All refinements were performed using the JANA2006 software.46 All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were theoretically generated.
The structure of Ru-5 was solved (intrinsic phasing method) and refined using the Bruker SHELXTL Software Package with anisotropic full-matrix least-squares refinement on F2. Hydrogen atom positions were calculated for all CH hydrogen atoms but thermal parameters were allowed to refine isotropically. The NH hydrogen atoms were located in the difference maps and then refined with isotropic thermal parameters and distance restraints (0.91 Å at 100 K).
For Os-5 and Os-7, X-ray single crystal intensity data were collected on a Nonius Kappa-CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). Temperature was controlled by an Oxford Cryostream cooling system (Oxford Cryostat). The strategy for the data collections was evaluated using the Bruker Nonius “Collect” program. Data were scaled and reduced using DENZO-SMN software.48 Absorption correction was performed using SADABS.49
The structures of Os-5 and Os-7 were solved by direct methods and refined employing full-matrix least-squares with the program SHELXL-97 refining on F2.50 Figures were produced using the program PovRay and graphic interface X-seed.51 All non-hydrogen atoms were refined anisotropically.
For Os-5, all hydrogen atoms were placed in idealized positions and refined in riding models with Uiso assigned to be 1.2 or 1.5 times those of their parent atoms and the distance of C–H constrained ranging from 0.95 Å to 0.99 Å and N–H 0.88 Å. The structure was refined to an R factor of 0.0266.
For Os-7, all hydrogen atoms, except H2, were placed in idealised positions and refined in riding models with Uiso assigned the values to be 1.2 or 1.5 times Ueq of the atoms to which they are attached and the constraint distances of C–H ranging from 0.95 Å to 1.00 Å. The hydrogen H2 was located in the difference Fourier maps and refined with bond length constraint d(N2–H2) = 0.88 Å. The structure was refined to an R factor of 0.0374. Selected crystal data and structure refinement parameters are listed in Table 5.
| Ru-1 | Ru-3 | Ru-5 | Os-5 | Os-7 | |
|---|---|---|---|---|---|
| Formula | C28H29Cl2N3ORu | C28H28Cl3N3ORu | C28H28Cl2IN3ORu | C28H28Cl2IN3OOs | C29H31Cl2N3O2Os |
| Formula weight | 595.53 | 629.97 | 721.40 | 810.56 | 714.67 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21 | P21/c | P21/c | P21/n | P21/c |
| a (Å) | 8.9756(16) | 9.3205(14) | 6.5071(11) | 9.6159(6) | 8.9203(2) |
| b (Å) | 15.207(3) | 15.822(5) | 18.214(3) | 15.6469(10) | 15.7392(4) |
| c (Å) | 18.917(3) | 18.804(5) | 22.628(4) | 18.6189(12) | 19.7232(4) |
| β (°) | 103.018(2) | 103.675(18) | 91.422(2) | 104.1850(10) | 99.676(2) |
| V (Å3) | 2515.7(8) | 2694.3(12) | 2681.0(8) | 2716.0(3) | 2729.71(11) |
| Z | 4 | 4 | 4 | 4 | 4 |
| D c (g cm−3) | 1.572 | 1.5527 | 1.787 | 1.982 | 1.739 |
| μ (mm−1) | 0.863 | 0.906 | 1.963 | 6.055 | 4.899 |
| θ range for data collection (°) | 2.329 to 27.079 | 3.018 to 28.79 | 1.435 to 27.262 | 1.70 to 28.50 | 2.46 to 27.50 |
| Limiting indices | −11 < h < 11 | −12 < h < 12 | −8 < h < 8 | −12 < h < 12 | −11 < h < 11 |
| −19 < k < 19 | −20 < k < 21 | −23 < k < 23 | −20 < k < 20 | −20 < k < 20 | |
| −24 < l < 24 | −25 < l < 25 | −29 < l < 29 | −25 < l < 24 | −25 < l < 25 | |
| No. of reflexions measured | 29412 |
29607 |
31278 |
84625 |
12294 |
| No. of unique reflexions | 11009 |
6414 | 6003 | 6859 | 6261 |
| R int | 0.032 | 0.0817 | 0.0371 | 0.071 | 0.017 |
| No. of parameters | 644 | 328 | 331 | 328 | 342 |
| R 1 | 0.0433 | 0.0415 | 0.0282 | 0.0266 | 0.0374 |
| wR2 | 0.1088 | 0.0647 | 0.0719 | 0.0462 | 0.0750 |
| Goodness of fit on F2 | 1.066 | 1.05 | 1.06 | 1.06 | 1.510 |
Electrochemistry
The electrochemical analyses were performed using a WaveNow potentiostat (Pine Research). The solvent was anhydrous dichloromethane stored over molecular sieves and the supporting electrolyte was 0.1 M tetrabutylammonium perchlorate. Measurements were carried out using a 2 mm diameter glassy carbon working electrode, a Pt wire auxiliary electrode and a Ag/AgCl reference electrode separated from the working solution by a glass frit. Differential pulse measurements used a 10 mV increment. Oxygen was excluded during measurement by a flow of nitrogen gas. All potentials are reported versus the ferrocene/ferrocenium potential (0.400 V vs. NHE) as an internal standard.Determination of in vitro antiplasmodial activity
Three strains of Plasmodium falciparum were used in this study: the chloroquine sensitive strains D10 and NF54 and the chloroquine resistant strain Dd2. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen.52 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler.53 The samples were tested in triplicate on two separate occasions; HL1–8, Ru-1–Ru-8 and Os-1–Os-3, Os-5 and Os-7, were tested against NF54 (CQS) and Dd2 (CQR) strains and HL1, Ru-1 and Os-1, HL9, Ru-HL9and Os-HL9 and L10, Ru-10 and Os-10 were tested against D10 (CQS) and Dd2 (CQR) strains. The test samples were prepared as 20 mg ml−1 stock solutions in 100% DMSO. Samples were tested as a suspension if not completely dissolved. Stock solutions were stored at −20 °C. Further dilutions were prepared on the day of the experiment. Chloroquine diphosphate (CQ) was used as the reference drug in all experiments. A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Samples were tested at a starting concentration of 10 μg ml−1, which was then serially diluted 2-fold in complete medium to give 10 concentrations with the lowest concentration being 0.02 μg ml−1. The same dilution technique was used for all samples. Samples were also tested at a starting concentration of 1000 ng ml−1, which was then serially diluted 2-fold in complete medium to give 10 concentrations; with the lowest concentration being 2 ng ml−1. Chloroquine was tested at a starting concentration of 100 ng ml−1. The highest concentration of solvent to which the parasites were exposed had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.Summary and conclusions
Eight new ligands, HL2–9, and the previously known HL1, which contain a 7-chloroquinoline framework functionalized with a salicylaldimine moiety in position 4, have been synthesized. These ligands and the related ligand HL10, in which the salicylaldimine moiety is replaced by an amine-(N-methyl) imidazolyl chelating unit, have been used to prepare the ruthenium complexes [Ru(η6-cym)(L1–8)Cl] (Ru-1–Ru-8, cym = p-cymene) and the previously known [Ru(η6-cym)(L10)Cl]Cl (Ru-10), in which the ligands coordinate to the metal via the bidentate chelating functionality. In the case of ligand HL9, spectroscopic evidence indicates that the metal coordinates to the quinoline nitrogen of the ligand to form [Ru(η6-cym)(HL9)Cl2] (Ru-9) presumably due to the steric hindrance of the tert-butyl group in 2-position on the salicylaldiminate moiety. Furthermore, the direct osmium analogues Os-1–Os-3, Os-5, Os-7, Os-9, and Os-10 were synthesized and characterized.All ligands and metal complexes were (or have previously been) evaluated in vitro for antimalarial activity against both chloroquine sensitive and chloroquine resistant strains of P. falciparum. Ligands HL1–8 exhibited antiplasmodial activities on par with chloroquine. The corresponding ruthenium complexes did also exhibit antiplasmodial activities, but at IC50 concentrations that were consistently higher than the corresponding ligands. Two complexes, Ru-5 and Ru-6, did not show any activity against a chloroquine resistant strain. However, the resistance indices for the active ruthenium complexes were better than that of chloroquine. The osmium analogues that were investigated gave antiplasmodial activities that were similar, but in most cases worse, than their ruthenium congeners. A weak correlation of antiplasmodial activity to the electronic influence of the substituent in para position on the salicylaldimine moiety in the ligands HL1–8 could be detected, with more electron-withdrawing substituents giving higher activity; however, this correlation is not borne out in the analogous metal complexes.
Acknowledgements
This research has been funded by the Swedish International Development Cooperation Agency (SIDA) and the Lund University Graduate School for Pharmaceutical Science (FLÄK, http://www.flak.lu.se). Financial support from the University of Cape Town, the National Research Foundation (NRF) of South Africa and the South African Medical Research Council (MRC) is gratefully acknowledged.References
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- Bioorganometallics: Biomolecules, Labeling, Medicine, ed. G. Jaouen, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- C. Biot and D. Dive, Top. Organomet. Chem., 2010, 32, 155–193 CrossRef CAS.
- C. Biot and D. Dive, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer, 2010, pp. 155–194 Search PubMed.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715–5727 CrossRef CAS.
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450–3492 CrossRef CAS PubMed.
- WHO Malaria Report 2014, http://www.who.int/malaria/publications/world_malaria_report_2014/en/.
- D. Das, A. P. Phyo, J. Tarning, D. Ph, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, T. Herdman, S. S. An, S. Yeung, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef PubMed.
- I. M. Hastings, P. G. Bray and S. A. Ward, Science, 2002, 298, 74–75 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J.-F. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- C. S. K. Rajapakse, A. Martínez, B. Naoulou, A. A. Jarzecki, L. Suárez, C. Deregnaucourt, V. Sinou, J. Schrével, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sánchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- R. A. Sánchez-Delgado, M. Navarro, H. A. Pérez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095–1099 CrossRef PubMed.
- A. Martínez, C. S. K. Rajapakse, R. A. Sánchez-Delgado, A. Varela-Ramirez, C. Lema and R. J. Aguilera, J. Inorg. Biochem., 2010, 104, 967–977 CrossRef PubMed.
- L. Glans, A. Ehnbom, C. de Kock, A. Martínez, J. Estrada, M. Haukka, P. J. Smith, R. A. Sánchez-Delgado and E. Nordlander, Dalton Trans., 2012, 41, 2764–2773 RSC.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS PubMed.
- A. F. A. Peacock, A. Habtemariam, S. A. Moggach, A. Prescimone, S. Parsons and P. J. Sadler, Inorg. Chem., 2007, 46, 4049–4059 CrossRef CAS PubMed.
- S. H. van Rijt, A. J. Hebden, T. Amaresekera, R. J. Deeth, G. J. Clarkson, S. Parsons, P. C. McGowan and P. J. Sadler, J. Med. Chem., 2009, 52, 7753–7764 CrossRef CAS PubMed.
- A. Dorcier, W. H. Ang, S. Bolaño, L. Gonsalvi, L. Juillerat-Jeanneret, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
- G. Mühlgassner, C. Bartel, W. F. Schmid, M. a. Jakupec, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2012, 116, 180–187 CrossRef PubMed.
- B. Boff, C. Gaiddon and M. Pfeffer, Inorg. Chem., 2013, 52, 2705–2715 CrossRef CAS PubMed.
- W. F. Schmid, R. O. John, G. Mühlgassner, P. Heffeter, M. A. Jakupec, M. Galanski, W. Berger, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 6343–6355 CrossRef CAS PubMed.
- L. K. Filak, G. Mühlgassner, M. A. Jakupec, P. Heffeter, W. Berger, V. B. Arion and B. K. Keppler, J. Biol. Inorg. Chem., 2010, 15, 903–918 CrossRef CAS PubMed.
- R. K. Rath, M. Nethaji and A. R. Chakravarty, Polyhedron, 2002, 21, 1929–1934 CrossRef CAS.
- J. A. K. Howard, G. Ilyashenko, H. A. Sparkes and A. Whiting, Dalton Trans., 2007, 2108–2111 RSC.
- J. A. K. Howard, G. Ilyashenko, H. A. Sparkes, A. Whiting and A. R. Wright, Adv. Synth. Catal., 2008, 350, 869–882 CrossRef CAS PubMed.
- W. I. Sundquist, D. P. Bancroft and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 1590–1596 CrossRef CAS.
- H. D. Flack, Helv. Chim. Acta, 2003, 86, 905–921 CrossRef CAS PubMed.
- B. Dalhus and C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 715–719 CAS.
- L. Fábián and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 94–103 Search PubMed.
- I. Bernal and S. Watkins, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 216–221 CAS.
- P. Govender, F. Edafe, B. C. E. Makhubela, P. J. Dyson, B. Therrien and G. S. Smith, Inorg. Chim. Acta, 2014, 409, 112–120 CrossRef CAS PubMed.
- H. Brunner, T. Zwack, M. Zabel, W. Beck and A. Bo, Organometallics, 2003, 1741–1750 CrossRef CAS.
- Y. Fu, R. Soni, M. J. Romero, A. M. Pizarro, L. Salassa, G. J. Clarkson, J. M. Hearn, A. Habtemariam, M. Wills and P. J. Sadler, Chem. – Eur. J., 2013, 19, 15199–15209 CrossRef CAS PubMed.
- Y. Fu, A. Habtemariam, A. M. B. H. Basri, D. Braddick, G. J. Clarkson and P. J. Sadler, Dalton Trans., 2011, 40, 10553–10562 RSC.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158–1167 RSC.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- H. Werner and K. Zenkert, J. Organomet. Chem., 1988, 345, 151–166 CrossRef CAS.
- R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2005, 24, 4343–4346 CrossRef CAS.
- K. Yearick, K. Ekoue-Kovi, D. P. Iwaniuk, J. K. Natarajan, J. Alumasa, A. C. de Dios, P. D. Roepe and C. Wolf, J. Med. Chem., 2008, 51, 1995–1998 CrossRef CAS PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 Search PubMed.
- CrysAlisPro, version 1.171.35.19, Agilent Technologies, Yarnton England, 2011 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 306–315 Search PubMed.
- G. M. Sheldrick, SADABS version 2.05, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrochemical details, Hammett's σ-plots, 1H-NMR assignment of Ru-HL9 and Os-HL9, and additional structure drawings of Ru-1. CCDC 992172, 1005351, 1005352, 1408709 and 1408710. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02410b |
| This journal is © The Royal Society of Chemistry 2015 |