
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium(II) complexes with highly basic imidazolin-2-imines and their reactivity toward small bio-molecules†
Jovana
Bogojeski
a,
Jeroen
Volbeda
b,
Matthias
Freytag
b,
Matthias
Tamm
*b and
Živadin D.
Bugarčić
*a
aDepartment of Chemistry, Faculty of Science, University of Kragujevac, R. Domanovića 12, P. O. Box 60, 34000 Kragujevac, Serbia. E-mail: bugarcic@kg.ac.rs; Fax: +381 (0)34335040; Tel: +381 (0)34300262
bInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: m.tamm@tu-bs.de; Fax: +49-531-391-5387; Tel: +49-531-391-5309
First published on 2nd September 2015
Abstract
A series of novel Pd(II) complexes with chelating mono(imidazolin-2-imine) and bis(imidazolin-2-imine) ligands were synthesized. The crystal structures of [Pd(DMEAImiPr)Cl2] and [Pd(DPENImiPr)Cl2] were determined by X-ray diffraction analysis. The reactivity of the six Pd(II) complexes, namely, [Pd(en)Cl2], [Pd(EAImiPr)Cl2], [Pd(DMEAImiPr)Cl2], [Pd(DPENImiPr)Cl2], [Pd(BLiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2], were investigated. Spectrophotometric acid–base titrations were performed to determine the pKa values of the coordinated water molecules in [Pd(en)(H2O)2]2+, [Pd(EAImiPr)(H2O)2]2+, [Pd(DMEAImiPr)(H2O)2]2+, [Pd(DPENImiPr)(H2O)2]2+, [Pd(BLiPr)(H2O)2]2+ and [Pd(DACH(ImiPr)2)(H2O)2]2+. The substitution of the chloride ligands in these complexes by TU, L-Met, L-His and Gly was studied under pseudo-first-order conditions as a function of the nucleophile concentration and temperature using stopped-flow techniques; the sulfur-donor nucleophiles have shown better reactivity than nitrogen-donor nucleophiles. The obtained results indicate that there is a clear correlation between the nature of the imidazolin-2-imine ligands and the acid–base characteristics and reactivity of the resulting Pd(II) complexes; the order of reactivity of the investigated Pd(II) complexes is: [Pd(en)Cl2] > [Pd(EAImiPr)Cl2] > [Pd(DMEAImiPr)Cl2] > [Pd(DPENImiPr)Cl2] > [Pd(BLiPr)Cl2] > [Pd(DACH(ImiPr)2)Cl2]. The solubility measurements revealed good solubility of the studied imidazolin-2-imine complexes in water, despite the fact that these Pd(II) complexes are neutral complexes. Based on the performed studies, three unusual features of the novel imidazolin-2-imine Pd(II) complexes are observed, that is, good solubility in water, very low reactivity and high pKa values. The coordination geometries around the palladium atoms are distorted square-planar; the [Pd(DMEAImiPr)Cl2] complex displays Pd–N distances of 2.013(2) and 2.076(2) Å, while the [Pd(DPENImiPr)Cl2] complex displays similar Pd–N distances of 2.034(4) and 2.038(3) Å. The studied systems are of interest because little is known about the substitution behavior of imidazolin-2-imine Pd(II) complexes with bio-molecules under physiological conditions.
Introduction
The chemical behavior, in solution, of structurally analogous Pt(II) and Pd(II) complexes is very similar, which has advanced the progress of research in the area of the development of new Pd(II) complexes.1 However, palladium(II) complexes react ca. 104–105 times faster than their platinum(II) analogues,1 so the selectivity of such complexes in the binding of bio-molecules is limited, which leads to low anti-tumor activity. The reaction rate of Pd(II) complexes can be slowed down and side reactions can be prevented through the introduction of inert sterically hindered ligands into the coordination sphere of the complex. A common practice in anticancer drug development is to modify leading structures in a suitable way that maximizes cytotoxic potency toward cancer cells.2–4Nowadays, cisplatin and its analogues are among the most effective chemotherapeutic agents in clinical use for the treatment of different types of cancers.2–5 However, the advantages and drawbacks of the widely used platinum-based anticancer drug cisplatin prompted a search for analogous transition metal complexes.3,4,6 To overcome the disadvantages of cisplatin, a huge number of metal ion complexes, among which are the Pd(II) complexes, were synthesized and tested.7–9
The chemistry of imidazolin-2-imine ligands has attracted attention because of the characteristic ability of imidazolines for effective acquisition and stabilization of a positive charge, which leads to the pronounced basic properties of nitrogen donor atoms and the formation of highly stable nitrogen–metal bonds.10 These features make imidazolin-2-imines the ideal ancillary ligands for applications in homogeneous catalysis. Over the past 10 years, this has led to the synthesis and application of a significant number of their metal complexes (from main group elements to lanthanides and actinides) in coordination chemistry and homogeneous catalysis.10–12
We anticipated that the great electron donating capacity and bulkiness of the mono- and bis(imidazolin-2-imine) ligands would result in Pd(II) complexes with greatly reduced reactivity; these complexes should in turn have increased potential as anti-tumor agents. Fig. 1 shows the structures of the Pd(II) complexes used in this study.
 | ||
| Fig. 1 Structures of the investigated Pd(II) complexes, along with their abbreviations. | ||
Results and discussion
Ligand synthesis
The ligands were synthesized in analogy to the procedure described in a previous publication.13 Stirring one equivalent of 2-chloro-1,3-diisopropyl-4,5-dimethylimidazolium salt, six equivalents of KF, three equivalents of triethylamine and an appropriate amount of different amines in acetonitrile overnight at room temperature gave the desired ligand salts (Scheme 1). The deprotonation of the imidazolin-2-imine ligand salts was performed by stirring with KOtBu in THF for a couple of days at room temperature, with exception of the [DPEN(ImiPrH)NH2][BF4] ligand salt, which was deprotonated by heating a THF solution overnight with NaNH2 at 40 °C. | ||
| Scheme 1 Synthesis of bidentate imidazolin-2-imine ligands starting from 2-chloro-1,3-diisopropyl-4,5-dimethylimidazolium tetrafluoroborate. | ||
All imidazolin-2-imine ligands (Scheme 1), together with the corresponding dicationic tetrafluoroborate salts, were characterized by 1H, 13C, and 19F NMR, elemental analysis and ESI-MS spectroscopy. These analytical methods confirmed the formation of the imidazolin-2-imines.
Complex synthesis
The complexes shown in Fig. 1 were synthesized by stirring equimolar amounts of [Pd(COD)Cl2] and the imidazolin-2-imine ligands in THF. The Pd(II) complexes [Pd(EAImiPr)Cl2], [Pd(DMEAImiPr)Cl2], [Pd(DPENImiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2] were characterized by 1H and 13C NMR spectroscopy, elemental analysis and ESI-MS mass spectrometry. For the complexes [Pd(DMEAImiPr)Cl2] and [Pd(DPENImiPr)Cl2], single crystals suitable for X-ray analysis were also obtained.The 1H NMR as well as 13C NMR spectra of the [Pd(EAImiPr)Cl2] and [Pd(DMEAImiPr)Cl2] complexes display a set of signals for the imidazolin moiety significantly shifted compared to the free ligand. In addition, the methyl groups of the isopropyl substituents afford two doublets in the 1H NMR spectrum. This indicates hindered rotation along the imine C–N bond on the NMR timescale at room temperature, which has previously been observed for related complexes bearing both chiral and achiral ligands.10–13 The achiral Pd(II) complex, [Pd(BLiPr)Cl2],14 shows just one doublet in the 1H NMR spectra for the methyl groups of the isopropyl substituents. The [Pd(DPENImiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2] exhibit four doublets in the 1H NMR spectrum, which is expected considering that ligands DPENImiPr and DACH(ImiPr)2 show diastereotopic signals for the methyl substituents on the isopropyl groups combined with a hindered rotation around the imine C–N bond in the Pd(II) complex.
The mass spectrum of [Pd(DACH(ImiPr)2)Cl2] in the m/z range of 400–700 includes main peaks at m/z = 471 (1+), 575 (2+), and 611 (1+), which correspond to [(DACH(ImiPr)2)H]+, [Pd(DACH(ImiPr)2)]2+ and [Pd(DACH(ImiPr)2)Cl]+ and represent fragments of the [Pd(DACH(ImiPr)2)Cl2] complex. Furthermore, Fig. S1† shows the isotopic pattern at m/z = 611.28, which belongs to the [Pd(DACH(ImiPr)2)Cl2] complex, without one chloride ion, i.e. [Pd(DACH(ImiPr)2)Cl]+, and its corresponding simulated pattern.
Preparation and structure of [Pd(DMEAImiPr)Cl2]
The reaction of DMEAImiPr with [Pd(COD)Cl2] (COD = 1,5-cyclooctadiene) in THF afforded the complex [Pd(DMEAImiPr)Cl2] as a deep-red solid. Red crystals suitable for X-ray diffraction analysis were isolated from a dichloromethane/n-hexane solution, and the molecular structure of [Pd(DMEAImiPr)Cl2] is shown in Fig. 2. The molecule crystallizes in the orthorhombic space group Pnma, with one co-crystallized molecule of CH2Cl2 per asymmetric unit and one located on a crystallographic mirror plane. The ligand is coordinated to the palladium atom in a chelating, bidentate fashion with an N1–Pd–N2 bite angle of 83.08(9)°. The Pd–N bond lengths are 2.013(2) and 2.076(2) Å for Pd–N1 and Pd–N2, respectively, indicating a stronger coordination of the more basic imine nitrogen to the Pd atom. The imine bond length of 1.349(4) Å (N1–C3) is long for a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond, indicating a pronounced negative charge on the imine nitrogen. This is also confirmed by the ρ-value of 0.998; the ρ-value is defined as ρ = 2a/(b + c), with (a) denoting the exo- and (b) and (c) the endo-cyclic bond lengths within the guanidine CN3 moiety. Values approaching unity indicate complete charge delocalization within the CN3 fragment.15 The electron-donating capacity of the imidazolin-2-imine is also reflected in the different Pd–Cl bond lengths, with the Pd–Cl1 bond length (2.3390(7) Å) trans to the imine nitrogen being significantly longer than the Pd–Cl2 bond length (2.2951(7) Å) trans to the tertiary amine. These structural parameters are comparable to those observed for [Pd(BLiPr)Cl2] and the PdCl2 complex of tetramethylethylenediamine.14,16
N double bond, indicating a pronounced negative charge on the imine nitrogen. This is also confirmed by the ρ-value of 0.998; the ρ-value is defined as ρ = 2a/(b + c), with (a) denoting the exo- and (b) and (c) the endo-cyclic bond lengths within the guanidine CN3 moiety. Values approaching unity indicate complete charge delocalization within the CN3 fragment.15 The electron-donating capacity of the imidazolin-2-imine is also reflected in the different Pd–Cl bond lengths, with the Pd–Cl1 bond length (2.3390(7) Å) trans to the imine nitrogen being significantly longer than the Pd–Cl2 bond length (2.2951(7) Å) trans to the tertiary amine. These structural parameters are comparable to those observed for [Pd(BLiPr)Cl2] and the PdCl2 complex of tetramethylethylenediamine.14,16
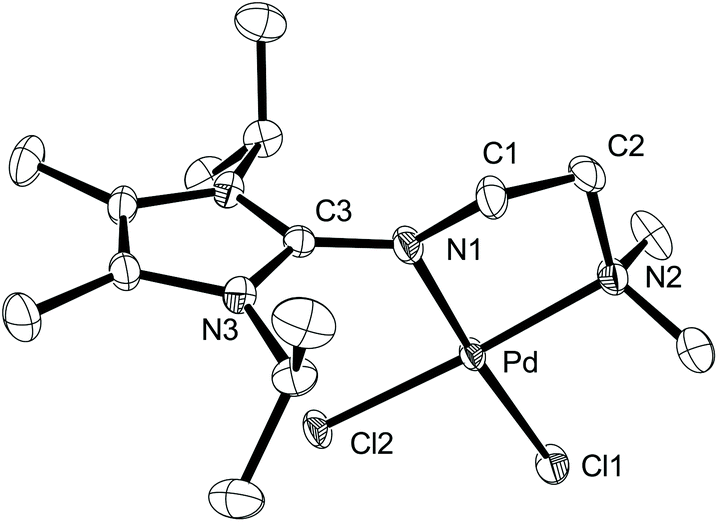 | ||
| Fig. 2 ORTEP drawing of [Pd(DMEAImiPr)Cl2]·CH2Cl2 with thermal displacement parameters drawn at 50% probability. The alternative disordered positions of C1 and C2, hydrogen atoms and a co-crystallized molecule of CH2Cl2 were omitted for clarity. Selected bond distances [Å] and angles [°]: Pd–N1 2.013(2), Pd–N2 2.076(2), Pd–Cl1 2.3390(7), Pd–Cl2 2.2951(7), N1–C3 1.349(4), N3–C3 1.352(2); N1–Pd–N2 83.08(9), Cl1–Pd–Cl2 92.91(7). CCDC 1407330. | ||
Preparation and structure of [Pd(DPENImiPr)Cl2]
The reaction of DPENImiPr with [Pd(COD)Cl2] in THF afforded the complex [Pd(DPENImiPr)Cl2] as an orange-red solid. Red crystals suitable for X-ray diffraction analysis were isolated from an acetone/n-hexane solution, and the molecular structure of [Pd(DMEAImiPr)Cl2] is shown in Fig. 3. The compound crystallizes in the orthorhombic space group P212121 with four independent complexes, five molecules of acetone and one molecule of hexane per asymmetric unit. The ligand is coordinated to the palladium atom in a chelating, bidentate fashion with an N1–Pd–N2 bite angle of 82.69(14)°. The Pd–N bond lengths are virtually identical at 2.034(4) and 2.038(3) Å for Pd–N1 and Pd–N2, respectively. The imine C15–N1 bond length of 1.367(5) Å is long for a CN double bond and the corresponding ρ-value of 1.004 indicates a strong negative charge on the exocyclic imine nitrogen atom. Again, the Pd–Cl2 (2.3395(10) Å) distance trans to the imine nitrogen is longer than the Pd–Cl bond which is trans to the primary amine Pd–Cl1 (2.3193(10) Å).
 | ||
| Fig. 3 ORTEP drawing of one of the four molecules of [Pd(DPENImiPr)Cl2] in [Pd(DPENImiPr)Cl2]·acetone with thermal displacement parameters drawn at 50% probability. Hydrogen atoms and a co-crystallized molecule of acetone were omitted for clarity. Selected bond distances [Å] and angles [°]: Pd–N1 2.034(4), Pd–N2 2.038(3), Pd–Cl1 2.3193(10), Pd–Cl2 2.3395(10), N1–C15 1.367(5), N3–C15 1.363(5), N4–C15 1.359(6); N1–Pd–N2 82.69(14), Cl1–Pd–Cl2 91.85(4). CCDC 1407331. | ||
Solubility of the imidazolin-2-imine Pd(II) complexes
Despite the fact that the prepared Pd(II) complexes are neutral, UV-Vis spectrophotometric measurements showed that they have good solubility in water (see Table 1). The solubility of the complexes is about four times greater than that observed for cisplatin and ca. twice that of oxaliplatin. This high solubility is promising for the intended application of these complexes as anti-tumor agents. To this day, cisplatin is the most widely used metal complex based cytostatic in the world.2–5 One of the drawbacks of cisplatin as an anti-tumor drug is its low solubility in aqueous media, therefore an increase in water solubility is an important goal in the design of any new metallo-drug. A higher water solubility can also result in a decreased toxicity of metallo-drugs, as is the case for oxaliplatin, which has less nephrotoxicity and a higher water solubility than cisplatin.2–5 Herein, we show that the introduction of different imidazolin-2-imines affords Pd(II) complexes which are significantly more water soluble than cisplatin.pKa Determination of the aqua Pd(II) complexes
It is well established that the low chloride concentration of approximately 4 mM in living cells causes Pt(II) and Pd(II) dichloride complexes to form diaqua complexes upon entering the cell. These diaqua complexes then bind to the DNA.17 There is a correlation between the pKa values of the coordinated water molecules and the electronic structure of the complexes and accordingly the reactivity of the complexes. Therefore, the pKa values of the complexes in aqueous solution were determined. This was done via UV-vis spectrophotometric pH titration with NaOH as a base in the pH range between 2 and 12. Each pKa titration was performed twice and the average of both values was taken. Fig. 4 (see also Fig. S2–S4 of the ESI†) shows a plot of the absorbance versus pH at specific wavelengths, which was used to determine the pKa values of the coordinated water molecules. The data were fitted using a nonlinear least-squares procedure, as shown in the inset of Fig. 4. The overall process can be presented by eqn (1) and (2). The data obtained for the pKa values are summarized in Table 2. | (1) |
 | (2) |
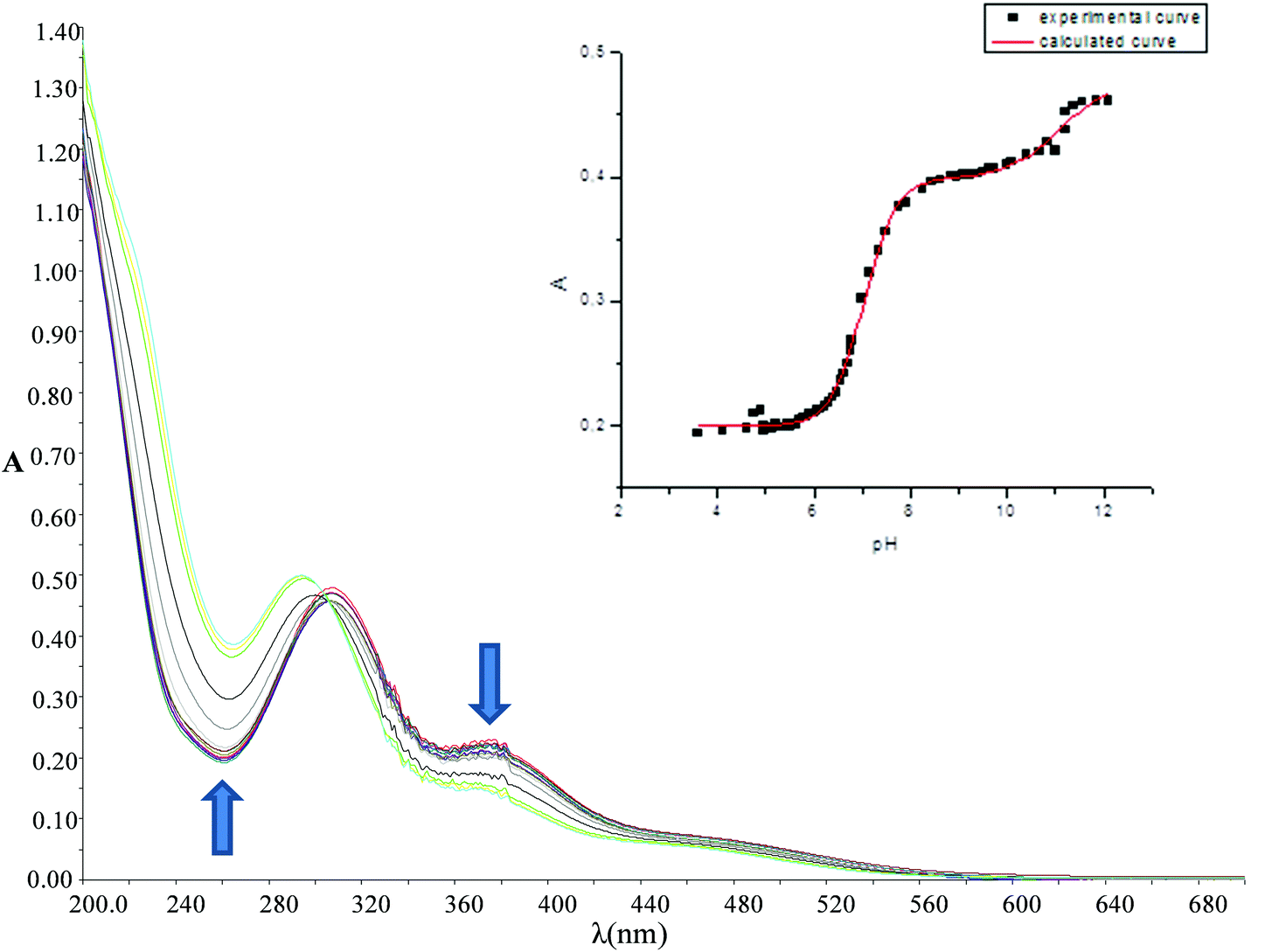 | ||
| Fig. 4 UV-vis spectra recorded for 0.1 mM [Pd(DPENImiPr)(H2O)2]2+ in the pH range 2 to 12 at 25 °C. Insert: plot of absorbance vs. pH at 260 nm (experimental curve and calculated curve). | ||
| pKa1 | pKa2 | |
|---|---|---|
| [Pd(en)(H2O)2]2+ (ref. 18) | 5.60 ± 0.05 | 7.30 ± 0.05 |
| [Pd(EAImiPr)(H2O)2]2+ | 5.45 ± 0.10 | 7.62 ± 0.20 |
| [Pd(DMEAImiPr)(H2O)2]2+ | 5.75 ± 0.10 | 8.28 ± 0.10 |
| [Pd(DPENImiPr)(H2O)2]2+ | 7.17 ± 0.20 | 11.21 ± 0.10 |
| [Pd(BLiPr)(H2O)2]2+ (ref. 14) | 6.18 ± 0.05 | 10.07 ± 0.05 |
| [Pd(DACH(ImiPr)2)(H2O)2]2+ | 7.56 ± 0.20 | 8.39 ± 0.20 |
Table 2 shows that there is a significant difference between pKa1 and pKa2 values. The data for [Pd(en)(H2O)2]2+ (pKa1 = 5.6, pKa2 = 7.3)18 represent typical values for complexes containing sp3-hybridized amines.19 In comparison, Pd(II) complexes with imidazolin-2-imine ligands give higher pKa values, in agreement with the electron-rich nature of the palladium centers in these complexes, in which deprotonation of the aqua ligand and formation of a negatively charged hydroxo-ligand are disfavored. The complexes with mono(imidazolin-2-imine) ligands, [Pd(EAImiPr)(H2O)2]2+, [Pd(DMEAImiPr)(H2O)2]2+ and [Pd(DPENImiPr)(H2O)2]2+, have two different types of donors, viz. an imidazolin-2-imine moiety and an sp3-hybridized primary or tertiary amine (–NR2) unit. In these cases, it can be assumed that the first aqua ligand to be deprotonated would be that trans to the less electron donating amine donor.
The pH in healthy human cells lies between 7.3 and 7.4.20 As the pKa1 values listed in Table 2 are lower than 7.3, the formation of Pd(II) monohydroxo species can be expected. Conversely, the pH in cancer cells is lower (around 6.2 to 6.9),20 suggesting that the hydrolyzed [Pd(DPENImiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2] complexes will mainly exist as diaqua species while other complexes will exist as monohydroxo species. Formation of monohydroxo species and diaqua complexes is good because Pd–OH species are more substitution-inert but Pd–OH can also act as a base, deprotonating a H–N site in nucleobases and binding to the N atom, while diaqua species can readily bind to DNA.
Kinetic studies
To get an idea of the robustness of the Pd(II) complexes in living tissue, the substitution reactions of Pd(II) complexes with selected nucleophiles (Fig. 5) was investigated. The change in absorbance was followed, at suitable wavelengths, as a function of time at 310 K and pH ≈ 7.2. The proposed reaction pathways for all the observed substitution processes are presented in Scheme 2. The substitution reactions of all Pd(II) complexes proceed in two successive reaction steps that are both dependent on the nucleophile concentration. | ||
| Fig. 5 Structures of the investigated nucleophiles, along with the abbreviations used. | ||
 | ||
| Scheme 2 Nu = TU, L-Met, L-His and Gly. | ||
The entering ligands, L-Met, L-His and Gly, are essential amino acids; therefore, the metal complex (as an antitumor active drug) on its way from the injection to the diseased tissue will encounter them. On the basis of many investigations in the field of Pt(II) and Pd(II) complexes, it is known that both ions can easily form a bond with thioethers (such as L-Met).1,14,17 Pt–sulfur(thioether) adducts have been postulated to be a drug reservoir for platinum and may act as intermediate platinum complexes that can be transformed into Pt–DNA adducts,17 which makes the study of substitution reactions with L-Met very important. Thiourea was selected because it is a ligand with high solubility, neutral character and good nucleophilicity. Also, thiourea combines the ligand properties of thiolates as π donors21 and thioethers as σ donors and π acceptors.22 Also, thiourea is used as a protective and rescue agent to prevent side effects which are caused by Pt(II) antitumor drugs.23,24 Therefore, the investigation of the interactions of the selected nucleophiles and Pd(II) complexes is important for the development of potential antitumor drugs.
The substitution reactions of square-planar metal complexes can proceed according to two parallel pathways.25 One involves the formation of a solvent-coordinated complex, e.g. a diaqua complex, followed by rapid substitution of the coordinated solvent by the entering nucleophile (solvolytic pathway), whilst the other involves a direct nucleophilic attack by the entering nucleophile. To suppress the solvolytic pathway, a 30 mM NaCl solution was added (see the ESI, Fig. S5†). The rate constants for substitution could be determined, under pseudo-first-order conditions, from a plot of the linear dependence of kobsdversus the total nucleophile concentration, according to eqn (3) and (4). The slope of the line represents k1 or k2, whilst the intercept represents k−1[Cl−] or k−2[Cl−]. Plots of kobsd1,2versus nucleophile concentration led to a linear dependence with no meaningful intercept for all complexes and both substitution steps. The results are summarized in Tables 3 and 4.
| kobsd1 = k1[Nu] + k−1[Cl−] ≈ k1[Nu] | (3) |
| kobsd2 = k2[Nu] + k−2[Cl−] ≈ k2[Nu] | (4) |
| First reaction step | TU, k1 [M−1 s−1] | l-Met, k1 [M−1 s−1] | Gly, k1 [M−1 s−1] | l-His, k1[M−1 s−1] |
|---|---|---|---|---|
| [Pd(en)Cl2] | (6.95 ± 0.10) × 105 | (5.05 ± 0.10) × 105 | (1.02 ± 0.10)× 104 | (2.87 ± 0.20)× 105 |
| [Pd(EAImiPr)Cl2] | (1.25 ± 0.20) × 105 | (5.54 ± 0.20) × 104 | (1.45 ± 0.10) × 103 | (2.78 ± 0.10) × 103 |
| [Pd(DMEAImiPr)Cl2] | (6.78 ± 0.20) × 103 | (4.5 ± 0.10) × 103 | (1.28 ± 0.10) × 103 | (2.00 ± 0.20) × 103 |
| [Pd(DPENImiPr)Cl2] | (5.13 ± 0.20) × 103 | (2.53 ± 0.20) × 103 | (8.52 ± 0.20) × 102 | (1.48 ± 0.20) × 103 |
| [Pd(BLiPr)Cl2] | (9.20 ± 0.20) × 102 | (5.15 ± 0.20) × 102 | (2.93 ± 0.20) × 102 | (1.67 ± 0.20) × 102 |
| [Pd(DACH(ImiPr)2)Cl2] | (1.64 ± 0.20) × 102 | (1.51 ± 0.20) × 102 | 97.70 ± 0.10 | (1.35 ± 0.10) × 102 |
| Second reaction step | TU, k2 [M−1 s−1] | l-His, k2 [M−1 s−1] |
|---|---|---|
| [Pd(en)Cl2] | (1.53 ± 0.20) × 104 | (1.03 ± 0.10) × 104 |
| [Pd(EAImiPr)Cl2] | (8.52 ± 0.10) × 102 | 70.20 ± 0.10 |
| [Pd(DMEAImiPr)Cl2] | (4.22 ± 0.10) × 102 | 43.80 ± 0.10 |
| [Pd(DPENImiPr)Cl2] | (2.80 ± 0.20) × 102 | 39.20 ± 0.20 |
| [Pd(BLiPr)Cl2] | (1.84 ± 0.10) × 102 | 24.30 ± 0.10 |
| [Pd(DACH(ImiPr)2)Cl2] | 70.90 ± 0.10 | 19.20 ± 0.10 |
As an example, the kinetic traces for [Pd(DMEAImiPr)Cl2] including the necessary time scales for both reaction steps are shown in Fig. 6.
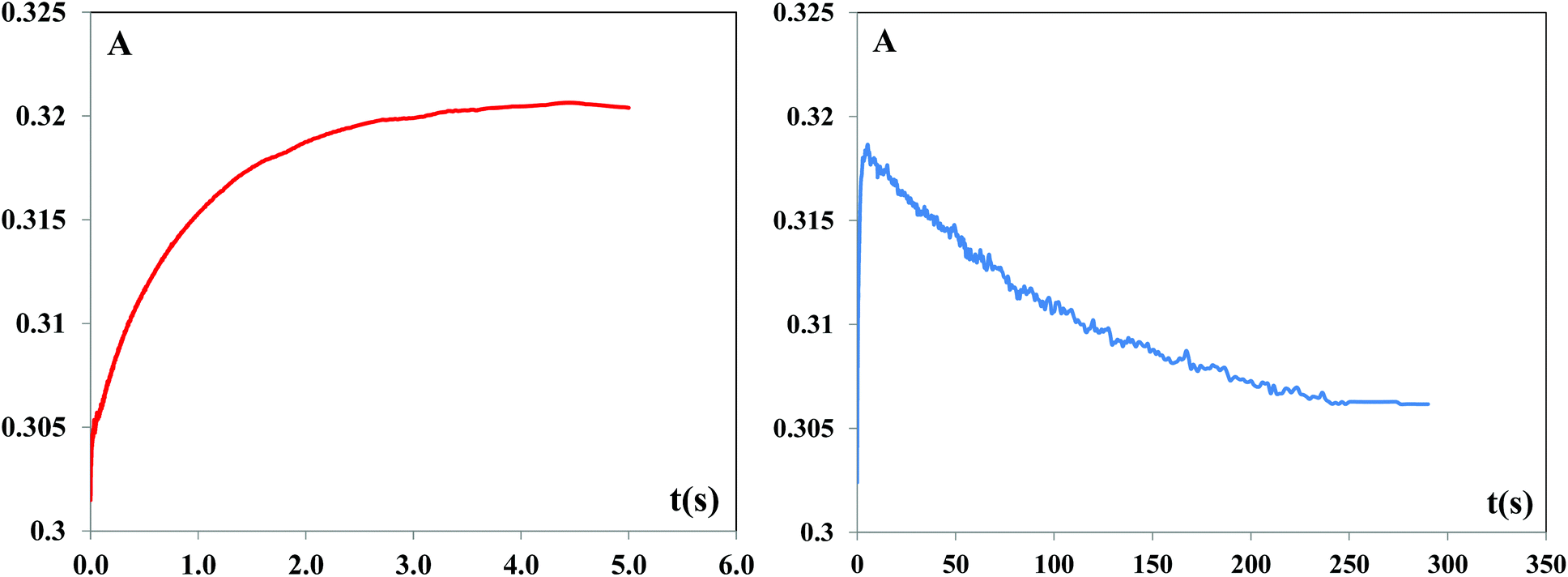 | ||
| Fig. 6 Absorbance–time traces for the reaction between [Pd(DMEAImiPr)Cl2] and TU (2 × 10−4 M); the left graph presents absorbance–time traces for the first substitution step, the right graph presents absorbance–time traces for both substitution steps at pH 7.2 and 310 K in 25 mM Hepes buffer and 30 mM NaCl. | ||
Fig. 7 shows the dependence of kobsd on nucleophile concentration for the [Pd(DPENImiPr)Cl2] and [Pd(BLiPr)Cl2] complexes (see also the ESI, Fig. S6–S11†).
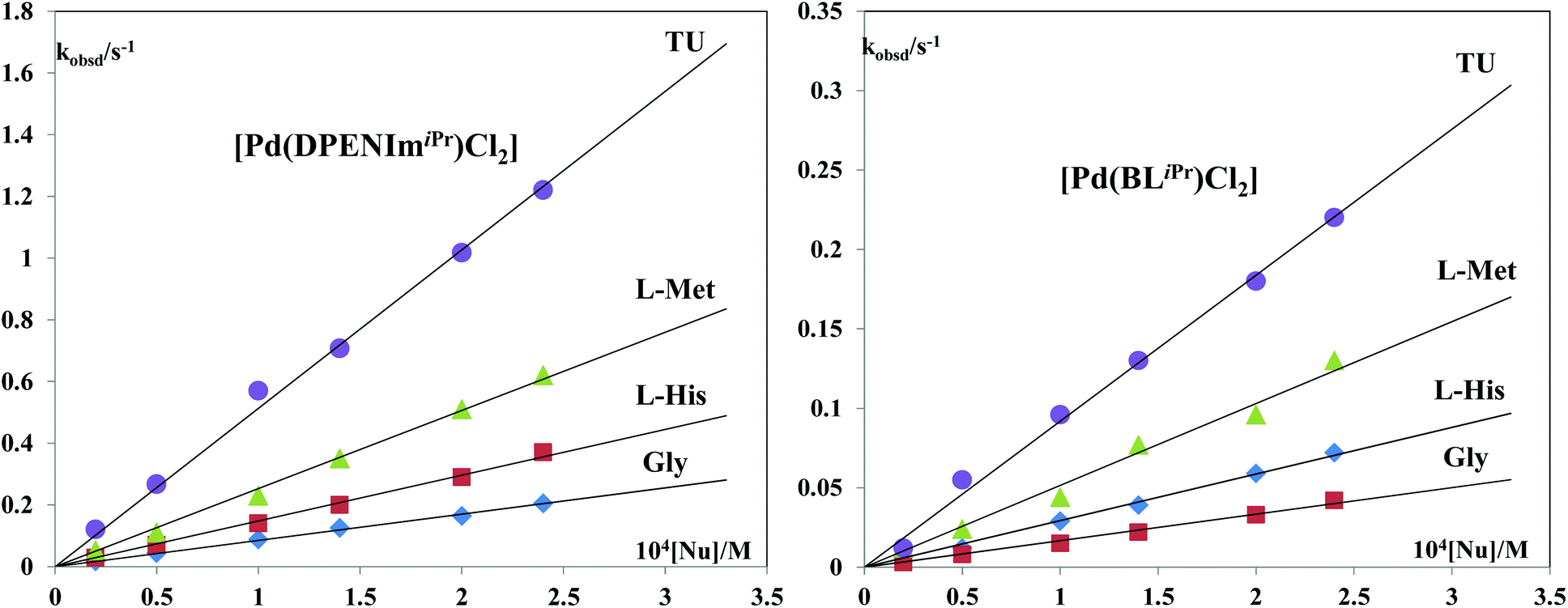 | ||
| Fig. 7 Pseudo-first-order rate constants plotted as a function of nucleophile concentration for the first step of the substitution reactions of the [Pd(DPENImiPr)Cl2] and [Pd(BLiPr)Cl2] complexes with TU, L-Met, L-His and Gly at pH = 7.2 and 310 K in 25 mM Hepes buffer and 30 mM NaCl. | ||
The order of reactivity of the investigated Pd(II) complexes for both reaction steps is (Table 3):

As expected, [Pd(en)Cl2] is the most reactive of the investigated complexes; the values obtained for this complex are in line with those found for similar complexes.1 The introduction of one imidazolin-2-imine moiety into [Pd(EAImiPr)Cl2] results in a decrease in reactivity. Compared to [Pd(en)Cl2], the first reaction step is between five and 100 times slower. The second substitution is even more impaired and proceeds up to 1000 times slower. It appears that the strong electron donating capacity of the imidazolin-2-imine results in an electron rich Pd(II) center, which is less electrophilic and thus less susceptible to attack by bio-relevant donor molecules. [Pd(DMEAImiPr)Cl2] shows a further reduction in reactivity, the first substitution being between 10 and 100 times slower than that in [Pd(en)Cl2]. This is likely caused by an increase in electron donating and steric hindrance due to the exchange of the primary amine for a tertiary amine compared to [Pd(EAImiPr)Cl2]. [Pd(DPENImiPr)Cl2] is the least reactive of the investigated complexes bearing ligands with a single imidazolin-2-imine moiety. Its reactivity for the first substitution is reduced roughly 100 fold compared to [Pd(en)Cl2] for all the tested donor molecules. Introduction of ligands bearing two imidazolin-2-imine moieties into [Pd(BLiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2] reduces the reactivity even further with both complexes showing a first substitution reaction that proceeds between 100 and 1000 times slower than that for [Pd(en)Cl2].
For all the investigated complexes, the second substitution step is significantly slower than the first substitution. In case of the reference complex [Pd(en)Cl2], this difference is the least pronounced. For [Pd(EAImiPr)Cl2], [Pd(DMEAImiPr)Cl2] and [Pd(DPENImiPr)Cl2], it can be assumed that the first substitution would take place next to the less sterically hindered side of the ligand viz. next to the amine donor. Therefore, the next substitution would have to take place next to the bulky imidazolin-2-imine donor, rendering it less facile. In addition, the first substitution would result in a less electrophilic Pd(II) center, reducing the reaction rate for the second substitution.
Interestingly, the rate constants which were obtained for the substitution reactions of the studied imidazolin-2-imine Pd(II) complexes are in line with the rate constants determined for substitution reactions of aqua Pt(II) complexes (Table S1, ESI†).26 This confirms that the investigated complexes have decreased reactivity, since it is known that Pd(II) complexes react much faster than Pt(II) complexes.1 The low reactivity of the investigated complexes raises the possibility that they might find biological application.
The rate constants, k1, presented in Table 3, indicate that the used nitrogen and sulfur donor ligands are good entering ligands in the substitution reactions with the investigated Pd(II) complexes.
The order of reactivity of the investigated nucleophiles for the first reaction step is: TU > L-Met > L-His > Gly, Table 3. This is the expected order of reactivity, as sulfur-donor nucleophiles react faster with Pd(II) complexes than nitrogen-donor nucleophiles. The Pd(II) ion is a soft acid and will easily form bonds with a soft base such as sulfur.
The data for the second substitution step show that TU reacts faster than L-His. Kinetic traces for reactions with L-Met and Gly gave fits with a double exponential function. The constants, kobsd1 and kobsd2, were plotted against the concentration of the entering L-Met or Gly nucleophiles. For kobsd1, a linear dependence on the nucleophile concentration was observed for all the complexes studied; kobsd2 was found to be independent of the L-Met or Gly concentration, suggesting a chelate formation process as presented in Scheme 3 and Fig. 8.
 | ||
| Scheme 3 The second step of the substitution reaction of the investigated Pd(II) complexes with L-Met and Gly. | ||
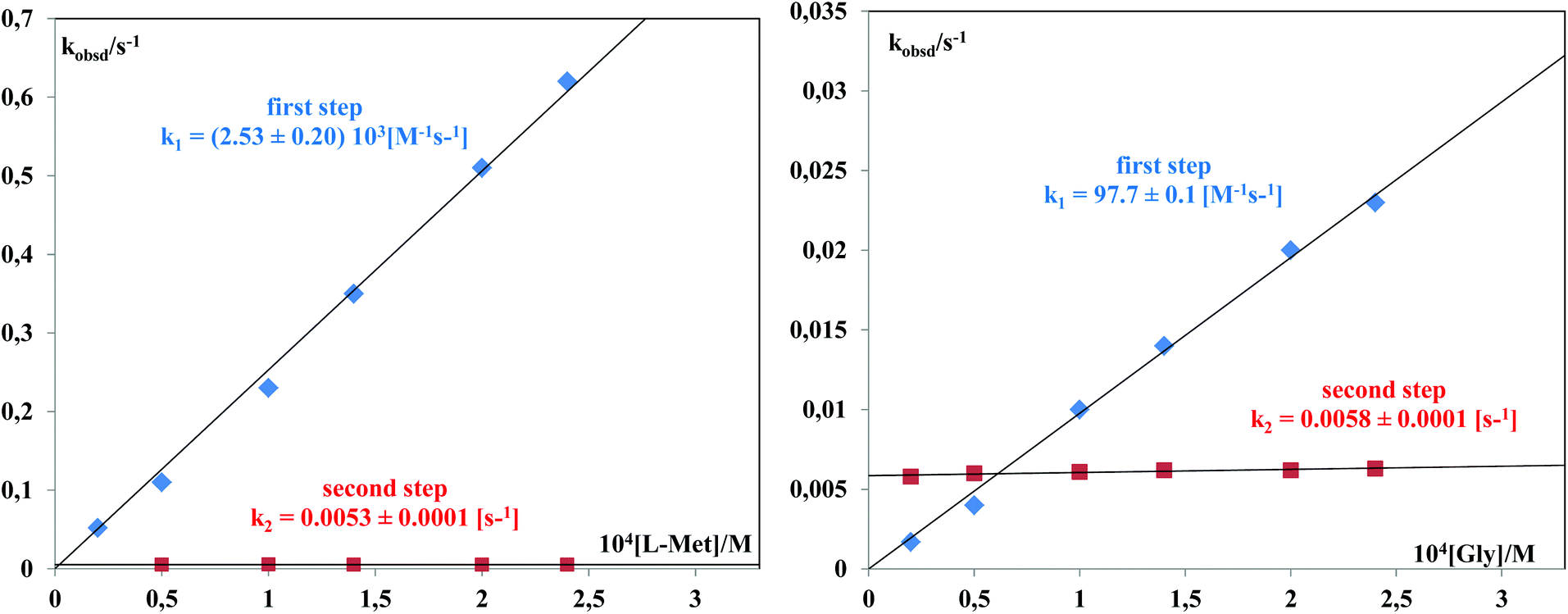 | ||
| Fig. 8 Plots of kobsdversusL-Met concentration for the [Pd(DPENImiPr)Cl2] complex and plots of kobsdversus Gly concentration for the [Pd(DACH(ImiPr)2)Cl2] complex (pH = 7.2, 310 K, 25 mM Hepes buffer, 30 mM NaCl). | ||
The substitution reactions of the investigated Pd(II) complexes with L-Met proceed with a nucleophilic attack by the sulfur donor of the thioether group, and subsequently a six-membered ring (see Scheme 3) is formed by substitution with the nitrogen donor of the amine group. The acid dissociation constants of free L-Met are: pKCOOH = 2.28,27 pKNH3+ = 9.2,27 so ring-closure must involve deprotonation of the amine group (Scheme 3). Ring-closure and formation of a six-membered ring also occur in the substitution reactions of Pt(II) complexes and L-Met.17,28 In the substitution reaction between Gly and the investigated Pd(II) complexes, the formation of a five-membered ring is confirmed. The nucleophilic attack occurs via the nitrogen donor of the amine group and then via the oxygen from the carboxyl group (Scheme 3). A similar ring-closure between Pd(II) or Pt(II) complexes and Gly was observed in earlier publications.28,29
To confirm that the second step is chelation, the kinetics were studied with the Pd(II) complexes in excess instead of L-Met or Gly. This would mean that a two-step reaction can only occur if ring closure is involved. The obtained kinetic traces for such reactions gave fits to a double exponential function. Similar values for the rate constants were obtained compared to those observed in the experiments with L-Met or Gly added in excess (see the ESI Fig. S13 and 14†). All this indicates that ring closure occurs.
Activation parameters
The activation parameters ΔH≠ and ΔS≠ (Table S2†) were calculated using the Eyring equation for the reactions with TU (see Fig. 9 and S12, ESI†) for the first and second reaction steps. The activation parameters support an associative mechanism for each of these reactions which is supported by the significantly negative activation entropies. This suggests that the activation process in the studied systems is strongly dominated by bond formation.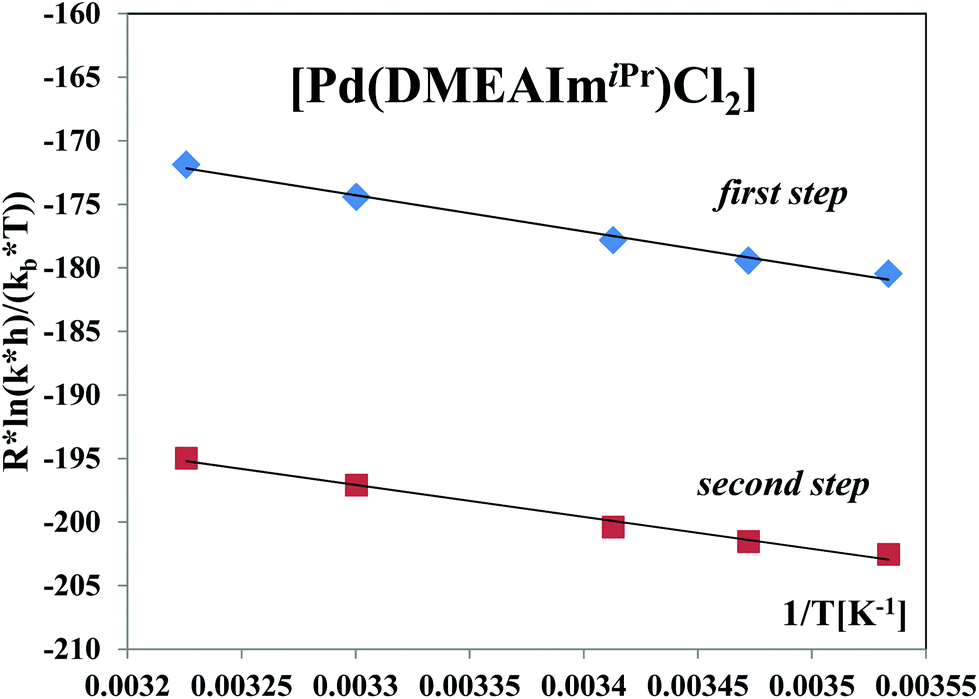 | ||
| Fig. 9 Eyring plots for the two reaction steps of the [Pd(DMEAImiPr)Cl2] complex with TU at pH = 7.2 and 310 K in 25 mM Hepes buffer and 30 mM NaCl. | ||
Conclusion
In conclusion, we were able to use novel imidazolin-2-imine ligands for the synthesis of a series of new Pd(II) complexes. For [Pd(DMEAImiPr)Cl2] and [Pd(DPENImiPr)Cl2], X-ray structures could be determined. Solubility measurements showed that the imidazolin-2-imine Pd(II) complexes have good solubility in water. Spectrophotometric pH titration experiments showed two pKa values for the diaqua complexes of all the investigated Pd(II) compounds. A clear correlation between the structure of the imidazolin-2-imine and the determined pKa values was found. The pKa values of the more electron rich complexes (i.e. those bearing a more electron-donating ligand) were significantly higher than those of the less electron rich complexes. Kinetic experiments were performed with selected small bio-molecules (i.e. thiourea, L-methionine, L-histidine and glycine) under pseudo-first-order conditions as a function of the nucleophile concentration and temperature using stopped-flow techniques. These measurements showed that mono(imidazolin-2-imine) Pd(II) complexes react faster than bis(imidazolin-2-imine) Pd(II) complexes. This can be attributed to both electronic and steric effects of the imidazolin-2-imine ligands. Two reaction steps are observed that belong to the displacement of both chloride ligands. We found that the reactivity of the Pd(II) complexes decreases as follows: [Pd(en)Cl2] > [Pd(EAImiPr)Cl2] > [Pd(DMEAImiPr)Cl2] > [Pd(DPENImiPr)Cl2] > [Pd(BLiPr)Cl2] > [Pd(DACH(ImiPr)2)Cl2]. The sulfur-donor ligands TU and L-Met react faster than nitrogen-donor ligands L-His and Gly. Both L-methionine and glycine react by undergoing a ring closure as the second substitution step. The mechanism of the substitution reactions is associatively supported by the negative values of ΔS≠.The coordination of imidazolin-2-imine ligands to Pd(II) gives three benefits with regard to the utilization of such complexes as metallo-drugs. The first is an increase of the solubility of these Pd(II) complexes, as the low solubility of neutral Pd(II) and Pt(II) complexes, such as cisplatin, is one of the major disadvantages in their application as cytostatics. Secondly, imidazolin-2-imine Pd(II) complexes react more slowly than most of the known Pd(II) complexes; their reactivity is such that it lies in the same range as that of aqua Pt(II) complexes. The reduced reactivity of Pd(II) complexes should lead to a better selectivity of such complexes in the binding of bio-molecules. Lastly, the introduction of imidazolin-2-imines leads to an increase of the pKa values of the coordinated water molecules in these complexes. For [Pd(DPENImiPr)Cl2] and [Pd(DACH(ImiPr)2)Cl2], the pKa values are such that it can be assumed that they will exist mostly as diaqua species at the pH of tumor cells. These combined advantages should lead to a more selective attack of these complexes at the DNA of tumor cells as a final target in drug delivery.
Experimental
Chemicals and solutions
Thiourea, L-methionine, L-histidine, glycine, ethylenediamine, N,N-dimethylethylenediamine, (1R,2R)-(−)-1,2-diaminocyclohexane, (1S,2S)-(−)-1,2-diphenylethylenediamine, NaBF4, KF, NaNH2, KOtBu and PdCl2 were obtained from Acros Organics or Sigma Aldrich, and were used without further purification. Hepes buffer (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) was obtained from Sigma Aldrich. All the other chemicals were of the highest purity commercially available and were used without further purification. Ultra-pure water was used in all experiments. Nucleophile stock solutions were prepared shortly before use by dissolving the chemicals.Preparation of the ligands and complexes
All reactions were performed in a glove box under a dry argon atmosphere (MBraun 200B) or on a high-vacuum line using standard Schlenk techniques, unless noted otherwise. Commercial grade solvents were purified by the use of a solvent purification system from MBraun GmbH and stored over molecular sieves (4 Å) under a dry argon atmosphere.All the syntheses of the ligands started from 2-chloro-1,3-diisopropyl-4,5-dimethylimidazolium tetrafluoroborate, which was prepared according to a literature procedure.30
1H NMR (400 MHz; CDCl3): δ = 5.12 (bs, 1H, N![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ), 4.78 (sept, 2H, JHH 7.0 Hz, C(Me)2), 3.22 (bt, 2H, JHH 6.0 Hz, CNHC2CH2), 2.59 (t, 2H, JHH 6.0 Hz, CH2C2NMe2), 2.26 (s, 6H, N(C3)2), 2.25 (s, 6H, CC3), 1.54 (d, 12H, JHH 7.0 Hz, CH(C3)2) ppm.
), 4.78 (sept, 2H, JHH 7.0 Hz, C(Me)2), 3.22 (bt, 2H, JHH 6.0 Hz, CNHC2CH2), 2.59 (t, 2H, JHH 6.0 Hz, CH2C2NMe2), 2.26 (s, 6H, N(C3)2), 2.25 (s, 6H, CC3), 1.54 (d, 12H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 MHz; CDCl3): δ = 144.1 (N2![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) NH), 122.9 (Me), 58.7 (CH2H2NMe2), 49.9 (HMe2), 46.7 (CNHH2CH2), 45.4 (N(H3)2), 21.4 (CH(H3)2), 10.1 (CH3) ppm.
NH), 122.9 (Me), 58.7 (CH2H2NMe2), 49.9 (HMe2), 46.7 (CNHH2CH2), 45.4 (N(H3)2), 21.4 (CH(H3)2), 10.1 (CH3) ppm.
19F NMR (188 MHz; CDCl3): δ = −152.08 (B![[F with combining low line]](https://www.rsc.org/images/entities/i_char_0046_0332.gif) 4) ppm.
4) ppm.
Anal. Calcd for (C15H31BF4N4)·0.10(CHCl3) C: 49.53; H: 8.56; N: 15.30. Found: C: 49.43; H: 8.80; N: 15.64.
ESI-HRMS: [M − BF4]+: Calcd: 267.25487; Found: 267.25446 (Δ: 0.41 mmu).
1H NMR (400 MHz; C6D6): δ = 4.43 (sept, 2H, JHH 7.0 Hz, CMe2), 3.73 (t, 2H, JHH 7.3 Hz, CNC2CH2), 2.66 (t, 2H, JHH 7.3 Hz, CH2C2NMe2), 2.25 (s, 6H, N(C3)2), 1.71 (s, 6H, CC3), 1.20 (d, 12H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 MHz; C6D6): δ = 150.3 (N2N), 115.9 (Me), 64.8 (CH2H2NMe2), 49.0 (CNH2CH2), 46.6 (N(H3)2), 46.3 (HMe2), 21.3 (CH(H3)2), 10.9 (CH3) ppm.
Anal. Calcd for (C15H30N4) C: 67.62; H: 11.35; N: 21.03. Found: C: 67.36; H: 11.16; N: 20.67.
1H NMR (400 MHz; CDCl3): δ = 4.85 (sept, 2H, JHH 7.0 Hz, C(Me)2), 3.16 (t, 2H, JHH 6.0 Hz, CNHC2CH2), 3.01 (t, 2H, JHH 6.0 Hz, CH2C2NH2), 2.32 (bs, 2H, N2), 2.25 (s, 6H, CC3), 1.54 (d, 12H, JHH 7.0 Hz, CH(C3)2), 1.53 (bs, 2H, N2) ppm.
13C NMR (100 MHz; CDCl3): δ = 144.1 (N2NH), 123.0 (Me), 51.4 (CNHH2CH2), 50.0 (H(Me)2), 41.5 (CH2H2NH2), 21.4 (CH(H3)2), 10.1 (C(H3) ppm.
19F NMR (188 MHz; CDCl3): δ = −152.08 (B4) ppm.
Anal. Calcd for (C13H27BF4N4)·0.10(CHCl3) C: 46.53; H: 8.08; N: 16.57. Found: C: 46.78; H: 8.03; N: 16.55.
ESI-HRMS: [M-BF4]+: Calcd: 239.22357; Found: 239.22316 (Δ: 0.41 mmu).
1H NMR (400 MHz; C6D6): δ = 4.51 (sept, 2H, JHH 7.0 Hz, C(Me)2), 3.61 (t, 2H, JHH 5.4 Hz, CNC2CH2), 3.01 (bt, 2H, JHH 5.3 Hz, CH2C2NH2), 1.68 (s, 6H, CC3), 1.28 (bs, 2H, N2), 1.20 (d, 12H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 HMz; C6D6): δ = 151.1 (N2N), 115.8 (Me), 53.0 (CNH2CH2), 46.7 (CH2H2NH2), 46.1 (H(Me)2), 21.3 (CH(H3)2), 10.8 (C(H3) ppm.
Anal. Calcd for (C13H26N4) C: 65.50; H: 10.99; N: 23.50. Found: C: 65.67; H: 10.99; N: 23.36.
The synthesis and characterization of [DPEN(ImiPrH)NH2][BF4] (5) were performed according to a literature procedure.13
1H NMR (300 MHz; CDCl3): δ 7.18–6.90 (m, 10H, HAr), 4.70 (sept, 2H, JHH 7.0 Hz, CMe2), 4.50 (d, 1H, JHH 9.0 Hz, NH2HC(Ph)CH), 3.85 (d, 1H, JHH 9.0 Hz, CNHC(Ph)CH), 1.82 (s, 6H, CC3), 1.65 (bs, 2H, N2), 1.10 (d, 6H, JHH 7.0 Hz, CH(C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)2), 0.94 (d, 6H, JHH 7.0 Hz, CH(C3)2) ppm.
3)2), 0.94 (d, 6H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 MHz; CDCl3): δ 154.1 (N2N), 139.1 (ipso-Ar(CHNH2)), 132.6 (ipso-![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) Ar(CHN), 124.7 (Ar), 122.2 (Ar), 121.1 (Ar), 123.5 (m-Ar(CHNH2)), 123.3 (m-Ar(CHNH), 123.0 (Ar), 118.2 (Me) 70.3 (CNHH(Ph)CH), 57.1 (NH2HH(Ph)CH), 46.2 (HMe2), 20.0 (CH(H3)2), 19.5 (CH(H3)2), 9.8 (CH3) ppm.
Ar(CHN), 124.7 (Ar), 122.2 (Ar), 121.1 (Ar), 123.5 (m-Ar(CHNH2)), 123.3 (m-Ar(CHNH), 123.0 (Ar), 118.2 (Me) 70.3 (CNHH(Ph)CH), 57.1 (NH2HH(Ph)CH), 46.2 (HMe2), 20.0 (CH(H3)2), 19.5 (CH(H3)2), 9.8 (CH3) ppm.
Anal. Calcd for (C25H34N4) C: 76.88; H: 8.77; N: 14.35. Found: C: 76.01; H: 8.98; N: 14.75.
The synthesis and characterization of the ligands BLiPr (7), DACH(ImiPr)2 (8) were performed according to literature procedures.13,31
Preparation of the complexes
The complexes [Pd(en)Cl2] and [Pd(BLiPr)Cl2] were synthesized according to published procedures.14,32
1H NMR (300 MHz; CDCl3): δ = 5.40 (sept, 2H, JHH 7.0 Hz, C(Me)2), 4.13 (t, 2H, JHH 4.9 Hz, CNC2CH2), 2.68 (t, 2H, JHH 5.0 Hz, CH2C2NH2), 2.04 (s, 6H, CC3), 1.60 (d, 6H, JHH 7.0 Hz, CH(C3)2), 1.47 (d, 6H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 HMz; CDCl3): δ = 152.7 (N2N), 118.6 (Me), 55.2 (CNH2CH2), 48.6 (CH2H2NH2), 47.4 (H(Me)2), 22.4 (CH(H3)2), 21.9 (CH(H3)2), 10.9 (C(H3) ppm.
Anal. Calcd for (C13H26Cl2N4Pd) C: 37.56; H: 6.30; N: 13.48. Found: C: 36.99; H: 6.19; N: 13.33.
1H NMR (300 MHz; CDCl3): δ = 5.30 (sept, 2H, JHH 7.0 Hz, CMe2), 2.73 (t, 2H, JHH 7.3 Hz, CNC2CH2), 2.35 (t, 2H, JHH 7.3 Hz, CH2C2NMe2), 2.75 (s, 6H, N(C3)2), 2.05 (s, 6H, CC3), 1.65 (d, 6H, JHH 7.0 Hz, CH(C3)2), 1.52 (d, 6H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 MHz; CDCl3): δ = 152.6 (N2N), 120.7 (Me), 68.2 (CH2H2NMe2), 53.7 (CNH2CH2), 51.3 (N(H3)2), 48.8 (HMe2), 22.7 (CH(H3)2), 22.0 (CH(H3)2), 10.09 (CH3) ppm.
Anal. Calcd for (C15H30Cl2N4Pd) C: 40.60; H: 6.81; N: 12.63. Found: C: 40.59; H: 6.78; N: 12.26.
1H NMR (300 MHz; CDCl3): δ 7.38–7.10 (m, 10H, HAr), 5.31 (sept, 2H, JHH 7.0 Hz, CMe2), 4.98 (d, 1H, JHH 9.0 Hz, NH2HC(Ph)CH), 4.21 (d, 1H, JHH 9.0 Hz, CNHC(Ph)CH), 2.04 (d, 3H, JHH 7.0 Hz, CH(C3)2), 1.99 (s, 6H, CC3), 1.85 (s, 2H, N2), 1.72 (d, 3H, JHH 7.0 Hz, CH(C3)2), 1.58 (d, 3H, JHH 7.0 Hz, CH(C3)2), 0.79 (d, 3H, JHH 7.0 Hz, CH(C3)2), ppm.
13C NMR (100 MHz; CDCl3): δ 156.2 (N2N), 139.8 (ipso-Ar(CHNH2)), 132.9 (ipso-Ar(CHN), 125.1 (Ar), 122.9 (Ar), 121.8 (Ar), 124.2 (m-Ar(CHNH2)), 123.9 (m-Ar(CHNH), 123.5 (Ar), 119.5 (Me) 72.1 (CNHH(Ph)CH), 59.3 (NH2HH(Ph)CH), 48.4 (HMe2), 47.2 (HMe2), 22.2 (CH(H3)2), 21.5 (CH(H3)2), 21.0 (CH(H3)2), 20.6 (CH(H3)2), 10.8 (CH3), 10.5 (CH3) ppm.
Anal. Calcd for (C25H34Cl2N4Pd) C: 52.87; H: 6.03; N: 9.87. Found: C: 52.97; H: 6.14; N: 10.01.
1H NMR (300 MHz; CDCl3): δ = 5.45 (sept, 2H, CMe2), 5.28 (sept, 2H, CMe2), 3.82 (bs, 4H, CH2C(NC)C(NC)CH2), 2.72–2.60 (m, 2H, C2), 2.15 (s, 6H, CC3), 2.05 (s, 6H, CC3) 2.10–1.92 (m, 2H, C2), 1.89–1.81 (m, 2H, C2), 1.78–1.69 (m, 2H, C2), 1.62 (d, 6H, JHH 7.0 Hz, CH(C3)2), 1.52 (d, 6H, JHH 7.0 Hz, CH(C3)2), 1.43 (d, 6H, JHH 7.0 Hz, CH(C3)2), 1.08 (d, 6H, JHH 7.0 Hz, CH(C3)2) ppm.
13C NMR (100 MHz; CDCl3): δ = 153. (N2N), 117.8 (Me), 65.7 (CH2H(NC)H(NC)CH2), 50.1 (HMe2), 49.3 (HMe2), 36.9 (H2), 26.5 (H2), 23.7 (CH(H3)2), 22.5 (CH(H3)2), 21.6 (CH(H3)2), 21.0 (CH(H3)2), 13.1 (CH3), 12.3 (CH3) ppm.
Anal. Calcd for (C28H50Cl2N6Pd) C: 51.89; H: 7.78; N: 12.97. Found: C: 51.26; H: 7.93; N: 12.01.
ESI-HRMS: [M − Cl]+: Calcd: 611.28; Found: 611.14 (Δ: 0.41 mmu).
Preparation of aqua complexes
The aqua complexes of Pd(II) complexes were prepared starting from the corresponding chlorido complexes. The conversion was performed by the addition of the corresponding amount of AgClO4 to a solution of the chloride complex and stirring for 5 h at 50 °C. The white precipitate that formed (AgCl) was filtered off using a Millipore filtration unit, and the solutions were diluted. Great care was taken to ensure that the resulting solution was free of Ag+ ions and that the chlorido complexes had been completely converted into the aqua form. Since it is well known that perchlorate ions do not coordinate to Pd(II) and Pt(II) in aqueous solution,33 pH titrations were studied in perchlorate medium.Instrumentation and measurements
NMR spectra were recorded on Bruker DPX 200, DRX 400 and AV 300 devices. Chemicals shifts (δ) are reported in ppm and referenced to tetramethylsilane (1H, 13C) and trichlorofluoromethane (19F). Coupling constants (J) are reported in hertz (Hz) and splitting patterns are indicated as s (singlet), d (doublet), t (triplet), sept (septet), bs (broad signal) and m (multiplet). Elemental analyses (C, H, N) were performed by combustion and gas chromatographic analysis with an Elementar Vario MICRO elemental analyzer. High resolution electron spray ionization (ESI) mass spectroscopy was performed on a Finnigan MAT 95 XL trap device. pH measurements were carried out using a Mettler Delta 350 digital pH meter with a resolution of ±0.01 mV, with a combination glass electrode. This electrode was calibrated using standard buffer solutions of pH 4, 7 and 9 obtained from Sigma. Kinetic measurements of the Pd(II) complex were carried out on an Applied Photophysics SX.18MV stopped-flow instrument coupled to an on-line data acquisition system. The temperature was controlled throughout all kinetic experiments to ±0.1 °C. All kinetic measurements were performed under pseudo-first-order conditions, i.e., at least a 10-fold excess of the nucleophile was used. UV-Vis spectra were recorded on a PerkinElmer Lambda 35 double-beam spectrophotometer equipped with thermostated 1.00 cm quartz Suprasil cells.Determination of the pKa values of the Pd(II) complexes
Spectrophotometric pH titrations of the solutions of the complexes were performed with NaOH as a base at 298 K. To avoid absorbance corrections due to dilution, a large volume (300 mL) of the complex solution was used in the titration. The change in pH from 2 to approximately 3 was achieved by the addition of known amounts of crushed pellets of NaOH. The consecutive pH changes were obtained by adding drops of saturated solutions of NaOH, 1 or 0.1 M, using a micropipette. To avoid contamination released by the pH electrode, it was necessary to take 2 mL aliquots from the solution into narrow vials for the pH measurements. The aliquots were discarded after the measurements. The total reversibility of the titration could be achieved by subsequent addition of HClO4.The titration data for the complexes were fitted to the following eqn (5) for the determination of both pKa values34–36 and the obtained data are presented in Table 2.
| y = a + (b − a)/(1 + 2.718 × ((x − pKa1/m)) + (c − b)/(1 + 2.718 × ((x − pKa2)/n)) | (5) |
The parameter a represents the value of the absorbance at the beginning of the titration, b represents the absorbance during the titration and c is the absorbance at the end of the titration. The parameters m and n are used to optimize the titration curve. In this equation y represents the absorbance value and x refers to the pH.
Solubility measurements
The concentrations of saturated solutions of the studied Pd(II) complexes were determined by UV-vis spectrophotometry. Therefore the specific absorptivity of the compounds in water was determined first. This was measured using five dilution series (5, 10, 30, 40, 50 mM) of the studied complexes and then the calibration curve was calculated using the Lambert–Beer law. The slope of the curve gave specific absorptivity.The required quantity of water solution was added to the 5 ml volumetric flask. The solution was heated up to 298 K. A previously weighed quantity of Pd(II) complexes was added to the volumetric flask until the saturation point occurs. Stirring was continued up to 7 hours at 298 K. The sample was filtered through a 0.20 μm membrane filter. A measured quantity of the filtered sample was transferred into another volumetric flask and made further dilutions. The absorbance was measured using UV-vis spectrophotometry. The same process was repeated two times.
Kinetic measurements
The substitution reactions of the Pd(II) complex with the nucleophiles: TU, L-Met, L-His and Gly were studied spectrophotometrically by following the change in absorbance at suitable wavelengths as a function of time. Spectral changes resulting from the mixing of the complex and nucleophile solutions were recorded over the wavelength range 200 to 400 nm to establish a suitable wavelength at which kinetic measurements could be performed.Substitution reactions were initiated by mixing equal volumes of the complex and ligand solutions directly in the stopped-flow instrument and followed for at least eight half-lives. The substitution process was monitored as the change in absorbance with time under pseudo-first-order conditions. The observed pseudo-first-order rate constants, kobsd, were calculated as the average value from four to six independent kinetic runs using the program OriginPro 8. Experimental data are reported in Tables S3–S26 (ESI†).
X-ray diffraction studies
Data were recorded at 100(2) K using an Oxford Diffraction Nova A diffractometer with monochromated Cu Kα radiation. The structures were refined anisotropically using the SHELXL-97 program.37 Hydrogen atoms were either (i) located and refined isotropically (NH2), (ii) included as idealized methyl groups allowed to rotate but not tip or (iii) placed geometrically and allowed to ride on their attached carbon atoms. Special features: [Pd(DMEAImiPr)Cl2]·2CH2Cl2 crystallizes as half a molecule on a crystallographic mirror plane perpendicular to the five-membered ring. The ethylene bridge is disordered across the mirror plane. The structure of [Pd(DPENImiPr)Cl2]·5/4C3H6O·1/4C6H14 contains four ordered molecules of acetone, one disordered molecule of acetone and one disordered molecule of hexane in the asymmetric unit. The latter two were removed mathematically using the program SQUEEZE.38 Derived parameters such as the formula weight correspond to four additional molecules of acetone and four molecules of hexane per cell.| [Pd(DMEAImiPr)Cl2]·2CH2Cl2 | [Pd(DPENImiPr)Cl2]·5/4·C3H6O·1/4·C6H14 | |
|---|---|---|
| Empirical formula | C17H34Cl6N4Pd | C30.25H45Cl2N4O1.25Pd |
| Formula weight | 613.58 | 662.00 |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pnma | P212121 |
| a/Å | 16.5015(4) | 14.8839(3) |
| b/Å | 19.3631(5) | 28.4936(9) |
| c/Å | 8.2474(2) | 30.6466(9) |
| Volume [Å3] | 2635.21(11) | 12997.1(6) |
| Z | 4 | 16 |
| Reflections collected | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 762 762 |
158785 |
| Independent reflections | 2857 [Rint = 0.0391] | 26899 [Rint = 0.0654] |
| ρ C/g cm−3 | 1.547 | 1.345 |
| μ/mm−1 | 11.366 | 6.331 |
| R(Fo) [I > 2σ(I)] | 0.0246 | 0.0376 |
| R w (Fo2) | 0.0612 | 0.0890 |
| Goodness of fit on (F2) | 1.071 | 1.052 |
| Flack parameter | — | 0.005(4) |
| Δρ/e Å–3 | 0.450/−0.806 | 0.895/−1.145 |
Abbreviations
| En | Ethylendiamine |
| EAImiPr | 2-(1,3-Diisopropyl-4,5-dimethylimidazolin-2-imine)ethan-1-amine |
| BLiPr | 1,2-Bis(1,3-diisopropyl-4,5-dimethylimidazolin-2-imino)ethane |
| DMEAImiPr | 2-(1,3-Diisopropyl-4,5-dimethylimidazolin-2-imine)ethan-1-dimethylamine |
| DPENImiPr | 2-(1,3-Diisopropyl-4,5-dimethylimidazolin-2-imine)-1,2-diphenylethan-1-amine |
| DACH(ImiPr)2 | N,N′-(Cyclohexane-1,2-diyl)bis(1,3-diisopropyl-4,5-dimethylimidazolin-2-imine) |
| TU | Thiourea |
| L-Met | L-Methionine |
| L-His | L-Histidine |
| Gly | Glycine |
Acknowledgements
The authors gratefully acknowledge financial support from the Ministry of Education, Science and Technological Development Serbia, project no. 172011 and the Deutsche Forschungsgemeinschaft (DFG).References
- Ž. D. Bugarčić, J. Bogojeski and R. van Eldik, Coord. Chem. Rev., 2015, 292, 91 CrossRef PubMed.
- Cisplatin, Chemistry and Biochemistry of Leading Antitumor Drugs, ed. B. Lippert, Wiley-VCH, Zürich, 1999 CrossRef CAS PubMed; D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS PubMed; S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 24, 2845 CrossRef PubMed; H. Zorbas and B. K. Keppler, ChemBioChem, 2005, 6, 1157 CrossRef PubMed.
- Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, 2011, ch. 1–4 Search PubMed.
- P. E. N. Barry and J. P. Sadler, Chem. Commun., 2013, 49, 5106 RSC.
- L. Ronconi and J. P. Sadler, Coord. Chem. Rev., 2007, 251, 1633 CrossRef CAS PubMed.
- B. Rosenberg, L. V. Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS PubMed.
- A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337 CrossRef CAS.
- E. Gao, C. Liu, M. Zhu, H. Lin, Q. Wu and L. Liu, Anticancer Agents Med. Chem., 2009, 9, 356 CrossRef CAS.
- E. Gao, M. Zhu, H. Yin, L. Liu, Q. Wu and Y. Sun, J. Inorg. Biochem., 2008, 102, 1958 CrossRef CAS PubMed.
- X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116 CrossRef CAS PubMed.
- T. Glöge, D. Petrović, C. G. Hrib, C. Daniliuc, E. Herdtweck, P. G. Jones and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2303 CrossRef PubMed.
- T. K. Panda, S. Randoll, C. G. Hrib, P. G. Jones, T. Bannenberg and M. Tamm, Chem. Commun., 2007, 5007 RSC; T. K. Panda, D. Petrovic, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2008, 361, 2236 CrossRef CAS PubMed; T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462 CrossRef PubMed; A. G. Trambitas, T. K. Panda, J. Jenter, P. Roesky, C. G. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435 CrossRef PubMed; T. K. Panda, C. G. Hrib, P. G. Jones and M. Tamm, J. Organomet. Chem., 2010, 695, 2768 CrossRef PubMed; M. Tamm, A. G. Trambitas, C. G. Hrib and P. G. Jones, Terrae Rarae, 2010, 7, 1 Search PubMed; A. G. Trambitas, J. Yang, D. Melcher, C. G. Daniliuc, P. G. Jones, Z. Xie and M. Tamm, Organometallics, 2011, 30, 1122 CrossRef; A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753 CrossRef PubMed.
- J. Volbeda, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2014, 422, 158 CrossRef CAS PubMed.
- J. Bogojeski, R. Jelić, D. Petrović, E. Herdtweck, P. G. Jones, M. Tamm and Ž. D. Bugarčić, Dalton Trans., 2011, 40, 6515 RSC.
- (a) J. Börner, U. Flörke, K. Huber, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362–2376 CrossRef PubMed; (b) V. Raab, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Org. Chem., 2003, 68, 8790–8797 CrossRef CAS PubMed; (c) A. Neuba, R. Haase, M. Bernard, U. Flörke and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2008, 634, 2511–2517 CrossRef CAS PubMed.
- R. C. Boyle, J. T. Mague and M. J. Fink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m40–m41 CAS.
- Ž. D. Bugarčić, J. Bogojeski, B. Petrović, S. Hochreuther and R. van Eldik, Dalton Trans., 2012, 41, 12329–12345 RSC.
- T. Rau and R. van Eldik, Interactions of metal ions with nucleotides, nucleic acids, and their constituents, ed. A. Sigel and H. Sigel, in Metal Ions in Biological Systems, Marcel Dekker, New York, 1996, 339 Search PubMed.
- S. J. Barton, K. J. Barnham, A. Habtemariam, R. E. Sue and R. J. Sadler, Inorg. Chim. Acta, 1998, 273, 8 CrossRef CAS.
- Y. Xu, J. Cui and D. Puett, Cancer Bioinformatics, Springer, 2014, p. 209 Search PubMed.
- M. T. Ashby, Comments Inorg. Chem., 1990, 10, 297 CrossRef CAS PubMed.
- S. G. Murray and F. R. Hartley, Chem. Rev., 1981, 81, 365 CrossRef CAS.
- J. Reedijk, J. Chem. Commun., 1996, 801 RSC.
- J. H. Burchenal, K. Kalaher, K. Dew, L. Lokys and G. Gale, Biochimie, 1978, 60, 961 CrossRef CAS.
- Inorganic Reaction Mechanism, ed. M. L. Tobe and J. Burgess, Longman, England, 1999, p. 70, p. 364 Search PubMed.
- N. Summa, W. Schiessl, R. Puchta, N. van Eikema Hommes and R. van Eldik, Inorg. Chem., 2006, 45, 2948 CrossRef CAS PubMed.
- Proteins, Amino Acids and Peptides as Ions and Dipolar Ions, ed. E. J. Cohn and J. T. Edsall, Reinhold Publishing Corporation, New York, 1943 Search PubMed.
- N. Summa, T. Soldatović, L. Dahlenburg, Ž. D. Bugarčić and R. van Eldik, JBIC, J. Biol. Inorg. Chem., 2007, 12, 461 CrossRef CAS PubMed.
- B. Petrović, Ž. D. Bugarčić and R. van Eldik, Dalton Trans., 2008, 807 RSC.
- R. A. Kunetskiy, S. M. Polyakova, J. Vavrík, I. Císarová, J. Saame, E. R. Nerut, I. Koppel, I. A. Koppel, A. Kütt, I. Leito and I. M. Lyapkalo, Chem. – Eur. J., 2012, 18, 3621 CrossRef CAS PubMed.
- D. Petrović, T. Bannenberg, S. Randoll, P. G. Jones and M. Tamm, Dalton Trans., 2007, 2812 RSC.
- H. Hohmann and R. van Eldik, Inorg. Chim. Acta, 1990, 174, 87 CrossRef CAS.
- T. G. Appleton, J. R. Hall, S. F. Ralph and C. S. M. Thompson, Inorg. Chem., 1984, 23, 3521 CrossRef CAS.
- T. Soldatović, S. Jovanović, Ž. D. Bugarčić and R. van Eldik, Dalton Trans., 2012, 41, 876 RSC.
- A. Mambanda, D. Jaganyi, S. Hochreutther and R. van Eldik, Dalton Trans., 2010, 39, 3595 RSC.
- (a) A. Hofmann and R. van Eldik, Dalton Trans., 2003, 2979 RSC; (b) H. Erturk, A. Hofmann, R. Puchta and R. van Eldik, Dalton Trans., 2007, 2295 RSC; (c) H. Erturk, J. Maigut, R. Puchta and R. van Eldik, Dalton Trans., 2008, 2759 RSC; (d) H. Erturk, R. Puchta and R. van Eldik, Eur. J. Inorg. Chem., 2009, 1334 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structure from Diffraction Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. Spek, Part of the PLATON suite, University of Utrecht, Netherlands, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. The CCDC number for [Pd(DMEAImiPr)Cl2] is 1407330 and the CCDC for [Pd(DPENImiPr)Cl2] is 1407331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02307f |
| This journal is © The Royal Society of Chemistry 2015 |