
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural diversity of late transition metal complexes with flexible tetra-NHC ligands†
Daniel T.
Weiss
a,
Philipp J.
Altmann
a,
Stefan
Haslinger
a,
Christian
Jandl
a,
Alexander
Pöthig
b,
Mirza
Cokoja
c and
Fritz E.
Kühn
*a
aChair of Inorganic Chemistry/Molecular Catalysis, Department of Chemistry, Catalysis Research Center, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
bDepartment of Chemistry, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany
cDepartment of Chemistry, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany
First published on 20th July 2015
Abstract
The synthesis of copper, gold, nickel, palladium, platinum, and iron complexes with open chain tetra-N-heterocyclic carbene (NHC) ligands via transmetalation using silver NHC complexes is presented. The obtained complexes show differing coordination geometries depending on both ligand structure and metal. While the complexes of the coinage metals form di- or tetranuclear structures, the group 10 metal complexes exhibit a distorted square planar coordination geometry at the metal centers. In the case of iron an enhanced flexibility of the ligand – caused by a longer alkyl bridge – leads to octahedral complexes with a sawhorse-type coordination by the tetracarbene ligand and two cis acetonitrile ligands. To the best of our knowledge, this is the first known example of a tetracarbene ligand in sawhorse-type coordination within an octahedral coordination sphere. The remaining cis-labile sites are prone to exchange reactions as shown by addition of trimethylphosphine.
Introduction
During the past 20 years, the detailed examination of N-heterocyclic carbenes as ligands for transition metals unveiled a very rich coordination chemistry with an increasing variety of applications,1–5 including homogeneous catalysis,6–9 medicinal chemistry,10–12 and photoluminescent materials.13 Polydentate NHC ligands were found to stabilize high-valent iron species, which appeared to act as intermediates in catalytic oxidations and aziridations.14–20Only few examples for tetra-NHC ligand motifs are known to date. They exhibit either macrocyclic or acyclic structures. Depending on the linkage of the NHC subunits, macrocyclic tetra-NHC ligands are able to chelate metal ions with coordination numbers of four or higher.19,21–29 In case of rigid and bulky linkers like arene units, only coinage metal complexes are known.30–32 Acyclic tetra-NHC ligands were limited to rigid, substituted arene entities, which are unable to chelate single metal ions.33–36 Therefore, reports on mononuclear metal complexes with tetra-NHC ligands are limited to macrocyclic systems.19–29,37 In addition, the accessible coordination modes for tetra-NHC ligands are limited, as they coordinate generally in an equatorial fashion, with the only exceptions being a tetrahedral CoII complex and a trigonal prismatic FeIV tetrazene complex.20,23 To overcome the structural rigidity that is responsible for this limitation, two open chain tetra-NHC ligands L1 and L2 with alkyl linkers of different lengths have been reported by our group together with the respective silver(I) complexes 1 and 2 (Fig. 1).38
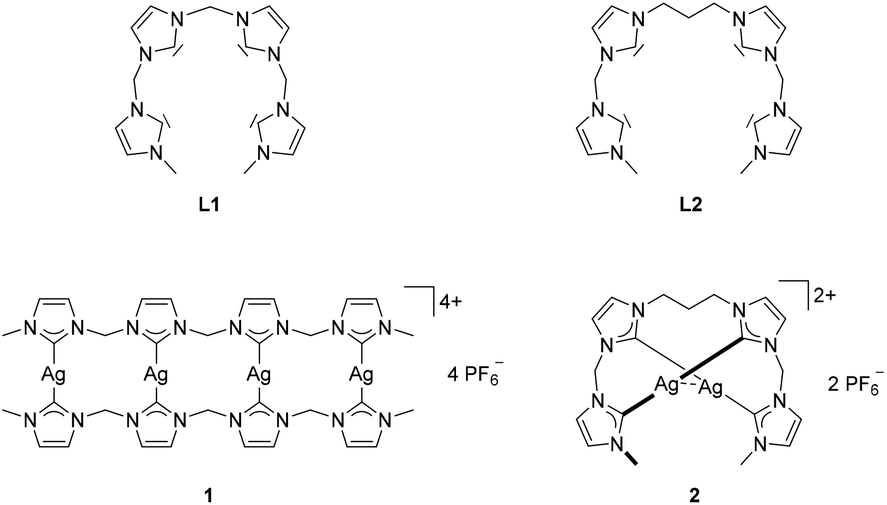 | ||
| Fig. 1 Structures of flexible, open chain tetra-NHC ligands L1, L2 and the respective silver complexes 1 and 2.38 | ||
In this work the use of 1 and 2 as versatile transmetalation agents is demonstrated to access a range of d-block metal complexes. By utilizing a flexible ligand structure, new coordination modes of tetra-NHC complexes are targeted. Single crystal X-ray diffraction in combination with NMR spectroscopy is used for the identification of coordination geometries in both solid state and solution.
Results and discussion
Transmetalation to coinage metals
In analogy to previous studies,30,33,34 transmetalation reactions to Cu(I) and Au(I) starting from silver tetra-NHC complexes 1 and 2 were performed with CuICl or AuI(SMe2)Cl as precursors, respectively (Scheme 1).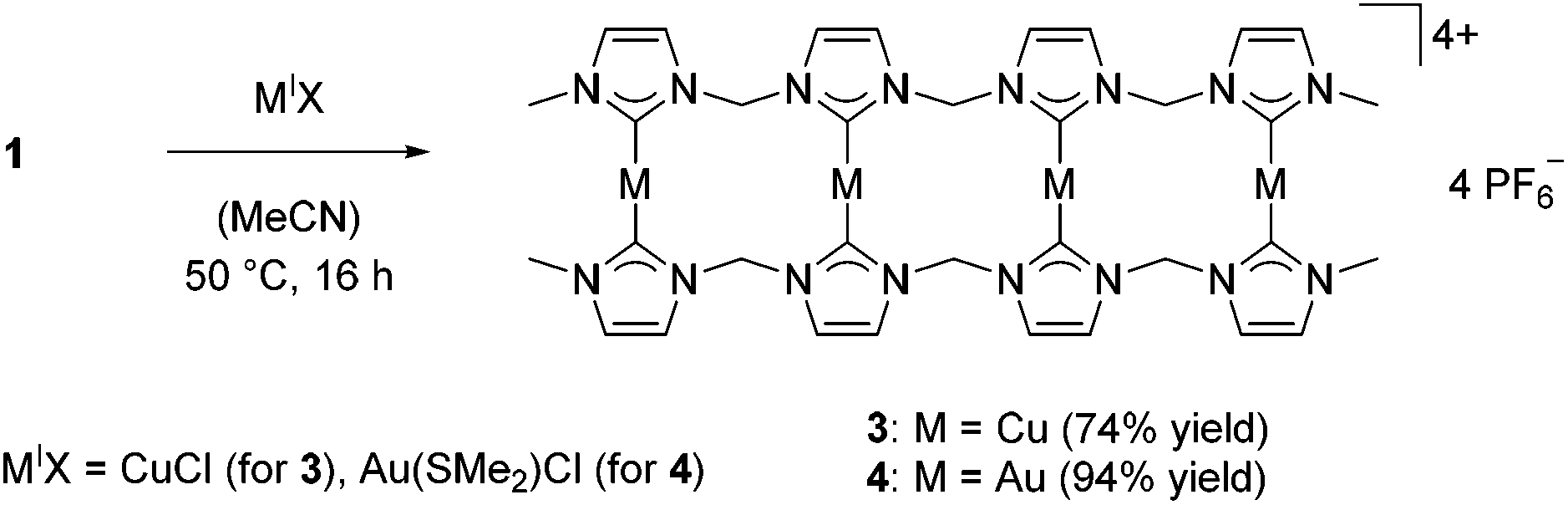 | ||
| Scheme 1 Transmetalation to coinage metals starting from silver complex 1. | ||
While copper(I) complex 3 has to be handled under inert conditions, gold(I) complex 4 shows high stability towards air and moisture. Both complexes were obtained as colorless solids in high yields. ESI-MS data for 4 confirms the dimeric, tetranuclear structure with m/z = 1894.60. In analogy to 1,38 variable temperature (VT) 1H NMR spectra of coinage metal complexes 3 and 4 reveal the existence of different conformers in solution (see Fig. S3 and S6 in the ESI†). For complex 3, an elevation of the temperature from −40 °C to 0 °C results in peak broadening, while a further increase in temperature to 70 °C leads to sharp signals. This indicates fast interconversion of the conformers which no longer can be resolved on an NMR time scale. In case of 4, VT NMR experiments yield similar results (compare ESI†); however the conformations are frozen already at room temperature compared to −40 °C for 3. Again, elevation of the temperature to up to 75 °C leads to a broadening and a subsequent peak sharpening. In addition, all expected signal sets for terminal methyl groups, methylene bridges, and imidazolium backbone protons are observed for complexes 3 and 4. These findings are supported by 13C NMR spectra recorded at 70 °C, which feature similar broad signals for 1 and 3–4 (ESI†). Due to the broadness of the peaks and the low intensity of carbene–carbons, not all expected signals are detected.
Single crystal X-ray diffraction of 3 confirmed the dimeric, tetranuclear structure in solid state, which is in accord with silver complex 1. Two additional solvent molecules are coordinated to Cu2 and Cu3, resulting from the crystallization of 3 from an acetonitrile solution (Fig. 2). The coordinated solvent molecules can be removed under high vacuum. The acetonitrile ligands are coordinated to the inner copper ions Cu2 and Cu3 which were found to be triply-coordinated with C–Cu–C angles of 137° and C–Cu–N angles of 112°. The outer copper ions Cu1 and Cu4 are coordinated almost linearly. The Cu1–Cu2–Cu3 and Cu2–Cu3–Cu4 angles are in the range of 132° and 133°, which is substantially smaller than the corresponding angle in 1 (145°). Unfortunately, no single crystals of 4 could be obtained.
 | ||
| Fig. 2 ORTEP style representation of the cationic fragment of 3 with ellipsoids shown at a 50% probability level. Hydrogen atoms and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu1–Cu2 3.0211(5), Cu2–Cu3 2.8570(5), Cu1–C19 1.919(3), Cu2–C6 1.960(3), Cu2–N17 2.071(2), C19–Cu1–C2 172.47(12), C23–Cu2–C6 137.03(11), C23–Cu2–N17 111.86(10), Cu3–Cu2–Cu1 132.224(15). | ||
In contrast to 1, silver complex 2 exhibits a monomeric, dinuclear structure with linear coordination of the coinage metal ions due to the longer and more flexible propylene bridge of ligand L2.38 Silver complex 2 was treated with CuCl and Au(SMe2)Cl to yield complexes 5 and 6, respectively (Scheme 2). As observed for 3 and 4, copper complex 5 is only stable under inert conditions while gold complex 6 is air stable. 1H NMR spectroscopy confirms the structural similarity of 2 and 5–6 as all spectra exhibit very similar signal patterns with the only major difference being the signals assigned to the methylene groups at 6.0 to 6.5 ppm and 3.7 to 4.1 ppm (Fig. 3). The broader signals in case of 5 indicate a higher flexibility compared to 2 and 6. However, 13C NMR spectra show a frozen conformation of 5 as solely sharp and defined signals are obtained. The two differing sets of carbene species are observed at 178.50 and 176.56 ppm for 5 and at 185.96 and 182.31 ppm for 6.
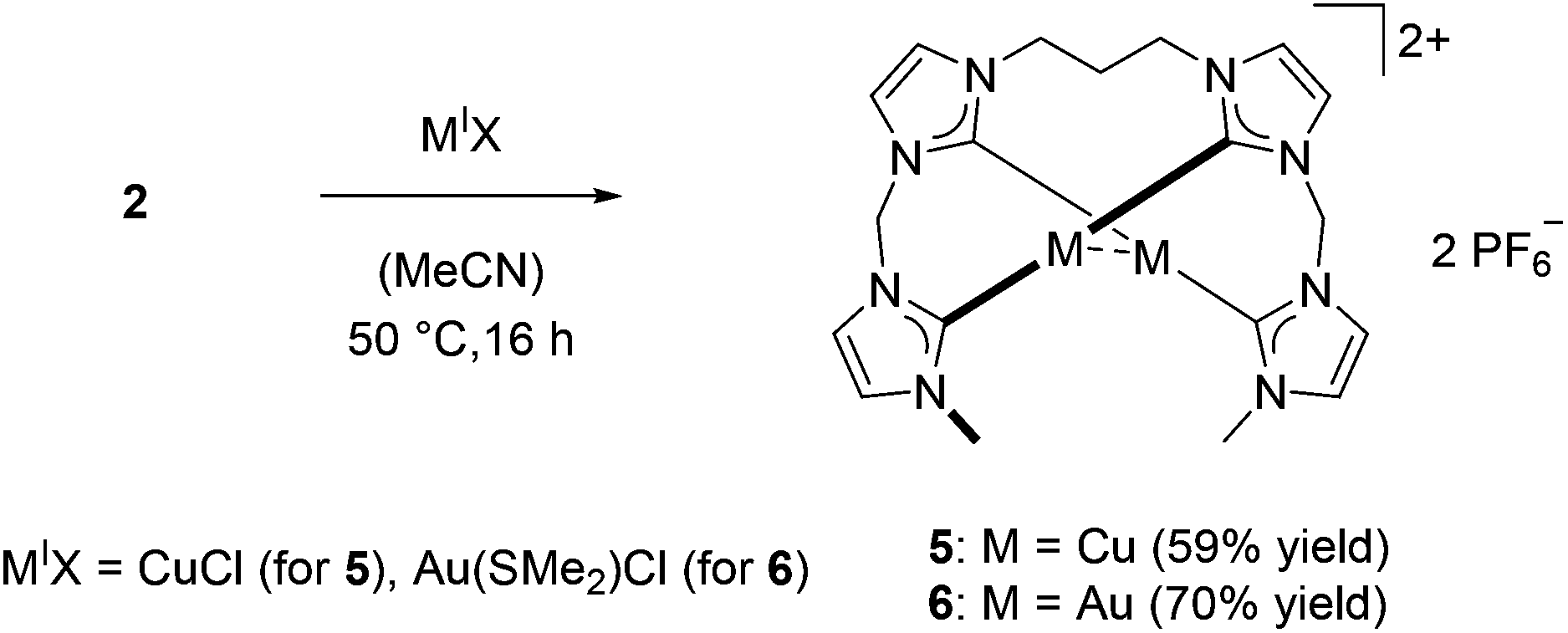 | ||
| Scheme 2 Transmetalation to coinage metals starting from silver complex 2. | ||
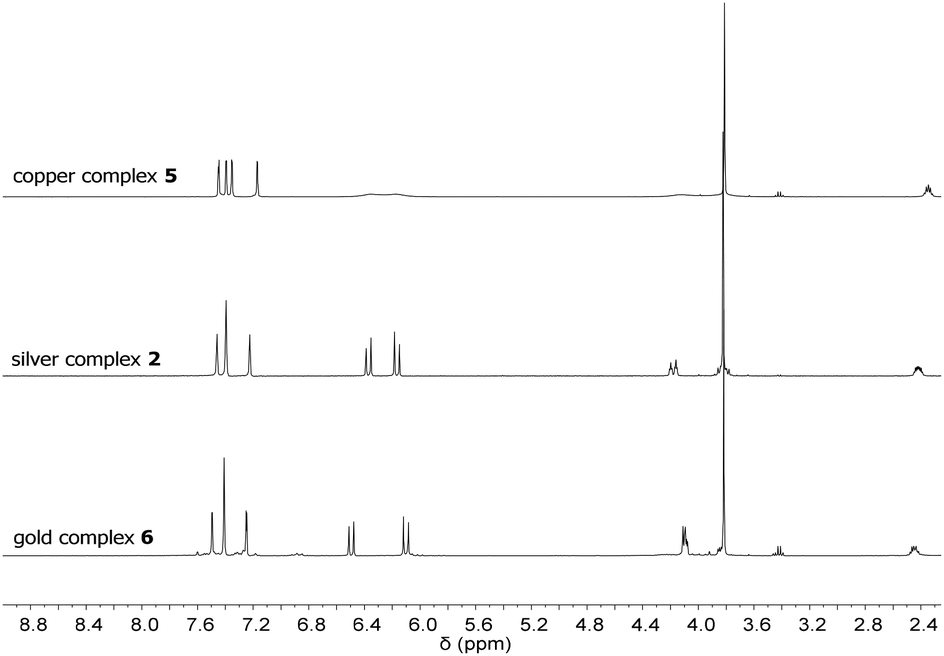 | ||
| Fig. 3 1H NMR spectra of copper complex 5, silver complex 2, and gold complex 6 at room temperature. | ||
The monomeric structure in solution was additionally confirmed by ESI-MS measurements of 6 (m/z 903.17). Due to the sensitivity of 5, ESI-MS data could not be collected. In solid state the monomeric structures of 5 and 6 were supported by single crystal X-ray diffraction, confirming the existence of a dinuclear metal–metal core in analogy to 2 (Fig. 4).
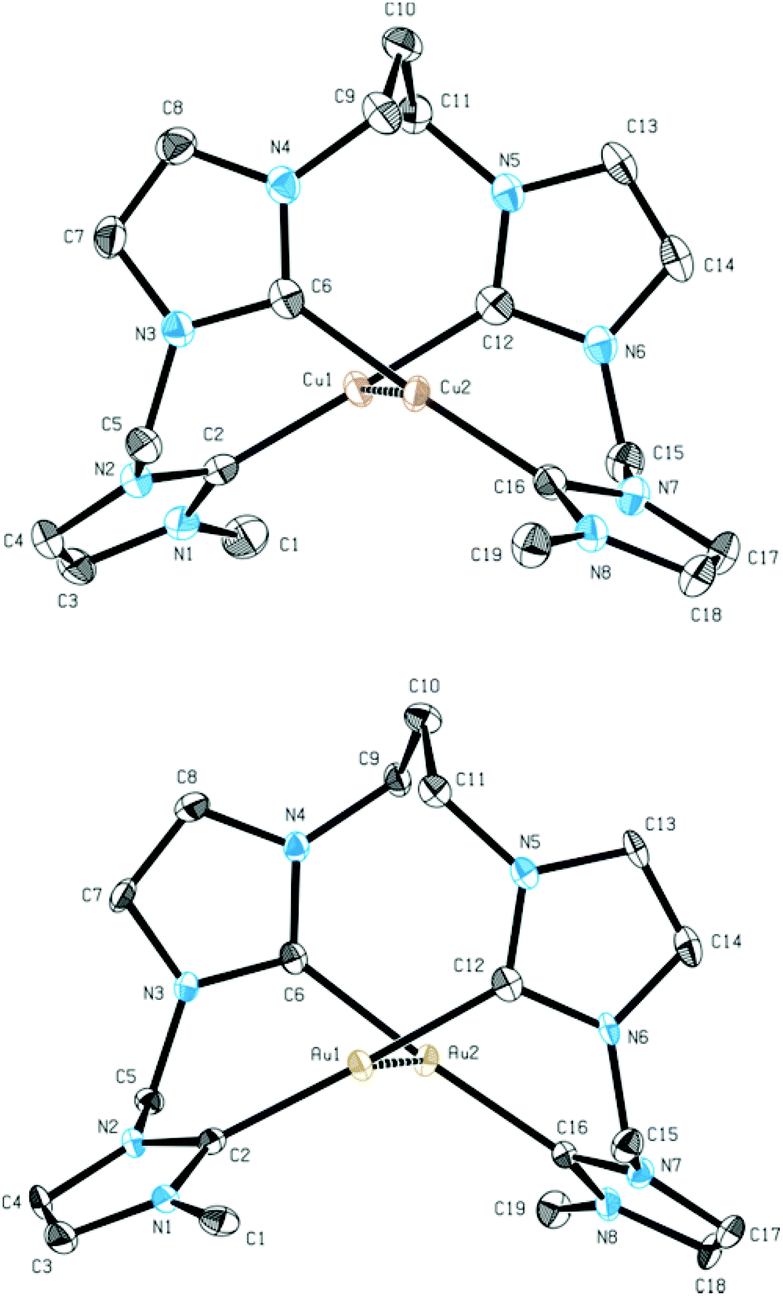 | ||
| Fig. 4 ORTEP style representation of the dicationic fragment of 5 (top) and 6 (bottom). Ellipsoids are shown at a 50% probability level. Hydrogen atoms and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: (5) Cu1–Cu2 2.6582(9), Cu1–C2 1.909(3), Cu2–C6 1.921(3), C2–Cu1–C12 177.88(11), C6–Cu2–C16 176.69(10); (6) Au1–Au2 2.9676(3), Au1–C2 2.008(4), Au2–C6 2.034(4), C2–Au1–C12 176.54(14), C6–Au2–C16 174.92(14). | ||
In comparison to the Ag–Ag distance of 2.9986 Å in 2, the Cu–Cu distance in 5 is 2.6582 Å. This finding is in agreement with previous results for the related macrocyclic tetra-NHC system reported by Murphy et al.22 The Au–Au distance in 6 is 2.9676 Å and comparable to the Ag–Ag distance in 2. Cu–C bonds in complex 5 are in the range of 1.909 to 1.921 Å while the copper ions are coordinated almost linearly with C–Cu–C angles of 177° and 178°. Au–C distances in 6 are in the range of 2.008 to 2.034 Å and again slightly shorter than the comparable bonds in 2. The planes, which are formed by the centered NHC rings are arranged in a nearly parallel fashion but tilted against the metal–metal axis by 74° to 75° for complex 5 and 72° to 73° for complex 6 compared to 71° for complex 2.
For all coinage metal complexes similar coordination modes are observed, with the flexibility of the ligand being the structure determining factor. For methylene-bridged ligand L1 tetranuclear structures are formed while propylene-bridged ligand L2 results in monomeric, dinuclear complexes.
Transmetalation to metals of the nickel group
Beside the coinage metals, the group 10 metals are interesting candidates for transmetalation reactions of silver NHC complexes.26,39 Using two equivalents of metal halides, the respective group 10 metal complexes were obtained in very good yields (>90%) for methylene-bridged tetra-NHC ligand L1 (Scheme 3). 1H and 13C NMR spectroscopic investigations in solution of compounds 7–9 suggest monomeric complexes and show mirror symmetry in the ligand. The characteristic signals for carbene carbons were found at 169.39 and 165.11 ppm for 7, at 169.28 and 164.18 ppm for 8, and at 161.20 and 155.78 ppm for 9, respectively. The monomeric structures in solution of 7–9 were confirmed by ESI mass spectrometry with monocationic fragments of m/z 538.53 (7), 586.59 (8), and 675.50 (9). Single crystal X-ray diffraction of complexes 7 and 9 confirmed the monomeric structure also in solid state (Fig. 5). Complexes 7 and 9 exhibit strongly distorted square planar geometries, which are typical for group 10 metal complexes with non-macrocyclic tetradentate polyNHC ligands.40 Coordination geometries within four-coordinated metal complexes can be compared with respect to the metal bond angles using a model by Houser et al.41 which was recently refined by the group of Kubiak.42 | ||
| Scheme 3 Synthesis of NiII (7), PdII (8), PtII (9) tetra-NHC complexes utilizing silver complex 1. | ||
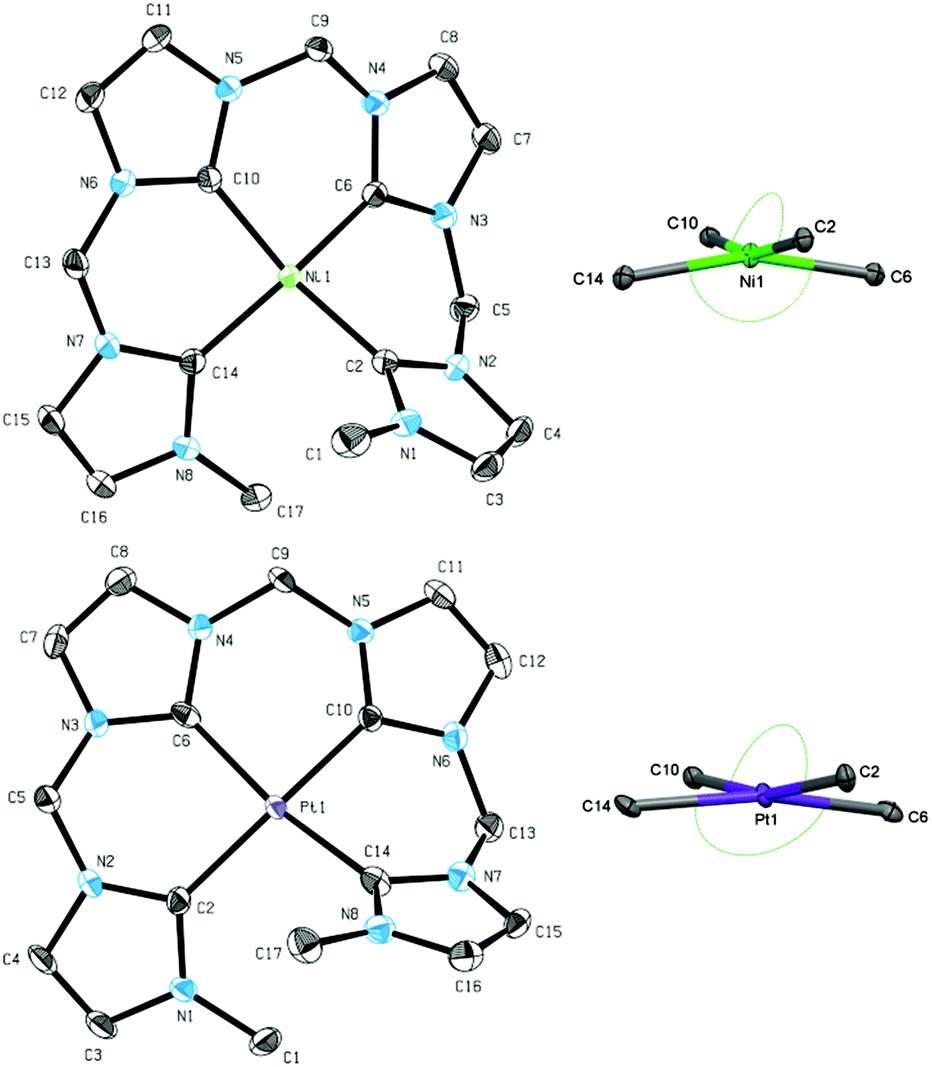 | ||
| Fig. 5 ORTEP style representation of the dicationic fragment of 7 (top left), the NiC4 core of 7 (top right), the dicationic fragment of 9 (bottom left), and the PtC4 core of 9 (bottom right) with ellipsoids shown at a 50% probability level. Hydrogen atoms and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: (7) Ni1–C2 1.896(2), Ni1–C6 1.871(2), Ni1–C10 1.888(2), Ni1–C14 1.910(2), C10–Ni1–C2 164.46(7), C6–Ni1–C14 162.15(7); (9) Pt1–C2 2.041(5), Pt1–C6 2.000(5), Pt1–C10 2.002(5), Pt1–C14 2.026(5), C2–Pt1–C10 166.0(2), C6–Pt2–C14 167.01(19). | ||
The resulting τ4 and τδ (ppm) values vary between 0.00 for an ideally square planar coordinated metal and 1.00 for a tetrahedral coordination. For complex 7 the values are determined with 0.23 which is slightly closer to square planar coordination compared to τ4 and τδ values for similar NiII(NCCN) systems (0.28 to 0.30).43 However, macrocyclic tetra-NHC complexes with group 10 metals feature τ values below 0.10, indicating a nearly ideal square planar coordination geometry.26τ values for 9 are calculated to 0.19, being slightly lower than in the case of nickel. Despite our best efforts, single crystals of 8 could not be obtained.
Elongation of the centered alkyl bridge from methylene to propylene causes a significant structural change for silver complexes.38 However, reacting silver complex 2 with one equivalent of metal halide leads to the formation of square planar-coordinated monomeric complexes for all metals of the nickel group similar to the results obtained with 1 as precursor (Scheme 4).
 | ||
| Scheme 4 Synthesis of NiII (10), PdII (11), PtII (12) tetra-NHC complexes starting from silver complex 2. | ||
In analogy to 7–9, 1H and 13C NMR spectroscopy in solution indicate a plane of symmetry for 10–12. Characteristic signals for carbene carbons are identified at 170.67 and 170.64 ppm for 10, at 169.88 and 169.51 ppm for 11, and at 163.65 and 162.23 ppm for 12. ESI-MS measurements confirm the results obtained by NMR spectroscopy with m/z 566.99 (10), 614.57 (11), and 703.49 (12).
Single crystal XRD of both 10 and 11 reveal a distorted square planar coordination of the metal, confirming the suggested structure also in solid state (Fig. 6). As seen for 7, metal–carbon distances are in the usual range of 1.888 Å to 1.927 Å for nickel complex 10 and 2.019 Å to 2.055 Å for palladium complex 11.26 Enhanced flexibility in the ligand leads to less distortion of the square planar structure, reflected in the τ4 and τδ values of 0.11 for 10 and 0.08 for 11, respectively. This finding contrasts results obtained for the introduction of a centered propylene bridge to an NCCN ligand. τ values of 0.23 to 0.31 still indicate a strongly distorted square planar coordination.44,45
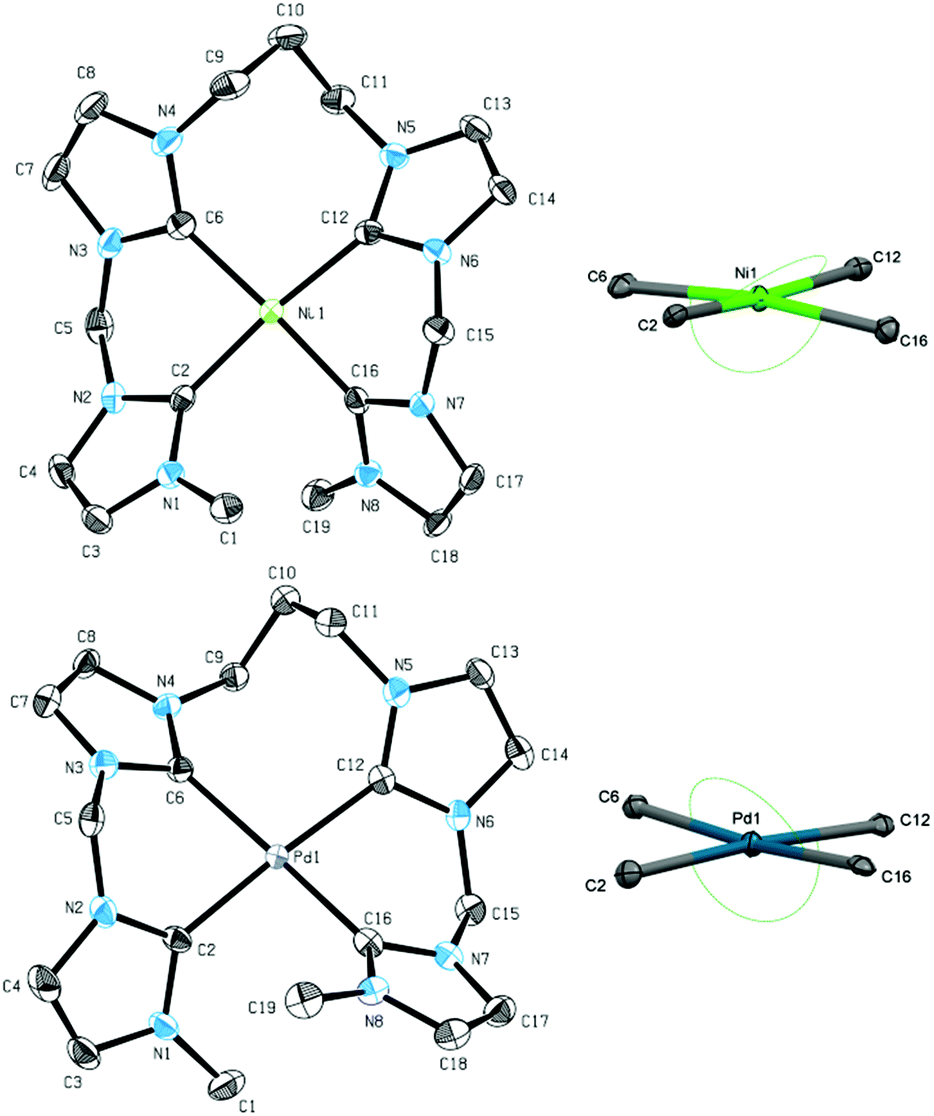 | ||
| Fig. 6 ORTEP style representation of the dicationic fragments of 10 (top left), the NiC4 core of 10 (top right), the dicationic fragment of 11 (bottom left), and the PdC4 core of 11 (bottom right) with ellipsoids shown at a 50% probability level. Hydrogen atoms, co-crystallized solvent molecules, and PF6− counter ions are omitted for clarity. In case of 11, the asymmetric unit contains two additional moieties. Selected bond lengths [Å] and angles [°]: (10) Ni1–C2 1.906(2), Ni1–C6 1.927(3), Ni1–C12 1.925(2), Ni1–C16 1.888(3), C2–Ni1–C12 171. 9(1), C16–Ni1–C6 172.4(1); (11) Pd1–C2 2.019(9), Pd1–C6 2.031(10), Pd1–C12 2.055(9), Pd1–C16 2.023(10), C2–Pd1–C12 175.7(4), C16–Pd1–C6 173.1(4). | ||
Transmetalation to iron
Previously, it has been shown that the length of the alkyl bridge of a tetradentate ligand influences the coordination mode of the ligand in octahedral FeII complexes.46 While a methylene bridged NCMeCN ligand coordinates in an equatorial fashion, a propylene bridge (NCPrCN) induces a conformational change towards a sawhorse-type coordination of the ligand with two cis acetonitrile ligands. Inspired by these findings, silver tetra-NHC complex 1 was reacted with two equivalents of FeBr2(THF)2 (Scheme 5). The monomeric FeII complex 13 (m/z 536.58) was isolated as orange powder in 75% yield. Fluxionality of the coordinated tetra-NHC ligand as indicated in Scheme 5 was monitored by 1H NMR spectroscopy (Fig. 7). Two different conformers are found at low temperature in acetonitrile solution (−40 °C). The main signal set indicates a sawhorse-type coordination of the tetra-NHC ligand and two cis acetonitrile ligands. A plane of symmetry is revealed by the second signal set, as the terminal methyl groups give only one signal at 3.5 ppm and the bridging methylene groups do not split up into doublets. These observations suggest an equatorial coordination of the ligand with two trans acetonitrile ligands. At room temperature the coalescence point is found in the 1H NMR spectra with a peak width of more than 0.2 ppm. Higher temperatures of 50 °C to 80 °C lead to a fast interconversion of the two coordination modes, as shown by the reappearance of sharp signals.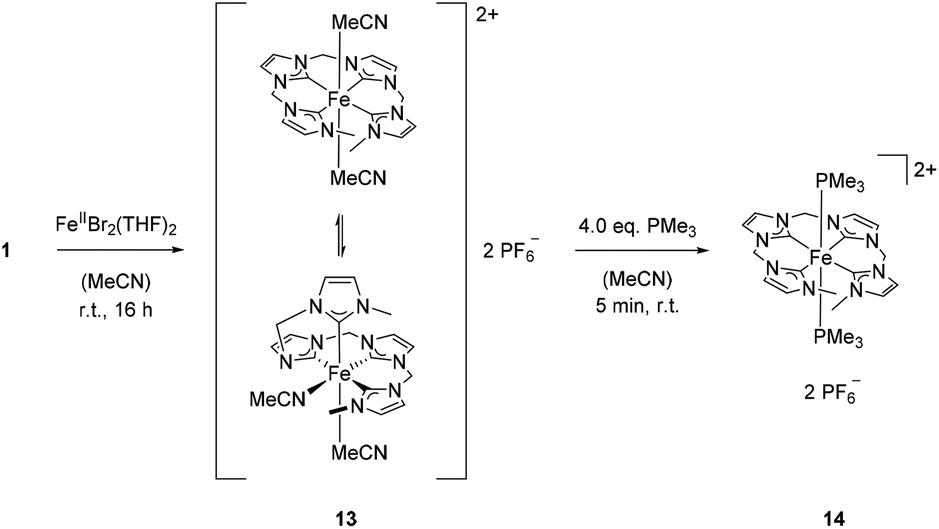 | ||
| Scheme 5 Synthesis of iron(II) complex 13 and subsequent reaction with PMe3 to form 14. | ||
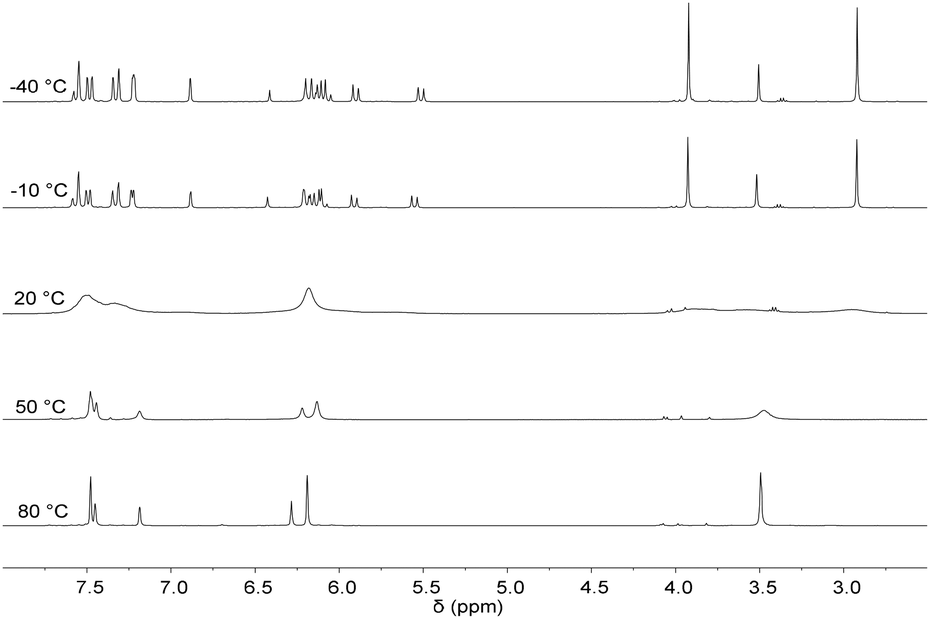 | ||
| Fig. 7 VT 1H NMR spectra of 13 in CD3CN from −40 °C to 80 °C. | ||
In order to inhibit interconversion, an irreversible exchange of the acetonitrile ligands by PMe3 was attempted.47 Upon addition of excess PMe3 to an acetonitrile solution of 13, di(PMe3) substituted iron(II) complex 14 can be isolated (Scheme 5). Compared to complex 13, 1H NMR recorded at room temperature exhibits sharp signals of only one species with a plane of symmetry as indicated by a single singlet signal for the terminal methyl groups and a single triplet signal for the methyl groups of the phosphine ligands. Integration of peak areas reveals the coordination of two phosphine ligands per iron center. Single crystal XRD of 14 confirms the spectroscopic findings in solution with two trans PMe3 ligands (Fig. 8). The tetra-NHC ligand is coordinated in an equatorial fashion with iron–carbon distances between 1.930 and 1.999 Å, which is comparable to those in literature for macrocyclic tetra-NHC ligands.26 The phosphine ligands are coordinated in the axial positions with a P1–Fe–P1a angle of 171° and P–Fe distances of around 2.27 Å. τ values could be calculated to 0.13 disregarding the axial phosphine ligands. This value is comparable to iron(II) systems with macrocyclic tetra-NHC ligands.26
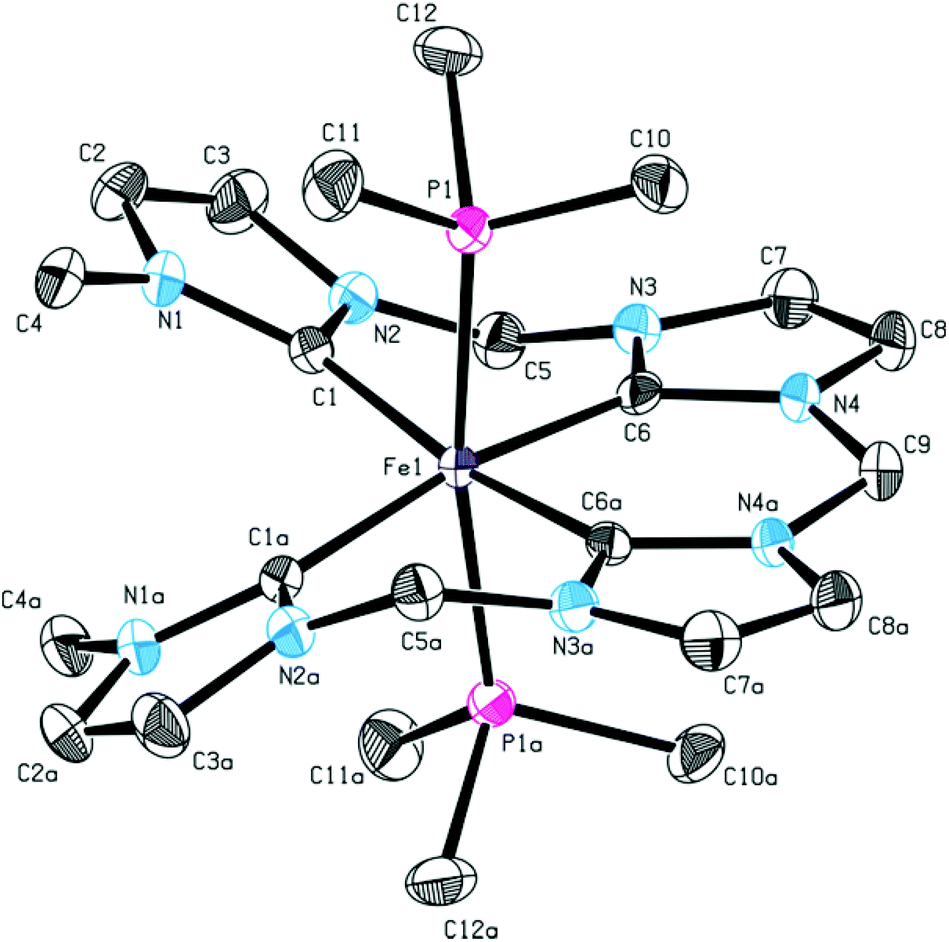 | ||
| Fig. 8 ORTEP style representation of the dicationic fragment of 14 with ellipsoids shown at a 50% probability level. Hydrogen atoms, co-crystallized solvent molecules, and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: Fe1–C1 1.999(4), Fe1–C6 1.930(4), Fe1–P1 2.2689(9), C6–Fe1–C6a 87.9(2), C6–Fe1–C1 89.53(13), C6–Fe1–C1a 170.42(14), P1–Fe1–P1a 170.57(5). | ||
Starting with complex 2, the synthesis of the iron(II) complex 15 was achieved in analogy to complex 13 (Scheme 6). Compared to 13, which shows fluxional behaviour in solution, 15 exhibits an inflexible octahedral coordination of the metal with two cis acetonitrile ligands and a sawhorse-type coordination of the tetra-NHC. Similar to the major species identified by low temperature 1H NMR spectroscopy of 13, an asymmetric set of signals was observed for 15.
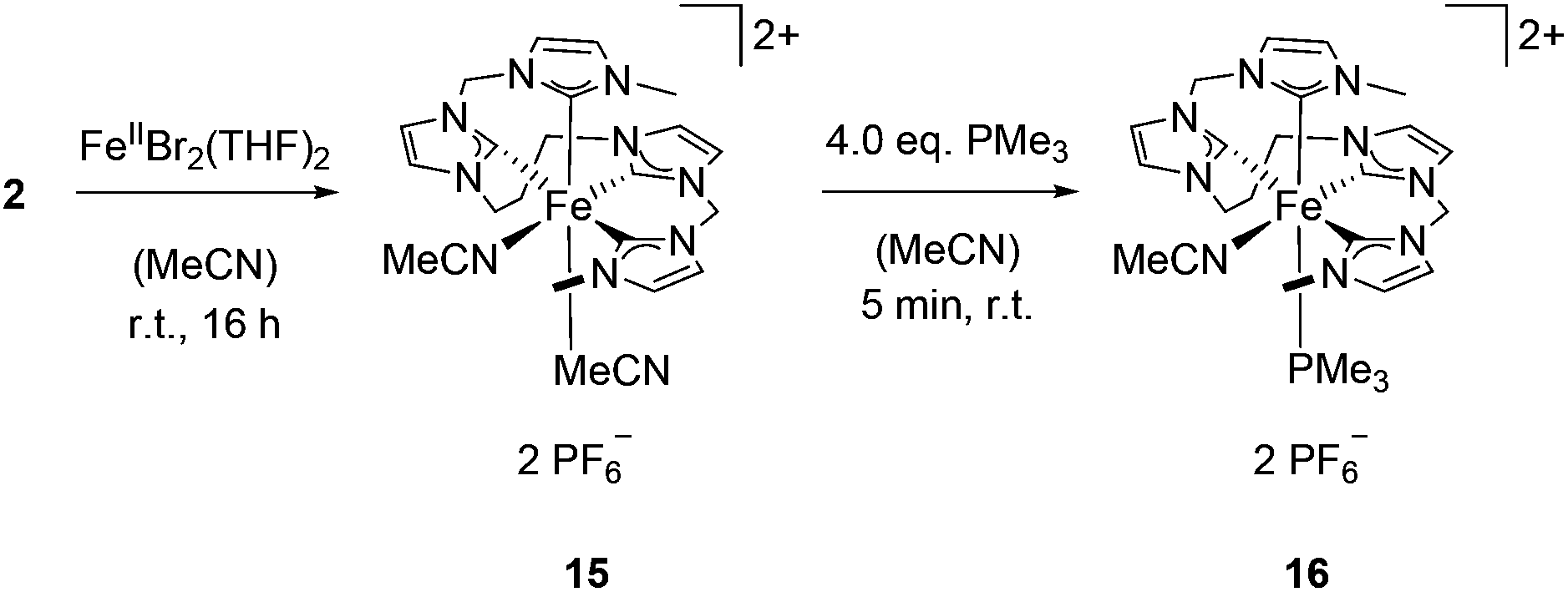 | ||
| Scheme 6 Synthesis of iron(II) complex 15 and subsequent reaction with PMe3 to yield 16. | ||
In addition, signals for the two methylene bridges with germinal coupling of 14 Hz and two independent signals for the different terminal methyl groups support a sawhorse-type coordination mode of the NHC ligand within the octahedral iron complex. With m/z 564.63, ESI-MS confirms the formation of a monomeric complex.
Reaction of 15 with an excess of PMe3 was performed (Scheme 6). Even with a fourfold excess of trimethylphosphine, only formation of mono(PMe3) substituted complex 16 is observed with the second acetonitrile ligand remaining coordinated. The sawhorse-type coordination mode of the tetra-NHC is retained upon introduction of the PMe3 ligand. The 1H NMR spectra of 15 and 16 only differ in the proton signals of trimethylphosphine. Single crystal XRD of 16 proves sawhorse-type coordination of the tetra-NHC ligand within a distorted octahedral-coordinated iron(II) complex featuring an acetonitrile ligand cis-positioned to the phosphine ligand (Fig. 9). Iron–carbon bond lengths are in the range of 1.936 to 1.996 Å while the Fe–P1 distance is 2.284 Å and therefore slightly larger than in complex 14. In case of 16, the τ4 value is calculated to 0.66 while the τδ value is 0.38.
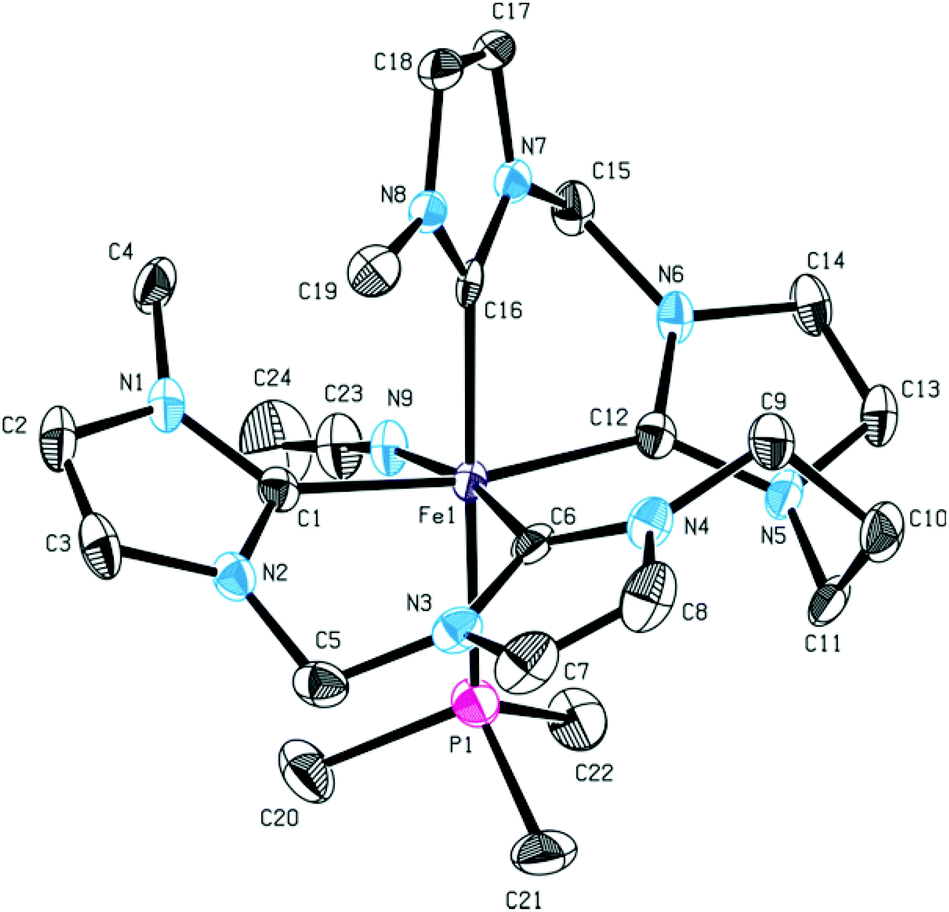 | ||
| Fig. 9 ORTEP style representation of the dicationic fragment of 16 with ellipsoids shown at a 50% probability level. Hydrogen atoms, co-crystallized solvent molecules, and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: Fe1–C1 1.972(3), Fe1–C6 1.936(3), Fe1–C12 1.996(2), Fe1–C16 1.965(2), Fe1–P1 2.2842(8), C16–Fe1–P1 173.98(7), C1–Fe1–C12 169.45(10), C6–Fe1–N9 169.97(10). | ||
Both values, within their scales, confirm a classical sawhorse-type coordination of the tetra-NHC ligand for the first time within an octahedral-coordinated metal complex.
Conclusion
The application of two silver complexes with flexible, open chain tetra-NHC ligands as versatile transmetalation agents was examined. A variety of d-block metal complexes has been synthesized and fully characterized. For coinage metals in the oxidation state +I tetranuclear, dimeric (L1) or dinuclear, monomeric complexes (L2) were obtained. Transmetalation to group 10 metals leads to monomeric, distorted square planar-coordinated complexes. Flexibility of the ligand system was demonstrated by formation of octahedrally coordinated iron complexes. For methylene-bridged tetracarbene ligand L1, the iron complex exhibits both sawhorse-type and equatorial coordination by the tetra-NHC with two additional acetonitrile ligands coordinated. In solution these conformers readily interconvert at room temperature, while replacing acetonitrile with PMe3 freezes the ligand in an equatorial coordination mode with two trans phosphine ligands. On the other hand, propylene-bridged tetracarbene ligand L2 forms a monomeric iron complex with sawhorse-type coordination of the ligand and two cis acetonitrile ligands. An excess of PMe3 only yields mono-substituted iron complex 16, retaining the sawhorse-type coordination of the ligand and an acetonitrile ligand cis to the phosphine. As shown for other ligand classes before, tetracarbene ligands can be designed according to specific demands; in this case, enhanced flexibility of the ligand leads to the first literature-known metal complex with a tetracarbene ligand coordinating in a sawhorse-type coordination mode with two cis-labile binding sites.Experimental section
General remarks
All chemicals were purchased from commercial suppliers and used without further purification. Anhydrous acetonitrile and diethyl ether were obtained from an MBraun solvent purification system, degassed by freeze–pump–thaw technique and stored over molecular sieves. All syntheses were performed using standard Schlenk technique if not stated otherwise. Silver complexes 1 and 2 were synthesized according to previously reported procedures.38 Liquid NMR spectra were recorded on a Bruker Avance DPX 400 and a Bruker DRX 400. Chemical shifts are given in parts per million (ppm) and the spectra were referenced by using the residual solvent shift as internal standards (acetonitrile-d3, 1H δ (ppm) 1.94, 13C δ (ppm) 118.26). MS-ESI analyses were performed on a Thermo Scientific LCQ/Fleet spectrometer by Thermo Fisher Scientific. Elemental analysis was obtained from the microanalytical laboratory of the Technische Universität München.Single crystal X-ray diffraction
For crystallization, diethyl ether was slowly diffused into acetonitrile solutions of compounds 3, 5, 6, 7, 9, 10, 11, 14, and 16, respectively. Data was collected on an X-ray single crystal diffractometer equipped with a CCD detector (Bruker APEX II, κ-CCD), a rotating anode (Bruker AXS, FR591) with MoKα radiation (λ = 0.71073 Å) and a Montel optic (3, 7) or a fine-focussed sealed tube with MoKα radiation (λ = 0.71073 Å) and a graphite monochromator (5, 6, 9, 10, 11, 14, 16) by using the APEX2 software package.48 The measurements were performed on a single crystal coated with perfluorinated ether. The crystal was fixed on the top of a glass fiber and transferred to the diffractometer. The crystal was frozen under a stream of cold nitrogen. A matrix scan was used to determine the initial lattice parameters. Reflections were merged and corrected for Lorenz and polarization effects, scan speed, and background using SAINT.49 Absorption corrections, including odd and even ordered spherical harmonics were performed using SADABS.49 Space group assignments were based upon systematic absences, E statistics, and successful refinement of the structures. Structures were solved by direct methods with the aid of successive difference Fourier maps, and were refined against all data using SHELXLE50 in conjunction with SHELXL-2014.51 Hydrogen atoms were assigned to ideal positions and refined using a riding model with an isotropic thermal parameter 1.2 times that of the attached carbon atom (1.5 times for methyl hydrogen atoms). If not mentioned otherwise, non-hydrogen atoms were refined with anisotropic displacement parameters. Full-matrix least-squares refinements were carried out by minimizing ∑w(Fo2 − Fc2)2 with SHELXL-9752 weighting scheme. Neutral atom scattering factors for all atoms and anomalous dispersion corrections for the non-hydrogen atoms were taken from International Tables for Crystallography.53 Images of the crystal structures were generated by PLATON.54Synthesis
Acknowledgements
D. T. W., P. J. A., S. H., and C. J. gratefully acknowledge support from the TUM Graduate School. D. T. W. is thankful to Bruno Dominelli and Dominik Reich for their valuable contributions.Notes and references
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS PubMed.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS PubMed.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS PubMed.
- D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610–641 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed.
- A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 CrossRef CAS PubMed.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- J. M. Smith and D. Subedi, Dalton Trans., 2012, 41, 1423–1429 RSC.
- W.-T. Lee, R. A. Juarez, J. J. Scepaniak, S. B. Muñoz, D. A. Dickie, H. Wang and J. M. Smith, Inorg. Chem., 2014, 53, 8425–8430 CrossRef CAS PubMed.
- J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 10515–10517 CrossRef CAS PubMed.
- S. Meyer, I. Klawitter, S. Demeshko, E. Bill and F. Meyer, Angew. Chem., Int. Ed., 2013, 52, 901–905 CrossRef CAS PubMed.
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed.
- S. A. Cramer, R. Hernandez Sanchez, D. F. Brakhage and D. M. Jenkins, Chem. Commun., 2014, 50, 13967–13970 RSC.
- F. E. Hahn, V. Langenhahn, T. Lügger, T. Pape and D. Le Van, Angew. Chem., Int. Ed., 2005, 44, 3759–3763 CrossRef CAS PubMed.
- R. McKie, J. A. Murphy, S. R. Park, M. D. Spicer and S.-z. Zhou, Angew. Chem., Int. Ed., 2007, 46, 6525–6528 CrossRef CAS PubMed.
- S. R. Park, N. J. Findlay, J. Garnier, S. Zhou, M. D. Spicer and J. A. Murphy, Tetrahedron, 2009, 65, 10756–10761 CrossRef CAS PubMed.
- H. M. Bass, S. A. Cramer, J. L. Price and D. M. Jenkins, Organometallics, 2010, 29, 3235–3238 CrossRef CAS.
- N. J. Findlay, S. R. Park, F. Schoenebeck, E. Cahard, S.-z. Zhou, L. E. A. Berlouis, M. D. Spicer, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2010, 132, 15462–15464 CrossRef CAS PubMed.
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087 RSC.
- H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160–2167 CrossRef CAS.
- S. A. Cramer, F. L. Sturgill, P. P. Chandrachud and D. M. Jenkins, Dalton Trans., 2014, 43, 7687–7690 RSC.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Chem. – Eur. J., 2008, 14, 10900–10904 CrossRef CAS PubMed.
- C. Radloff, H.-Y. Gong, C. Schulte to Brinke, T. Pape, V. M. Lynch, J. L. Sessler and F. E. Hahn, Chem. – Eur. J., 2010, 16, 13077–13081 CrossRef CAS PubMed.
- C. Schulte to Brinke, T. Pape and F. E. Hahn, Dalton Trans., 2013, 42, 7330–7337 RSC.
- A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed.
- A. Rit, T. Pape, A. Hepp and F. E. Hahn, Organometallics, 2010, 30, 334–347 CrossRef.
- B. N. Ahamed, R. Dutta and P. Ghosh, Inorg. Chem., 2013, 52, 4269–4276 CrossRef CAS PubMed.
- E. K. Bullough, M. A. Little and C. E. Willans, Organometallics, 2013, 32, 570–577 CrossRef CAS.
- C. Kupper, A. Schober, S. Demeshko, M. Bergner and F. Meyer, Inorg. Chem., 2015, 54, 3096–3098 CrossRef CAS PubMed.
- D. T. Weiss, S. Haslinger, C. Jandl, A. Pöthig, M. Cokoja and F. E. Kühn, Inorg. Chem., 2015, 54, 415–417 CrossRef CAS PubMed.
- I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642–670 CrossRef CAS PubMed.
- P. L. Chiu, C. L. Lai, C. F. Chang, C. H. Hu and H. M. Lee, Organometallics, 2005, 24, 6169–6178 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 CrossRef CAS PubMed.
- Z. Xi, X. Zhang, W. Chen, S. Fu and D. Wang, Organometallics, 2007, 26, 6636–6642 CrossRef CAS.
- V. S. Thoi and C. J. Chang, Chem. Commun., 2011, 47, 6578–6580 RSC.
- V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- A. Raba, M. Cokoja, S. Ewald, K. Riener, E. Herdtweck, A. Pöthig, W. A. Herrmann and F. E. Kühn, Organometallics, 2012, 31, 2793–2800 CrossRef CAS.
- S. Haslinger, J. W. Kück, E. M. Hahn, M. Cokoja, A. Pöthig, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2014, 53, 11573–11583 CrossRef CAS PubMed.
- APEX suite of crystallographic software, APEX 2, version 2008.4, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- SAINT, version 7.56a, SADABS, version 2008/1, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- SHELXLE: C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-2014, University of Göttingen, Göttingen, Germany, 2014 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
- A. J. C. Wilson, International Tables for Crystallography, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1992 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopic data and X-ray crystallographic. CCDC 1401411–1401419. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02386f |
| This journal is © The Royal Society of Chemistry 2015 |