
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structural characterisation of bismuth(III) hydroxamates and their activity against Helicobacter pylori†
Amita
Pathak
a,
Victoria L.
Blair
a,
Richard L.
Ferrero
b,
Peter C.
Junk
c,
Richard F.
Tabor
a and
Philip C.
Andrews
*a
aSchool of Chemistry, Monash University, Clayton, Melbourne, VIC 3800, Australia
bCentre for Innate Immunity and Infectious Diseases, Monash Institute of Medical Research, Clayton, Melbourne, VIC 3168, Australia
cSchool of Pharmacy and Molecular Sciences, James Cook University, Townsville, QLD 4811, Australia. E-mail: phil.andrews@monash.edu
First published on 9th September 2015
Abstract
Seven new bismuth(III) hydroxamate complexes derived from the hydroxamic acids N-methylfurohydroxamic acid (H-MFHA), N-benzoyl-N-phenylhydroxamic acid (H-BPHA), salicylhydroxamic acid (H2-SHA), benzohydroxamic acid (H2-BHA), and acetohydroxamic acid (H2-AHA) have been synthesized and characterized. The complexes formed are either tris-hydroxamato complexes containing only mono-anionic ligands, [Bi(H-SHA)3], [Bi(MFHA)3] and [Bi(BPHA)3]; mixed-anion complexes, [Bi(SHA)(H-SHA)] and [Bi(AHA)(H-AHA)]; and potassium bismuthate complexes, K[Bi(SHA)2] and K[Bi(BHA)2]. The solid-state structure of three complexes has been determined through single crystal X-ray diffraction; [Bi(MFHA)3]2·Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ and [Bi(BPHA)3]2·2EtOH. All the complexes and their parent acids were assessed for the bactericidal activity against three strains of Helicobacter pylori (26695, B128 and 251). Of the acids, only acetohydroxamic acid showed any activity at low concentrations (MIC 6.25 μg mL−1; 83.26 μM) while the others were not toxic below 25 μg mL−1. In contrast, their bismuth(III) complexes all showed excellent activity across all three strains (e.g. 0.28 μM for [Bi(H-SHA)3] to 6.01 μM for K[Bi(BHA)2] against strain 251) with only minor variations in activity being both ligand and composition dependant.
O, {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ and [Bi(BPHA)3]2·2EtOH. All the complexes and their parent acids were assessed for the bactericidal activity against three strains of Helicobacter pylori (26695, B128 and 251). Of the acids, only acetohydroxamic acid showed any activity at low concentrations (MIC 6.25 μg mL−1; 83.26 μM) while the others were not toxic below 25 μg mL−1. In contrast, their bismuth(III) complexes all showed excellent activity across all three strains (e.g. 0.28 μM for [Bi(H-SHA)3] to 6.01 μM for K[Bi(BHA)2] against strain 251) with only minor variations in activity being both ligand and composition dependant.
Introduction
Hydroxamic acids are a class of compound with general formula RCON(H)OH and are of significant biological importance. Principally known as strong polyfunctional iron chelators they also display antibacterial, anti-inflammatory and anti-asthmatic behaviour,1,2 and have been used in the design of therapeutic targets for cancer,3,4 Alzheimer's disease,5 haemochromatosis,6,7 and malaria.8 Being diprotic, deprotonation can occur at either the NH or OH functionality or at both, though they are much weaker acids than analogous carboxylic acids RC(O)OH. In addition to Fe(III), anions derived from hydroxamic acids are known to chelate to various metal ions, including heavy metals.1,9,10 They are biochemically significant in that they form constitutive parts of active sites of some enzymes.11 Their significance is also enhanced by their involvement in biological processes such as microbial iron transport,11 inhibition of the nickel-dependent urease enzymes12–15 and the zinc-dependent matrix metalloproteinases.16
The mode-of-action of metal compounds in exerting a bactericidal or bacteriostatic effect is under-researched and poorly understood. There are general known effects: generation of radical oxygen species and anti-oxidant depletion, interference in cell redox processes, enzyme and ATP inhibition, disruption of protein function, membrane impairment, and DNA damage.16,17 One of the key processes is ion mimicry, particularly of Fe(III). This is relevant for metals in the +III oxidation state, such as bismuth. The biological chemistry of Bi(III) intersects well with that of Fe(III), though Bi(III) is significantly more thiophilic.18 Bismuth(III) has a strong affinity for S-based ligands, such as glutathione and metallothionein, but also competes with ferric ions in vivo at N- and O-binding sites in biomolecules.19 for example with transferrin and lactoferrin in human serum.20 The few metallomic studies on bismuth relate almost exclusively to H. pylori,21 primarily because bismuth compounds (subsalicylate and citrate salts) are highly effective and used clinically in the treatment and eradication of H. pylori.22 Recent studies have begun to uncover mechanisms by which bismuth is toxic to the bacterium, including disrupting the enzymes urease and alcohol dehydrogenase, and interfering with Fe(III) regulating proteins.23
To survive, many bacteria rely on siderophores (polydentate O-binding molecules with carboxylate, catecholate and hydroxamate functional groups) as strong metal chelators to sequester and transport ferric ions.24 Targeting bismuth hydroxamates provides the potential to develop new antibacterial compounds and strategies – using siderophores as ‘trojan horses’ for the uptake of Bi(III) into a microbial cell.17
Since H. pylori does not use siderophores to take up iron, and instead utilizes host transferrin, lactoferrin and heme, our recent study on the strong bactericidal effect of bismuth(III) benzohydroxamates against H. pylori also suggests iron mimicking processes could be highly effective.25 In this study, the first on bismuth hydroxamates, we demonstrated that mono- and di-anionic hydroxamato complexes derived from benzohydroxamic acid (H2-BHA); [Bi2(BHA)3], [Bi(H-BHA)3] and [Bi(BHA)(H-BHA)], displayed nM activity against H. pylori, orders of magnitude greater that standard bismuth carboxylate based medications. The hydrolytic instability of the complexes was also demonstrated with the crystallization and structural elucidation of the first Bi34 oxido-cluster [Bi34O22(BHA)22(H-BHA)14(DMSO)6].26
In this paper we now report the results of our extended study into the formation, structure and bactericidal activity of a series of novel bismuth(III) hydroxamates derived from N-methylfurohydroxamic acid (H-MFHA), N-benzoyl-N-phenylhydroxamic acid (H-BPHA), salicylhydroxamic acid (H2-SHA), and acetohydroxamic acid (H2-AHA), shown in Fig. 1. In addition, a study of the K salt of the anionic benzohydroxamate bismuthate complex [Bi(BHA)2] is also now included. Three complexes have been structurally authenticated by single crystal X-ray diffraction; [Bi(MFHA)3]2·Me2CO (3·0.5Me2CO), {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ (4·(DMSO2)), and [Bi(BPHA)3]2·2EtOH (7·EtOH). The activity of the complexes towards three strains of H. pylori and their toxicity towards human fibroblast cells is also reported and discussed.
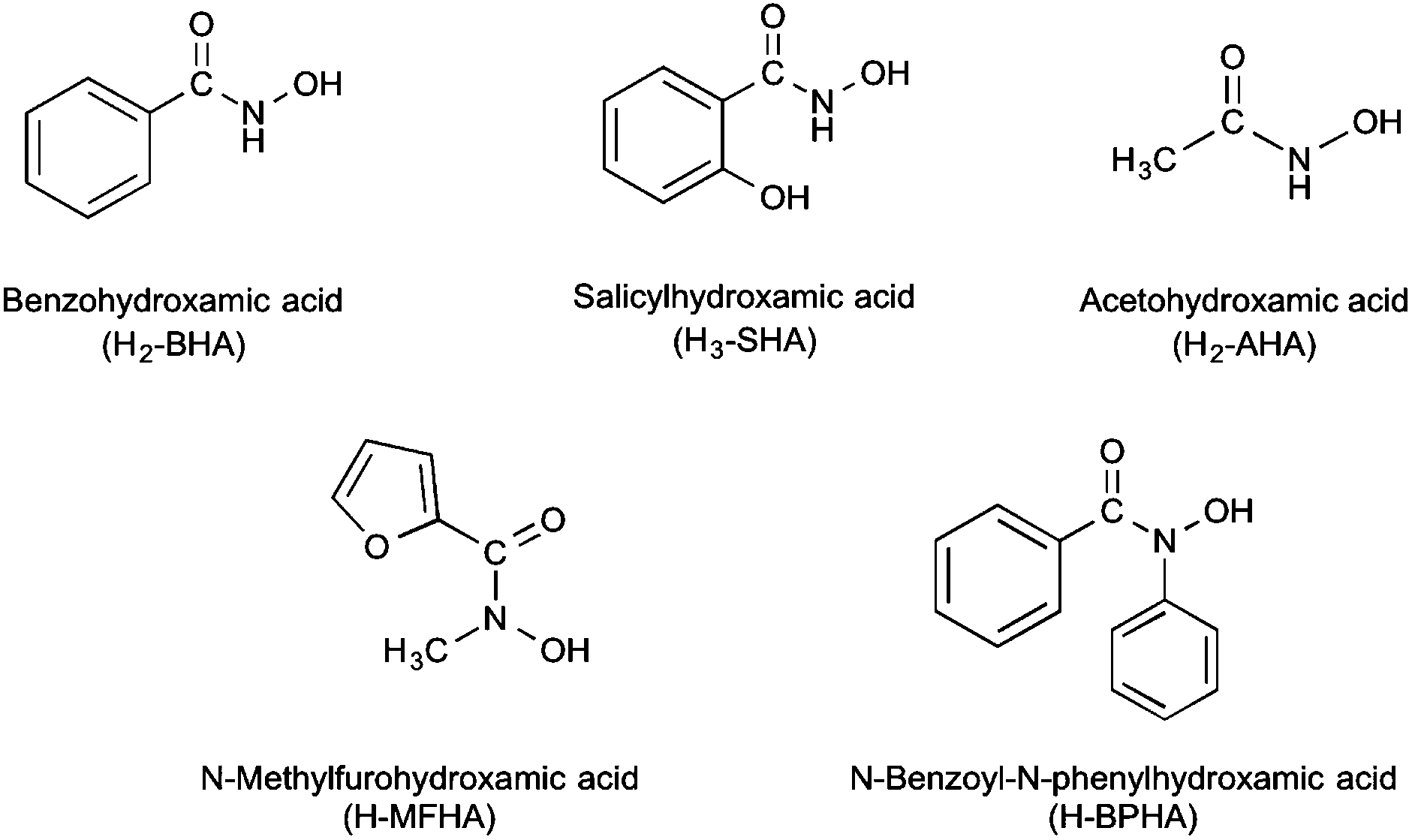 | ||
| Fig. 1 Hydroxamic acids employed in the formation of bismuth(III) complexes. | ||
Results and discussion
The five target hydroxamic acids (Fig. 1) were chosen to provide structural diversity in the composition of the Bi(III) complexes and also differences in possible binding modes and coordination environments. Acetohydroxamic acid is already known to inhibit the Ni-centred urease in H. pylori and so was an obvious target.27 The others provide varying protic sites for anion formation and donor atoms for intra- and inter-molecular bonding with the metal centre.The acids were treated with BiPh3 in a stoichiometric ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under both solvent-free (SF) and solvent-mediated (SM) conditions (Scheme 1). For the solvent free reaction the thermochemical profile of the protolysis reaction was first studied using differential scanning calorimetry (DSC) to establish whether the reaction proceeded and at which temperature. This DSC profiles indicated that solvent free reactions were possible with exotherms for N-methylfurohydroxamic acid (H-MFHA), salicyhydroxamic acid (H2-SHA) and benzohydroxamic acid (H2-BHA) at 100, 120 and 60 °C respectively. Grinding the acid together with crystalline BiPh3 and heating under a nitrogen atmosphere to just above the required temperature produced good yields of the target mono-anionic tris-hydroxamato complexes: [Bi(H-SHA)3] 1, [Bi(H-BHA)3] 2 and [Bi(MFHA)3] 3. We have reported previously on the formation and analysis of [Bi(H-BHA)3].26 Interestingly, N-benzoyl-N-phenylhydroxamic acid (H-BPHA, pKa 9.15) and acetohydroxamic acid (H2-AHA, pKa 9.40), were more problematic and no single isolable product could be obtained. Of the five hydroxamic acids these two have the highest reported pKa values. The DSC profiles showed exotherms at much higher temperatures. With (H2-AHA) and BiPh3 the component endothermic melting points are observed at 61 and 81 °C respectively, followed then by two exotherms at 193 and 200 °C. With (H-BPHA) and BiPh3 the melting endotherms are observed at 78 and 110 °C respectively, and then a large broad exotherm beginning at 167 °C and finishing at 220 °C. These high reaction temperatures most likely mean product formation by this method is not homogeneous and is accompanied by decomposition.
:1 under both solvent-free (SF) and solvent-mediated (SM) conditions (Scheme 1). For the solvent free reaction the thermochemical profile of the protolysis reaction was first studied using differential scanning calorimetry (DSC) to establish whether the reaction proceeded and at which temperature. This DSC profiles indicated that solvent free reactions were possible with exotherms for N-methylfurohydroxamic acid (H-MFHA), salicyhydroxamic acid (H2-SHA) and benzohydroxamic acid (H2-BHA) at 100, 120 and 60 °C respectively. Grinding the acid together with crystalline BiPh3 and heating under a nitrogen atmosphere to just above the required temperature produced good yields of the target mono-anionic tris-hydroxamato complexes: [Bi(H-SHA)3] 1, [Bi(H-BHA)3] 2 and [Bi(MFHA)3] 3. We have reported previously on the formation and analysis of [Bi(H-BHA)3].26 Interestingly, N-benzoyl-N-phenylhydroxamic acid (H-BPHA, pKa 9.15) and acetohydroxamic acid (H2-AHA, pKa 9.40), were more problematic and no single isolable product could be obtained. Of the five hydroxamic acids these two have the highest reported pKa values. The DSC profiles showed exotherms at much higher temperatures. With (H2-AHA) and BiPh3 the component endothermic melting points are observed at 61 and 81 °C respectively, followed then by two exotherms at 193 and 200 °C. With (H-BPHA) and BiPh3 the melting endotherms are observed at 78 and 110 °C respectively, and then a large broad exotherm beginning at 167 °C and finishing at 220 °C. These high reaction temperatures most likely mean product formation by this method is not homogeneous and is accompanied by decomposition.
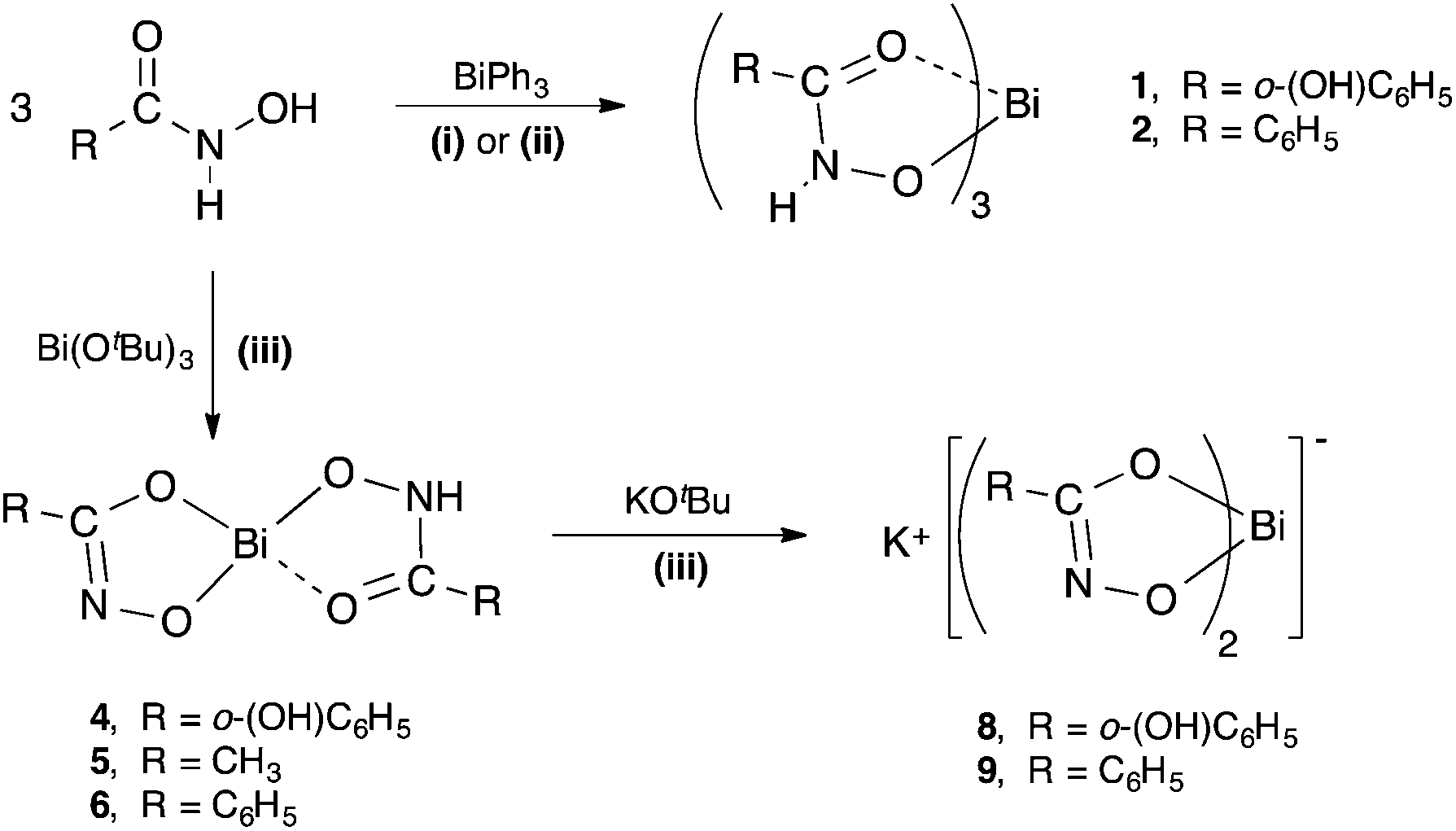 | ||
| Scheme 1 Synthetic pathways to bismuth(III) hydroxamate complexes 1, 2, 4–6, 8, and 9. (i) = solvent-free (80–120 °C); (ii) = solvent reflux in ethanol or toluene; (iii) = THF, −78 °C to RT. | ||
Analogous solvent-mediated reactions conducted in either ethanol or toluene at reflux over 24 h produced the same result. Yields for each reaction were marginally improved when conducted in toluene (58–63%) rather than ethanol (55–57%), but were both lower than for the equivalent solvent-free reactions (65–69%).
Bismuth tert-butoxide behaves as a stronger base than BiPh3 and was expected to react with the hydroxamic acids in a less selective manner. Reactions were conducted under inert conditions by adding a dry THF solution of Bi(OtBu)3 to the hydroxamic acid in THF cooled to −78 °C. The reaction mixture was then allowed to warm slowly to room temperature overnight. All volatiles were then removed in vacuo and the remaining product washed with dry EtOH, which was subsequently removed by filtration. In the absence of any knowledge of the hydrolytic stability of the resultant Bi(III) hydroxamates analyses were conducted using inert atmosphere protocols. The compounds were subsequently exposed to air and moisture and re-analysed.
The diprotic hydroxamic acids, (H2-SHA), (H2-AHA) and (H2-BHA), resulted in mixed-anion complexes: [Bi(SHA)(H-SHA)] 4, [Bi(AHA)(H-AHA)] 5, and [Bi(BHA)(H-BHA)] 6. Notably, the third accessible proton on the salicylate anion was not removed. The monoprotic acids (H-MFHA) and (H-BPHA) resulted in the tris-hydroxamato species; [Bi(MFHA)3] 3 and [Bi(BPHA)3] 7 (Scheme 2).
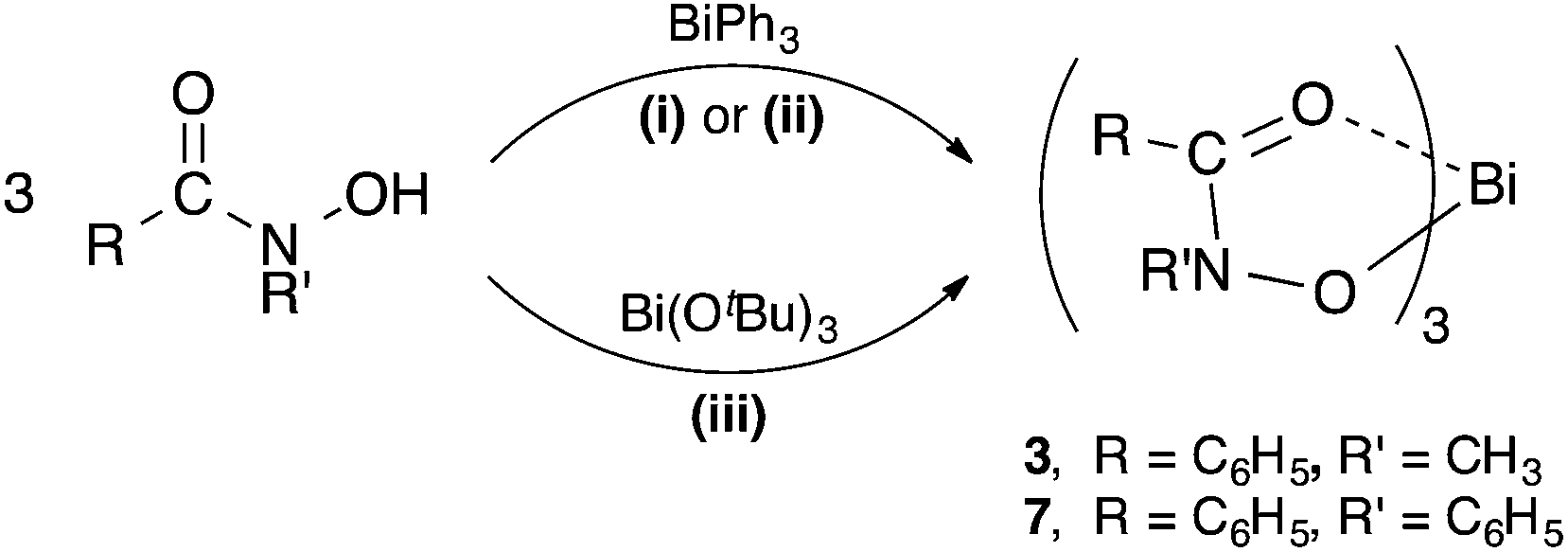 | ||
| Scheme 2 Synthesis of bismuth(III) hydroxamate complexes 3 and 7. (i) = solvent-free (80–120 °C); (ii) = solvent reflux in ethanol or toluene; (iii) = THF, −78 °C to RT. | ||
The dianionic complexes retain an acidic N–H on one of the ligands. In targeting more hydrophilic bismuthate complexes of form K[BiL2], each dianionic complex was treated with one equivalent of K(OtBu) using reaction conditions similar to those under which they were initially formed. This resulted in the formation and isolation of two novel potassium bismuthate complexes; K[Bi(SHA)2] 8 and K[Bi(BHA)2] 9. The reaction with Bi(AHA)(H-AHA)] 5 produces only an insoluble compound which does not analyse as the pure target complex but as a mixture.
Compound characterisation
The solution state structures of complexes 1–7 were studied by 1H-NMR and 13C-NMR spectroscopy in d6-DMSO as solvent. A summary of key chemical shifts for the acids and bismuth(III) complexes are presented in Table 1. In the 1H NMR spectra of the acids the NH and OH proton resonances are averaged, giving broad singlets. The formation of the hydroxamato bismuth(III) complexes is confirmed by the absence of the chemical shifts associated with one or both of these hydroxamic OH and/or NH protons following deprotonation with BiPh3 or Bi(OtBu)3. In those complexes where there is a residual NH proton, the signal for this is observed at a higher frequency relative to the averaged signal observed for the acid. Thus, in 1 and 4 the NH proton is observed at δ 11.96 and 12.20 respectively (from δ 11.66), while in 5 and 6 they are found at δ 11.62 (from 9.71) and δ 12.20 (from 10.14) respectively. The interesting spectrum is that of 2 in which two distinct proton signals are observed at δ 11.14 and 9.02, each integrating to 0.5H. This could result from a configurationally stable tautomerism in the hydroxamato anion, PhC(O)N(H)–O− and PhC(–OH)N–O−. In the absence of solid-state structural information it can only be speculated that this results from the ligand being more labile but with a slow interconversion rate caused by binding with the Bi atom.
| N–H | O–H |
C(X) |
|
|---|---|---|---|
| H2-SHA | 11.66 (2H) | 166.2 | |
| [Bi(H-SHA)3] 1 | 11.96 | — | 166.3 |
| [Bi(SHA)(H-SHA)] 4 | 12.21 | — | 166.4 |
| K[Bi(SHA)2] 8 | — | — | 170.2 |
| H2-BHA | 10.14 (2H) | 164.2 | |
| [Bi(H-BHA)3]212 | 11.14 (0.5H) | 9.02 (0.5H) | 164.6 |
| [Bi(BHA)(H-BHA)]216 | 12.48 | — | 165.3 |
| K[Bi(BHA)2] 9 | — | — | 167.8 |
| H2-AHA | 9.71 (2H) | 167.0 | |
| [Bi(AHA)(H-AHA)] 5 | 11.62 | — | 164.4 |
| (H-MFHA) | — | 10.24 | 158.3 |
| [Bi(MFHA)3] 3 | — | — | 153.7 |
| H-BPHA | — | 10.69 | 167.8 |
| [Bi(BPHA)3] 7 | — | — | 163.8 |
The main structural diagnostic in the 13C NMR spectra for the various complexes would be the presence of CO or CN. Unfortunately in d6-DMSO at 25 °C these resonances appear indistinguishable, presenting only as a single averaged signal. However, some general observations can be made. Where the CO is retained on deprotonation of the acid – in 3 and 7 – there is a low frequency shift for the carbonyl C of ca. 4–5 ppm. Though this is less pronounced in 1 and 2 and is perhaps expected in 2 because of the appearance of suspected tautomeric forms. In those complexes where the only form of the ligand is a dianion – in 8 and 9 – the rearrangement to a CN bond results in a noticeable high frequency shift for the new imino C of ca. 3–4 ppm. Complexes 4, 5 and 6 contain both a dianionic and mono-anionic ligand and show only minor shifts away from the carbonyl C chemical shift (+0.2 ppm in 4, −2.4 ppm in 5, and +0.9 ppm in 6) reflecting competing influences of CO vs. CN.
A summary of the key IR absorbances relating to each bismuth complex and comparing them with the parent hydroxamic acid is provided as Table S1 in the ESI.† Complexes 1, 2, 4, 5 and 6 show evidence of a residual NH bond in the hydroxamate anions, while absorbances relating to the acid OH stretch disappears for each and every complex. The shift to lower wavenumber for the NH stretching band is more pronounced in the mixed-anion hydroxamato complexes (4, 5, and 6) than in the mono-anionic tris-hydroxamato complexes (1 and 2).
In the four hydroxamate complexes proposed to have residual CO bonds after anion formation (1, 2, 3 and 7) the IR spectra indicate coordination of the carbonyl O atom to Bi, as evidenced by the prominent shift to lower wavenumbers for each. Each of these complexes also shows an absorbance for the C–N bond which shifts slightly to a lower wavenumber (ca. 10–20 cm−1). Indicative of dianion formation and rearrangement, the spectra of complexes 4, 5, 6, 8 and 9 show the appearance of pronounced absorbances for CN bonds at or close to 1595 cm−1 for each of these complexes.
Taking the IR and NMR data together along with the solid-state structures of 3, 4 and 7 described below, we have assigned the compound compositions provided in Schemes 1 and 2 above.
X-ray crystallography
Of the seven new bismuth(III) hydroxamate complexes, single crystals suitable for X-ray diffraction studies were obtained from solutions of 3, 4 and 7 in acetone, DMSO and ethanol/acetone respectively. Complexes 3·0.5Me2CO and 7·EtOH contain two crystallographically independent molecules without intermolecular interactions in the asymmetric unit. A summary of the crystallographic data for all three compounds is given in the Experimental section.
In both complexes 3·0.5Me2CO and 7·EtOH the parent hydroxamic acids have been deprotonated only at the hydroxyl group to give the tris-hydroxamato complexes {[Bi(MFHA)3]2·Me2CO} (Fig. 2) and {[Bi(BPHA)3]2·2EtOH} (Fig. 3), with the mono-anionic hydroxamate ligands bonding to the Bi(III) atoms in a bi-dentate fashion through the carbonyl and hydroxamato O atoms to form 5-membered BiO2NC chelate rings. The Bi–O bond lengths show asymmetry with the average covalent bond length [2.2417 Å (3); 2.237 Å (7)] distinctly shorter than the coordinative (dative) Bi–O carbonyl length [2.4523 Å (3); 2.496 Å (7)]. In each asymmetric unit of 3·0.5Me2CO and 7·EtOH the Bi(III) center is formally six coordinate while dimerization of two monomeric units, through the O atom of an adjacent hydroxamate ligand [Bi–O: 2.881(15) Å (3); 2.779(16) Å (7)], increases the coordination number to seven giving an overall distorted pentagonal bipyramidal geometry.
 | ||
| Fig. 2 Molecular structure of {[Bi(MFHA)3]2·Me2CO)} (3·0.5Me2CO) Only one of two independent molecules is shown; hydrogen atoms and uncoordinated solvent molecules are omitted for clarity. Thermal ellipsoids at 40% probability. Symmetry operator ′: 1 − x, 2 − y, 1 − z. Selected bond lengths (Å) and angles (°): Bi(1)–O(1), 2.2586(15); Bi(1)–O(2), 2.4459(16); Bi(1)–O(4), 2.2918(15); Bi(1)–O(5), 2.4734(15); Bi(1)–O(7), 2.1747(16); Bi(1)–O(8), 2.4373(15); N(1)–O(1), 1.372(2); N(1)–C(2), 1.373(3); N(2)–O(4), 1.368(2); N(2)–C(8), 1.317(3); N(3)–O(7), 1.383(2); N(3)–C(14), 1.333(3); C(2)–O(2), 1.273(3); C(8)–O(5), 1.270(3); C(4)–O(8), 1.262(3); O(1)–Bi(1)–O(2), 67.48(5); O(4)–Bi(1)–O(5), 66.88(5); O(7)–Bi(1)–(O8), 69.31(5); Bi(1)–O(1)–Bi(1)′, 81.28(8); O(1)–Bi(1)–O1)′, 98.72(9). | ||
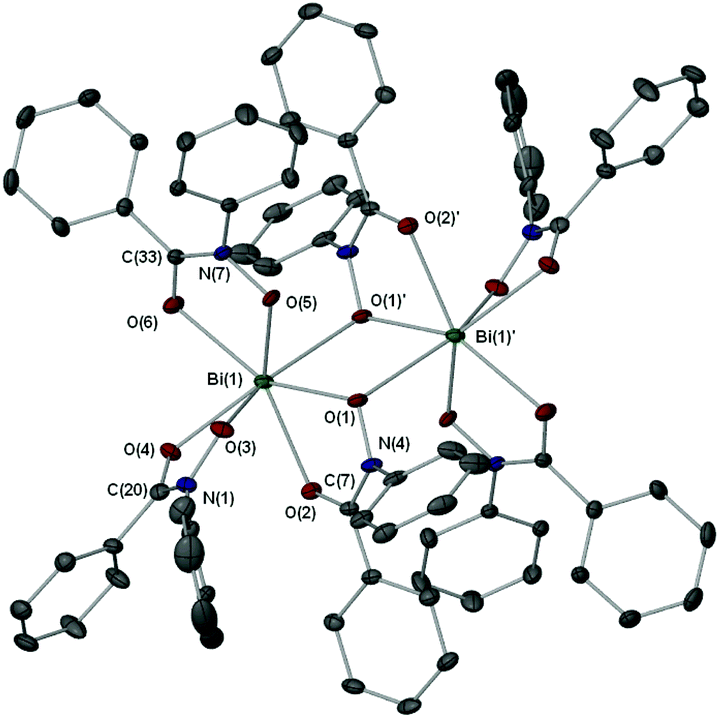 | ||
| Fig. 3 Molecular structure of {[Bi(BPHA)3]2·2EtOH} (7·EtOH). Hydrogen atoms and uncoordinated solvent molecules are omitted for clarity. Thermal ellipsoids at 40% probability. Symmetry operator ′: −x, 1 − y, −z. Selected bond lengths (Å) and angles (°): Bi(1)–O(1), 2.2804(19); Bi(1)–O(2), 2.5151(18); Bi(1)–O(3), 2.195(2); Bi(1)–O(4), 2.4994(18); Bi(1)–O(5), 2.2364(17); Bi(1)–O(6), 2.474(2); Bi(1)–O(1)′, 2.779(16); N(1)–O(3), 1.388(3); N(1)–C(20), 1.251(3); N(4)–O(1), 1.390(3); N(4)–C(7), 1.342(3); N(7)–O(5), 1.373(3); N(7)–C(33), 1.326(3); O(1)–Bi(1)–O(2), 66.07(6); O(3)–Bi(1)–O(4), 67.71(7); O(5)–Bi(1)–O(6), 68.00(6); O(1)–Bi(1)–O(1)′, 72.30(6); Bi(1)–O(1)–Bi(1)′, 107.70(8). | ||
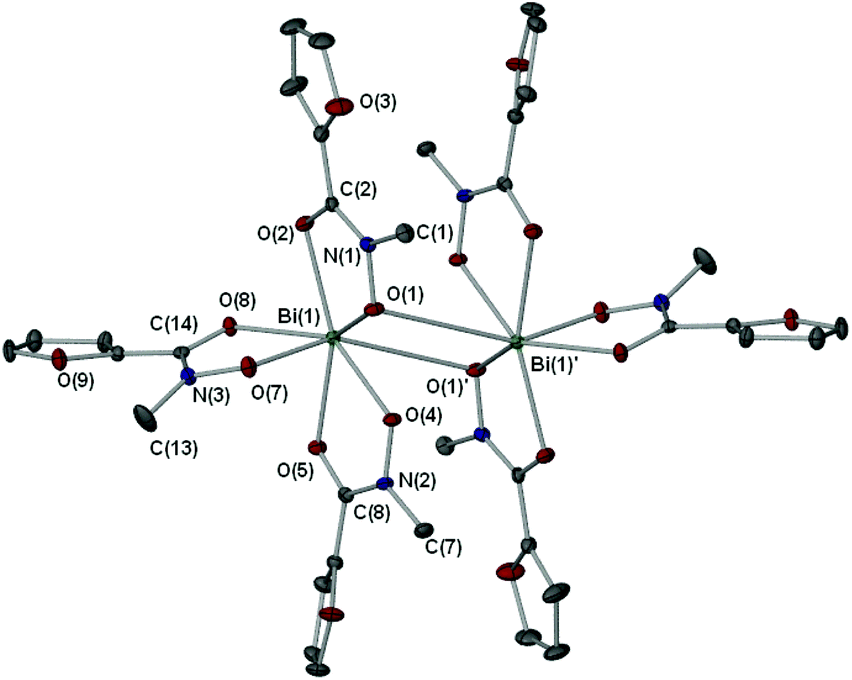
X-ray crystallographic studies on crystals grown from a DMSO solution of compound 4 confirmed the mixed mono- and di-anionic hydroxamato complex {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ [4(DMSO)1.5·0.5DMSO] as shown in Fig. 4. The asymmetric unit consists of two crystallographically independent molecules, essentially isostructural, which differ only by the number of coordinated DMSO solvent molecules (Bi(1) has one DMSO and Bi(2) has two, as shown in Fig. 4), for brevity only Bi(2) will be discussed in detail.
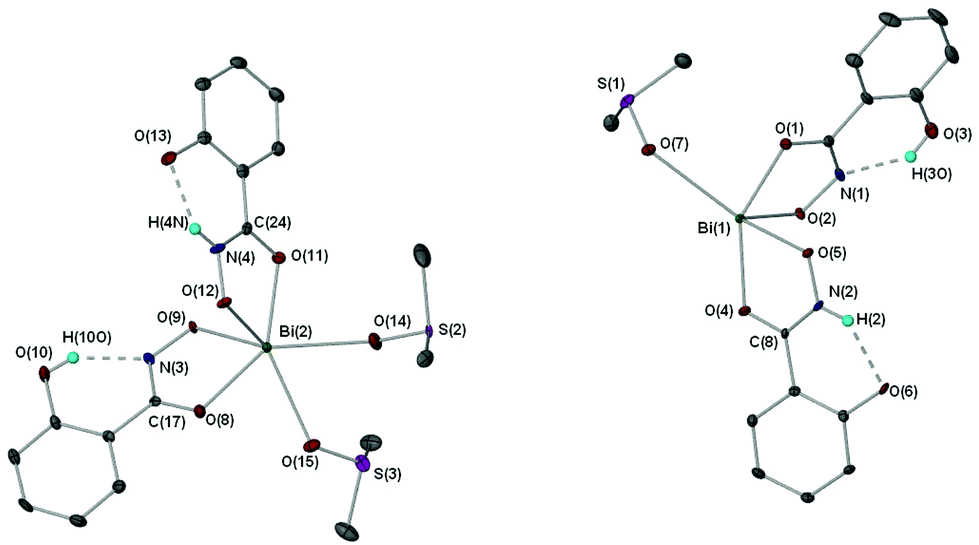 | ||
| Fig. 4 Molecular structure of {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ [4(DMSO)1.5·0.5DMSO]. Only one of two independent molecules is shown; hydrogen atoms (except NH) and uncoordinated solvent molecules are omitted for clarity. Thermal ellipsoids at 40% probability. Selected bond lengths (Å) and angles (°): Bi(2)–O(8), 2.346(3); Bi(2)–O(9), 2.268(3); Bi(2)–O(11), 2.284(3); Bi(2)–O(12), 2.231(3); Bi(2)–O(14), 2.748(3); Bi(2)–O(15), 2.710(6); N(3)–O(9), 1.382(4); N(3)–C(17), 1.319(5); N(4)–O(12), 1.390(4); N(4)–C(24), 1.297(5); C(17)–O(8), 1.275(5); C(24)–O(11), 1.288(5); O(13)–H(4N), 1.896(5); O(8)–Bi(2)–O(9), 69.79(10); O(11)–Bi(2)–O(12), 70.18(10). | ||
In the asymmetric unit of complex [4(DMSO)1.5·0.5DMSO] the Bi(III) center bonds to one doubly deprotonated hydroxamate ligand and one mono-deprotonated hydroxamate ligand through the carbonyl and hydroxamate O atoms with the remainder of the coordination sphere filled by coordinating DMSO solvent molecules giving an overall distorted octahedral geometry for Bi(2). The hydrogen atom H(4N) in [4(DMSO)1.5·0.5DMSO], which was placed in a calculated position confirmed by other analytical technique (see Experimental section), makes a hydrogen-bond contact with the hydroxyl group on the phenyl ring of the ligand [N(4H)–O(13) 1.896(5) Å]. The C–O bond distances of the carbonyl groups [C(17)–O(8) 1.275(5) Å (SHA2−); C(24)–O(11) 1.288(5) Å (H-SHA)] and C–N bonds [C(17)–N(3) 1.319(5) Å (SHA2−); C(24)–N(4) 1.297(5) Å (H-SHA)] show little variation for the expected single versus double bond characteristics of the di- and mono-anionic ligands respectively. This most likely arises from an averaging of the bonds through the crystal owing to the possibility of the calculated NH proton being located on either of the ligands at one time.
The full coordination environment of the Bi(III) centre in [4(DMSO)1.5·0.5DMSO] is raised to seven through formation of a linear polymeric chain (Fig. 5). The Bi–O bonds that propagate the polymeric chain [Bi(2)–O(9)′ 2.605(3) Å] are significantly longer than those in the asymmetric unit being more indicative of a dative Bi–O bond length.
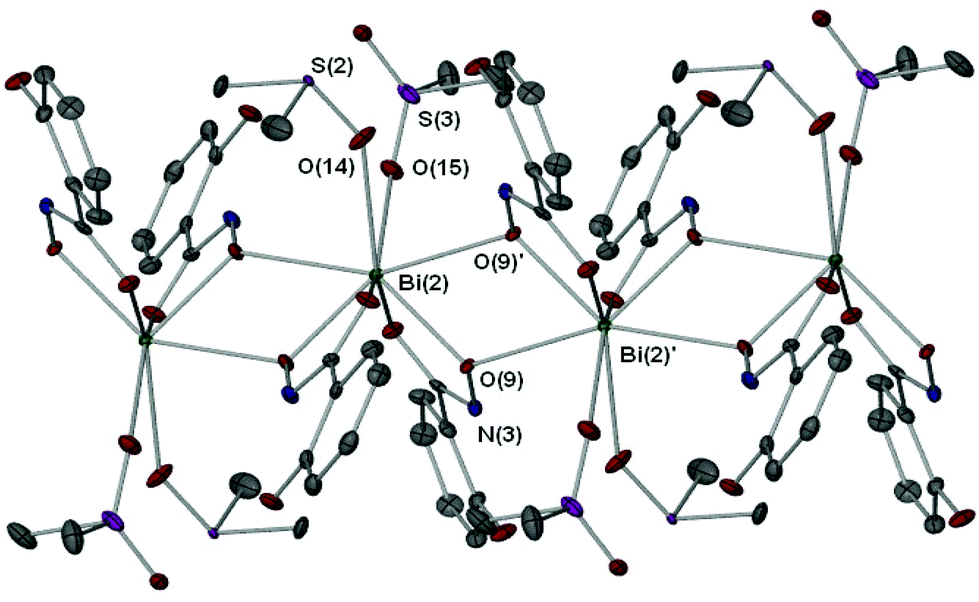 | ||
| Fig. 5 Extended polymeric section of [4(DMSO)1.5·0.5DMSO] with selected atom labeling. Symmetry operator ′: 1 − x, 2 − y, 1 − z. Selected bond lengths (Å) and angles (°): Bi(2)–O(9)′ 2.605(3); Bi(2)–O(9)–Bi(2)′, 112.77(6); O(9)–Bi(2)–O(9)′, 67.23(8). | ||
Recently we reported the structure of the first bismuth hydroxamate complex.26 Interestingly, this proved to be a unique Bi34 oxido-cluster [Bi34O22(BHA)22(H-BHA)14(DMSO)6] 10 resulting from the slow hydrolysis of [Bi2(BHA)3] in DMSO/toluene solution. Complex [4(DMSO)1.5·0.5DMSO] is best compared with 10 since both feature mono- and di-anionic hydroxamate ligands bound to the Bi(III) atoms. But unlike 10, dissolution of complex 4 in DMSO does not result in hydrolysis to give polynuclear oxido-clusters, perhaps due to the presence of the hydroxyl group on the phenyl ring allowing through hydrogen-bond stabilization resistance to hydrolysis and formation of a more simple polymeric solid state.
Complexes 3·0.5Me2CO and 7·EtOH display similar hydroxamate O,O bi-dentate coordination modes to the triphenyl antimony(V) complexes [SbPh3(H-BHA)MeO]29 and [SbPh3(BPHA)Cl]210.28 However, unlike 3·0.5Me2CO and 7·EtOH the dimers in complexes 9 and 10 are formed through weak hydrogen-bond interactions, which also serve to facilitate their overall polymeric chain structure.
Hydrolysis and oxido-cluster formation
From our initial study we reported that the dinuclear bismuth(III) benzohydroxamate complex [Bi2(BHA)3]∞ undergoes slow hydrolysis in a DMSO/toluene solvent mixture to give crystals of the unique {Bi34} oxido-cluster [Bi34O22(BHA)22(H-BHA)14 (DMSO)6].26 This result indicated that the bismuth(III) hydroxamates as a compound class could be prone to hydrolysis. Hydrolytic decomposition of bismuth complexes with O-bound ligands is a well known phenomenon. For alkoxides the close pKa values of H2O and the alcohol (conjugate acid) coupled with the strong Lewis acidity of the Bi3+ cation make the process to polynuclear oxido-clusters thermodynamically favourable. However, it also can occur with carboxylates and sulfonates,29–32 though with these the process is ligand dependent and does not occur for every compound in these two classes.To investigate the stability of the bismuth(III) hydroxamates complexes they were taken up in DMSO based solvent mixtures and allowed to stand for several weeks. The only crystals which appeared were consistent with those already described above (3, 4 and 7), with the others producing fine precipitates. To provide solutions of sufficient dilution to prevent short-term crystallisation or precipitation each complex was taken up in DMSO solution to give 2.0 mM concentration. These solutions were then allowed to stand undisturbed over several weeks or months until some fine precipitate began to appear. The same solutions were examined both by Electrospray Mass Spectrometry (Table S2 ESI†) and Atomic Force Microscopy. Sample preparation details for AFM are provided in the Experimental section.
The AFM height images are shown in Fig. 6 and demonstrate the presence of particle-like features in dried samples. The details for each sample are listed in Table 2.
 | ||
| Fig. 6 AFM height images of bismuth clusters dried onto mica substrates, where values refer to approximate cluster sizes obtained from analysis of the AFM image: (a) K[Bi(BHA)2] 9: 3.5 nm; (b) [Bi(BPHA)3] 7: 4 nm; (c) [Bi(H-SHA)3] 1: <0.5 nm; (d) [Bi(AHA)(H-AHA)] 5: 5 nm; (e) [Bi(MFHA)3] 3: 1.8 nm; (f) [Bi(BHA)(H-BHA)] 6: <0.5 nm. The white scale bar in each image represents 200 nm. | ||
| Complex | Code | Height (nm) | Sample age (weeks) |
|---|---|---|---|
| [Bi(H-SHA)3] 1 | a | <0.5 | 4 |
| [Bi(MFHA)3] 3 | b | 1.8 | 16 |
| [Bi(AHA)(H-AHA)] 5 | c | 5 | 4 |
| Bi(BHA)(H-BHA)] 6 | d | <0.5 | 8 |
| [Bi(BPHA)3] 7 | e | 4 | 24 |
| K[Bi(BHA)2] 9 | f | 3.5 | 4 |
In some samples, such as K[Bi(BHA)2] 9, the presence of discrete, unaggregated particles is clear. However, for other samples, such as [Bi(BPHA)3] 7, individual particles appear to be lower in number, with clear evidence of surface aggregates. This is most likely an effect of the drying procedure used in sample preparation for AFM, where strong lateral capillary forces tend to favour aggregation. It is unlikely that this reflects the structure in solution/dispersion.
For comparison, from single crystal X-ray diffraction the diameter of the [Bi34O22(BHA)22(H-BHA)14 (DMSO)6] oxido cluster is 3.05 nm at its widest point.26 The bismuth subsalicylate oxido-cluster [Bi38O44(HSal)26(Me2CO)16(H2O)2] is 2.37 nm wide.28 The smaller clusters [Bi9O7(HSal)13(Me2CO)5]29 and [Bi22O26(OSiMe2tBu)14]33 are both 1.72 nm at their widest point, and so the diameter is as dependant on the ligand as on the relative number of Bi atoms in the cage core. This would suggest that images which show particle sizes with diameters <0.5 nm the bismuth hydroxamato complexes (1 and 6) show little signs of cluster formation or particle aggregation. The mass spectra of both 1 and 6 display highly charged low molecular mass oxido-species, e.g. [Bi4O2L2(OH)2(H2O)4]4+, which would be <0.5 nm, with no evidence of larger cages.
From the AFM images it is likely that complexes 3 (1.8 nm) and 9 (3.5 nm) undergo hydrolysis to give polynuclear oxido-clusters. Complex 3 shows the presence of both {Bi6} and {Bi9} oxido-clusters; [Bi6O6L2(MeOH)(DMSO)2]4+ and [Bi8O6L2(OH)6(MeOH)(H2O)20]4+, which would match the 1.8 nm particles in the AFM image. Unfortunately, complex 9, on dilution of the solution with MeOH in preparation for recording the mass spectrum, fully precipitated and so the spectrum only gave peaks relating to the ligand with no Bi containing species evident. Heights of 5 nm for 5 and 4 nm for 7 likely result from the aggregation of smaller oxido-clusters. The mass spectrum of 5 displays m/z values corresponding to [Bi3L2(OH)(H2O)]4+ and [Bi3OL(OH)(MeOH)2]4+, while that of 7 indicates species containing {Bi4} and {Bi6} species, e.g. [Bi6O4L4(H2O)2(MeOH)4]4+. It may be expected that drying samples for AFM imaging would induce additional aggregation for clusters that experience attractive interactions, and this may explain the surprisingly large size of the aggregates seen for 5 and 7.
Activity against Helicobacter pylori
We have reported previously on our investigations into the antibacterial activity of the bismuth(III) benzohydroxamate complexes [Bi(H-BHA)3] 2 and [Bi(BHA)(H-BHA)] 6.26 Both complexes showed remarkably low MIC values against three laboratory strains of H. pylori: B128, 251 and 26695, ranging from 0.05–0.78 μg mL−1 (0.08 to 1.26 μM) for 2 and 0.09–1.56 μg mL−1 (0.19 to 3.24 μM) for 6. These compare favorably with other classes of bismuth(III) complexes which typically have higher MIC values: bismuth (III) carboxylates and thiobenzoates with MIC values of 6.25 μg mL−134,35 and thioxoketones, MIC 3.12 μg mL−1.36 They are an orders of magnitude more active than the current bismuth drugs used to treat H. pylori, bismuth subsalicylate (BSS) MIC 12.5 μg mL−1 and colloidal bismuth subcitrate (CBS) MIC 12.5 μg mL−1.37–39 The only complexes which have shown comparably low MIC values are bismuth(III) amino-arenesulfonates which displayed MIC values against the three H. pylori strains of MIC 0.05 μg mL−1 (0.06 μM), for example [Bi(O3S-(o-AB))3] from ortho-aminobenzenesulfonic acid ((o-AB)SO3H).40 Thus, the motivation to exam the broader family of hydroxamato complexes.
The minimum inhibitory concentration (MIC) of bismuth complexes 1, 3–5 and 7–9 and their parent hydroxamic acids were all assessed for their antibacterial activity against H. pylori strains 26695, B128 and 251 using the Agar Diffusion method and with compound concentrations ranging from 25 to 0.024 μg mL−1. The results are summarised in Table 3. The table shows that the result initially observed for the bismuth benzohydroxamate complexes is replicated across the whole range of different bismuth hydroxamate complexes, illustrating the effective powerful and bactericidal activity of this family of complexes.
| Compound | H. pylori strain | ||
|---|---|---|---|
| MIC μg mL−1 (μM) | |||
| 26695 | B128 | 251 | |
| H2-SHA | >25 | ||
| [Bi(H-SHA)3] 1 | 0.19 (0.28) | 0.39 (0.59) | 0.19 (0.28) |
| [Bi(SHA)(H-SHA)] 4 | 0.39 (0.76) | 0.78 (1.52) | 0.39 (0.76) |
| K[Bi(SHA)2] 8 | 0.78 (1.41) | 1.56 (2.83) | 0.78 (1.41) |
| H2-BHA | >25 | ||
| [Bi(H-BHA)3]212 | 0.09 (0.14) | 0.05 (0.08) | 0.78 (1.26) |
| [Bi(BHA)(H-BHA)]216 | 0.19 (0.39) | 0.09 (0.19) | 1.56 (3.24) |
| K[Bi(BHA)2] 9 | 1.56 (3.00) | 3.12 (6.01) | 3.12 (6.01) |
| H2-AHA | 6.25 (83.26) | ||
| [Bi(AHA)(H-AHA)] 5 | 0.78 (2.2) | 1.56 (4.58) | 1.56 (4.58) |
| H-MFHA | >25 | ||
| [Bi(MFHA)3] 3 | 1.56 (2.47) | 1.56 (2.47) | 1.56 (2.47) |
| H-BPHA | >25 | ||
| [Bi(BPHA)3] 7 | 0.39 (0.41) | 0.39 (0.41) | 0.39 (0.41) |
As expected, acetohydroxamic acid (H2-AHA) shows moderately good activity against all three strains of the bacterium (MIC of 6.25 μg mL−1 (83.26 μM)) since it is already known to effectively inhibit the nickel containing urease essential for the lifecycle of H. pylori.27 Its bismuth complex [Bi(AHA)(H-AHA)] 5 shows a magnification in this activity, exhibiting an MIC of 0.78 μg mL−1 (2.2 μM) for the 26695 strain and 1.56 μg mL−1 (4.58 μM) for strains B128 and 251, suggesting a synergistic effect of the metal and ligand.
Surprisingly, the other hydroxamic acids showed no activity towards the H. pylori strains below 25 μg mL−1. In contrast their bismuth(III) complexes are highly effective at very low concentrations showing some marginal variations in MIC within the overall class of compound.
Interestingly, there is a clear pattern within the family of compounds for both the salicylhydroxamates and benzohydroxamates in which the mono-anionic tris-hydroxamato complexes are the most active compounds, followed by the mixed anion complexes, with the di-anionic hydroxamato bismuthate complexes being marginally less effective. This is illustrated by tracking activity against the 251 strain which appears in the order: [Bi(H-SHA)3] (0.28 μM) < [Bi(SHA)(H-SHA)] (0.76 μM) < K[Bi(SHA)2] (1.41 μM) and [Bi(H-BHA)3] (0.78 μM) < [Bi(BHA)(H-BHA)] (1.56 μM) < K[Bi(BHA)2] (3.12 μM). The structural features of the complexes suggests this pattern possibly reflects a reduction in their lipophilicity as we move towards a more ionic species.
There appears to be little effect on the overall activity from N-substitution since the complexes [Bi(MFHA)3] 3 and [Bi(BPHA)3] 7 display similar activities to the other analogous mono-anionic tris-hydroxamato complexes: 2.47 μM and 0.41 μM for 3 and 7 respectively against the 251 strain, though it would suggest N-Ph promotes greater bactericidal activity rather than N-Me substitution.
Mammalian cell toxicity
All the bismuth complexes were assessed for their toxicity towards human skin fibroblast cells. The only compound which showed any cytotoxic effect up to 100 μM was 2, at which concentration it proved to be lethal to 94% of the cells, but only 5% at 12.5 μM. All the other bismuth complexes were non-toxic up to 100 μM, as were the hydroxamic acids themselves. The results are presented in the ESI.†Conclusion
In this study seven new bismuth(III) hydroxamate complexes were synthesized and fully characterized: tris-hydroxamato complexes containing only mono-anionic ligands; [Bi(H-SHA)3] 1, [Bi(MFHA)3] 3 and [Bi(BPHA)3] 7, mixed-anion complexes; [Bi(SHA)(H-SHA)] 4 and [Bi(AHA)(H-AHA)] 6, and bismuthate complexes; K[Bi(SHA)2] 8 and K[Bi(BHA)2] 9. The composition of each complex was established through NMR and FT-IR studies, with the solid-state structure of three of the complexes being determined through single crystal X-ray diffraction; [Bi(MFHA)3]2·Me2CO, {[Bi(SHA)(H-SHA)(DMSO)2][Bi(SHA)(H-SHA)(DMSO)]·DMSO}∞ and [Bi(BPHA)3]2·2EtOH. Stability studies using and comparing ES-MS and AFM indicated that most of the complexes undergo slow hydrolysis over several weeks in solution, generating either both low molecular weight oxido species (1, 5–7) and larger polynuclear cages (3, 9). Both techniques proved wanting in definitive characterisation but appeared at least complementary in indicating the onset and extent of hydrolysis.
All the complexes and their parent acids were assessed for the bactericidal activity against three strains of Helicobacter pylori (26695, B128 and 251). Of the acids, only acetohydroxamic acid showed any activity at low concentrations (MIC 6.25 μg mL−1; 83.26 μM) while the others were found to be non-toxic below 25 μg mL−1. In contrast, the bismuth(III) hydroxamato complexes all showed excellent activity across all three strains of H. pylori (e.g. 0.28 μM for [Bi(H-SHA)3] to 6.01 μM for K[Bi(BHA)2] against strain 251) with only minor variations in activity being both ligand and composition dependant. These results suggest that the synergistic combination of Bi(III) metal with hydroxamate ligands has the potential to generate new and effective ‘trojan horse’ type antibacterial agents.
Experimental section
Bismuth tert-butoxide, Bi(OtBu)3, was synthesized through a metathesis reaction from the treatment of BiCl3 with potassium tert-butoxide (KOtBu)3 in dried THF at 0 °C and subsequently the residue is extracted with pentane in a soxhelette extractor.41 Triphenyl bismuth (BiPh3) was synthesized according to a literature procedure.42 Acetohydroxamic acid, salicylhydroxamic acid, N-methylfurohydroxamic acid, N-benzoyl-N-phenyl hydroxamic acid and benzohydroxamic acid were purchased from Sigma-Aldrich Co. and TCI America, were used without further purification. 1H-NMR spectra were recorded using either a Bruker DRX 400 MHz or Bruker DPX 300 MHz spectrometer as solutions in d6-DMSO. Multiplicities are denoted as singlet (s), doublet (d), triplet (t), multiplet (m) and broad (b), or combination where necessary. 13C NMR spectra were recorded using either a Bruker DRX 400 MHz or DPX 300 MHz spectrometer as solutions in d6-DMSO. Infrared Spectra (IR) as pure solid were recorded on a Agilent Technologies Cary 630 FTIR. IR absorptions (Vmax) are reported in units of wavenumbers (cm−1).Mass spectra were recorded on a Micromass Platform Electrospray mass spectrometer at cone voltages as specified using a DMSO/methanol or methanol solution as the mobile phase. The ion peaks (m/z) and their assignments are listed. Elemental Analysis was performed by the Chemical & MicroAnalytical Services Pty Ltd, Belmont, Victoria, Australia. Melting points were measured on a Stuart Scientific SMP3 melting point apparatus. All the solvents were purified before use. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Pentane and ether were distilled from sodium. Ethanol and methanol were dried over magnesium turnings prior to use and stored over 4 Å MS under N2. All molecular sieves were dried at 120 °C and allowed to cool under vacuum before use. All reactions requiring anhydrous conditions were conducted with oven-dried glassware under an atmosphere of dry nitrogen using a vacuum/nitrogen line and Schlenk techniques.
Atomic force microscopy (AFM) height images were made using a Nanowizard 3 AFM from JPK in AC (intermittent contact) mode. This instrument incorporates capacitive sensors to ensure accurate reporting of height, z, and x–y lateral distances. The cantilevers used were from Bruker (model NCHV), and possessed nominal resonant frequencies of 340 kHz and spring constants of ∼40 N m−1 respectively. Images were obtained with a force set-point <1 nN. Post acquisition, images were ‘flattened’ to arrange the scan lines in a plane within the JPK image analysis software, and no further manipulations were made. The height images shown convert the normal (z) heights acquired by the AFM z-piezoelectric drive capacitive feedback loop to a colour for ease of presentation. The height scales of the images are (a–f): 8.8, 6.6, 1.1, 12.5, 11.0 and 1.3 nm. Cluster heights were estimated by taking line profiles across each image and calculating the average feature height. Due to tip convolution, lateral dimensions of small objects tend to be exaggerated in AFM imaging, whereas heights are accurately reported by the instrument.
Bacterial strains and culture conditions
H. pylori strains 251, B128, and 26695 were routinely cultured on horse blood agar (HBA) or in brain heart infusion broth (BHI), supplemented with either 7.5% (v/v) fresh horse blood or 10% (v/v) FCS,43 respectively. Culture media were further supplemented with 155 μg L−1 polymyxin B, 6.25 μg L−1 vancomycin, 3.125 μg L−1 trimethoprim, and 1.25 μg L−1 amphotericin B.Determination of the minimum inhibitory concentration
The MICs of bismuth complexes 1–7 were determined by the agar dilution technique. All bismuth complexes were dissolved in DMSO to give clear, colorless solutions of known concentration. H. pylori cultures were incubated in BHI for 18 h, shaking at 140 rpm at 37 °C under micro aerobic conditions. Bacteria were pelleted, washed in phosphate-buffered saline, then resuspended in BHI.44 Each suspension was adjusted to give a fairly accurate density of 106 bacteria per mL. Aliquots (10 mL) of these suspensions were then streaked onto HBA plates containing doubling dilutions of the different concentrations of bismuth compounds, ranging in concentration from 0.024 to 25 μg mL−1. Each compound was tested along with the free ligands in equal concentrations. The MICs of the different compounds were determined by assessment of the plates after incubation for 3–5 days at 37 °C.X-ray data
Crystallographic data for compound 7 was obtained on an OXFORD Gemini Ultra equipped with an OXFORD Cryosystems 700 Cryostream and cooled to 123(1) K. Data was collected with monochromatic (graphite) Mo Kα radiation (λ = 0.71073 Å) and processed using the CrysAlisProv 1.171.34.345 software; Lorentz, polarization and absorption corrections (multi-scan) were applied. Crystallographic data for compounds 3 and 4 were obtained on a Bruker X8 APEXII CCD diffractometer46 equipped with an OXFORD Cryosystems 700 Cryostream and cooled to 123(1) K. Data was collected with monochromatic (graphite) Mo Kα radiation (λ = 0.71073 Å) and processed using the Bruker Apex2 v2012.2.0 software; Lorentz, polarization and absorption corrections (multi-scan – SADABS)47 were applied. Compounds 3, 4 and 7 were solved and refined with SHELX-97.48 All non-hydrogen atoms were refined with anisotropic thermal parameters unless otherwise indicated and hydrogen atoms were placed in calculated positions using a riding model with C–H = 0.95–0.98 Å and Uiso(H) = xUiso(C), x = 1.2 or 1.5. Data for 3, 4 and 7 has been deposited with the Cambridge Crystallographic Database with CCDC number 1046081, 1046082 and 1046083 respectively.
Synthesis and characterization of bismuth(III) hydroxamates 1–9
[Bi(H-SHA)3], 1. Solvent reflux: in a Schlenk flask and under inert conditions, a mixture of triphenyl bismuth (0.22 g, 0.5 mmol) and salicylhydroxamic acid (0.23 g, 1.5 mmol) in ethanol or toluene (10 mL) was heated to reflux for 24 hours. This resultant mixture was allowed to cool to room temperature before the precipitate was collected by filtration. Any excess triphenyl bismuth was removed by washing the precipitate with small aliquots (5 mL) of ethanol. The precipitate was then dried under vacuum.
Solvent-free: in a Schlenk flask and under inert conditions, a solid mixture of triphenyl bismuth (0.22 g, 0.5 mmol) and salicylhydroxamic acid (0.23 g, 1.5 mmol) was heated at 120 °C for 1 hour. On cooling unreacted triphenyl bismuth was removed by washing the precipitate with small amounts of ethanol and toluene (5 mL). The precipitate was then dried under vacuum.
Yields: 0.21 g, 63% (toluene reflux); 0.19 g, 57% (ethanol reflux); 0.23 g, 69% (solvent-free). Melting point: 225–226 °C (decomp.). FT-IR (cm−1): 661 (Vs, ring in plane bend), 744 (m, CH-out of plane bend), 909 (Vs, NO), 1248 (m, C–O), 1440 (m, C–N), 1561 (m, CO), 1596 (w, CN), 3066 (brd, Phe-OH), 3281 (brd, NH). 1H NMR (400 MHz, d6-DMSO): δ 6.83 (t, 3H, Hd), 6.91 (d, 3H, Hc), 7.28 (bs, 3H, Hb), 7.66 (d, 3H, Ha), 9.89 (s, 0.5H, Hi), 11.96 (s, 3.8H, Hh). 13C NMR (100 MHz, d6-DMSO): 116.3 (Cd), 118.5 (Cc), 127.2 (Ca), 131.6 (Cf), 157.6 (Ce), 166.3 (Cg). Elemental analysis; (BiC21H18N3O9), Calc (Found): C 37.91 (37.83), H 2.73 (2.90), N 6.32 (6.27) %. Mass spectrum, (ESI+): 513.1 [BiL2]+, 722.0 [BiL3 + K(H2O)]+, (ESI−) 152.2 [L]−, 510.9 [BiL2]−; (where LH2 = H2-SHA).
Bi(SHA)(H-SHA)] 4. A solution of bismuth tert-butoxide (0.21 g, 0.5 mmol) in THF (10 mL) was added under inert atmosphere to a clear solution of salicylhydroxamic acid (0.23 g, 1.5 mmol) in THF (20 mL) at −80 °C. The reaction mixture was then allowed to warm slowly to room temperature overnight. Removal of volatiles under vacuum left a light yellow coloured solid, which was washed with dry ethanol to remove any unreacted acid and/or bismuth butoxide. Crystals suitable for X-ray diffraction were grown from DMSO solution.
Yield: 0.20 g, 78%. Melting point: 206–208 °C (decomp.). FT-IR (cm−1): 745 (m, CH-out of plane bend), 910 (Vs, NO), 1244 (m, C–O), 1445 (m, C–N), 1565 (m, CO), 1593 (m, CN), 3070 (brd, Ph(OH)), 3229 (brd, NH). 1H NMR (300 MHz, d6-DMSO): δ 6.80 (t, 2H, Hb), 6.91 (d, 2H, Hd), 7.27 (bs, 2H, Hc), 7.65 (d, 2H, Ha), 9.35 (s, 0.4H, Hi), 12.21 (s, 2.8H, Hh). 13C NMR (100 MHz, d6-DMSO): 116.1 (Cd), 118.4 (Cc), 127.1 (Ca), 131.2 (Cf), 157.4 (Ce), 166.4 (Cg). Elemental analysis; (BiC14H11N2O6 + C4H8O), Calc (Found): C 37.00 (36.79), H 3.38 (3.28), N 5.14 (4.79) %. Mass spectrum, (ESI+): 513.2 [Bi(LH)(L) + H]+, 535.1 [Bi(LH)(L) + Na]+, (ESI−) 152.1 [L]−, 510.9 [BiL2]−; (where LH2 = H2-SHA).
Crystal data for 4: Bi2N4O16C37H49; Mr = 1352.00; triclinic; space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 7.7836(4), b = 12.4644(7), c = 23.1300(11); α = 87.484(2); β = 85.407(2); γ = 84.557(2); V = 2225.2(2)Å3; Z = 2, reflections collected/unique: 46278/12791 (Rint = 0.0434); R1 values (I > 2σ(I)) = 0.0331; wR(F2) values (I > 2σ(I)) = 0.0744; R1 values (all data) = 0.0462; wR(F2) values (all data) = 0.0789; GOF = 1.046.
; a = 7.7836(4), b = 12.4644(7), c = 23.1300(11); α = 87.484(2); β = 85.407(2); γ = 84.557(2); V = 2225.2(2)Å3; Z = 2, reflections collected/unique: 46278/12791 (Rint = 0.0434); R1 values (I > 2σ(I)) = 0.0331; wR(F2) values (I > 2σ(I)) = 0.0744; R1 values (all data) = 0.0462; wR(F2) values (all data) = 0.0789; GOF = 1.046.
K[Bi(SHA)2] 8. The reaction was carried out under nitrogen atmosphere using pre-dried solvents. A solution of potassium tert-butoxide (0.11 g, 1.0 mmol) in THF (10 mL) was added to a solution of [Bi(SHA)(H-SHA)] 4 (0.51 g, 1.0 mmol) in THF (10 mL) at −80 °C. The reaction mixture was then allowed to warm slowly to room temperature overnight. All volatiles were removed under vacuum leaving behind a pale yellow precipitate, which was washed with ethanol.
Yield: 0.32 g, 63%. Melting point: 194–196 °C (decomp.). FT-IR (cm−1): 664 (Vs, ring in plane bend), 751 (m, CH-out of plane bend), 910 (NO), 1251 (m, C–O), 1593 (m, CN), 3056 (brd, Ph(OH)). 1H NMR (300 MHz, d6-DMSO): δ 6.63 (bs, 2H, Hb), 6.74 (bs, 2H, Hd), 7.08 (bs, 2H, Hc), 7.60 (d, 2H, Ha), 8.38 (s, 0.4H, Hi), 13.06 (s, 1.5H, Hh). 13C NMR (100 MHz, d6-DMSO): 114.7 (Cb), 117.3 (Cd), 121.3 (Cc), 126.4 (Ca), 128.6 (Cf), 157.4 (Ce), 170.2 (Cg). Elemental analysis; (BiC14H10KN2O6 + 1/2(C4H8O)), Calc (Found): C 32.77 (33.11), H 2.41 (2.83), N 4.78 (5.10) %. Mass spectrum, (ESI+): 551.0 [(Bi(L2)K) + H]+, (ESI−): 152.0 [L]−, 510.9 [BiL2]−; (where LH2 = H2-SHA).
K[Bi(BHA)2] 9. All manipulations were carried out under nitrogen atmosphere with predried solvents. A solution of potassium tert-butoxide (0.11 g, 1.0 mmol) in THF (10 mL) was added to a solution of bismuth benzohydroxamate [Bi(BHA)(H-BHA)] 6 (0.48 g, 1.0 mmol) in THF (10 mL) at −80 °C and stirred overnight. The reaction mixture was then allowed to warm slowly to room temperature overnight. All volatiles were removed under vacuum leaving behind a yellow precipitate which was washed with ethanol.
Yield: 0.34 g, 67%. Melting point: 304–305 °C (decomp.). FT-IR (cm−1): 679 (Vs, ring in plane bend), 773 (m, CH-out of plane bend), 898 (NO), 1306 (m, C–O), 1442 (m, C–N), 1561 (w, CO), 1596 (m, CN). 1H NMR (300 MHz, d6-DMSO): δ 7.21 (m, 6H, Hb, c, d), 7.82 (bs, 4H, Ca, e). 13C NMR (75 MHz, d6-DMSO): 126.1 (Ca, e), 126.7 (Cb, d), 127.6 (Cc), 137.4 (Cf), 167.8 (Cg). Elemental analysis; (BiC14H10KN2O4), Calc (Found): C 32.44 (32.67), H 1.94 (2.17), N 5.40 (5.22) %. Mass spectrum, (ESI+): 481.0 [BiL2]+, 519.0 [Bi (L)(LH) + K]+, 541.0 [(Bi (L2)K) + Na]+, (ESI−): 136.0 [L]−, 478.9 [BiL2]−; (where LH2 = H2-BHA).
[Bi(AHA)(H-AHA)] 5. All manipulations were carried out under nitrogen atmosphere with predried solvents. A 10 mL THF solution of bismuth tert-butoxide (0.21 g, 0.5 mmol) was added to a solution of acetohydroxamic acid (0.11 g, 1.5 mmol) in THF (20 mL) at −80 °C. The reaction mixture was allowed to warm to room temperature overnight. Removal of all volatiles under vacuum left a white solid which was washed with dry ethanol to remove any unreacted acid and/or bismuth butoxide.
Yield: 0.13 g, 73%. Melting point: 231–232 °C (decomp). FT-IR (cm−1): 1084 (Vs, NO), 1280 (m, C–O), 1313 (m, C–N), 1425 (m, CH3 def.), 1534 (m, CO), 1595 (m, CN), 2916 (m, CH3–alkyl strech), 3163 (brd, NH). 1H NMR (300 MHz, d6-DMSO): δ 1.77 (s, 6H, Ha), 11.62 (s, 1H, Hc). 13C NMR (75 MHz, d6-DMSO): 18.1 (Ca), 164.4 (Cb). Elemental analysis; (BiC4H7N2O4), Calc (Found): C 13.49 (13.43), H 1.98 (2.15), N 7.87 (7.82) %. Mass spectrum, (ESI+): 107.1 [L + H(MeOH)]+, 139.1 [L + H (MeOH)2]+, 357.1 2 [Bi(LH)(L) + H]+, 379.1 [Bi(LH)(L) + Na]+, (ESI−) 74.1 [L]−, 355.2 [BiL2]−; (where LH2 = H2-AHA).
[Bi(MFHA)3] 3. Solvent reflux: in a Schlenk flask and under inert conditions, a mixture of triphenyl bismuth (0.22 g, 0.5 mmol) and N-methylfurohydroxamic acid (0.21 g, 1.5 mmol) in ethanol or toluene (10 mL) was heated to reflux for 24 hours. This mixture was then allowed to cool to room temperature before the precipitate was collected by filtration. An remaining triphenyl bismuth was removed by washing the precipitate with small amounts of ethanol (5 mL). The precipitate was then dried under vacuum.
Solvent-free: in a Schlenk flask and under inert conditions, a mixture of triphenyl bismuth (0.22 g, 0.5 mmol) and N-methylfurohydroxamic acid (0.21 g, 1.5 mmol) was heated at 100 °C for 1 hour. This remaining solid was allowed to cool to room temperature and any remaining triphenyl bismuth was removed by washing with small amounts of ethanol (5 mL) and toluene (5 mL). The precipitate was then dried under vacuum.
A 10 mL THF solution of N-methylfurohydroxamic acid (0.21 g, 1.5 mmol) was added to a solution of bismuth tert-butoxide (0.21 g, 0.5 mmol) in THF (20 mL) at −80 °C. The reaction mixture was left to reach room temperature while stirring overnight. Removal of solvent under vacuum left a light yellow solid, which was washed with ethanol to remove any unreacted acid and/or bismuth butoxide.
Crystals suitable for X-ray diffraction were grown from acetone or ethanol solution.
Yield: 0.19 g, 60% (toluene reflux); 0.17 g, 55% (ethanol reflux), 0.21 g, 67% (solvent-free), 0.30 g, 95% (in THF). Melting point: 165–167 °C (decomp.). FT-IR (cm−1): 959 (Vs, NO), 1401 (Vs, C–N) 1552 (m, CO), 2931 (m, CH sym. CH3). 1H NMR (300 MHz, d6-DMSO): δ 3.58 (s, 9H, Ha), 6.64 (q, 3H, Hd), 6.93 (dd, 3H, Hc), 7.88 (q, 3H, He). 13C NMR (100 MHz, d6-DMSO): 42.4 (Ca), 111.7 (Cd), 114.9 (Cc), 144.9 (Ce), 146.0 (Cb), 153.7 (Cg). Elemental analysis; (BiC18H18N3O9), Calc (Found): C 34.35 (34.52), H 2.88 (3.01), N 6.68 (6.54) %. Mass spectrum, (ESI+): 488.9 [BiL2]+, 651.9 [BiL3 + Na]+ (ESI−) 140.1[L]−; (where LH = H-MFHA).
Crystal data for 3: Bi4N12O37C75H78; Mr = 2575.41; triclinic; space group: P; a = 10.8304(3), b = 11.6831(4), c = 17.0183(5); α = 83.5330(10); β = 75.7080(10); γ = 83.6910(10); V = 2065.90(11)Å3; Z = 1, reflections collected/unique: 67867/12227 (Rint = 0.0327); R1 values (I > 2σ(I)) = 0.0190; wR(F2) values (I > 2σ(I)) = 0.0386; R1 values (all data) = 0.0268; wR(F2) values (all data) = 0.0405; GOF = 1.029.
[Bi(BPHA)3] 7. The reaction was carried out under nitrogen atmosphere using predried solvents. A 10 mL THF solution of N-phenylbenzohydroxamic acid (0.32 g, 1.5 mmol) was added to a solution of bismuth tert-butoxide (0.21 g, 0.5 mmol) in THF (20 mL) at −80 °C. The reaction was allowed to reach room temperature while stirring overnight. Removal of all volatiles under vacuum left a yellow solid which was washed with ethanol to remove any unreacted acid and/or bismuth butoxide. Crystals suitable for X-ray diffraction were grown from acetone or ethanol solution.
Yield: 0.29 g, 68%. Melting point: 148–149 °C (decomp.). FT-IR (cm−1): 691 (Vs, ring in plane bend), 766 (m, CH-out of plane bend), 916 (Vs, NO), 1420 (Vs, C–N), 1530 (m, CO). 1H NMR (300 MHz, d6-DMSO): δ 7.05 (m, 6H, Hi, i′), 7.17 (m, 9H, Hj, h, h′), 7.26 (bs, 12H, He, e′, f, f′), 7.33 (m, 3H, Hd). 13C NMR (75 MHz, d6-DMSO): 126.7 (Ci,i′), 127.7 (Ch,h′), 128.1 (Cj), 128.3 (Cf, f′), 129.1 (Ce, e′), 129.8 (Cd), 134.1 (Cc), 143.7 (Cb), 163.8 (Cg). Elemental analysis; (BiC39H30N3O6), Calc (Found): C 55.19 (55.26), H 3.58 (3.70), N 4.97 (4.93) %. Mass spectrum, (ESI+): 286.0 [LH + H (THF)]+, 494.0 [BiL + H (THF)]+; (where LH = C13H11O2N).
Crystal data for 7: Bi2N6O13C80H66; Mr = 1737.35; monoclinic; space group: P121/c1; a = 14.1095(8), b = 18.8537(11), c = 15.4923(7); β = 99.868(4); V = 4060.2(4) Å3; Z = 2, reflections collected/unique: 27627/12635 (Rint = 0.0363); R1 values (I > 2σ(I)) = 0.0319; wR(F2) values (I > 2σ(I)) = 0.0672; R1 values (all data) = 0.0469; wR(F2) values (all data) = 0.0711; GOF = 1.032.
Acknowledgements
We thanks the Australian Research Council (DP110103812) and Monash University for financial support. We also thank Dr Lukasz Kedzierski (Walter and Eliza Hall Medical Research Institute, Melbourne) for the toxicity testing of the compounds on human fibroblast cells.References
- E. M. Muri, M. J. Nieto, R. D. Sindelar and J. S. Williamson, Curr. Med. Chem., 2002, 9, 1631–1653 CrossRef CAS.
- A. S. Reddy, M. S. Kumar and G. R. Reddy, Tetrahedron Lett., 2000, 41, 6285–6288 CrossRef CAS.
- W. P. Steward and A. L. Thomas, Expert Opin. Invest. Drugs, 2000, 9, 2913–2922 CrossRef CAS PubMed.
- W. P. Steward, Cancer Chemother. Pharmacol., 1999, 43(Suppl), S56–S60 CrossRef CAS.
- J. L. Domingo, Reprod. Toxicol., 1998, 12, 499–510 CrossRef CAS.
- I. Turcot, A. Stintzi, J. Xu and K. N. Raymond, J. Biol. Inorg. Chem., 2000, 5, 634–641 CrossRef CAS.
- M. J. Miller, Chem. Rev., 1989, 89, 1563–1579 CrossRef CAS.
- Z. I. Cabantchik, Parasitol. Today, 1995, 11, 74–78 CrossRef CAS.
- R. Ge, Z. Chen and Q. Zhou, Metallomics, 2012, 4, 239–243 RSC.
- R. Codd, Coord. Chem. Rev., 2008, 252, 1387–1408 CrossRef CAS PubMed.
- S. Odake, K. Nakahashi, T. Morikawa, S. Takebe and K. Kobashi, Chem. Pharm. Bull., 1992, 40, 2764–2768 CrossRef CAS.
- K. Kobashi, S. Takebe, N. Terashima and J. i. Hase, J. Biochem., 1975, 77, 837–843 CAS.
- Z.-P. Xiao, Z.-Y. Peng, J.-J. Dong, R.-C. Deng, X.-D. Wang, H. Ouyang, P. Yang, J. He, Y.-F. Wang, M. Zhu, X.-C. Peng, W.-X. Peng and H.-L. Zhu, Eur. J. Med. Chem., 2013, 68, 212–221 CrossRef CAS PubMed.
- R.-C. Deng, W. Wei, X.-D. Wang and Z.-P. Xiao, Chin. New Drugs J., 2014, 18, 2189–2194 Search PubMed.
- C. A. Kontogiorgis, P. Papaioannou and D. J. Hadjipavlou-Litina, Curr. Med. Chem., 2005, 12, 339–355 CrossRef CAS.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- P. Domenico, R. J. Salo, S. G. Novick, P. E. Schoch, K. van Horn and B. A. Cunha, Antimicrob. Agents Chemother., 1997, 41, 1697–1703 CAS.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed.
- H. Sun and K. Y. Szeto, J. Inorg. Biochem., 2003, 94, 114–120 CrossRef CAS.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y.-T. Lai, G. Jiang and H. Sun, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- P. Malfertheiner and M. Selgrad, Curr. Opin. Gastroenterol., 2010, 26, 618 CrossRef PubMed.
- N. Yang and H. Sun, Coord. Chem. Rev., 2007, 251, 2354–2366 CrossRef CAS PubMed.
- J. B. Neilands, J. Biol. Chem., 1995, 270, 26723–26726 CrossRef CAS PubMed.
- O. Senkovich, S. Ceaser, D. J. McGee and T. L. Testerman, Infect. Immun., 2010, 78, 1841–1849 CrossRef CAS PubMed.
- A. Pathak, V. L. Blair, R. L. Ferrero, M. Mehring and P. C. Andrews, Chem. Commun., 2014, 50, 15232–15234 RSC.
- W. N. Fishbein and P. P. Carbone, J. Biol. Chem., 1965, 240, 2407–2414 CAS.
- Q. Wu, H. Yin, C. Yue, X. Zhang, M. Hong and J. Cui, J. Coord. Chem., 2012, 65, 2098–2109 CrossRef CAS PubMed.
- P. C. Andrews, G. B. Deacon, C. M. Forsyth, P. C. Junk, I. Kumar and M. Maguire, Angew. Chem., Int. Ed., 2006, 45, 5638–5642 CrossRef CAS PubMed.
- P. C. Andrews, G. B. Deacon, P. C. Junk, I. Kumar and J. G. MacLellan, Organometallics, 2009, 28, 3999–4008 CrossRef CAS.
- P. C. Andrews, M. Busse, G. B. Deacon, R. L. Ferrero, P. C. Junk, J. G. MacLellan and A. Vom, Dalton Trans., 2012, 41, 11798–11806 RSC.
- M. Busse, E. Border, P. C. Junk, R. L. Ferrero and P. C. Andrews, Dalton Trans., 2014, 43, 17980–17990 RSC.
- D. Mansfeld, M. Mehring and M. Schürmann, Angew. Chem., Int. Ed., 2005, 44, 245–249 CrossRef CAS PubMed.
- P. C. Andrews, R. L. Ferrero, P. C. Junk, I. Kumar, Q. Luu, K. Nguyen and J. W. Taylor, Dalton Trans., 2010, 39, 2861–2868 RSC.
- P. C. Andrews, R. L. Ferrero, P. C. Junk, J. G. Maclellan and R. M. Peiris, Aust. J. Chem., 2012, 65, 883–891 CrossRef CAS.
- P. C. Andrews, V. L. Blair, R. L. Ferrero, P. C. Junk, L. Kedzierski and R. M. Peiris, Dalton Trans., 2014, 43, 1279–1291 RSC.
- Y. Glupczynski, M. Delmee, C. Bruck, M. Labbe, V. Avesani and A. Burette, Eur. J. Epidemiol., 1988, 4, 154–157 CrossRef CAS.
- C. E. Haas, D. E. Nix and J. J. Schentag, Antimicrob. Agents Chemother., 1990, 34, 1637–1641 CrossRef CAS.
- C. A. McNulty, J. Dent and R. Wise, Antimicrob. Agents Chemother., 1985, 28, 837–838 CrossRef CAS.
- M. Busse, I. Trinh, P. C. Junk, R. L. Ferrero and P. C. Andrews, J. Chem., 2013, 19, 5264–5275 CrossRef CAS PubMed.
- D. Mansfeld, Synthese und Charakterisierung neuartiger Bismutsilanolate, Bismut-oxo-cluster und Bismutkoordinations-polymere, Technische Universität Chemnitz, Chemnitz, 2009 Search PubMed.
- D. H. R. Barton, N. Y. Bhatnagar, J.-P. Finet and W. B. Motherwell, Tetrahedron, 1986, 42, 3111–3122 CrossRef CAS.
- R. L. Ferrero, J.-M. Thiberge, M. Huerre and A. Labigne, J. Infect Immunol., 1998, 66, 1349–1355 CAS.
- J. Viala, C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott and R. L. Ferrero, Nat. Immunol., 2004, 5, 1166–1174 CrossRef CAS PubMed.
- CrysAlisPro v1.171.34.36, Oxford Diffraction Ltd (Agilent Technologies), Oxfordshire, UK, 2010 Search PubMed.
- Bruker Apex2 v2012.2.0, Bruker AXS, Madison, US, 2006 Search PubMed.
- G. M. Sheldrick, SADABS v2.30, University of Gottingen, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1046081–1046083. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02259b |
| This journal is © The Royal Society of Chemistry 2015 |