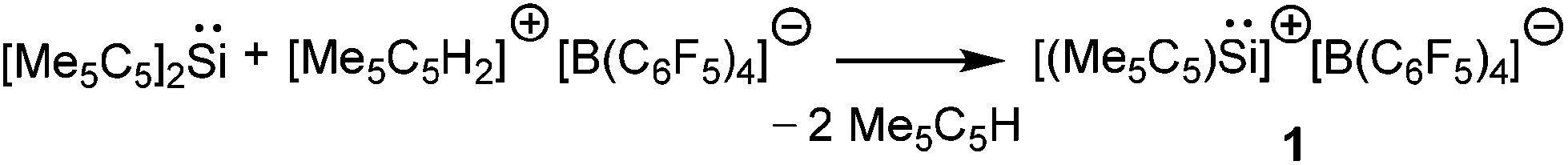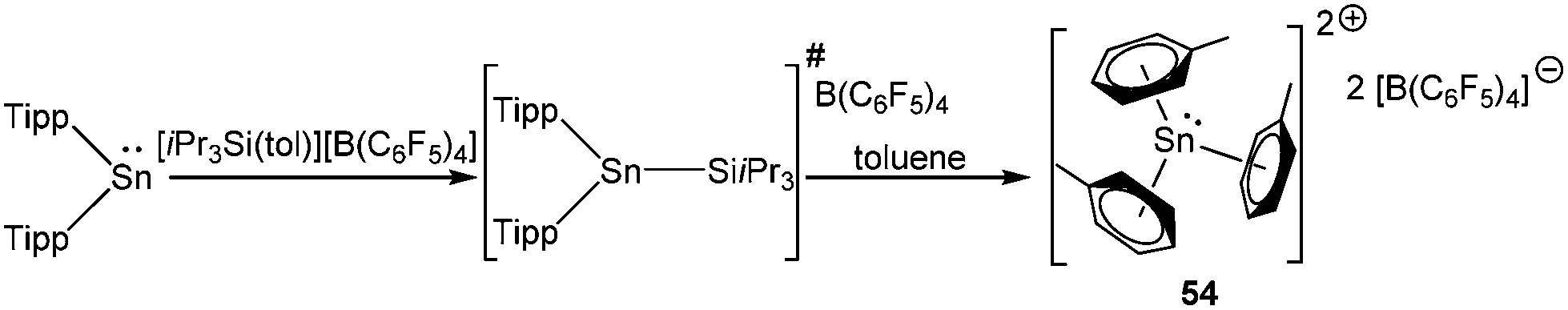DOI:
10.1039/C5DT01912E
(Perspective)
Dalton Trans., 2015,
44, 12903-12923
Cations and dications of heavier group 14 elements in low oxidation states†
Received
30th April 2015
, Accepted 28th May 2015
First published on 1st June 2015
Abstract
Cations and dications of heavier group 14 elements in their low oxidation state have received widespread attention in recent years. The journey started with the isolation of a series of cations of the composition [(C5Me5)E:]+ [E = Si–Pb], followed by the more recent isolation of a Ge(II) dication encapsulated within a cryptand, a carbodiphosphorane stabilized [GeCl]+ monocation with a two coordinate Ge atom, Si(II) cations and dications stabilized by N-heterocyclic carbenes (NHCs), which highlights the ongoing growth and interest in the chemistry of tetrel(II) cations. This is presumably because the central atom (E) in these compounds contains two or three unoccupied valence orbitals as well as holds a lone pair of electrons. Such an electronic description represents ambiphilicity, which is of great interest for catalysis. The successful synthesis of divalent group 14 cations requires new synthetic strategies based on the sterically demanding neutral or monoanionic ligands, utilization of counter anions, and solvents with low nucleophilicity in order to minimize the degree of interactions with the cations. An alternative approach for the realization of divalent cations of group 14 elements is their coordination to the transition metals. This synthetic approach was successfully applied for the isolation of a range of transition metal coordinated divalent cations of group 14 elements. Apart from arousing academic interest some of these cations have found application as activators in the Ziegler–Natta polymerization of alkenes.
 V. S. V. S. N. Swamy | V. S. V. S. N. Swamy was born in Andhra Pradesh, India in 1989. After receiving his MSc degree from Andhra University he joined the research group of Dr Sakya S. Sen, Inorganic Chemistry and Catalysis Division at the CSIR-National Chemical Laboratory, Pune, India. His research interest lies in the preparation and reactions of heavier group 14 elements in low oxidation states. |
 Shiv Pal | Shiv Pal was born in Jaunpur, India, in 1989. He obtained his M.Sc. degree from Banaras Hindu University in 2012. Currently he is working as a PhD student under the supervision of Dr Shabana Khan at the Indian Institute of Science Education and Research, Pune, India. His research interests mainly concern the synthesis and reactivity studies of low valent compounds of main group elements, especially of the carbon family. |
 Shabana Khan | Shabana Khan was born in Faizabad, India in 1979. She obtained her PhD degree from the Indian Institute of Technology, Delhi in 2008 under the supervision of Prof. Jai Deo Singh. After spending eight months as a senior scientist in Bharat Petroleum Corporation Ltd, India (R&D centre), she took up the Deutscher Akademischer Austausch Dienst fellowship and joined the research group of Prof. Herbert W. Roesky as a postdoctoral fellow. In 2011 she started her second post doc with Dr Manuel Alcarazo at the Max Planck Institute for Coal Research. Currently she is working as an assistant professor in the Department of Chemistry, Indian Institute of Science Education and Research, Pune, India. Her research interest includes the synthesis and application of low valent compounds in catalysis. |
 Sakya S. Sen | Sakya S. Sen was born in Kolkata, India, in 1983. After finishing his M.Sc. from IIT Kharagpur, India in 2006, he joined the research group of Prof. Herbert W. Roesky at the University of Göttingen, Germany and received the title “Dr rer. nat.” in 2010. Then he joined as a postdoctoral researcher, supported by the Alexander von Humboldt Foundation, in the laboratory of Prof. Holger Braunschweig at the University of Würzburg, Germany. Since 2014 Dr Sen has been working in CSIR-National Chemical Laboratory, Pune, India as a senior scientist. His research interests lie in main group chemistry with low-valent elements and bio-organometallic chemistry. |
1. Introduction
At the beginning of the 20th century, J. F. Norris and F. Kehrmann independently discovered that when triphenylmethanol and triphenylmethyl chloride were dissolved in concentrated H2SO4, the color of the solution changed from colorless to deep yellow.1 These observations along with von Baeyer's subsequent interpretation that the intense color was due to the ionization of the triphenylmethanol2 eventually paved the way for the first stable “carbocation” in chemistry, commonly known as the trityl cation. Following a series of debates over the nomenclature of this class of compounds, chemists finally accepted Olah's categorization of carbocations based on the valency of the charged carbon: carbenium ions (CR3+) and carbonium ions (CR5+).3 The last century has witnessed a remarkable progress in the isolation of various stable carbenium ions, in understanding their structural and bonding properties, and in the investigation of their reaction chemistry.
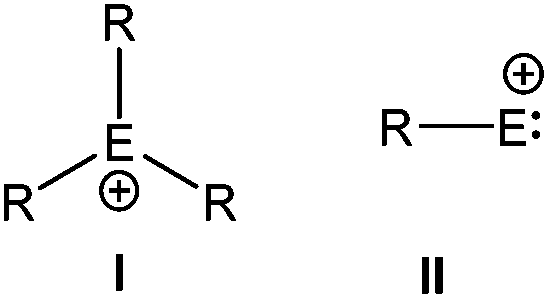
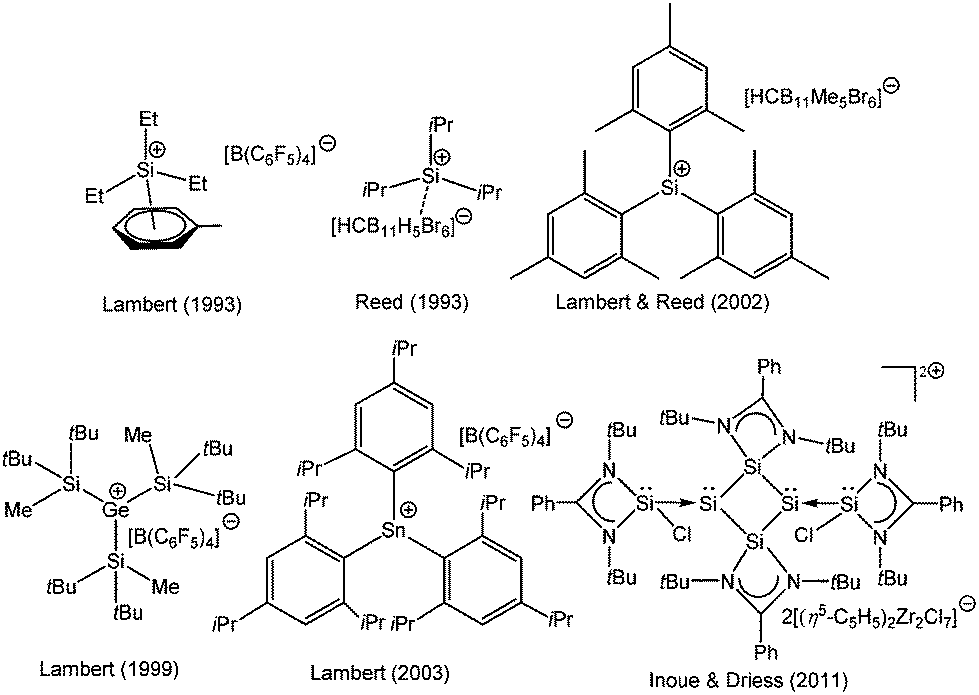
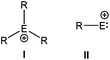
Considering the rich chemistry displayed by carbenium ions, the realization of their heavier analogs i.e. [R3E+ (E = Si–Pb)] has become one of the important areas of modern inorganic chemistry and is still an area of continuous investigation. However, in contrast to the thoroughly studied chemistry of carbenium ions, only a little is known about the chemistry of heavier group 14 cations. The cations of heavier group 14 elements can be classified into two distinct groups, based on the oxidation state of the atom (Scheme 1). The most common class of the cations is tetrylium ions (I) (group 14 is called the tetrel group), which possess six valence electrons and are considered as heavier analogues of carbenium ions (R3C+). The generation of such a cation is easier in the gas phase, which provides an environment devoid of possible interfering solvents. However, their generation in the condensed phase is very difficult due to the highly electrophilic nature of R3E+ and their propensity to react with any donor moiety, even with arenes. Therefore, bulky donor substituents are required to quench the electrophilicity of the cation as well as to shield the cationic center from any nucleophilic attack. With these new synthetic strategies as well as taking advantages of the low nucleophilicity of borates and carboranes as counter anions and toluene as a solvent, Lambert and Reed et al. independently isolated the first silylium ions [Et3Si(toluene)]+[B(C6F5)4]− and [iPr3Si]+[CB11H6Br6]−, respectively.4 However, the Si centers in these silylium cations adopted a markedly pyramidal geometry instead of the expected planar geometry and resonated at δ = 92.3 and 102.8 ppm in their respective 29Si NMR spectrum. The calculated gas phase chemical shift of Et3Si+ was 354.6 ppm,5 which was considerably downfield than the experimentally observed values. Consequently, the cationic character of Si atoms in these compounds was heavily questioned and eventually both Lambert and Reed agreed that neither [Et3Si(toluene)]+ nor [iPr3Si]+ were “bona fide” silylium cations.6 The state of affairs was similar to those of germanium and tin. The groups of Lambert and Kira independently reported the isolation of [nBu3Sn]+X− [X = B(C6F5)3H and B{(3,5-CF3)2C6H3}4] featuring a tri-coordinate Sn atom with 119Sn NMR of δ = 360 and 356 ppm.7 However, Edlund et al. proposed that the NMR data of the aforementioned tin cations corresponded to the arene bound Sn atoms as three-coordinate trigonal-planar R3Sn+ was computationally predicted to resonate at δ = 1500–2000 ppm.8 Thus, a lot of controversies were sparked and simultaneously a huge effort was dedicated for the isolation of three-coordinate trigonal planar tetrylium ions. This was finally accomplished in 2000s with the isolation of “bona fide” three coordinate trigonal planar silylium [{Mes3Si}+{CB11HMe5Br6}−, Mes = 2,4,6-Me3-C6H2],9 germylium [{(tBu2MeSi)3Ge+}{B(C6F5)4}−], and stannylium [{(tBu2MeSi)3Sn+}{B(C6F5)4}−],10 [{Tipp3Sn+}{B(C6F5)4}−], (Tipp = 2,4,6-iPr3-C6H2) cations (Chart 1).11 Subsequently, the advances in the syntheses of novel condensed-phase cations as well as the reactivity studies have become important research areas with mainly two fold objectives: (i) to study these exotic compounds in the solid state due to the paucity of previous reports of structurally characterized heavier analogues of carbenium ions and (ii) to explore these species as efficient Lewis acid catalysts. Hallmark studies by Lambert, Reed, Müller, Corriu, Belzner, Oestreich, and others not only led to the isolation of many more such derivatives but revealed them as potential Lewis acids in homogeneous catalysis.12 Their enhanced electrophilicity has already been exploited in Diels–Alder reactions,13 C–C bond formation,14 and C–F activation reactions,15 as well as in small molecule activation e.g. dihydrogen.16
 |
| | Scheme 1 Classes of cations featuring heavier group 14 elements. | |
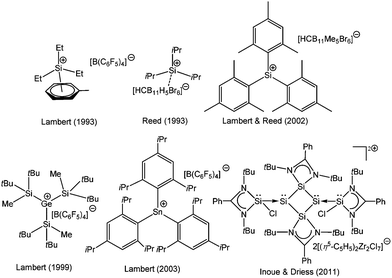 |
| | Chart 1 Selected examples of silylium, germylium, and stannylium cations. | |
Another distinctly different class of group 14 cations is tetrylium-ylidenes (II) (Scheme 1), which features a group 14 element with a lone pair. It can be imagined that a species of composition (RE:+), which combines the nucleophilic nature of carbene as well as the high electrophilicity of cations is of high synthetic importance as a potential noninnocent ligand in catalysis. However, the number of valence electrons in type II cations is only four, while there are six valence electrons in the type I cations. Thus, further loss of two electrons from the closed shell might imply even greater reactivity and more difficulty in isolation. It was obvious that like R3Si+, the laboratory realization of RSi+ (a derivative of HSi+) could only be accomplished through kinetic stabilization, but neither steric protection nor electronic stabilization may be adequate if only one substituent is attached to the Si center.
Increasing the coordination number of Si in RSi+ without perturbing the formal oxidation state can be achieved mainly by two ways: (i) taking advantage of the cyclopentadienyl type of ligands, which can undergo haptotropic shift depending on electronic requirements of the system; (ii) attach a neutral donor ligand to the cation. The donor moiety occupies the empty coordination site of the cation and thereby reduces its electrophilicity. Both these synthetic tricks have been successfully implemented. The elegant use of the pentamethylcyclopentadienyl (Cp*) ligand by Jutzi et al. allowed the isolation and characterization of the first Si(II) cation, [(η5-C5Me5)Si:]+.17 The concept of donor-stabilization has recently been used for the isolation of a series of highly reactive compounds which were otherwise inaccessible. For example, the groups of Roesky and Filippou showed that the reactivity of silylenes like SiCl2 or SiBr2 can be tamed when they are attached to an N-heterocyclic carbene.18 The N-donor stabilized amidinato chlorosilylene [PhC(NtBu)2SiCl] from the Roesky group has also enjoyed substantial attention in recent years.19 Similar N-donor stabilization afforded a unique cationic silyliumylidene ([LSi][B(C6F5)4] (L = CH[C(MeNAr)2], Ar = 2,6-iPr2-C6H3)) through protonation of the Si(II) center of the corresponding silylene, L1Si (L1 = CH[C(Me)(C = CH2)(NAr)2]).20 These results kick-started the chemistry of low-valent Si(II) cations although the existence of mono-coordinated Si(II) cations is yet to be established.
In contrast to carbon and silicon atoms, germanium, tin, and lead are more stable in their MII oxidation state as the stability increases with the increase of the principal quantum number. Going down the periodic table, the s–p separation increases with increasing nuclear charge. As a result s/p hybridization becomes more and more difficult, leading to the “inert s-pair effect”, in which only the p electrons are used in the bonding. Therefore, the isolation of LE:+ (E = Ge–Pb) is relatively simpler. To the best of our knowledge, the first mention of LE:+ (E = Ge–Pb) came from the group of Jutzi, who reported a series of nido-cluster type cations of the composition [(C5Me5)E:]+.21 Since then a number of germanium(II), tin(II), and lead(II) mono- and dications have been reported. A few of these compounds have been cited in recent review articles22,23 as well as in an excellent book by Lee and Sekiguchi, which primarily deals with “carbenium derivatives of heavier group 14 elements (R3E+)”.24 However, to our knowledge no efforts have been dedicated to compile only the cations of composition RE:+, where E is a heavier group 14 element in the +2 oxidation state. Moreover, several important accomplishments like [Cl–Si:]+, Si(II) dication, polyether ligated Ge(II) and Sn(II) mono- and dications etc. that deserve mentioning have been recently reported. However, the chemistry of cations and dications of heavier group 14 elements in low oxidation states is very far from being complete; there are still many challenges left. The practical application of these classes of compounds is still awaited. Recent studies showed that heavier carbenes have the potential to compete with transition metal complexes for single site small molecule activation, which is of significant importance in order to find cheap and green alternatives to transition metal complexes for this important class of reactions.25 Therefore, the utilization of the cations and dications of heavier group 14 elements in various catalytic transformations and small molecule activations seems to be attractive and promising. These arguments justify the requirement of a review at this juncture dedicated solely to tetrel(II) cations, which will extensively kindle further interest, directed towards its development. This present review will deal with the available synthetic routes for the preparation of cations of heavier group 14 elements in low oxidation states, starting from the compounds of the composition Cp*E+, which were the first to be prepared. We shall mainly concentrate on the cations stabilized by using bulky ligands. We shall also cover the transition metal supported EII (E = Si–Pb) cations. The literature coverage of this review is up to 2014. Because it is difficult to definitively conclude the formation of the cations by spectroscopy alone, we have chosen to mainly cover compounds which were structurally characterized by single crystal X-ray diffraction studies.
2. The ligands
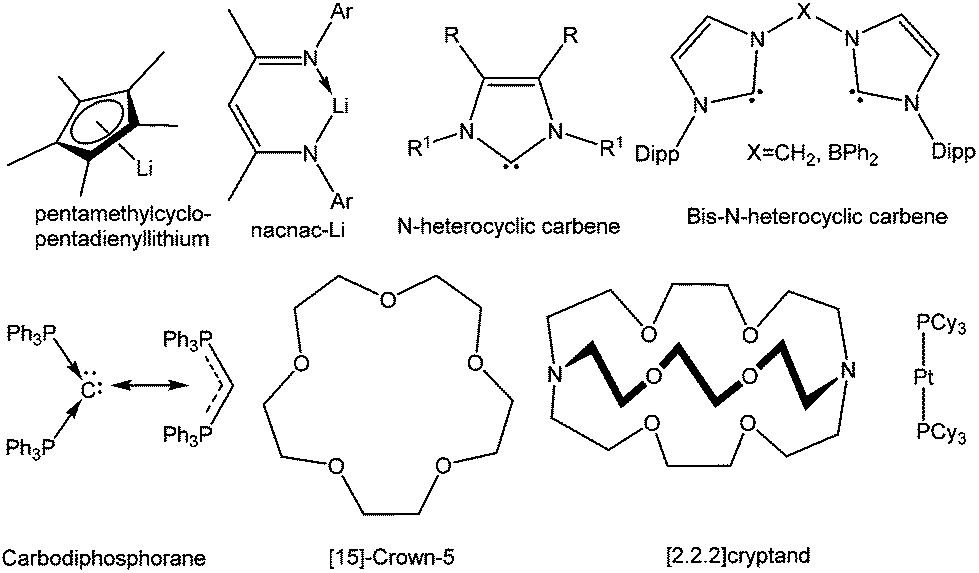
The isolation of the aforesaid compounds and various other remarkable subvalent compounds of silicon, germanium, tin, and lead would not have been feasible without the appropriate choice of ligands that afford thermodynamic stabilization through the donor sites and kinetic stabilization through bulky substituents (Chart 2). For decades, organometallic chemistry and to a lesser degree the study of main group elements, has focused on the study of complexes bearing the cyclopentadienyl ligand and its ring-substituted derivatives. A current trend in main group chemistry is the search for alternative ligand sets which are able to attenuate the reactivity of subvalent main group compounds and allow their isolation. Among them, β-diketiminato ligands have emerged as very versatile ligands in recent years.26 A major advantage of the β-diketiminato ligands is that the steric and the electronic environment of these ligands can be fine-tuned with minimum synthetic effort. This flexibility allows a degree of control over the chemistry at the metal center and can be attributed for the widespread use of β-diketiminato ligands for stabilizing the highly reactive species etc.
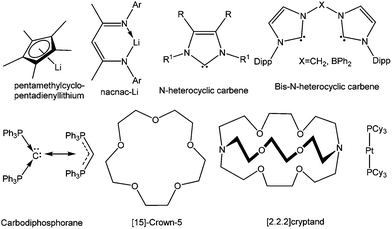 |
| | Chart 2 Selective ligands used for stabilizing cations of heavier group 14 elements in low oxidation states. | |
In an effort to explore the feasibility of isolating dications of heavier group 14 elements in the +2 oxidation state, synthetic chemists moved from the formally anionic ligands and focused on neutral ligands like N-heterocyclic carbenes (NHCs). Baines and coworkers introduced NHC for the isolation of the first Ge(II) dication coordinated by three IiPr (1,3-iPr2-imidazol-2-ylidene) groups (vide infra).27 Similar to β-diketiminato ligands, NHCs also offer high synthetic flexibility by varying the “wingtip” substituents (substituent at the nitrogen atom), which makes them one of the most sought after ligands in main group chemistry.28 Another leading motif over the last few years has been the linking of two NHCs by several bridging spacers to generate discrete multitopic bis-NHCs, which exhibit high affinities toward a broad range of metals.29 Complexes featuring such ligands are stabilized by the chelate effect and offer various possibilities for tuning their geometric and electronic properties.
The enormous success of NHCs prompted the investigation of other carbon-based neutral ligands. The groups of Alcarazo and Vidović used a divalent C(0) compound, commonly known as carbodiphosphorane [(PPh3)2C], for the realization of B,30a Ge,30b and P31 centered cations. The main advantage of these C(0) ligands over NHCs is that they are capable of acting as σ- as well as π-donors. A recent computational paper by Toner and Frenking proposed that the substitution of an NHC by a carbodiphosphorane in the Grubbs’ catalyst for alkene metathesis might lead to an enhanced reactivity.32
Macrocyclic ligands like cryptands, crown ethers, azamacrocycles etc. have been noted for their remarkable metal complexation properties through numerous weak donor sites. There are many examples of cryptand and crown ether coordinated s-block and d-block elements in the literature.33 Schmidbaur et al. found that such a macrocyclic ligand (cyclophane) can also stabilize germanium and tin cations.34 However, the use of macrocyclic ligands for realizing p-block cations has extensively increased since Baines’ report of a germanium(II) dication encapsulated in [2.2.2]cryptand.35 In the following years, a range of macrocyclic ligands like crown ethers, cryptands, azamacrocycles etc. were used to give rise to an array of cations and dications of germanium(II) and tin(II), which revealed an interrelation between the cavity size of the macrocycles and the stability of the E(II) dication or the [EX]+ (E = Ge and Sn) monocation (vide infra section 4.1.4).
It is only recently that the various research groups have started to exploit the coordination sphere of the electron rich coordinatively unsaturated transition metals such as W, Pt, Pd to stabilize germanium(II), tin(II), and lead(II) cations (vide infra). The precedent for this approach came from the isolation of an array of complexes featuring B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E (E = N and O),36 B
E (E = N and O),36 B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,37 SiMo,38 GeRe,39 SiO40 bonds, which had thus far been otherwise inaccessible. However, interpretation of the bonding situation in transition metal supported tetrel(II) cationic complexes is not very straightforward taking into consideration other possible canonical forms.
C,37 SiMo,38 GeRe,39 SiO40 bonds, which had thus far been otherwise inaccessible. However, interpretation of the bonding situation in transition metal supported tetrel(II) cationic complexes is not very straightforward taking into consideration other possible canonical forms.
3. Si(II) cations: synthesis, spectroscopic and structural elucidation, and reactivity
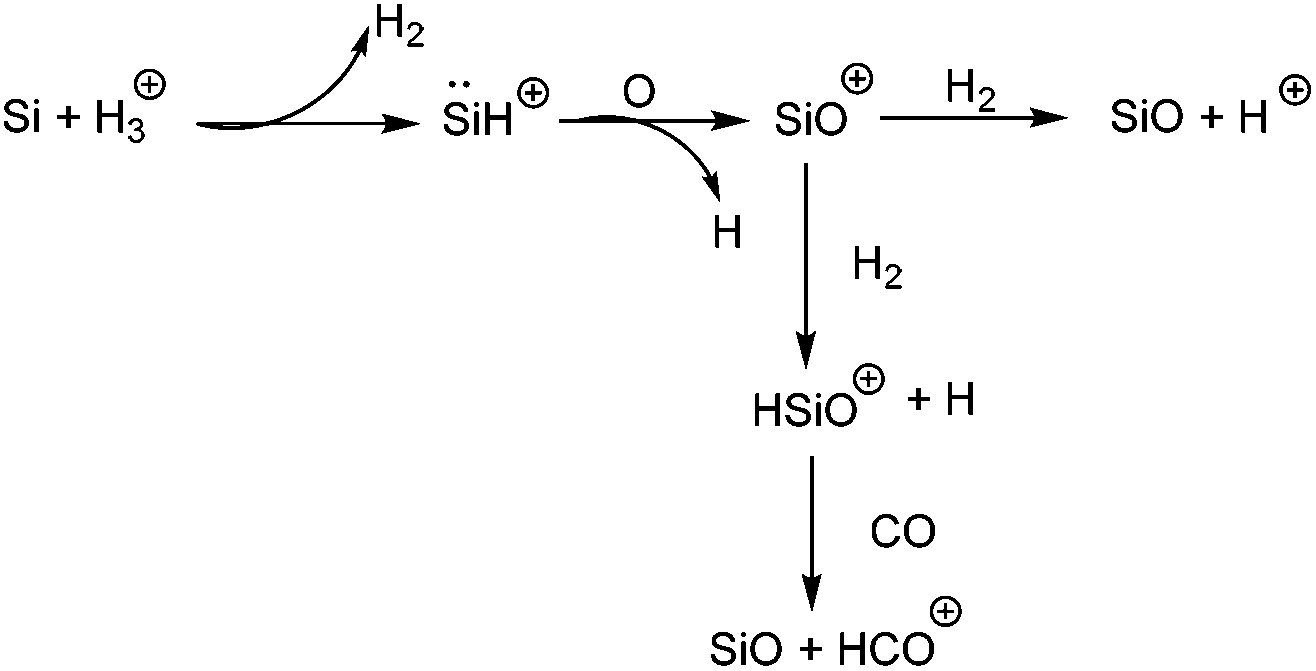
Silicon(II) cations have long been considered as a challenging target in synthetic inorganic chemistry. The laboratory identification on HSi+, the smallest possible Si(II) cation, was carried out by Douglas and Lutz,41 who observed five bands of the system in the emission spectrum. Following this, Grevesse and Sauval identified the presence of HSi+ in the solar photospheric spectrum through absorption spectroscopy.42 It is now also recognized that HSi+ is present in the interstellar space down to a large optical depth (the optical depth is just the amount of interstellar dust that the light must pass through), where it is rapidly converted to SiO by reacting with oxygen in the following sequence (Scheme 2).43
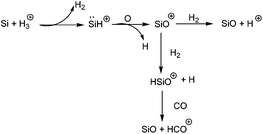 |
| | Scheme 2 Reactions of HSi+ in the interstellar medium leading to SiO. | |
3.1. Cyclic π-conjugated Si(II) cations
Kinetic stabilization of the labile RSi:+ core using the sterically demanding substituents and utilizing weakly coordinating counter-anions and solvents of low nucleophilicity was found to be a feasible strategy to furnish the Si(II) cations. Jutzi et al. employed the mono-anionic pentamethylcyclopentadienyl (Cp*) ligand for the isolation of the first Si(II) compound, decamethylsilicocene (Cp*2Si).44 Cp*2Si was initially treated with HBF4, which apparently led to the formation of the salt Me5C5Si+ BF4−. However, the latter instantly decomposed even at low temperatures with the liberation of BF3 and a polymeric product was obtained. Subsequently, Cp*2Si was reacted with [Me5C5H2]+[B(C6F5)4]− which gave rise to [η5-Me5C5Si]+[B(C6F5)4]− (1) with simultaneous formation of two equivalents of Me5C5H (Scheme 3).17 So, it is apparent that the selection of the proton source was very critical. The η5-coordination mode of the Cp* ring was reflected in the 1H NMR spectrum as five Me groups showed only one sharp singlet at δ = 2.23 ppm. The markedly upfield signal in the 29Si NMR (δ = −400.2 ppm) indicated the “π-complex” of a Si(II) atom. In the solid state structure, [η5-Me5C5Si]+ showed a weak interaction with the borate anion leading to a quasi-pentagonal-pyramidal geometry at the silicon atom. Ab initio calculations showed that the HOMO in 1 was formed by a π-interaction between the Cp* moiety and silicon whereas the lone-pair corresponded to HOMO−1. The HOMO–LUMO energy gap in 1 is 12.34 eV (MP2/TZVPP) indicating a strong acidic character of the cation. Such half-sandwich compounds like 1 possess six interstitial electrons (four electrons from the C5Me5+ ring and the Si+ cap donate two more electrons to complete the set of six interstitial electrons) and according to Jemmis and Schleyer, they can be best regarded as three dimensional aromatic compounds.45
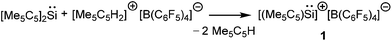 |
| | Scheme 3 Synthesis of [Cp*Si]+. | |
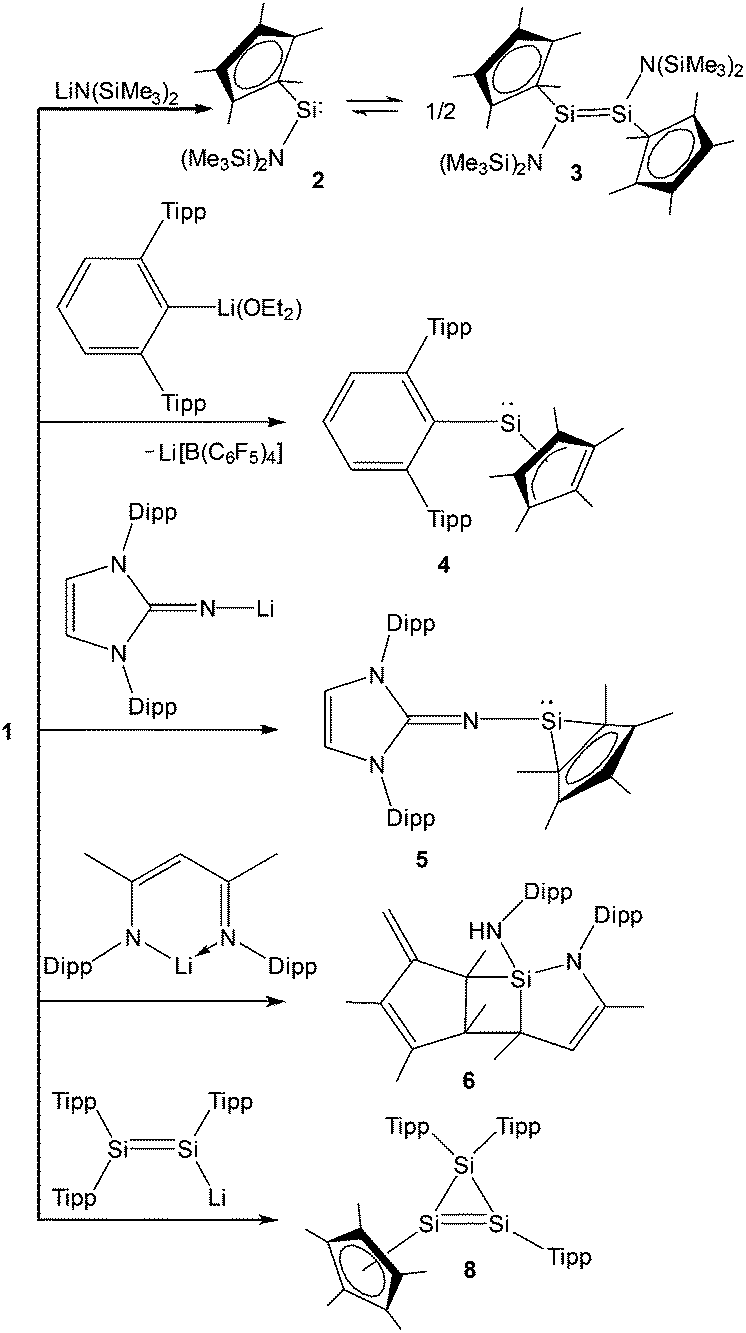
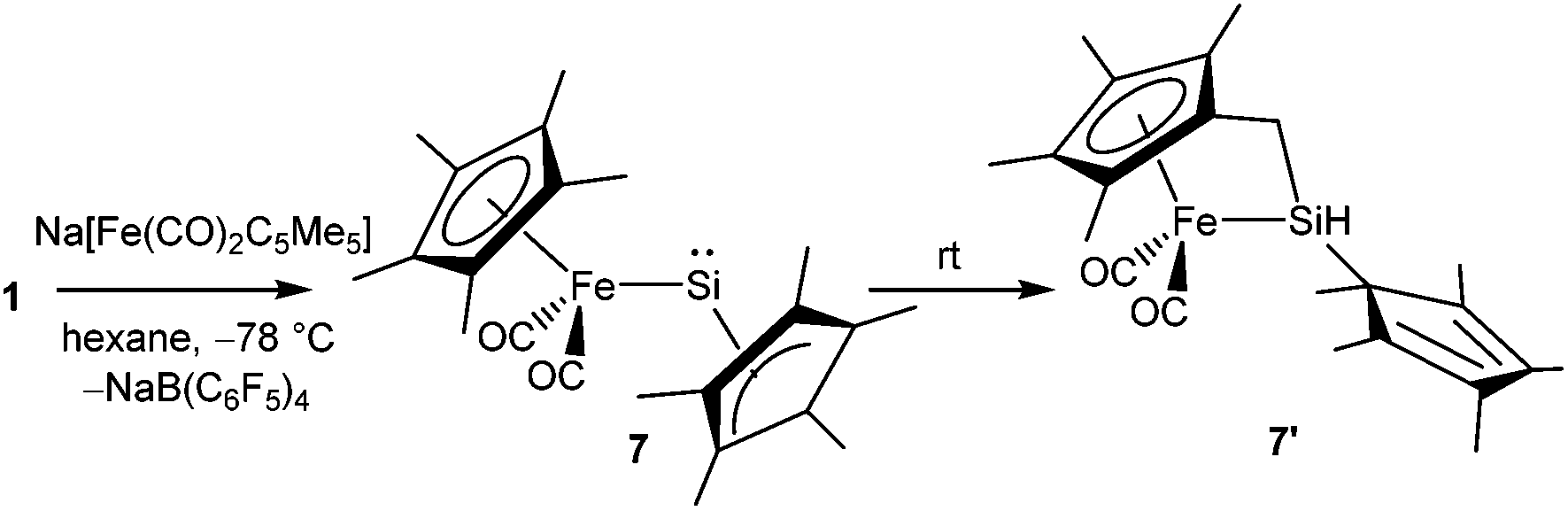
Following the synthesis of the [Cp*Si]+ cation, Jutzi, Scheschkewitz, and others reported many reactions taking advantage of 1 as a starting material, which are outlined in Scheme 4. The reaction of 1 with LiN(SiMe3)2 afforded silylene (Me5C5)SiN(SiMe3)2 (2) as a reactive intermediate, which dimerizes to give the disilene E-{(η1-Me5C5)[N(SiMe3)2]Si}2 (3).17 Later Jutzi et al. found that a rare reversible phase dependent dynamic equilibrium exists between silylene 2 and disilene 3.46 During crystallization colorless silylene 2 dimerized to yellow colored disilene 3. Again when 3 was dissolved in solvents, a colorless solution of 2 was obtained. This unusual behavior could be rationalized by steric strain in the solid disilene, flexibility in bonding modes of Cp* groups, low activation energy for the equilibrium process, and a small energy difference between 2 and 3. It should be noted here that dehydrochlorination of [(η1-Me5C5)SiHCl2] with KN(SiMe3)2 also led to disilene 3 in a higher yield.471 was further reacted with various lithium containing anions like Li(2,6-Tipp2-C6H3) (Tipp = 2,4,6-iPr3-C6H2) and Li[NC{N(Dipp)CH}2] (Dipp = 2,6-iPr2-C6H3) to obtain [(η3-C5Me5)(2,6-Tipp2-C6H3)]Si: (4) and [(C5Me5)(NC{N(Dipp)CH}2)]Si: (5), respectively, in salt elimination reactions.48 The former features a silylene with one σ-donor and one π-donor substituent attached to the Si(II) center. In contrast, the reaction of 1 with Li[HC(CMeNDipp)2] did not result in the putative Cp*[HC(CMeNDipp)2]Si: but a constitutional isomer 6.491 was also utilized as a stoichiometric source of silicon in the reaction with Na[Fe(η5-C5Me5)(CO)2], which gave rise to a ferrio-substituted silylene [Fe(η5-C5Me5)(CO)2{Si(η3-C5Me5)}] (7) at low temperatures (Scheme 5).50 However, under ambient conditions, the silylene fragment was inserted into one of the C–H bonds of the Cp* ring leading to a rearranged product 7′. The substitution reaction with the lithium disilenide [Tipp2SiSi(Tipp)(Li{dme}2)] led to straightforward access to the first cyclotrisilene with only carbon-based substituents (8) (Scheme 4).51
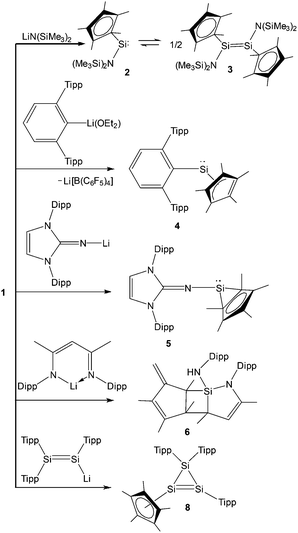 |
| | Scheme 4 Reactivity of [Cp*Si]+ towards various lithium containing reagents. | |
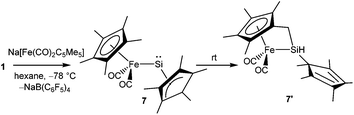 |
| | Scheme 5 Synthesis of iron-substituted silylene (7) from 1 and its rearrangement. | |
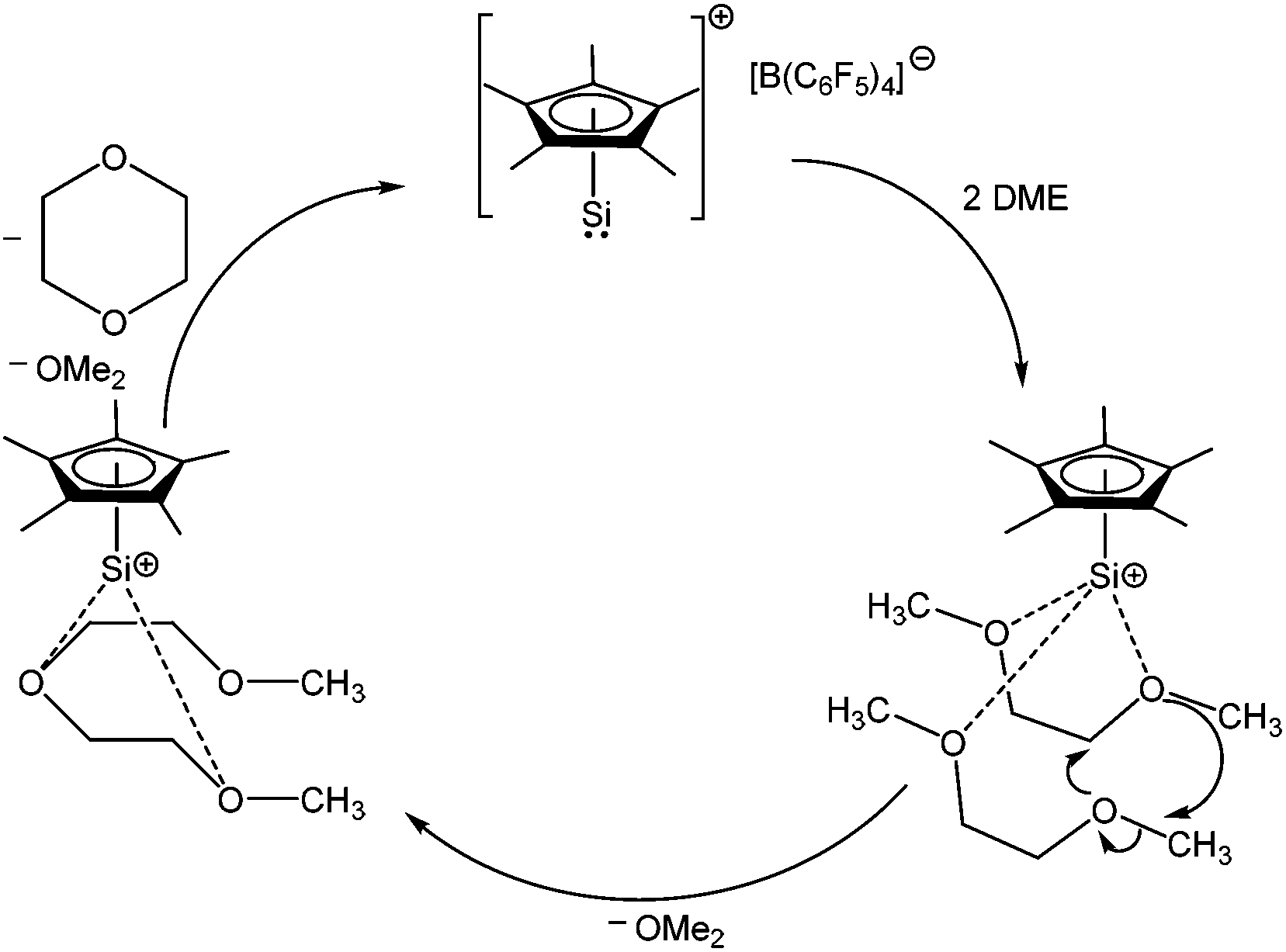
The study of compounds featuring low valent main group elements continues to be a worthwhile subject due to their anticipated application in metal free catalysis. Recently, 1 has been found to catalytically convert 1,2-dimethoxyethane (DME) to 1,4-dioxane and dimethyl ether (Scheme 6), which is a rare example of a metal free catalytic transformation.52Ab initio calculation suggests that the O→Si dative bond in the DME→1 complex is electrostatic in nature and the subsequent enhancement of positive charge at the silicon center facilitates the attack of another equivalent of DME. This catalytic process is found to be useful for a range of oligo(ethylene glycol)diethers, leading to 1,4-dioxane and dimethyl ether in each case.
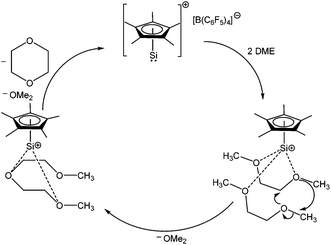 |
| | Scheme 6 Metal free catalytic cycle for conversion of DME to 1,4-dioxane. | |
3.2. Donor stabilized Si(II) cations
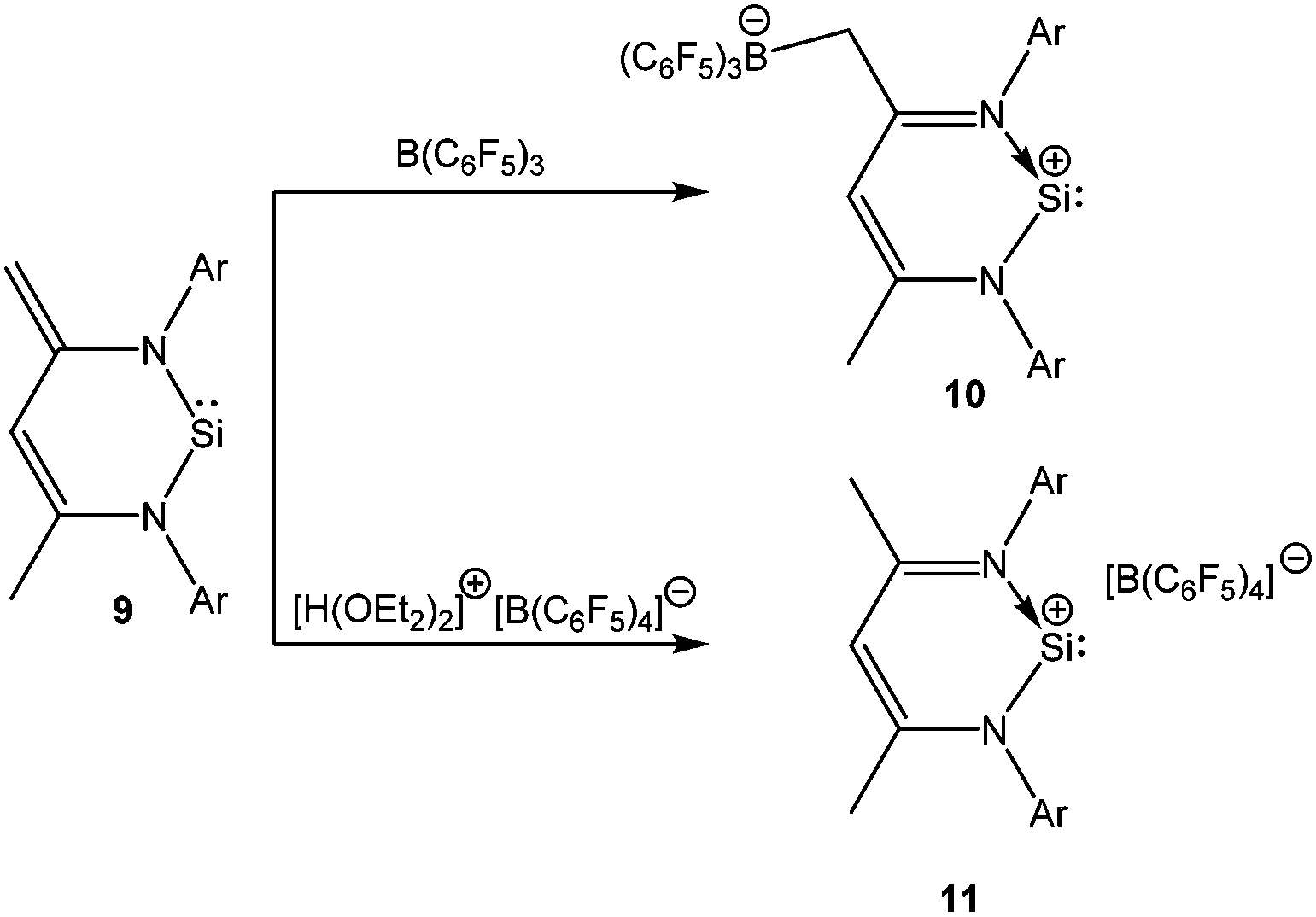
Stabilization of Si(II) cations can be achieved intra-molecularly by using N-donor ligands. Among various N-donor ligands, β-diketiminates with 2,6-disubsituted aryl groups on the nitrogen atoms have been recognized by many groups as a means of stabilizing low coordinate complexes. The journey of the β-diketiminato ligand in silicon chemistry began on 2006 with the synthesis of a unique zwitter-ionic N-heterocyclic silylene [CH{C(Me)(CCH2)(NAr)2}]Si: (Ar = 2,6-iPr2-C6H3) (9).53 Treatment of silylene 9 with B(C6F5)3 resulted in zwitter-ionic compound 10, whereas the same reaction with [H(OEt2)2]+[B(C6F5)4]− led to the formation of a separated ion pair ([LSi][B(C6F5)4] (L = CH(CMeNAr)2)) (11) (Scheme 7).20 It is noteworthy here that 10 readily decomposes when dissolved in CH2Cl2, whereas 11 showed no sign of decomposition in CH2Cl2.
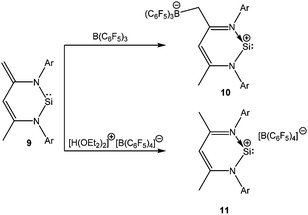 |
| | Scheme 7 Synthesis of β-diketiminato ligand supported Si(II) cations, 10 and 11. | |
Presented in Table 1 are selected NMR data for the related silicon(II) compounds. In general, more cationic charge density on the silicon center results in a more downfield shifted 29Si resonance. However, both 10 (δ = 40.5 ppm) and 11 (δ = 69.3 ppm) exhibit upfield shifted resonances compared to that of 9 (δ = 88.4 ppm) although the Si centers in 10 and 11 are more Lewis acidic in nature. This was presumably a consequence of a pπ–pπ interaction between the π-electron system of the β-diketiminate ligand and the Si atom. Moreover, the γ-H resonances of 10 (δ = 6.79 ppm) and 11 (δ = 6.92 ppm) also indicated the existence of aromatic ring current in the systems. X-ray studies on 10 and 11 showed that the six-membered SiC3N2 rings were planar and the endocyclic N–C bond lengths were shortened by 0.05 Å compared to those in 9. Such features were in accord with the aromatic nature of these systems, which was further confirmed by nuclear independent chemical shift (NICS) calculations (NICS (1): −3.9 ppm).
Table 1
29Si NMR values for related silylium ylidenes
| Compound |
Solvent |
29Si NMR (ppm) |
Ref. |
|
1
|
CD2Cl2 |
−400.2 |
17
|
|
10
|
THF-d8 |
40.5 |
20
|
|
11
|
CD2Cl2 |
69.3 |
20
|
|
12
|
CD2Cl2 |
−3.3 |
54
|
|
13
|
CD2Cl2 |
−58.4 |
55
|
|
14
|
CD3CN |
−89.9 |
56
|
|
15
|
CD3CN |
−55.3 |
56
|
|
16a
|
C6D6 |
8.3 (d, 1JP–Si 7.4 Hz; SiMe3), 234.5 (dt, 2JP–Si 219.4 Hz, 48.5 Hz, SiPt2) |
59
|
|
16b
|
THF-d8 |
0.01 (d, 1JP–Si 6.0 Hz, SiMe3), 187.8 (dt, 2JP–Si 43.4 Hz, 184.3 Hz, SiPd2) |
59
|
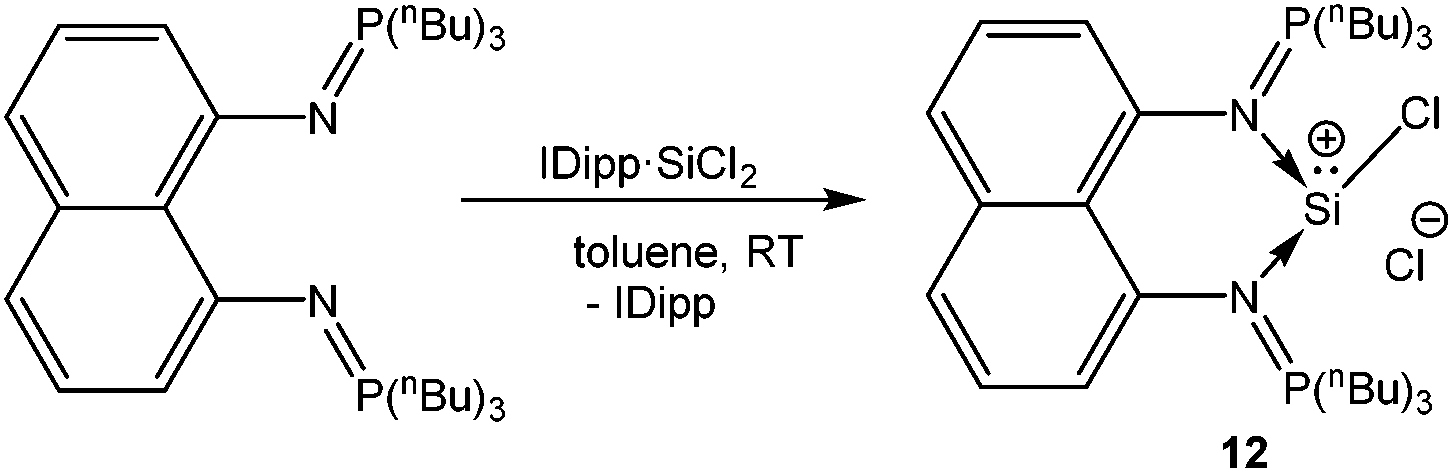
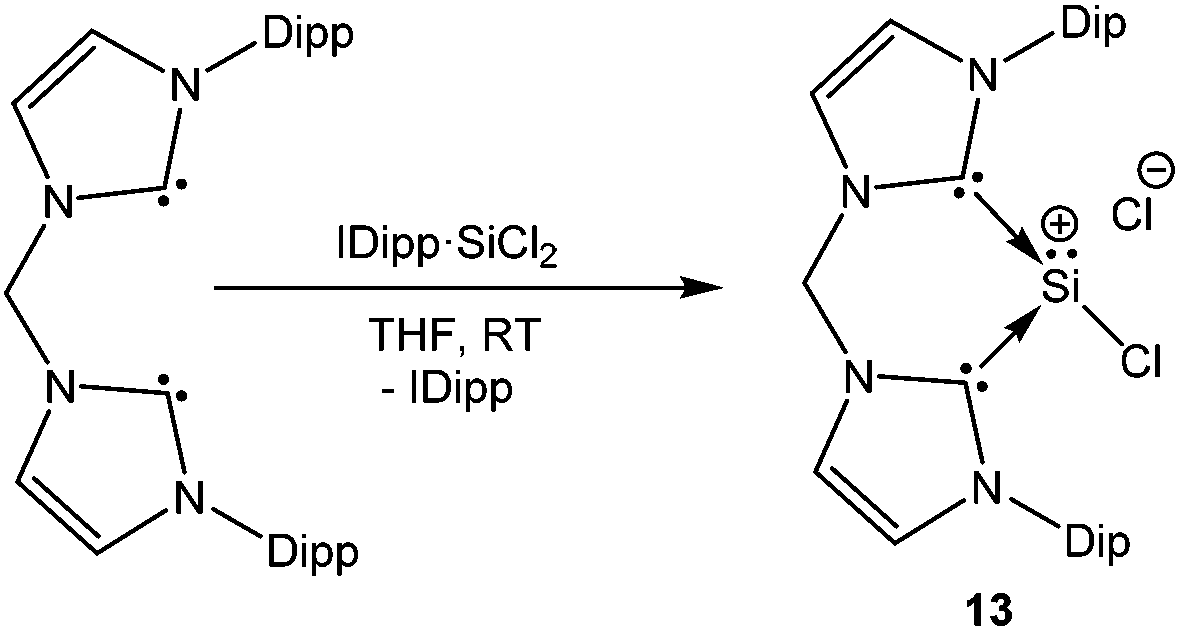
A chelating bis(iminophosphorane)ligand has been recently used by Driess et al. to obtain a chlorosilyliumylidene complex 12 through ligand exchange of the SiCl2 unit.54 NHC·SiCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18a was used as a stoichiometric source of SiCl2 and its reaction with the bis(iminophosphorane) ligand resulted in the ion pair with concomitant liberation of one equivalent of NHC (Scheme 8). Utilizing the same synthetic strategy, Driess et al. prepared another remarkable chlorosilyliumylidene derivative 13 by the reaction of the bidentate bis-NHC ligand with NHC·SiCl2 (Scheme 9).55 The 29Si NMR resonances due to the three-coordinate Si nuclei in 12+ and 13+ were observed at δ = −3.3 and −58.4 ppm, respectively. The upfield shift can also be attributed to the stronger electron donation from bis-NHC and iminophosphorane ligands. Alternatively, such shifts to a higher field may also indicate the decrease of the cationic character. Inspection of the frontier MOs revealed that the HOMO in 12+ is associated with the ten π-electrons from the naphthyl moiety and the nitrogen lone pair of the P–N ylide bonds and the lone pair on the silicon atom is depicted by the HOMO−1 orbital. This is in contrast to 13+ where the lone–pair of silicon constitutes the HOMO and the difference may be attributed to the stabilization of the Si lone pair by the π-system in 12+.
18a was used as a stoichiometric source of SiCl2 and its reaction with the bis(iminophosphorane) ligand resulted in the ion pair with concomitant liberation of one equivalent of NHC (Scheme 8). Utilizing the same synthetic strategy, Driess et al. prepared another remarkable chlorosilyliumylidene derivative 13 by the reaction of the bidentate bis-NHC ligand with NHC·SiCl2 (Scheme 9).55 The 29Si NMR resonances due to the three-coordinate Si nuclei in 12+ and 13+ were observed at δ = −3.3 and −58.4 ppm, respectively. The upfield shift can also be attributed to the stronger electron donation from bis-NHC and iminophosphorane ligands. Alternatively, such shifts to a higher field may also indicate the decrease of the cationic character. Inspection of the frontier MOs revealed that the HOMO in 12+ is associated with the ten π-electrons from the naphthyl moiety and the nitrogen lone pair of the P–N ylide bonds and the lone pair on the silicon atom is depicted by the HOMO−1 orbital. This is in contrast to 13+ where the lone–pair of silicon constitutes the HOMO and the difference may be attributed to the stabilization of the Si lone pair by the π-system in 12+.
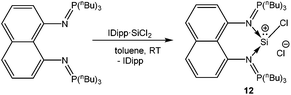 |
| | Scheme 8 Synthesis of [Cl–Si:]+derivative, 12. | |
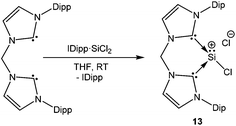 |
| | Scheme 9 Synthesis of [Cl–Si:]+derivative, 13. | |
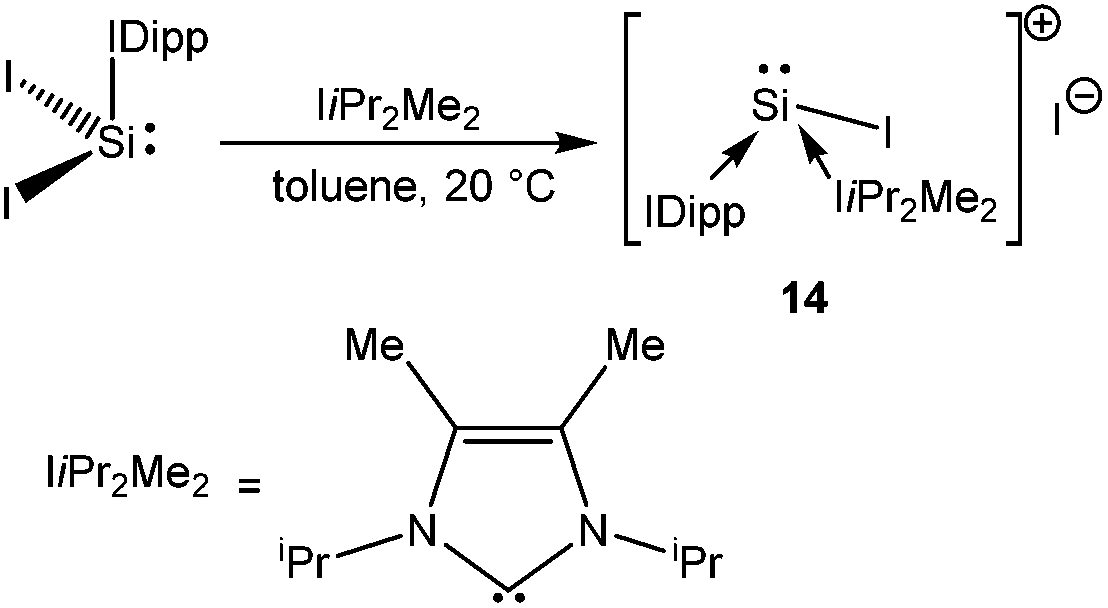
Unlike NHC stabilized SiCl2 and SiBr2, which were reported back in 2009,18 the synthesis of the first stable NHC stabilized diiodosilylene (IDipp·SiI2)56 (IDipp = 1,3-(2,6-iPr2-C6H3)2-imidazol-2-ylidene) has recently been accomplished by following the same synthetic protocol used for the isolation of IDipp·SiBr2.18b Following this, Filippou et al. have found that the reaction of IDipp·SiI2 with IiPr2Me2 (1,3-iPr2-4,5-Me2-imidazol-2-ylidene) resulted in the displacement of one iodide ligand leading to [(IDipp)(IiPr2Me2)·SiI]+I− (14) (Scheme 10), the first formal derivative of [I–Si:]+. The structure of 14 revealed a fixed orientation of one isopropyl C–H group pointing towards the Si(II) atom to maximize a Si⋯H–C anagostic interaction, as also supported by the 29Si–1H coupling constant (J = 10.4 Hz) and subsequent DFT calculations.
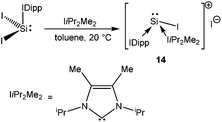 |
| | Scheme 10 Synthesis of [I–Si:]+ derivative, 14. | |
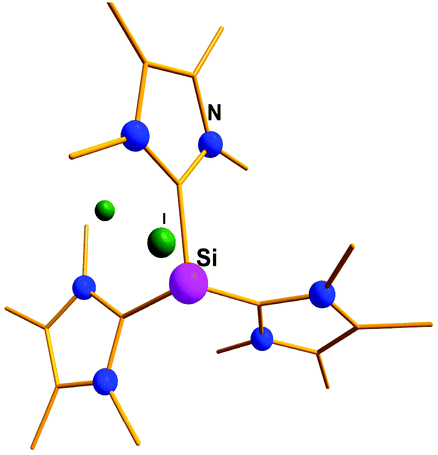
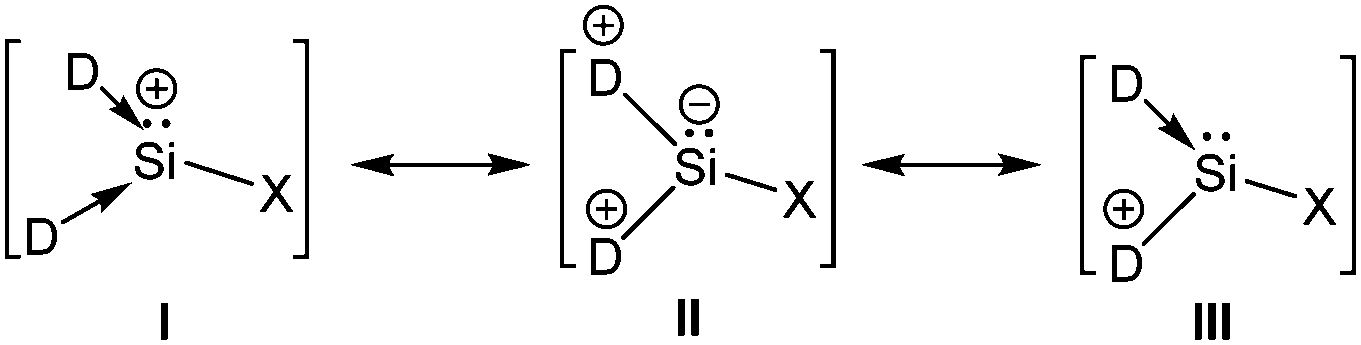
Addition of a less bulky NHC (IMe4 = 1,3,4,5-Me4-imidazol-2-ylidene) to IDipp·SiI2 led to the first Si(II) dication [(IMe4)3→Si:]2+I2− (15) (Scheme 11).56 The removal of the iodide anions from the Si center can be attributed to the steric bulk exerted by three NHCs. The Si center adopts a pyramidal propeller like conformation (Fig. 1), akin to the analogous Ge(II) dication (492+) published before by Baines and coworkers (vide infra).27 However, interpretation of the ionicity of 152+ as well as other donor stabilized Si(II) cations (12+–14+) is complicated by the fact that such stabilization takes place at the cost of the cationic character of the Si atom. Three possible canonical forms of 12+–14+ are outlined in Scheme 12. Two of the resonating structures oppose and diminish the cationic nature of the Si atom and indicate that the positive charge is distributed over the ligands. This was further manifested in the appearance of the 13C NMR signals of the carbene C in the relatively high field in these cations (13: δ = 161.6; 14: δ = 151.5 and 158.3; 15: δ = 150.7 ppm), which are closer to that of the imidazolium salt (IMe4H)Cl (δ = 136.9 ppm) than that of IMe4 (δ = 212.7 ppm). However, various charge calculations indicate the accumulation of positive charge on silicon atom. Therefore, perhaps it is safe to comment that all three resonating structures contribute to the overall bonding of these cations.
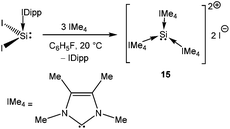 |
| | Scheme 11 Synthesis of Si dication derivative, 15. | |
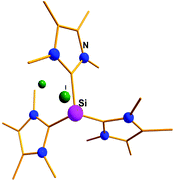 |
| | Fig. 1 Molecular structure of the Si dication, 15. | |
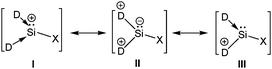 |
| | Scheme 12 Canonical forms of donor stabilized Si cations. | |
3.3. Transition metal supported Si(II) cations
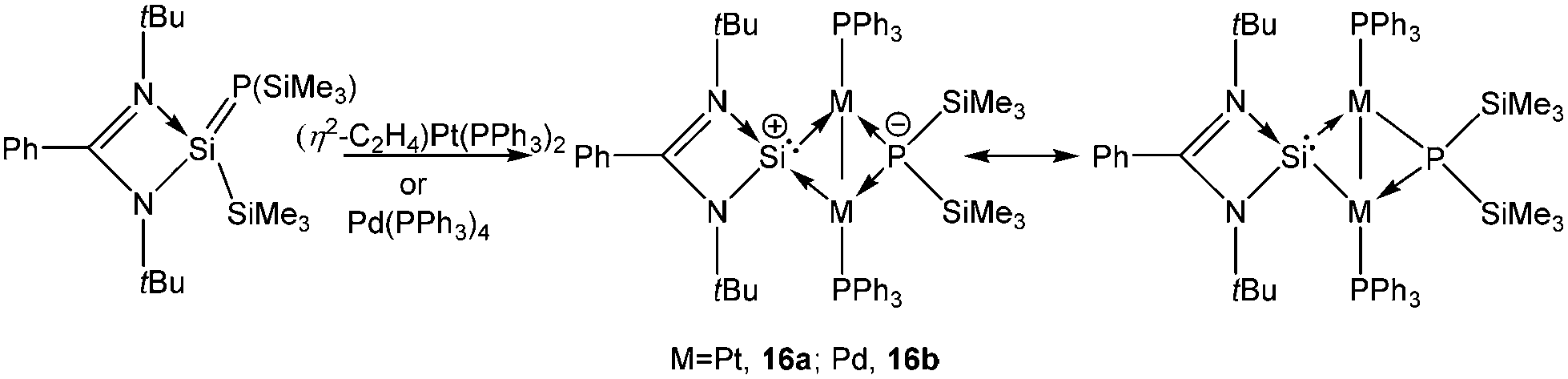
Driess and coworkers proposed that the reaction of [PhC(NtBu)2SiCl] with (η5-C5H5)ZrCl3 led to the formation of [PhC(NtBu)2Si]+[(η5-C5H5)2Zr2Cl7]−57 which spontaneously underwent a disproportionation reaction to afford the tetrasilacyclobutadiene dication (vide supraChart 1) along with various side products. All attempts to prepare [PhC(NtBu)2Si]+ have been unsuccessful thus far except for the work of So and coworkers who obtained 4-DMAP (4-DMAP = 4-dimethylaminopyridine) coordinated [PhC(NtBu)2Si]+ with a triflate anion.58 Recently, Inoue and coworkers isolated [PhC(NtBu)2Si]+ in the coordination sphere of Pd and Pt. A cationic transition metal complex, [LSi{M-(PPh3)}2P(SiMe3)2] (M = Pd (16a) and Pt (16b)) was prepared from the reaction of [PhC(NtBu)2Si(SiMe3) = P(SiMe3)] with Pd(PPh3)4 or [(η2-C2H4)Pt(PPh3)2] (Scheme 13).59 Determining the nature of the Pt–Si bonds is rather difficult. Two possible resonating structures are presented in Scheme 13. Natural population analysis (NPA) revealed a high fraction (+1.219) of overall positive charge to be accommodated on the Si center. NBO analysis also indicated the preference of a charge separated structure over a neutral one. However, it would not be erroneous to imagine that the real nature of this cation is somewhere between the two resonance extremes.
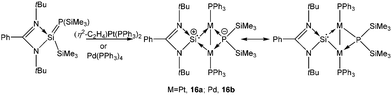 |
| | Scheme 13 Si(II) cation in the coordination sphere of a late transition metal. | |
4. Ge(II) and Sn(II) cations and dications
In contrast to silylenes, germylenes and stannylenes are less reactive due to the larger energy gap between their s- and p-orbitals. Therefore, the chemistry of Ge(II) and Sn(II) cations was developed rather much ahead of Si(II) cations. The major synthetic route leading to E(II) [E = Ge and Sn] cations is the dehalogenation of the corresponding germylenes and stannylenes. Of late, Reid, Driess, Roesky, Stalke, and their respective coworkers reported a series of Lewis base mediated ionization reactions of GeCl2 and SnCl2 to access Ge(II) and Sn(II) cations (vide infra). It should be emphasized at this point that the majority of Ge(II) and Sn(II) cations were stabilized using the same ligand sets and therefore we have decided to discuss the Ge(II) and Sn(II) cations together for the convenience of the readers. We shall divide the Ge(II) and Sn(II) cations in five different categories: (a) [LE:]+ (L = mono-anionic ligand), (b) [D→E–X]+ (D = Lewis bases except for macrocycles, X = halide), (c) [D→E]2+, (d) cationic polyether complexes of germanium(II) and tin(II), and (e) transition metal based germanium(II) and tin(II) cations.
4.1. Stable [LE:]+
4.1.1. Cations of Ge and Sn embedded within a cyclic system.
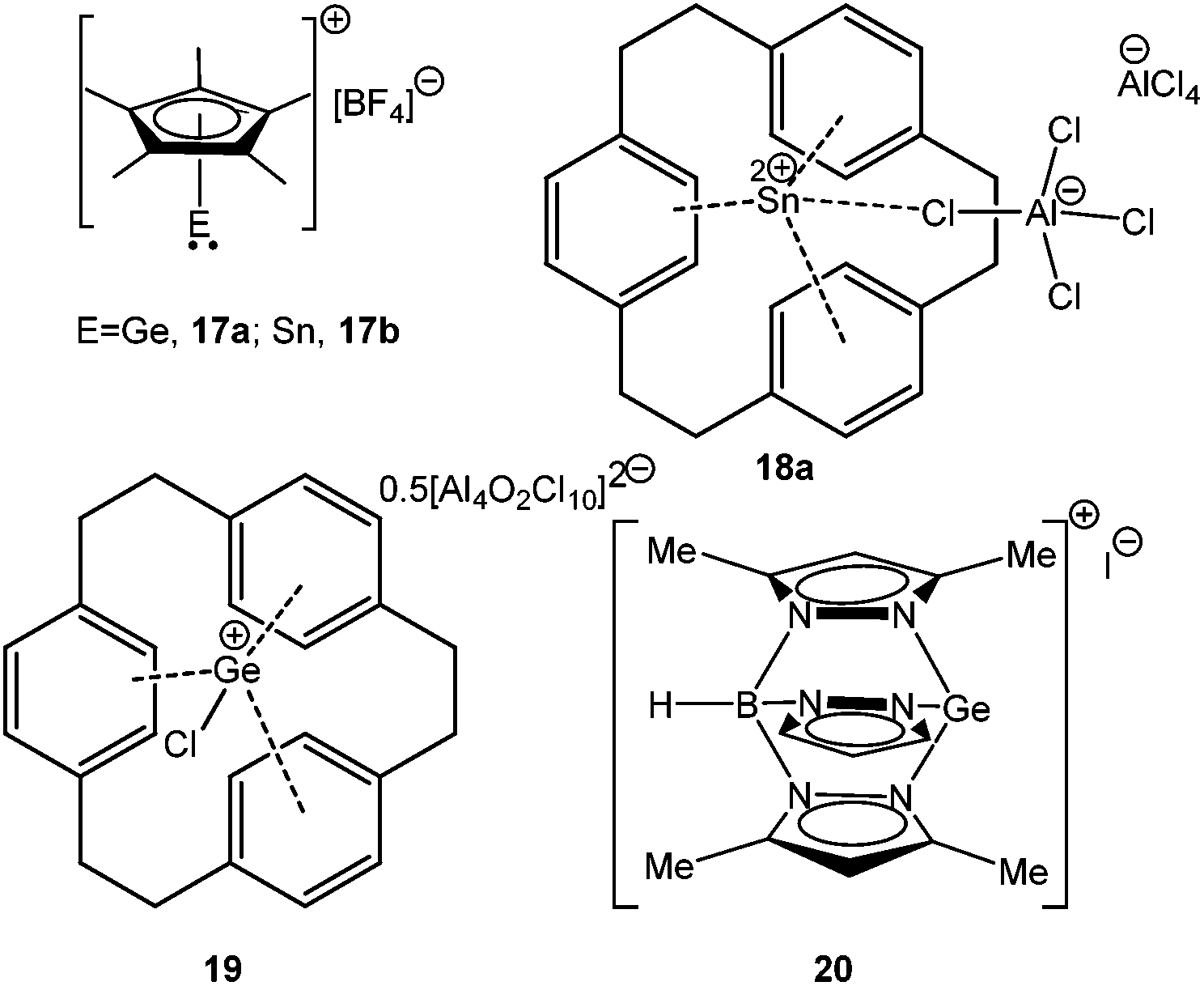
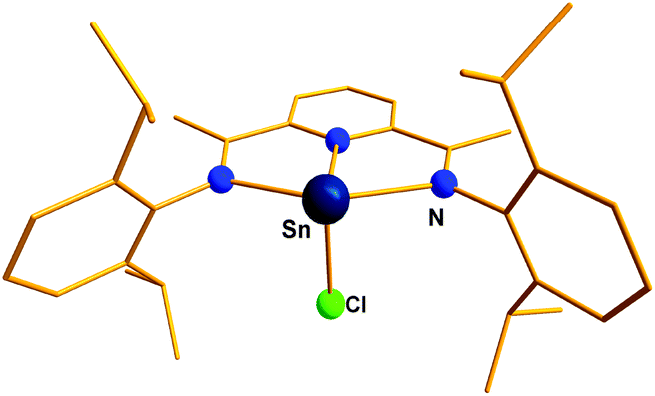
The first representative example of this type of cation, [(η5-C5Me5)E]+BF4− (E = Ge, 17a; Sn, 17b) (Scheme 14), was synthesized by Jutzi et al. by reacting the corresponding germylenes and stannylenes with HBF4. An X-ray crystal structure of [(η5-C5Me5)Sn]+BF4− revealed η5 coordination mode of the Cp* ligand.21a As expected, compound 17 functioned as a Lewis acid instead of a Lewis base, which was manifested in its adduct formation with pyridine and bipyridine.21a A few years later, Schmidbaur and coworkers reported two Sn(II) cations of compositions [{(C24H24)Sn(AlCl4)}][AlCl4−] (18a) and [{(C24H24)Sn(AlCl4)}][Al2Cl7−] (18b).34,60 The corresponding Ge(II) cation was found to have a Ge–Cl bond and during crystallization it reacted with H2O, leading to [(C24H24)GeCl]2(Al4O2Cl10) (19) (Scheme 14).34
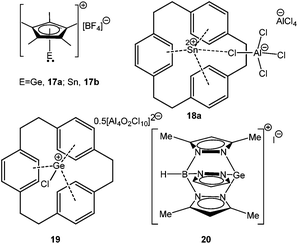 |
| | Scheme 14 Ge(II) and Sn(II) cations stabilized by pentamethylcyclopentadienyl, cyclophane, and pyrazole ligands. | |
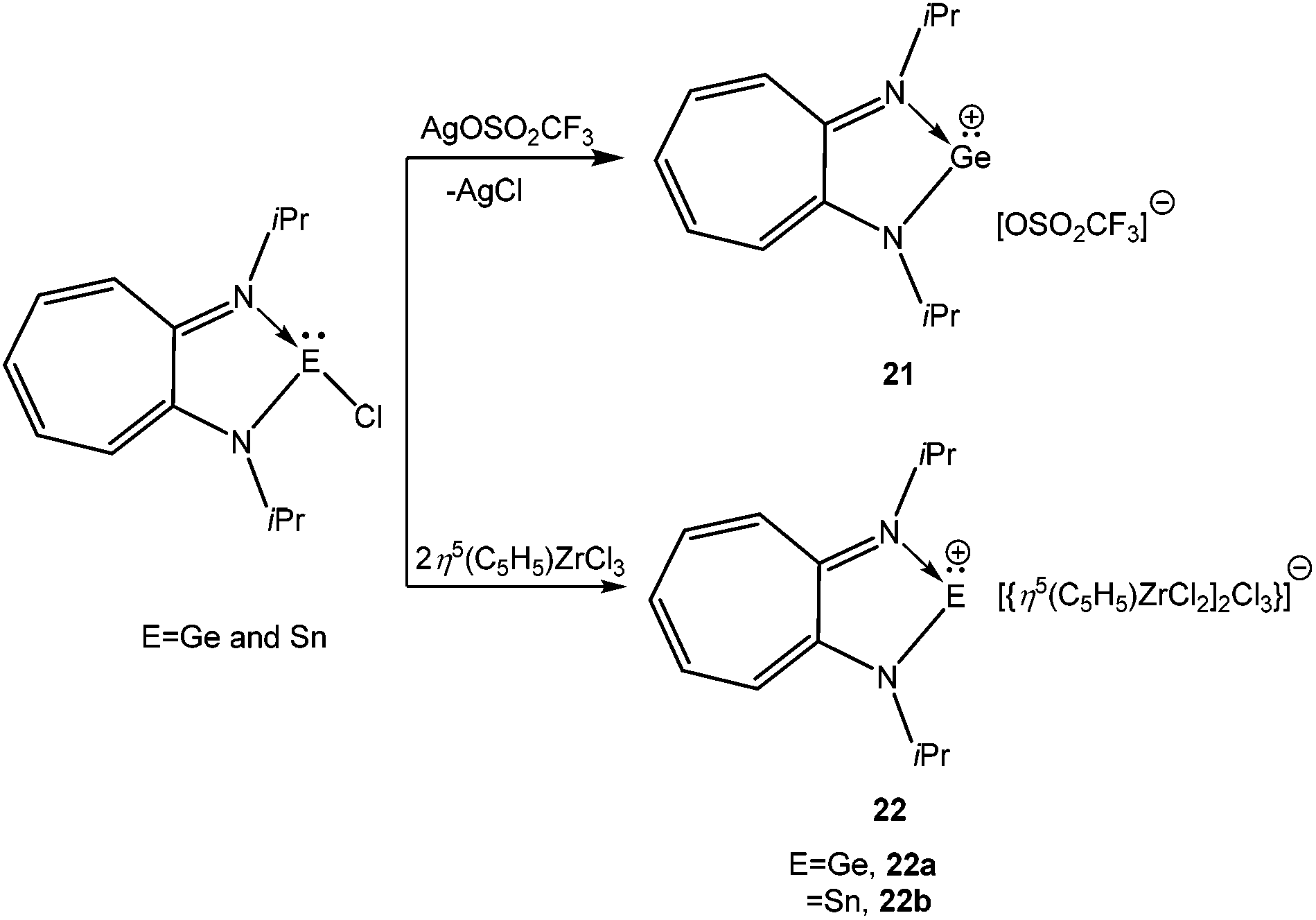
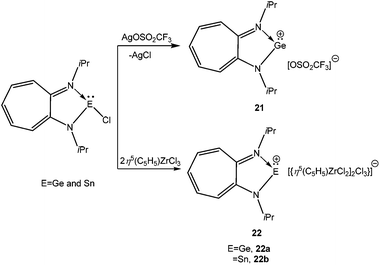
Reger and Coan reported the synthesis and structural elucidation of [HB(3,5-Me2pz)3Ge]+ (20+) with an iodide counter ion (Scheme 14).61 The shortest Ge⋯I distance is over 4 Å, clearly indicating the lack of a substantial covalent interaction between these atoms. The Ge(II) center adopted a pyramidal geometry with three neighboring nitrogen atoms coordinated to the Ge center. This result kick-started the use of N-donor ligands for the isolation of p-block cations. Following this, Dias et al. used bi-dentate, monoanionic amino-troponiminate (ATI) with a distinct 10π electron backbone for the isolation of Ge(II) and Sn(II) monocations. Substitution of the corresponding chloro precursor [(iPr2ATI)GeCl] with AgOSO2CF3 resulted in [(iPr2ATI)Ge][OSO2CF3] (21) (Scheme 15).62a The germanium atom was weakly bound to the oxygen atom of the triflate anion (Ge–O: 2.255(2) Å). In order to prepare a “free” Ge(II) cation, (iPr2ATI)GeCl was reacted with (η5-C5H5)ZrCl3, which serves as a chloride scavenger to result in [(iPr2ATI)Ge][(η5-C5H5)ZrCl2(μ-Cl)3ZrCl2(η5-C5H5)] (22a).62a A related Sn(II) cation, [(iPr2ATI)Sn][(η5-C5H5)ZrCl2(μ-Cl)3ZrCl2(η5-C5H5)] (22b) was earlier reported by the same group.62b However, the weak interactions between the cations and the terminal chloride atoms were found in the solid state structures of 22a,b.
 |
| | Scheme 15 Synthesis of Ge(II) and Sn(II) cations using the aminotroponiminate ligand. | |
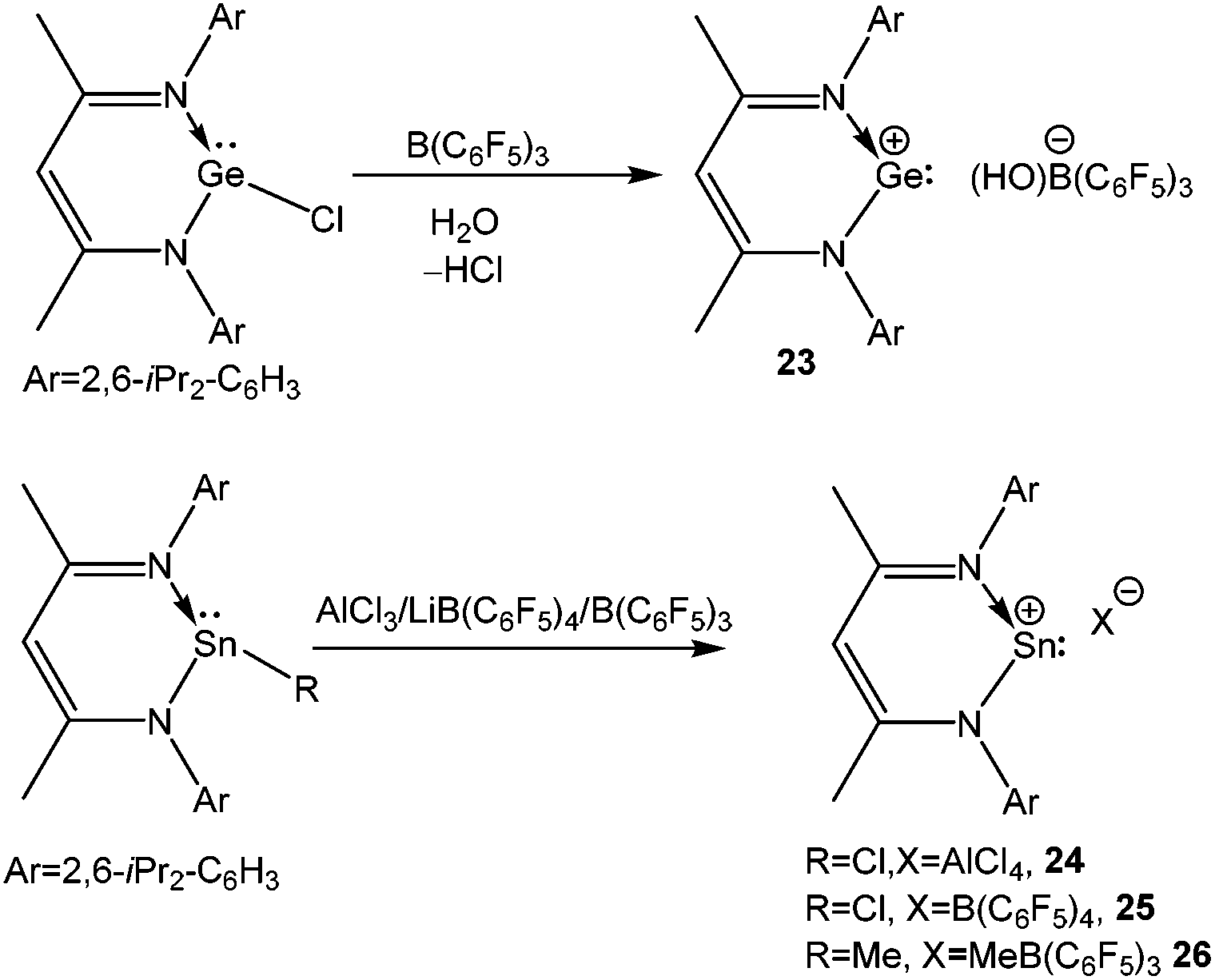
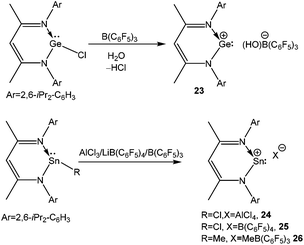
Following the synthesis of germylenemonochloride [LGeCl] [L = CH(CMeNAr)2; Ar = 2,6-iPr2C6H3] using the versatile mono-anionic bidentate β-diketiminate ligands,63 Power's group prepared a cyclic Ge(II) mono-cation [LGe][(HO)B(C6F5)3] (23) by reacting LGeCl and B(C6F5)3 in the presence of water (Scheme 16).64 The geometrical parameters of 23+ such as the decrease of C–N bond lengths (1.34av Å) with the concomitant increase of the Ge–N bond length (1.91av Å) and NICS(1) values (−2.4 ppm) were evocative of those observed for its silicon analogue 11, indicating the presence of a slight aromatic character in 23+.65 However, one must note here that the γ-H signal of 23+ appeared at a significantly higher field (δ = 4.23 ppm), than that of 11 (δ = 6.92 ppm).
 |
| | Scheme 16
β-Diketiminato ligand stabilized Ge(II) and Sn(II) cations. | |
The tin versions of 23+ with the [AlCl4]− (24), [B(C6F5)4]− (25), and [B(C6F5)3(Me)]− (26) counter-anions were later reported by Fulton and coworkers (Scheme 16).66 The 119Sn NMR chemical shifts for these cations are −626.7, 197.0, and −139.5 ppm, respectively (Table 2). Among them, only 25 displays a low-field 119Sn NMR shift from that of its precursor (−224 ppm). Unfortunately, 24 and 25 were not characterized by X-ray crystallography. The single crystals of 26 were only grown from a diethyl ether solution and in the solid-state structure of 26 one ether molecule was bound to the Sn atom, which explained the high-field chemical shift in the 119Sn NMR.
Table 2
119Sn NMR values for stannylium ylidenes and related compounds
| Compound |
Solvent |
119Sn NMR (ppm) |
Ref. |
|
18
|
|
— |
21a
|
|
19
|
|
— |
21a
|
|
22b
|
CDCl3 |
734 |
62b
|
|
24
|
CD2Cl2 |
−626.7 |
66
|
|
25
|
CD2Cl2 |
197.0 |
66
|
|
26
|
CD2Cl2 |
−139.5 |
66
|
|
32
|
CD2Cl2 |
46.3 (solid-state: 68) |
70
|
|
34
|
CD2Cl2 |
−30 |
70
|
|
39
|
CD2Cl2 |
249.71 |
30b
|
|
40
|
CD2Cl2 |
17.28(br) |
30b
|
|
44
|
THF-d8 |
−60.27 (SnCl3−) & −435.07 |
75
|
|
46
|
CDCl3 |
−73.2 (SnCl3−) and −330.4 |
76
|
|
54
|
CD3CN |
−1468 |
80
|
|
64a–c
|
|
Solid-state: −980 (Cl), −920 (Br), −810 (I) |
86b
|
|
65
|
|
Solid-state: −1533 |
86b
|
|
66
|
|
Solid-state: −1436 |
86a
|
|
67
|
|
Solid-state: −1457 |
86a
|
|
68
|
|
Solid-state: −840, −58 |
86a
|
|
69
|
|
Solid-state: −1578 |
86a
|
|
70
|
|
Solid-state: −1405 |
86a
|
|
71
|
|
Solid-state: −1721, −1706 |
86a
|
|
72
|
|
Not mentioned |
87
|
|
74
|
THF-d8 |
−8.4 |
89a
|
|
75
|
THF-d8 |
224.0 |
89b
|
|
76
|
THF-d8 |
249.9 |
89b
|
|
77
|
CDCl3 |
70.6 |
90
|
|
78 & 79 |
|
Not observed |
92
|
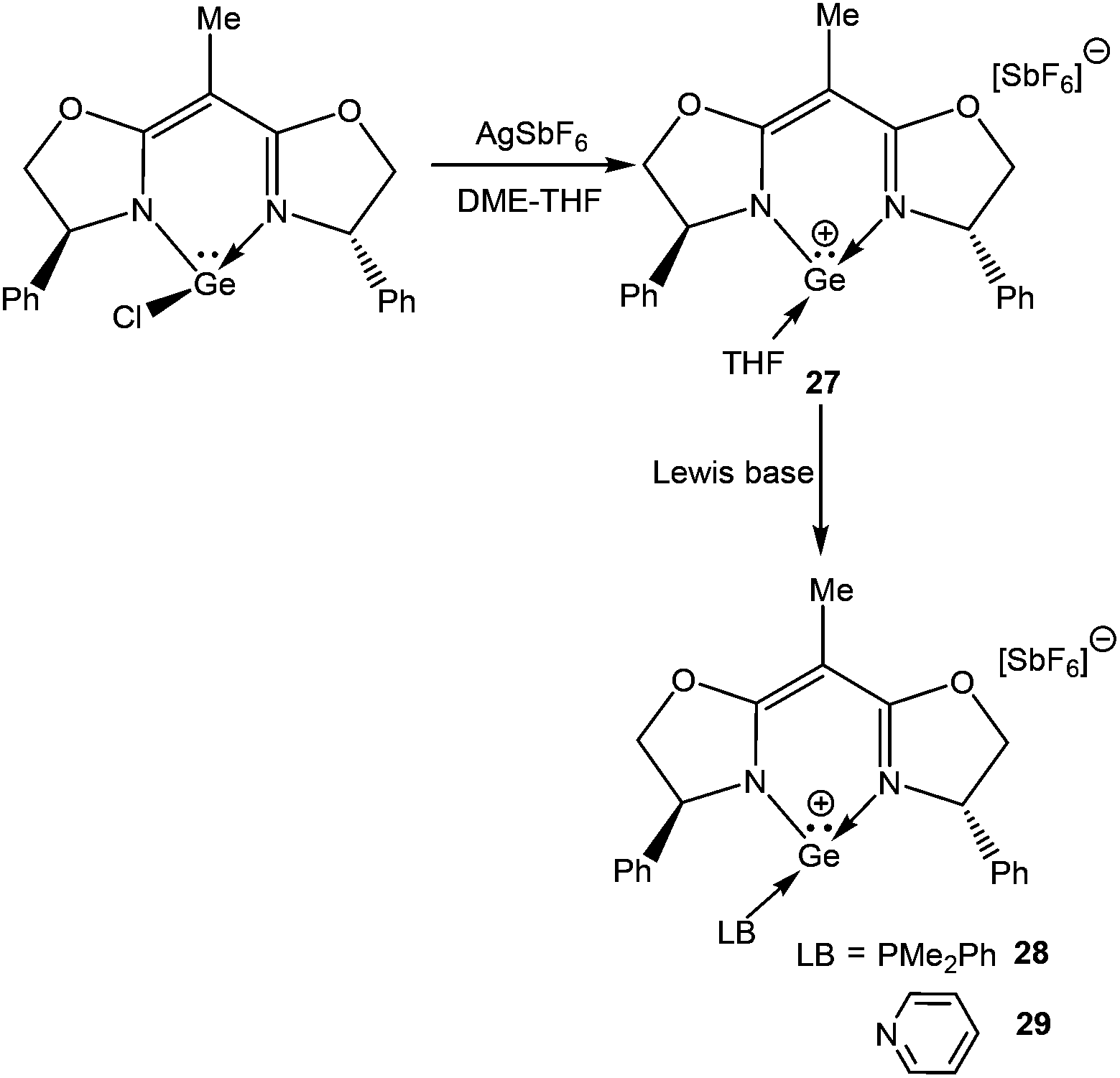
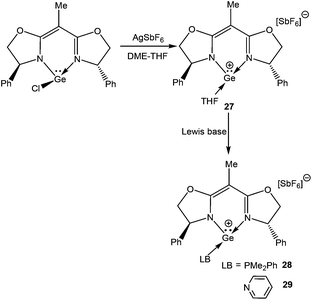
Mochida and coworkers introduced a chiral ligand, (1,1-bis[(4S)-4-phenyl-1,3-oxazolin-2-yl]ethane), popularly known as ({(S)-box-Ph}H) to isolate a germanium(II) cation.67 The motivation for this work presumably came from the use of the enantiomeric carbene as a ligand for metal complexes to catalyze asymmetric reactions.68 Abstraction of the chloride atom from the DME solution of the corresponding germylene with Ag[SbF6] in the presence of excess THF afforded the Ge(II) cation, [Ge((S)-box-Ph)(THF)](SbF6) (27) (Scheme 17). Substitution of the THF ligand with other Lewis bases like PMe2Ph and pyridine yielded [Ge((S)-box-Ph)(PMe2Ph)](SbF6) (28) and [Ge((S)-box-Ph)(py)](SbF6) (29), respectively. Inspection of the Ge–N bond lengths in these cations revealed that the average bond length increases on going from 27 to 29 [27: 1.928 (2) Å, 28: 1.933(3) Å, and 29: 1.939(4) Å]. This lengthening was assumed to be due to the increased D→Ge π donation from 27 to 29, resulting in less σ as well as π interactions, which in turn increases the Ge–N bond lengths.
 |
| | Scheme 17 Synthesis of Ge(II) cation, 27 and its derivatives. | |
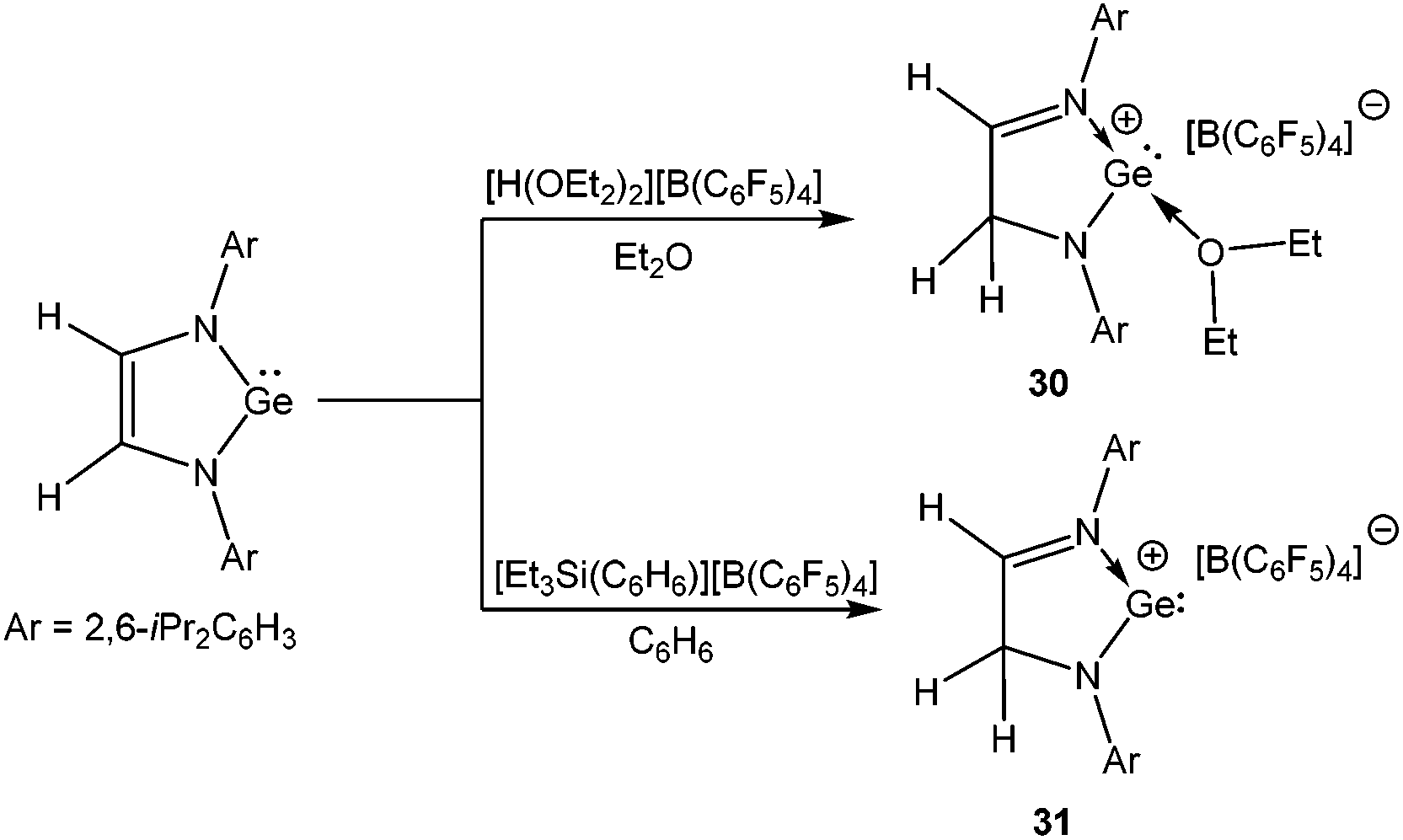
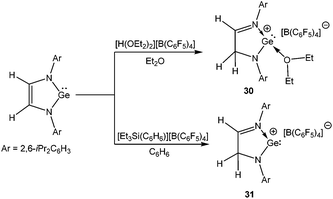
By adopting the same synthetic protocol that has been used for the synthesis of 11, Müller et al. isolated two more Ge(II) cations, 30 and 31 upon protonating 1,3-di(2,6-iPr2C6H3)-germaimidazol-2-ylidene with [H(OEt2)2]+[B(C6F5)4]− and [Et3Si(C6H6)]+[B(C6F5)4]− (Scheme 18).69 The protonation took place at the C-4 position instead of germanium and as a result the ligand underwent a shift from di-anionic to mono-anionic by rupturing the symmetry of the GeN2 moiety. The molecular structure of 30 revealed that one ether molecule is strongly coordinated to the Ge atom. Although exhibiting no interaction with the benzene solvent, 31 was weakly bound to the borate anion through the fluoride atoms. A detailed theoretical calculation was carried out by Müller et al. to understand why the protonation took place in the C-4 position instead of Ge. It was observed that the C-protonated product was more stable than the hypothetical Ge-protonated product by 68.3 kJ mol−1 (B3LYP/6-311G) and 44.8 kJ mol−1 (MP2), respectively. Another underlying factor responsible for this regioselective protonation is the formation of a C–H bond instead of a highly polarized Ge–H bond.
 |
| | Scheme 18 Ge cations 30 and 31. | |
4.1.2. Acyclic Ge(II) and Sn(II) cations.
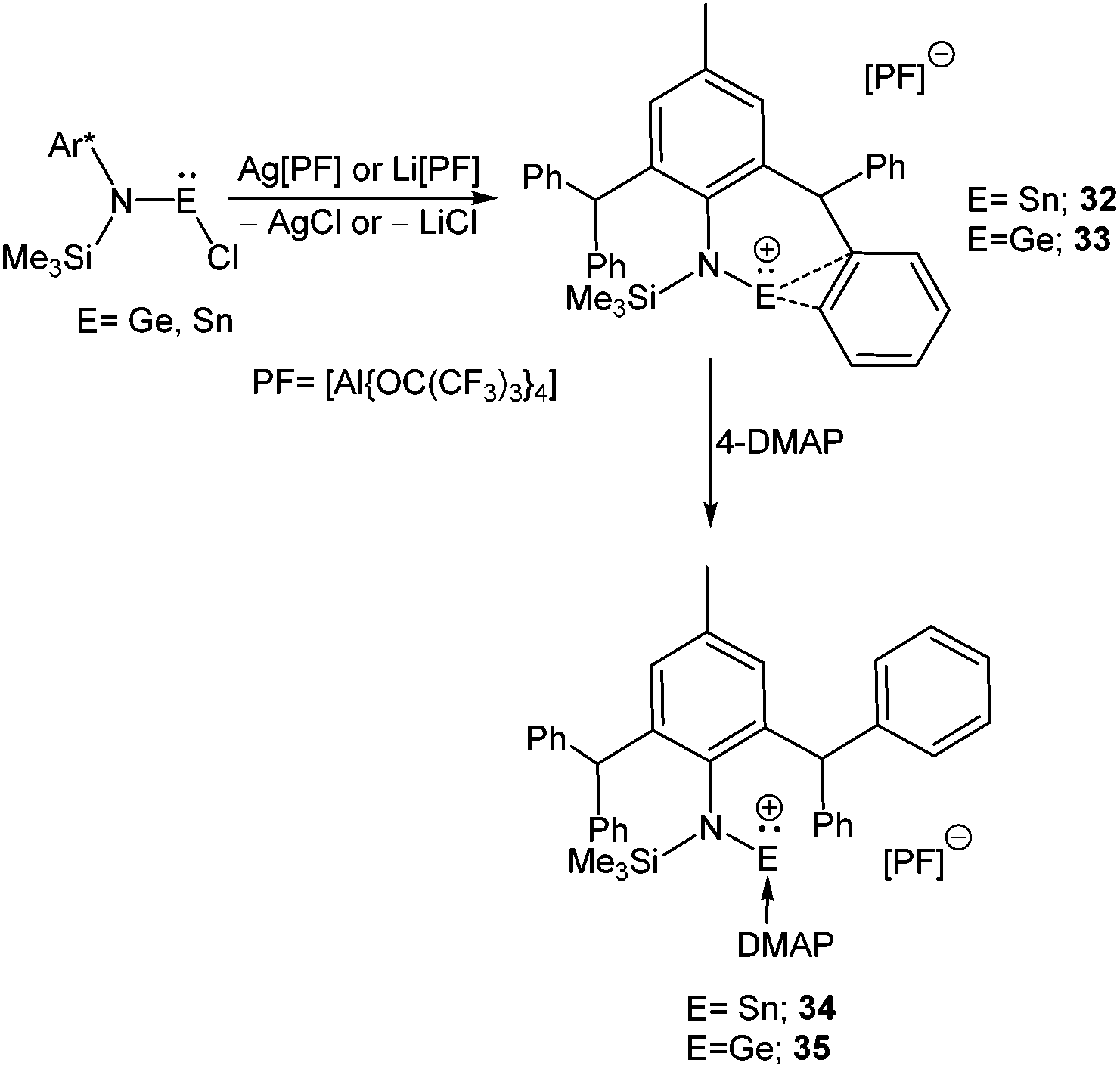
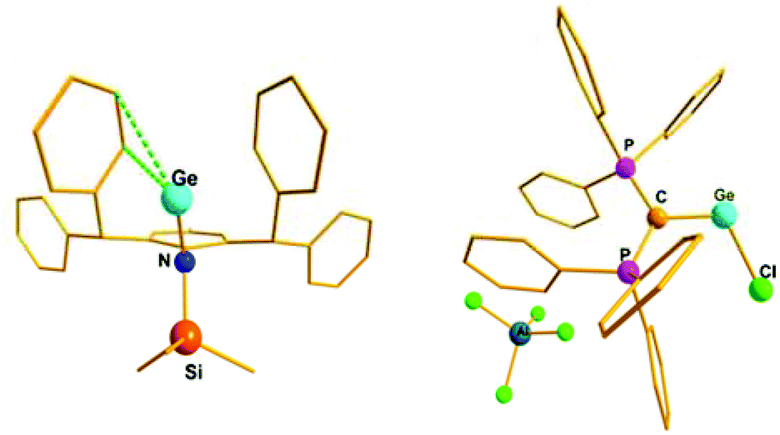
The paucity of mono-coordinated Ge(II) and Sn(II) cations over the di-coordinated ones can be attributed to the rigorous use of mono-anionic bidentate ligands, where the additional N-donor/donors quench the electrophilicity of these cations. It is only recently that Jones and Krossing obtained the first quasi mono-coordinate Sn(II) cation, [SnN(Ar*)(SiMe3)][PF] (32) using a combination of an extremely bulky amido ligand {N(Ar*)(SiMe3)}− [Ar* = (2,6-CHPh2-4-MeC6H2)] and a weakly coordinating anion PF = [Al{OC(CF3)3}4]− (Scheme 19).70 Synthesis of the analogous Ge(II) cation, [GeN(Ar*)(SiMe3)]+ (33+) was accomplished via a slightly modified synthetic route where a solution of LGeCl in DCM was slowly added to a PhF solution of Li[PF]. The molecular structures of both cations showed that there were no close contacts between anion and cation centers. However, an intramolecular η2 arene interaction to the metal centers was present in the solid state (Ge–C: 2.65 Å (mean) and Sn–C: 2.82 Å (mean)). This interaction was further reflected in the solution-state 119Sn NMR spectrum, where the signal appeared in quite a high-field (δ = 46.3 ppm) compared to that of the precursor, N(Ar*)(SiMe3)SnCl (δ = 173.7 ppm). The solid-state 119Sn MAS NMR spectrum (δ = 68 ppm) was also in good agreement with the solution state spectrum. The low temperature 13C{1H} NMR spectra of 32+ and 33+ exhibited 16 aryl signals, whereas room temperature NMR spectra showed only 12 signals for aryl carbons. The appearance of four extra signals at low temperatures corresponded to equivalent η2-arene interactions with the phenyl group of both C(H)Ph2 substituents (Fig. 2) that could not be distinguished at the room temperature NMR time scale. Consistent with this, DFT calculations revealed four C⋯E interactions rather than the two observed in the X-ray structures. The calculated Wiberg Bond Index (WBI) of the C⋯E contacts were 0.423 for 32+ and 0.283 for 33+, suggesting that the cations are arene stabilized. The +2 oxidation state of Sn was confirmed by Mössbauer spectroscopy, which shows a tin signal with an isomeric shift of δ = 3.369(4) mm s−1, a typical value for Sn(II) compounds.71
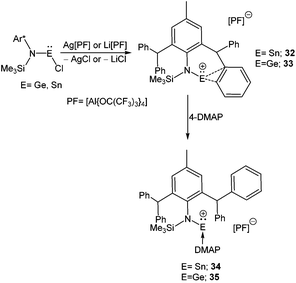 |
| | Scheme 19 Quasi-monocoordinate Ge and Sn cations, 32+ and 33+ and their adduct formation with 4-DMAP. | |
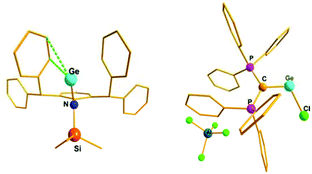 |
| | Fig. 2 Molecular structures of 33+ and 36+. | |
The electrophilic nature of 32+ and 33+ was observed upon addition of the N-donor ligand, 4-DMAP, which resulted in 4-DMAP coordinated Sn(II) (34+) and Ge(II) (35+) cations. The 4-DMAP coordination led to the displacement of C⋯E interactions in the latter, which is reflected in the longer C⋯E distances and higher 119Sn NMR resonance (δ = −30 ppm).
4.1.3. Cations featuring [E–Cl]+.
There has been much interest in the synthesis of cations featuring the [E–X]+ (E = Ge and Sn; X = halide) moiety. This can be achieved in two ways: (i) dehalogenation from the corresponding [D→EX2] compounds would lead to the synthesis of [D→EX]+ cations; (ii) Lewis base mediated ionization of EX2 represents an alternative synthetic route to give access to [E–X]+ cations.
4.1.3.1. Dehalogenation of [D→EX2] compounds.
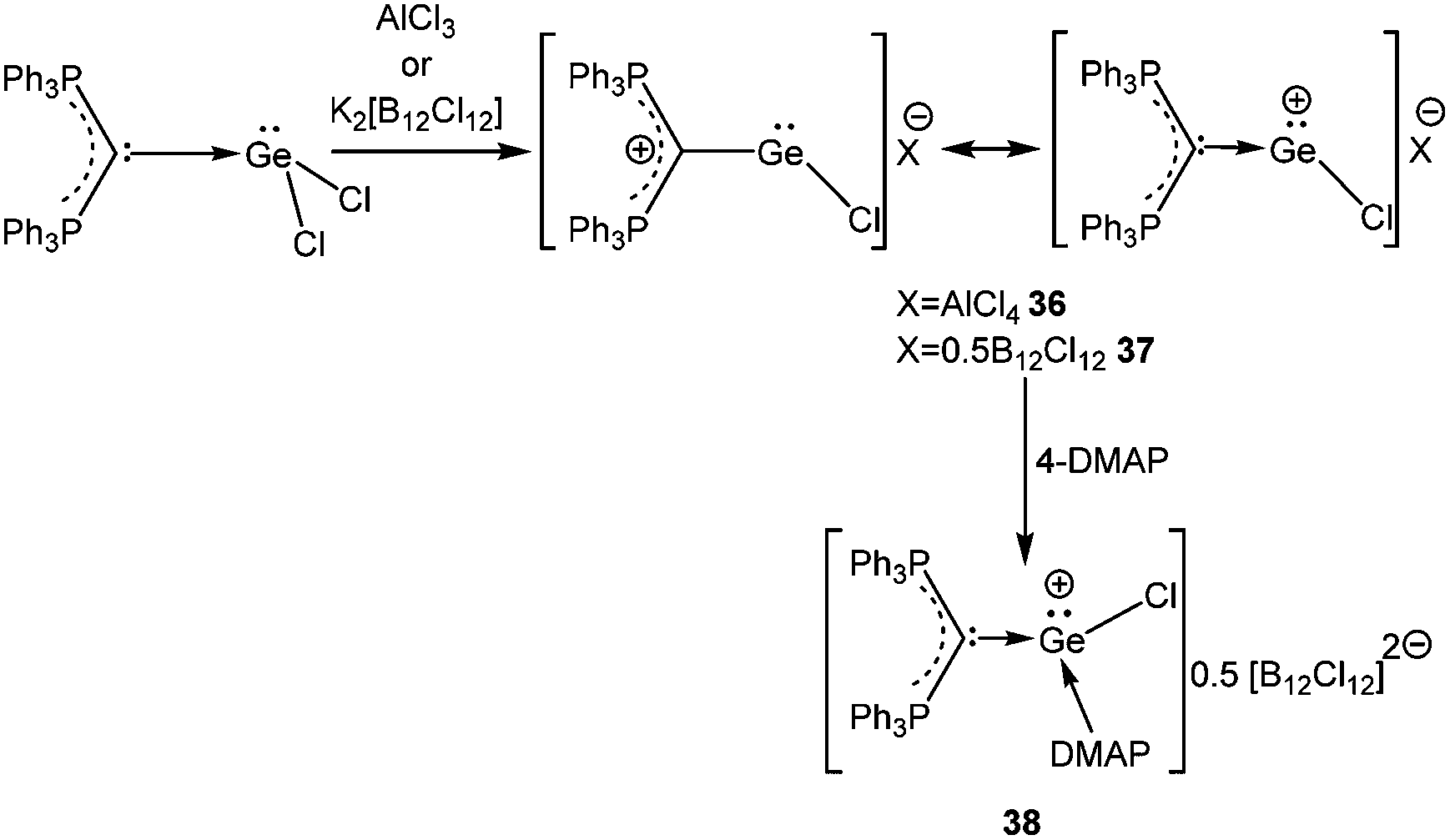
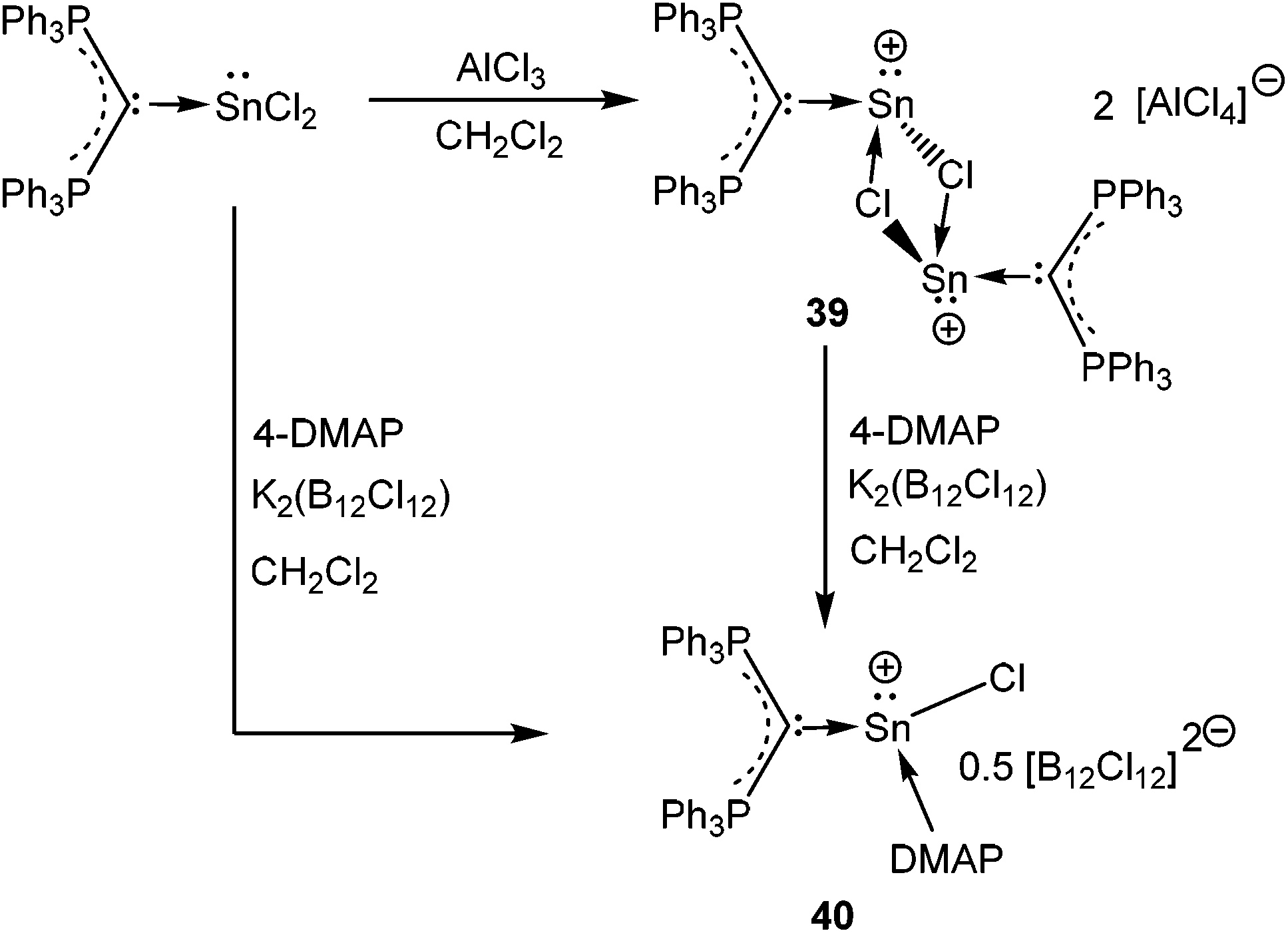
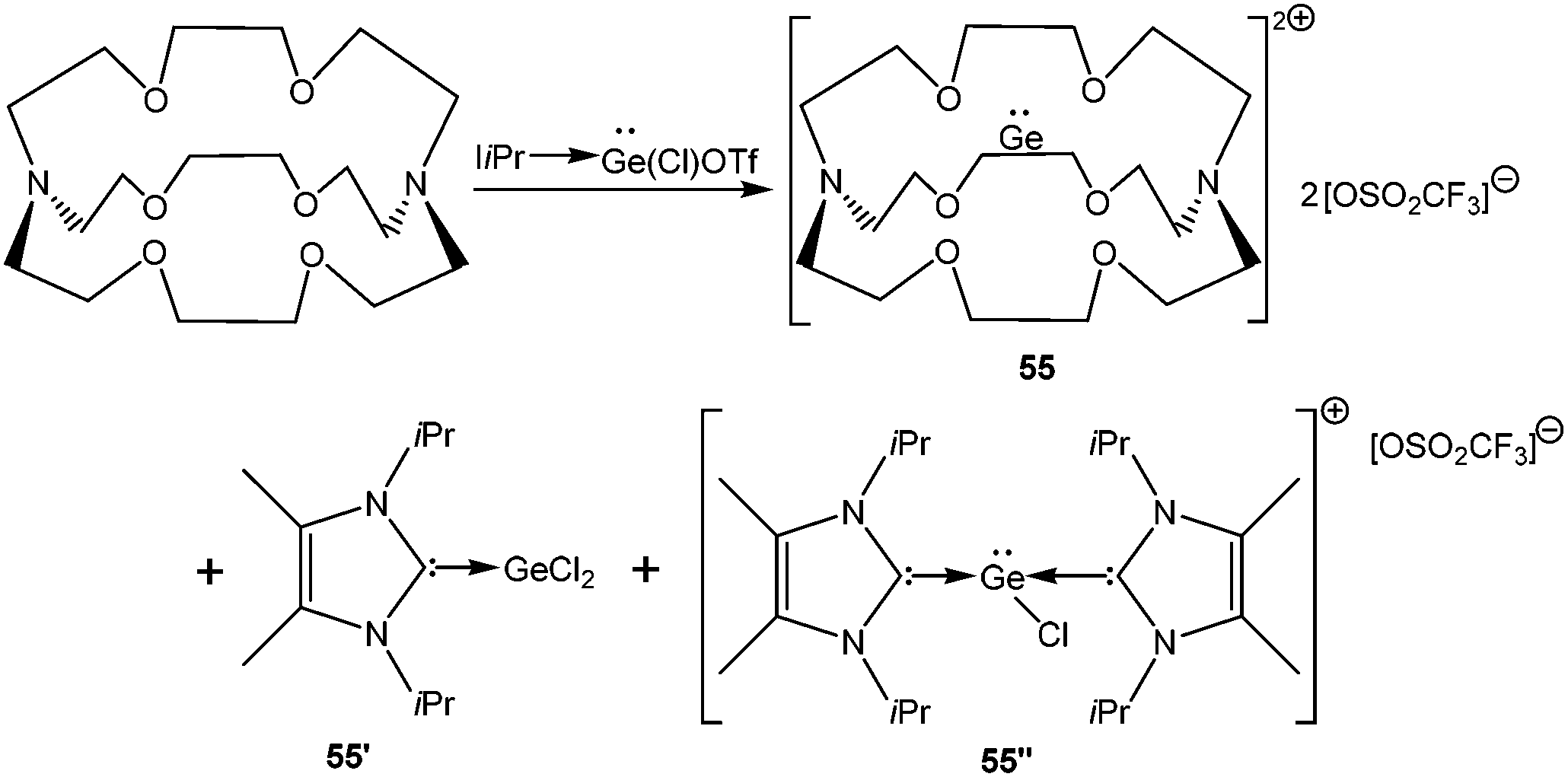
A starting point of our discussion on [D→EX]+ cations is a report from Baines’ group, who mentioned the generation of [D→Ge(Cl)←D]+ [D = IiPr] (55′′+) (Scheme 28, vide infra) with a [CF3SO3]− counter-anion as a side product during the reaction of IiPr with [2.2.2]cryptand.35 However, the cation was not structurally characterized. Nevertheless, the result suggested that IiPr (a strong σ-donor) ligand does not provide sufficient stabilization that allows the isolation of a reactive two-coordinate [Ge–Cl]+ cation. Replacement of IiPr by IDipp also failed to render the two-coordinate [Ge–Cl]+ moiety.30b,72 Finally, very recently, utilizing the concomitant σ- and π-donor capabilities of the sterically demanding carbodiphosphorane ligand Alcarazo et al. isolated the first two coordinate [(Ph3P)2C→GeCl]+ (36+) cation (Scheme 20).30b
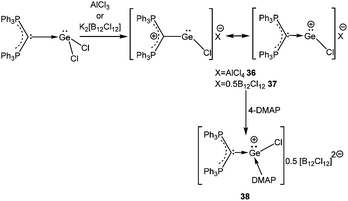 |
| | Scheme 20 Carbodiphosphorane stabilized Ge cation and its derivatives. | |
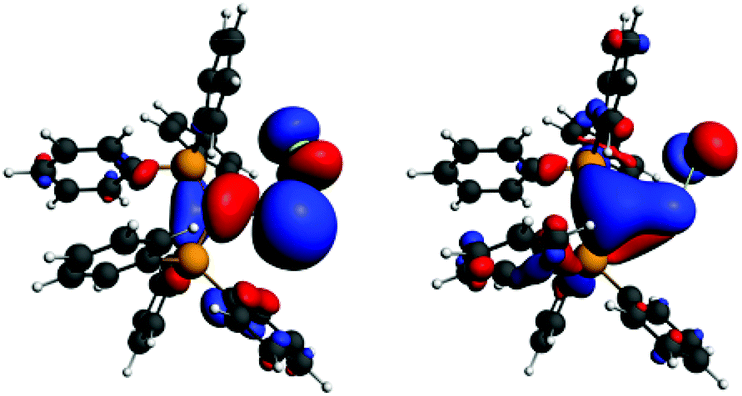
Interestingly, the P–C–Ge–Cl torsion angle in 36+ (Fig. 2) was only 8.18°, a feature that facilitated good overlap between the filled p orbital of C and the vacant orbital of Ge, leading to the shortening of the C–Ge distance (1.954(2) Å) than that in (Ph3P)2C·GeCl2 (2.063(2) Å). The WBI of the C–Ge bond in 36 (0.84) was considerably higher than that in (Ph3P)2C·GeCl2 (0.54), revealing further a significant strong π-donation from the central carbon of (Ph3P)2C to the germanium atom. The HOMO of 36+ represents mainly the σ-lone-pair while the HOMO−1 corresponds to a C–Ge π-bonding (Fig. 3). The LUMO of 36+ which was associated with a π* (C–Ge) orbital became populated upon addition of 4-DMAP leading to an increase of the C–Ge bond length (2.053(3) Å) and WBI value (0.64) in the corresponding Lewis adduct, 4-DMAP·[(Ph3P)2C(GeCl)]+ (38+).
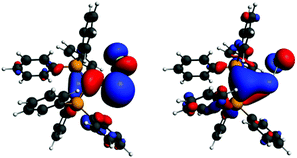 |
| | Fig. 3
Frontier orbitals of 36+ (left: HOMO, right: HOMO−1). Adapted from ref. 30b. | |
In contrast, the reaction of (Ph3P)2C·SnCl2 with an equivalent of AlCl3 did not yield the anticipated monomeric [(Ph3P)2C(SnCl)]+ cation and instead led to the formation of a chlorine bridged dimeric [(Ph3P)2C(SnCl)]22+ (39+) species (Scheme 21).30b This result indicates that the π(C→Sn) bond in 39+ is very weak, presumably due to a less-efficient overlap between the C(2p) and Sn(5p) orbitals. The dimeric structure was stabilized by the formation of chloride bridges between the Sn atoms to provide the extra electron density to the Sn center. The reaction of 4-DMAP with 39 resulted in a monomeric 4-DMAP·[(Ph3P)2C(SnCl)]+ adduct (40+).
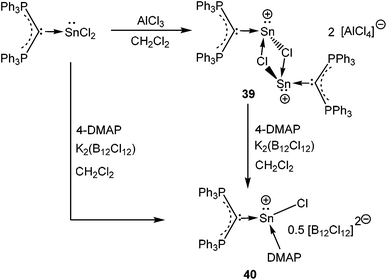 |
| | Scheme 21 Carbodiphosphorane stabilized Sn cation and its derivatives. | |
 |
| | Scheme 22 Lewis base mediated ionization of GeBr2 and GeCl2. | |
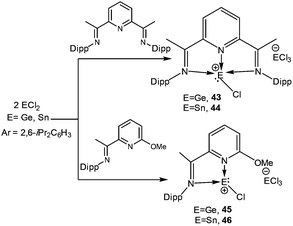 |
| | Scheme 23 Lewis base induced ionization of germanium and tin halides. | |
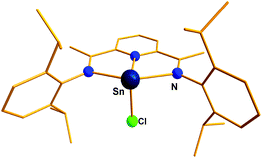 |
| | Fig. 4 Molecular structure of 44+. | |
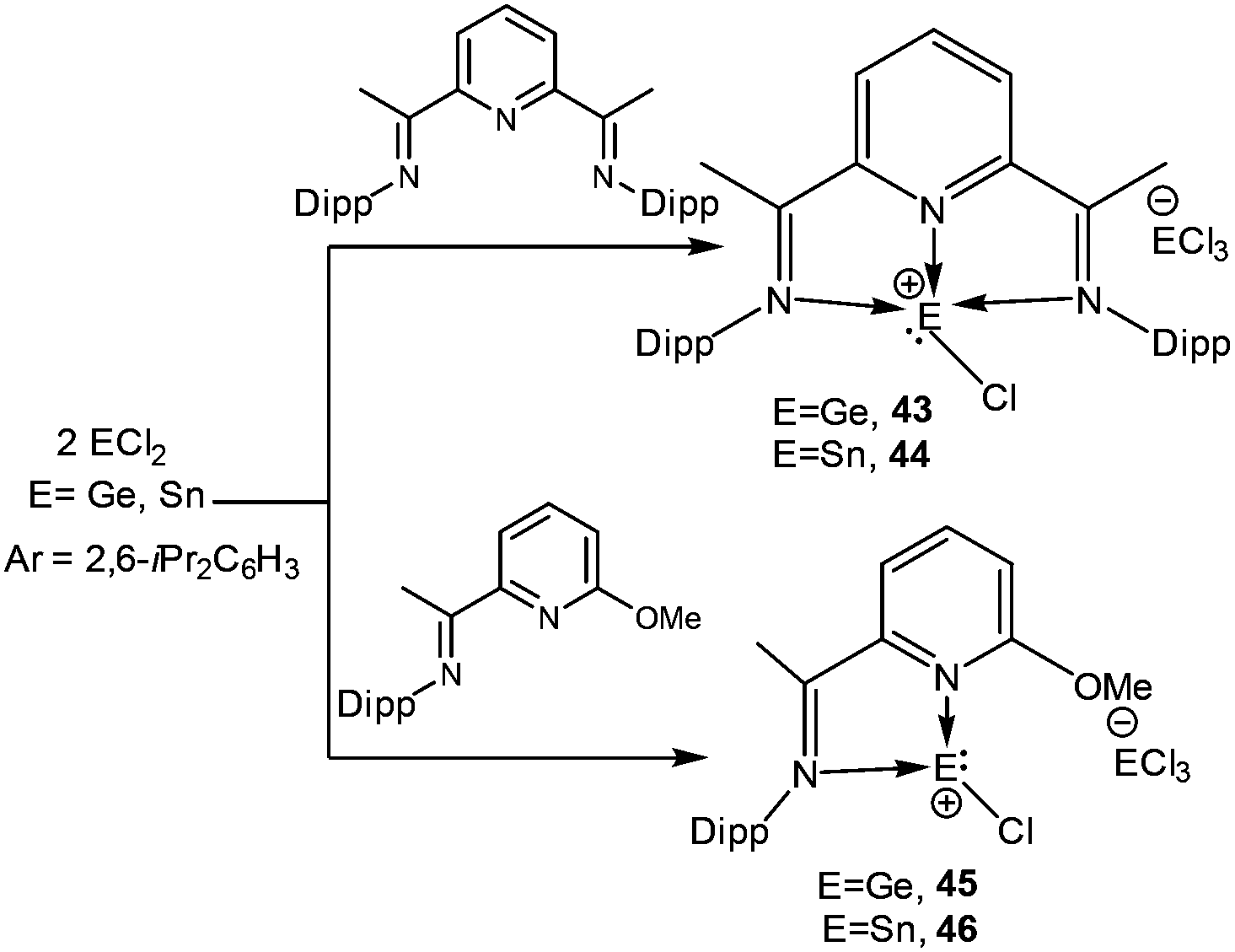
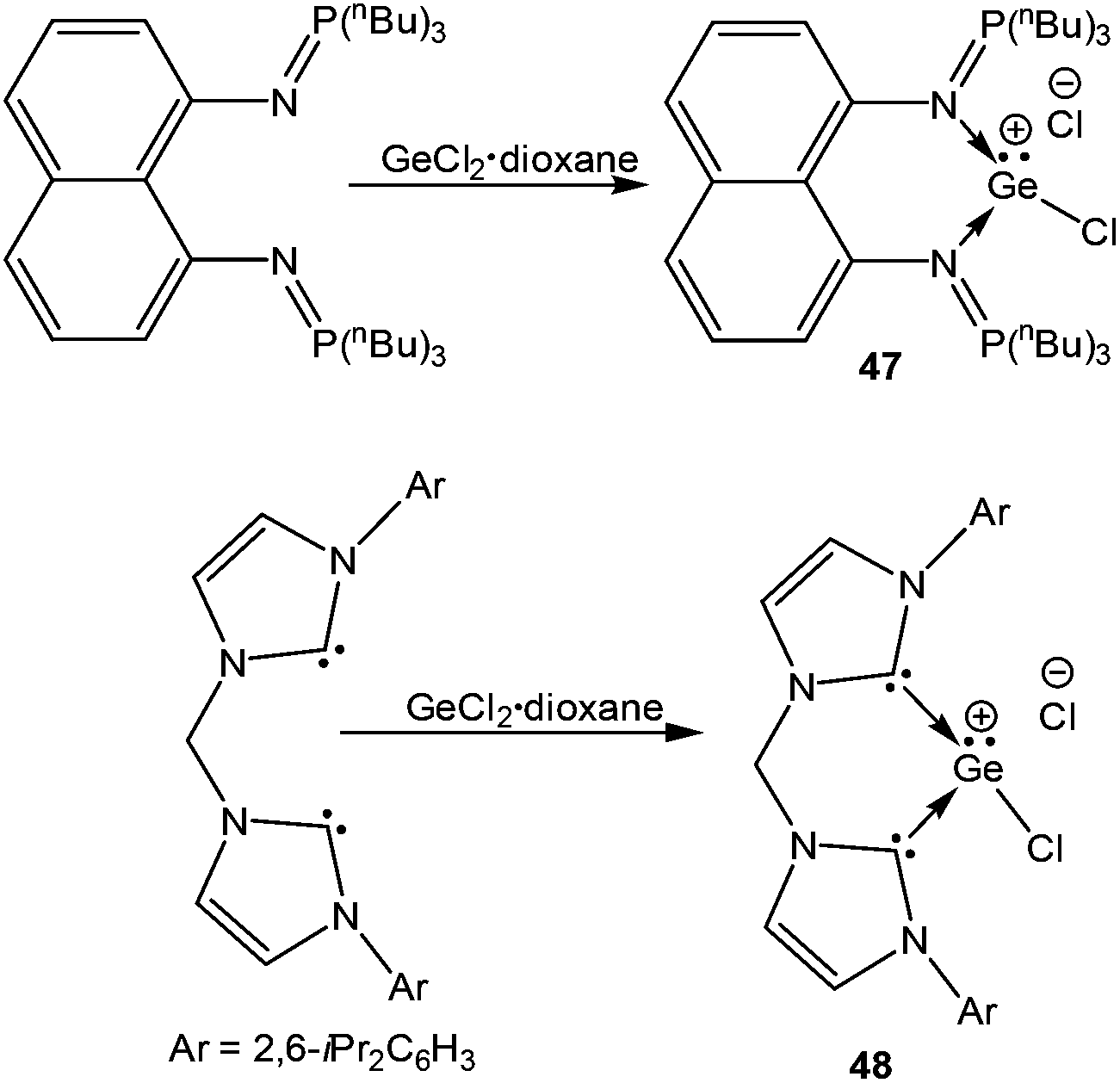
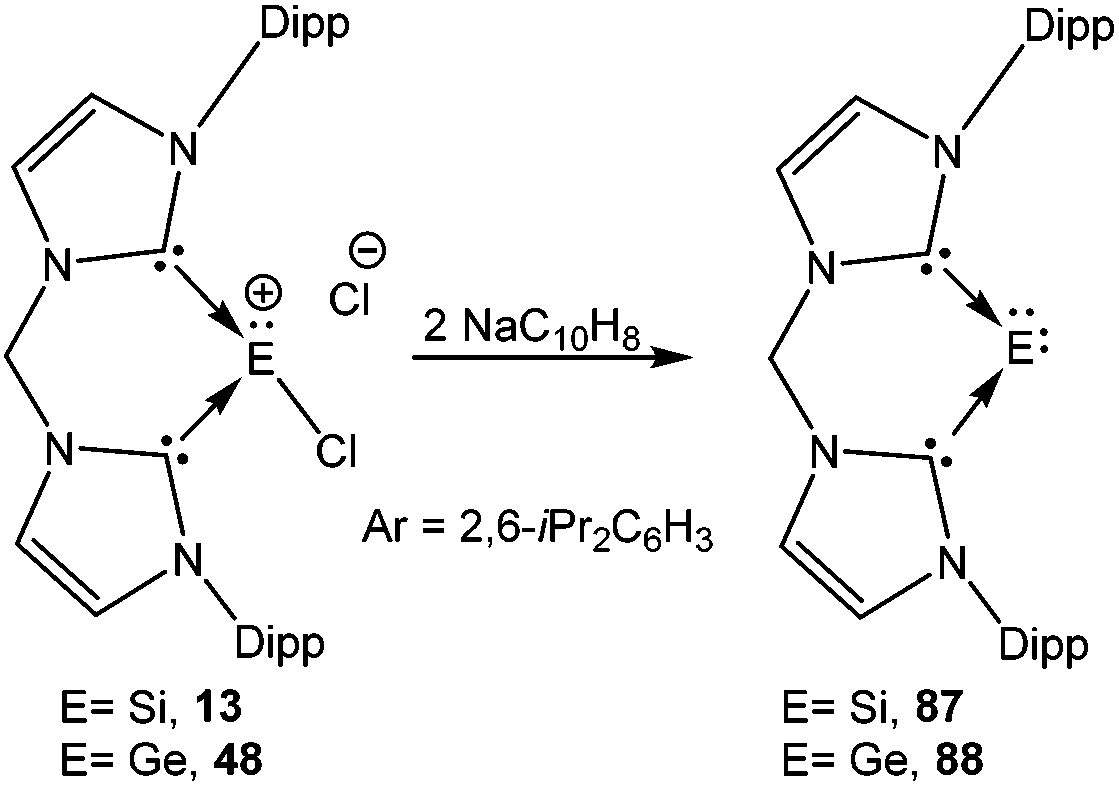
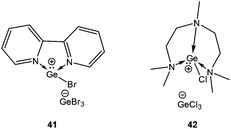
The germanium variants of 12 and 13, were reported by Driess and coworkers.77 Apparently, the reaction of GeCl2 with iminophosphorane and bis-NHC ligands led to the cleavage of one of the Ge–Cl bonds and thus afforded 4777a and 48 (Scheme 24).77b The Ge centers were well separated from the chloride anions (47: 6.83 Å and 48: 6.53 Å). The geometric features and the frontier molecular orbitals of 47+ and 48+ were almost identical to those mentioned earlier for their corresponding silicon analogs. The dative nature of the N→Ge and C→Ge bonds was further apparent from their respective WBI values (47+: 0.436 and 0.430; 48+: 0.608 and 0.611).
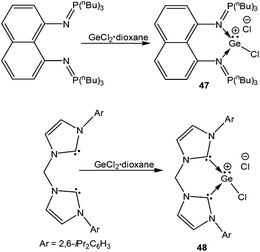 |
| | Scheme 24 Synthesis of the [Ge–Cl]+ cations 47 and 48. | |
4.1.4. Dications of germanium(II) and tin(II).
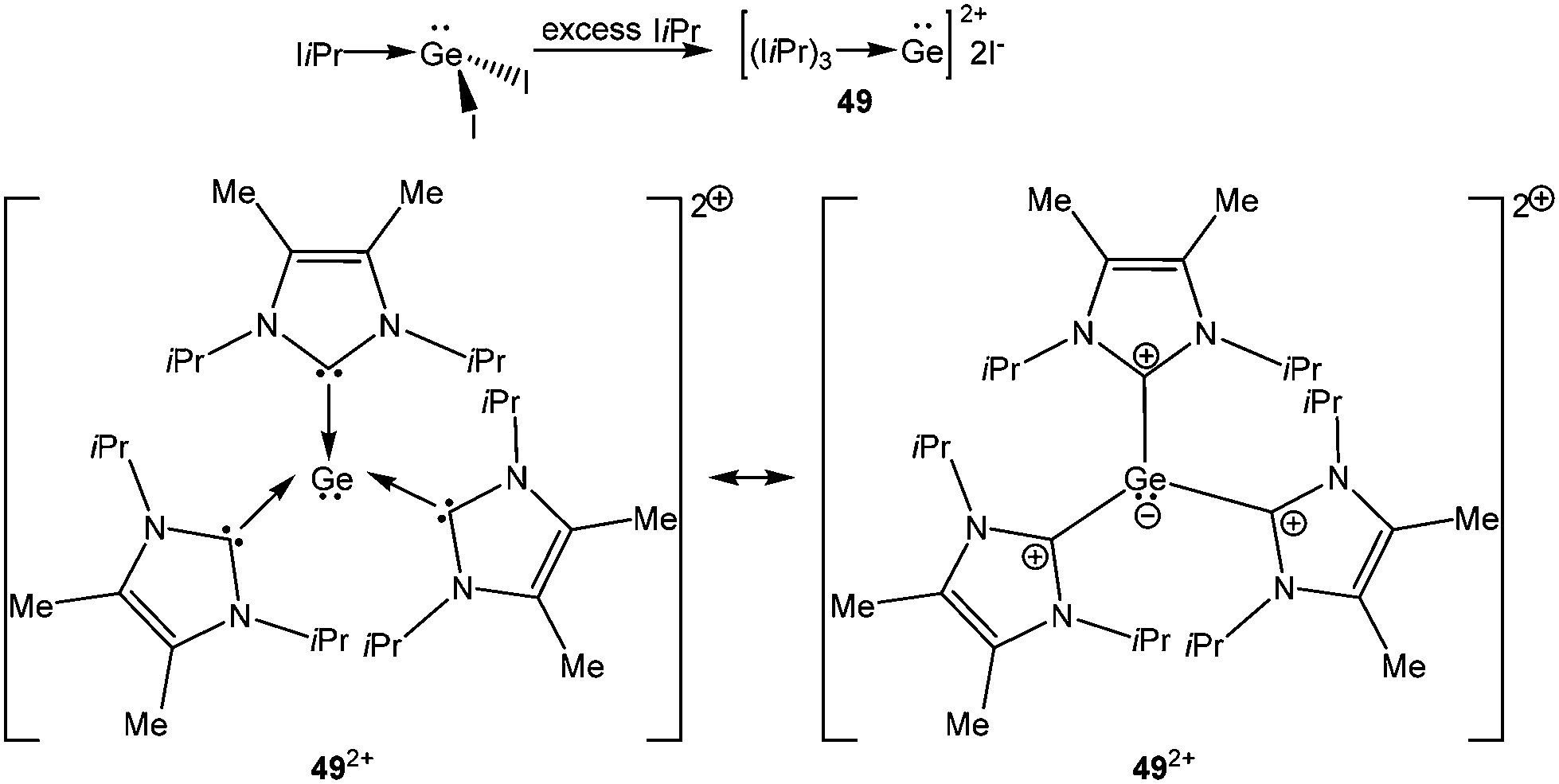
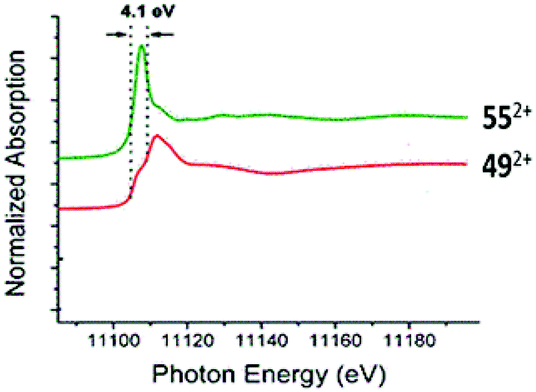
Significant advances have been made in the isolation of Ge(II) and Sn(II) dications which were earlier detected only by spectroscopic studies and considered as reactive intermediates. In particular, Baines and coworkers described the elegant use of different neutral ligands to accomplish the synthesis of Ge(II) dications.27 Utilizing the unique donor aptitudes of sterically demanding IiPr ligands, they prepared the first Ge(II) dication, 492+ (Scheme 25). All three C–Ge bonds are identical and 2.070(6) Å in length. The iodide counter-anion was located far away from the Ge center (closest Ge–I distance: 5.96 Å), but weakly bound to the CH3 protons with the I⋯H interaction of 3.11 Å. Like we showed in the case of donor stabilized Si(II) cations, two canonical forms of 492+ can be envisaged. No charge calculation (NPA: +0.64; Mulliken: +0.05, atomic polar tensor: +1.02) was either close to +2 or −1, so it is safe to say that the real bonding feature lies in between 492+ and 49′2+ and the positive charge on Ge was delocalized over three IiPr groups. Afterwards a Ge κ-edge XANES study on 492+ also revealed that the Ge center receives significant electron donation from NHC in 492+.78 The white line of 492+ (red line) appears as a shoulder and is broad. The decrease in intensity for the white line of 492+ is likely a result of electron donation from NHC to the Ge2+ center (Fig. 5).
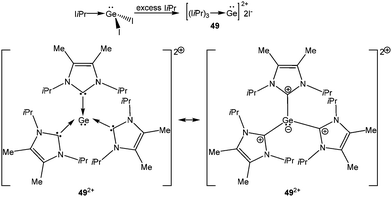 |
| | Scheme 25 Synthesis of the NHC stabilized Ge(II) dication, 492+. | |
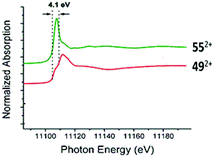 |
| | Fig. 5 Germanium κ-edge XANES of 552+ (green line) and 492+ (red line). Adapted from ref. 78. | |
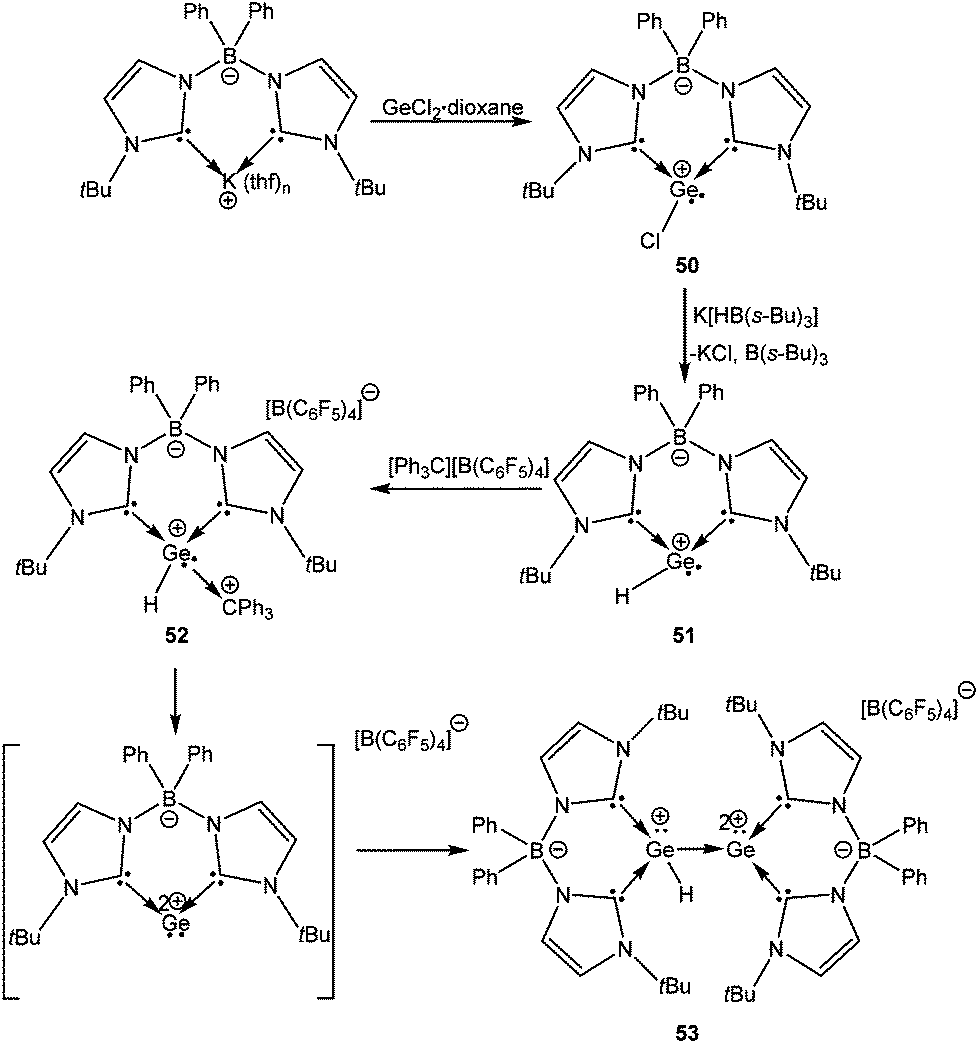
Another elegant variation of the NHCs is the linking of two NHCs using a borate spacer, which gives the ligand a certain degree of flexibility. This ligand has been recently used for the isolation of a compound featuring a Ge–Ge bond where one Ge atom is monocationic and another Ge atom is dicationic.79 The preparation of 53 is unique (the reaction sequence is shown in Scheme 26) and represents a tour de force in organometallic synthesis. A key step in this sequence was the generation of the zwitter-ionic Ge(II) cation (50) from a salt metathesis route using potassium bis(NHC)-borate and GeCl2·dioxane. The isolated Ge(II) cation was further derivatized to yield the zwitterionic Ge(II) cation with a [H–Ge:]+ moiety, 51, which upon reaction with [Ph3C]+[B(C6F5)4]− generated 52. Spontaneous elimination of the Ph3CH from the latter apparently led to a highly reactive Ge(II) dication, which underwent donor–acceptor stabilization with unreacted 51 to form an unprecedented compound with a [HGe:+→Ge:2+] motif (53). A deliberate synthetic route that involved the 1:1 reaction of 51 and 52 also led to compound 53 with concomitant elimination of Ph3CH.
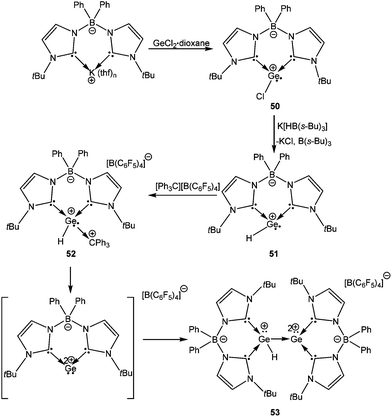 |
| | Scheme 26 Synthetic steps to access 53. | |
Müller and coworkers obtained a borate salt of tris toluene Sn(II) dication [Sn(C7H8)3]2+ (542+) from the serendipitous decomposition of a stannylium cation during its recrystallization in toluene.80 It was reported that the reaction of Tipp2Sn (Tipp = 2,4,6-iPr3-C6H2) with the silylarenium ion [iPr3Si(tol)]+[B(C6F5)4]− presumably generated the corresponding stannylium ion (Tipp2Sn(SiiPr3))+ initially, which in due course decomposed to furnish [Sn(C7H8)3][B(C6F5)4]2 (Scheme 27). No deliberate synthetic route was reported for 542+. A single crystal X-ray study of 542+ revealed that two toluene molecules were significantly closer to the Sn atom than the remaining third toluene molecule leading to an unsymmetrical coordination around the Sn atom. The large isomeric shift (4.14(1) mm s−1) confirmed the +2 oxidation state of the Sn center and indicated that the Sn(II) valence electrons reside in orbitals that are almost exclusively of 5s character. Computations showed significant charge transfer from the arene ring to the empty p orbitals of the Sn atom leading to accumulation of high positive charge (+1.28) on the Sn atom.
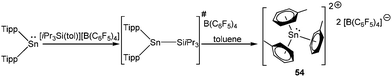 |
| | Scheme 27 Synthesis of Sn(II) dication, 542+ from stannylene. | |
4.1.4. Cationic polyether complexes of germanium(II) and tin(II).
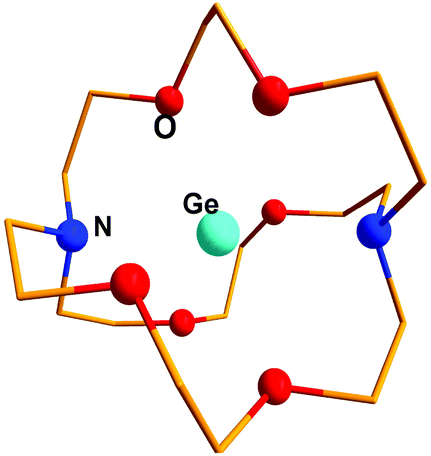
Crown ethers, cryptands, glymes etc. have been renowned for their remarkable metal complexation properties with s- and d-block elements.81 Baines and coworkers demonstrated successful implementation of the polyether ligation approach to stabilize a “naked” germanium(II) dication (552+) using an electron rich [2.2.2]-cryptand (Scheme 28).35,82 The Ge(II) center is encapsulated within the cryptand – presumably stabilized by numerous weak donor–acceptor interactions due to the presence of six oxygen and two nitrogen atoms in the cavity (Fig. 6). The anion OSO2CF3 exhibits no bonding interaction with Ge2+. The Ge–N and Ge–O distances (2.524(3) and 2.485(2) Å) indicate very weak interactions between Ge2+ with O- and N-donors, which is further reflected in their WBI values (Ge–N: 0.11 and Ge–O: 0.10). NPA calculation revealed the accumulation of +1.38 residual charge on the Ge center despite the donor–acceptor interaction in the cryptand. In 55, Ge2+ was protected not just from anions, but also from Lewis bases such as solvent molecules. The success of the method can be attributed to the cryptand's ability to encapsulate Ge2+ in three dimensions. A subsequent Ge κ-edge XANES study on 552+ revealed that the Ge center in 552+ is highly ionic (sharp intense white line) (Fig. 5).78 and can be best described as naked Ge2+ encapsulated within the cryptand cage.
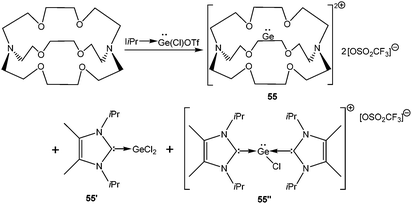 |
| | Scheme 28 [2.2.2]Cryptand stabilized Ge(II) dication, 552+. | |
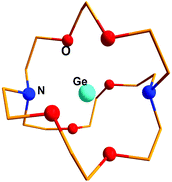 |
| | Fig. 6 Molecular structure of 552+. | |
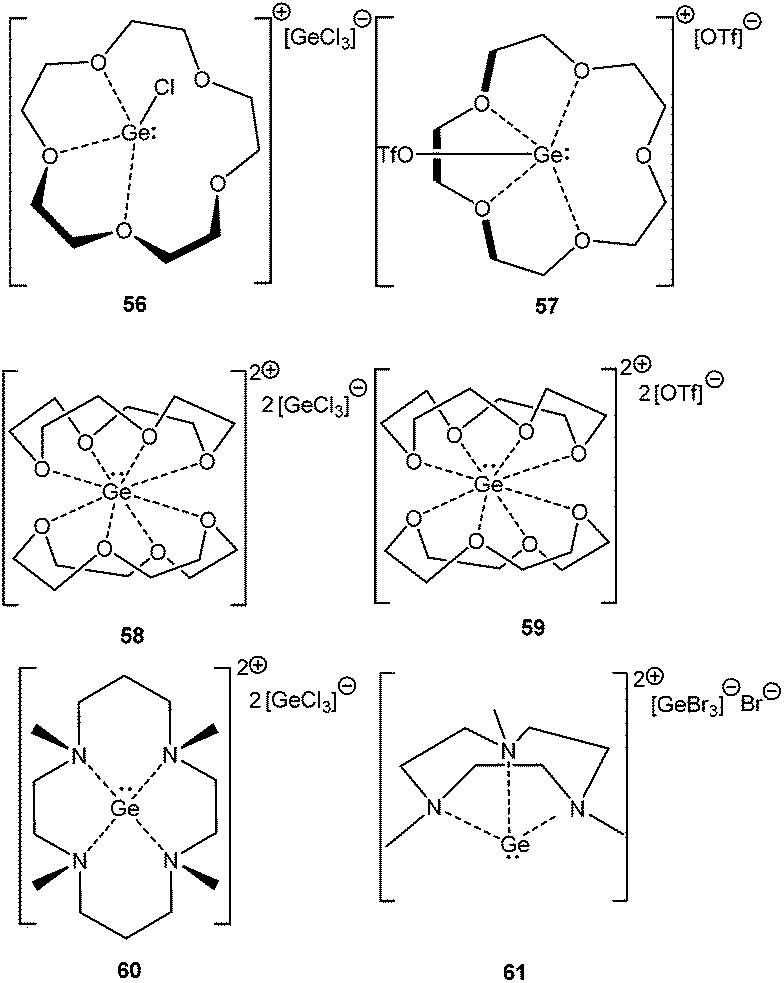
In a subsequent systematic study it was demonstrated that reacting differently sized crown ethers, such as [12]-crown-4, [15]-crown-5, and [18]-crown-6 with GeCl2·dioxane led to a range of Ge(II) mono- and dications (56–61) (Scheme 29).83 The structural properties of these cations were governed by the size of the crown ether employed. Ge2+ fits into the cavity of [15]crown-5 and [18]crown-6; while it formed a sandwich complex with two [12]crown-4 ligands. The structural properties can also be influenced by the substituent on the Ge atom. [15]-Crown-5 adopted a folded structure with the [GeCl]+ fragment and a planar conformation with [GeOTf]+. Related compounds with tacn (1,4,7-triazacyclononane) and cyclam (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) ligands have also been reported.84 Like in the cases of crown ethers, the size and the denticity of the azamacrocycles strongly influence the coordination geometry of the Ge2+ dications and the nature of the counter anion.
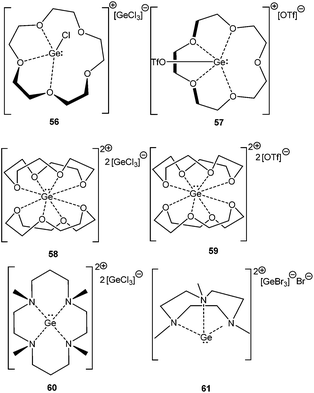 |
| | Scheme 29 Crown ether stabilized Ge cations and dications, 55–61. | |
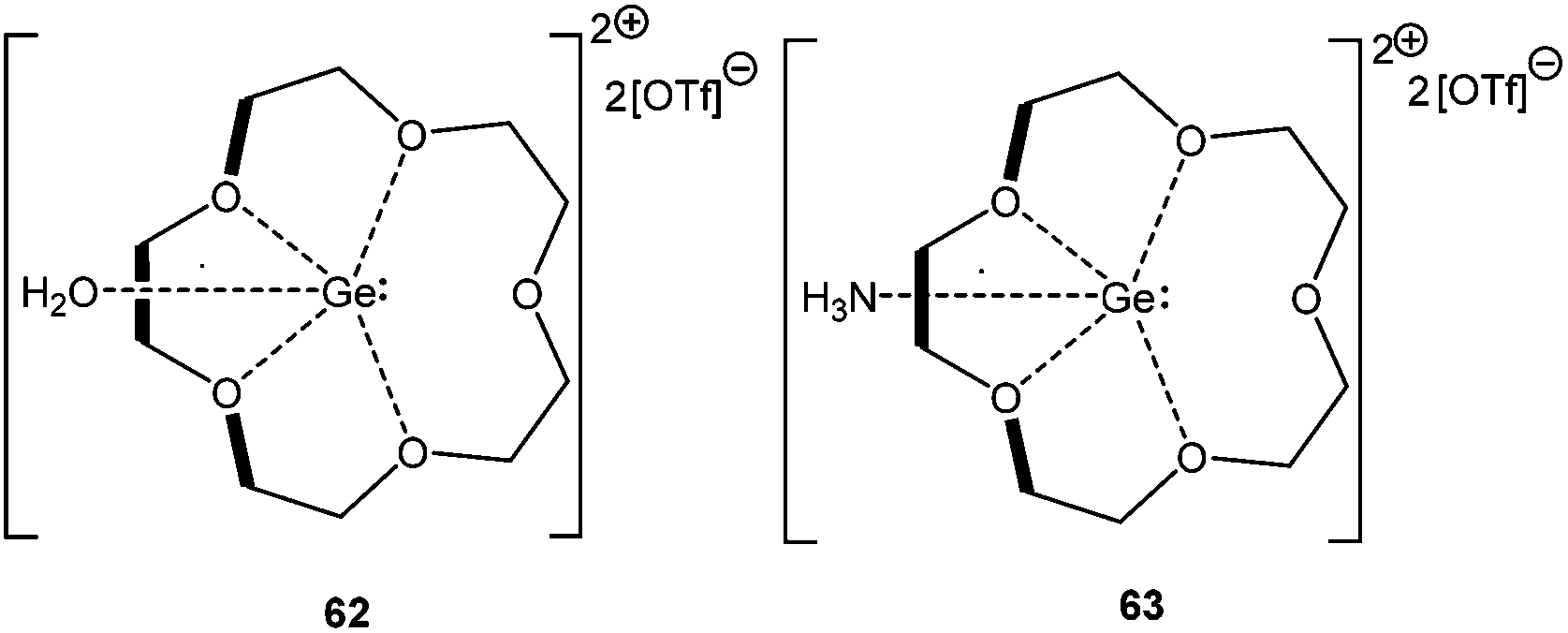
The reactivity of the Ge(II) crown ether cation towards H2O and NH3 has been recently demonstrated. Instead of oxidative addition, the formation of stable adducts like [([15]-crown-5) Ge·OH2][OTf]2 (62) and [([15]-crown-5)Ge·NH3][OTf]2 (63) was observed (Scheme 30).85 Subsequent computational studies showed that the purported oxidative addition products of water and ammonia are less stable than the adducts by 13 kJ mol−1 and 57 kJ mol−1, respectively, which can be attributed to the preferential formation of the O–H bond rather than the Ge–H bond. The hydrogen abstraction from 62 with a suitable base like pyridine, ammonia, NHC etc. was found to be feasible, as the formation of the corresponding conjugate acids was detected in the 1H NMR spectra. Such dehydrogenation would lead to multiply bound Ge–X [X = O or N] bonds, the formation of which was computationally supported with the increase of the electron density of the putative dehydrogenated variants at the bond critical points.
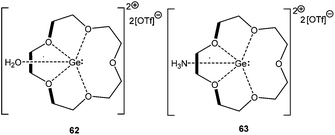 |
| | Scheme 30 Water and ammonia complexes of the Ge dication, 62–63. | |
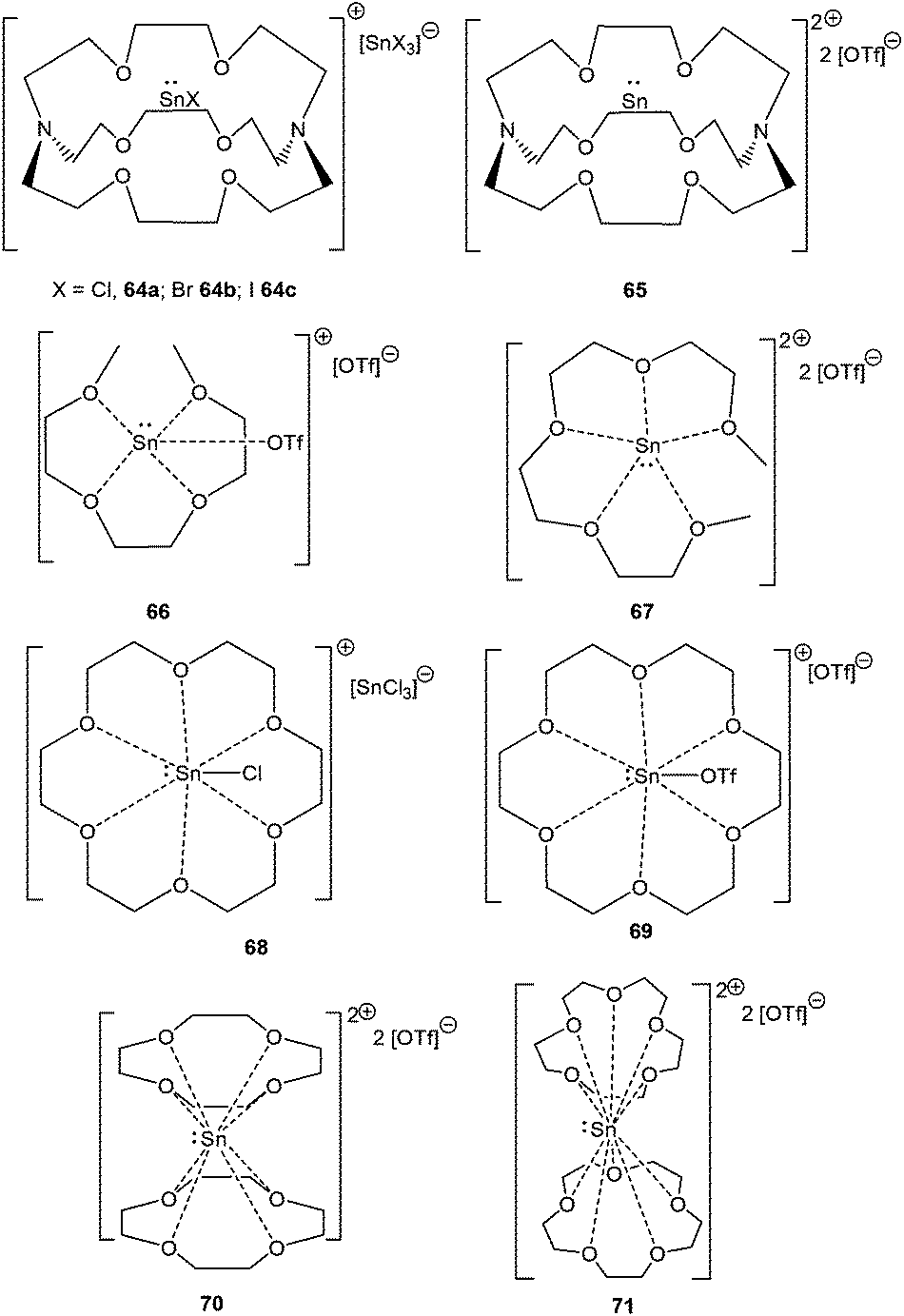
The groups of Baines and Macdonald recently published the cryptand and crown ether complexes of tin (Scheme 31).86 Unlike germanium, tin forms both mono-[cryptand[2.2.2]SnX][SnX3] (X = Cl; 64a, X = Br; 64b, X = I; 64c) and dicationic compounds [cryptand[2.2.2]Sn][OTf]2 (65) with [2.2.2]-cryptand depending on the Sn(II) precursor used in the reaction. The more flexible glyme ligands are also found to be suitable in isolating Sn(II) cations, as manifested in the isolation of [triglymeSn(OTf)]+ (66+) and [tetraglymeSn]2+ (672+) with triflate as the counter-anion. [18]-Crown-6 tends to form 1:1 adducts with Sn(II) precursors leading to 68 and 69, whereas the smaller crown ethers, [15]-crown-5 and [12]-crown-4, were not able to accommodate the tin atom within the crown ether cavity, and thereby resulted in 2:1 sandwich complexes with tin (70 and 71).
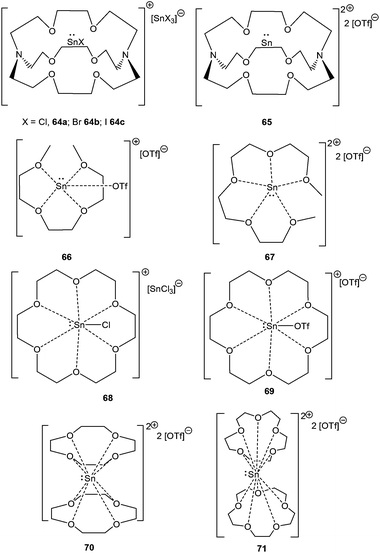 |
| | Scheme 31 Sn(II) poly ether cations, 64–71. | |
It was observed that crown ethers and glymes form Sn(II) dications with triflate precursors whereas [Sn–X]+ derivatives with the halide precursors. The difference in the reaction pattern can be attributed to the greater s-character of the Sn valence electrons in triflates than in halides. 119Sn solid state NMR data revealed that 119Sn nuclei are highly shielded in the triflate salt whereas considerably deshielded in the chloride salt, and thus support the formation of the resulting complexes. The +2 oxidation state of Sn(II) atoms in these polyether ligated cations was further reflected in Mössbauer spectroscopy, which revealed the pure 5s lone pair as the Sn valence orbital. The ligation of [15]crown-5 and [12]crown-4 to the Sn atom hardly perturbed its electronic configuration owing to the almost symmetrical Sn bonding environment in 70 and 71. In contrast, the coordination geometry around the Sn(II) centers in the [18]crown-6, triglyme, and tetraglyme complexes of Sn(II) is less symmetrical, leading to more perturbation in the valence electron as indicated by the increase of the quadrupolar splitting of the resonance in the respective Mössbauer spectrum. The effect of the poly-ether ligand and the substituent on Sn was also illustrated through cyclic voltammetry, where complexes in which the valence electrons of tin have more s-character need more energy to become oxidized. Besides, it also depends on the steric properties of the ligands around the tin atom as cyclic voltammetry data of 70 and 71 show no oxidation current.86a
4.1.5. Transition metal supported cations of germanium(II) and tin(II).
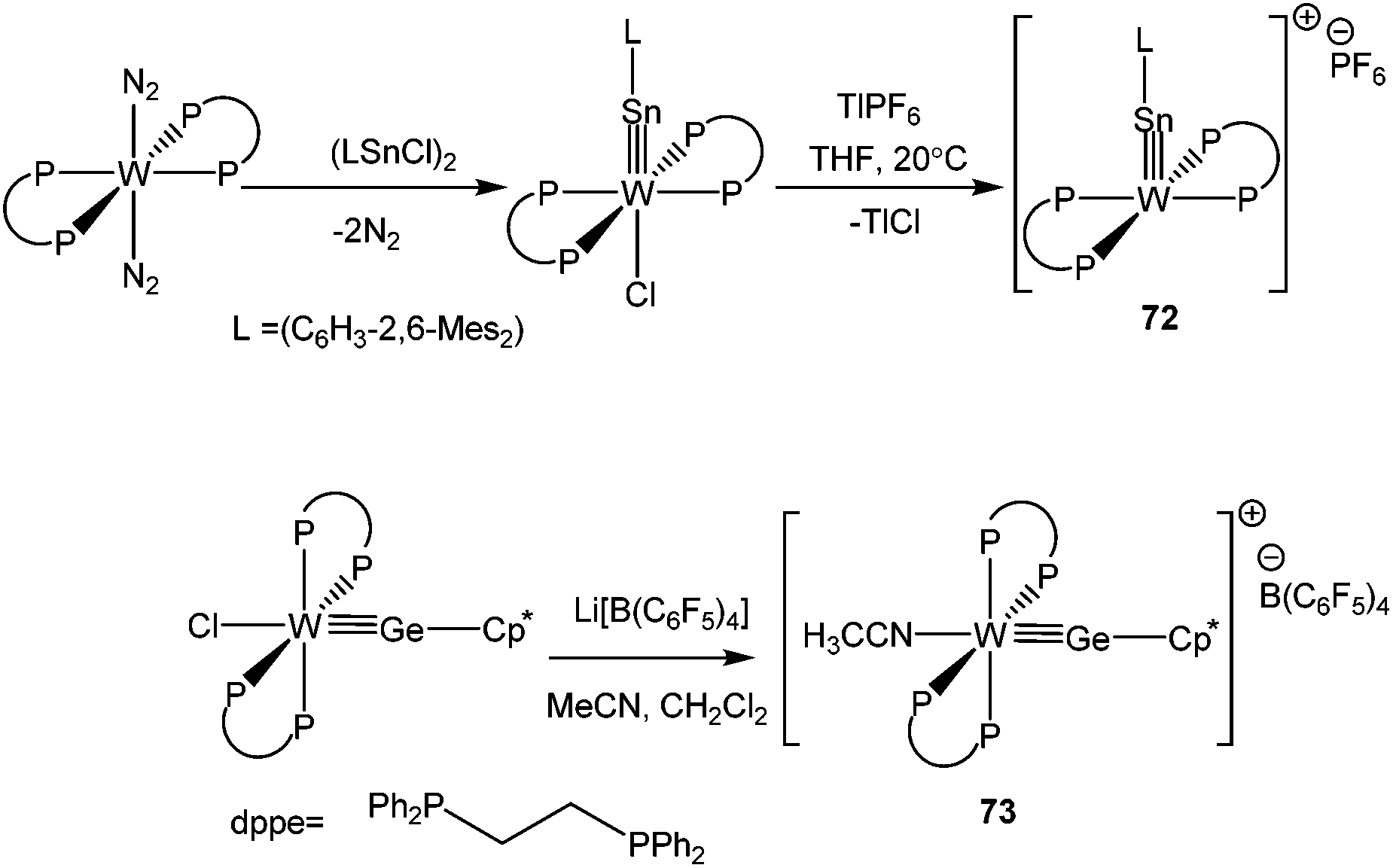
Now, we turn our discussion to another synthetic approach, where instead of a bulky organic ligand, an electron rich coordinatively unsaturated transition metal fragment was employed to stabilize Ge(II) and Sn(II) cations. Traditionally the construction of tetrel(II) cationic complexes follows two steps: (i) the oxidative addition/the adduct formation of the E–X bond to the transition metal and (ii) abstraction of the halides from the resulting transition metal–germanium/tin complexes. The idea of using transition metal fragments to stabilize p-block cations was put into practice by Filippou et al., who reported the isolation of trans-[(dppe)2WSn-C6H3-2,6-Mes2]+ (72) (dppe = 1,2-bis(diphenylphosphino)ethane).87 A sterically demanding and electron donating substituent was used to stabilize the cationic fragment. Since then a range of cationic complexes of germanium and tin incorporating various transition metals have been synthesized (vide infra). Their preparation and isolation still depends on the use of stabilizing substituents. So far, mainly Cp*, Mes* (Mes* = 2,4,6-tBu3-C6H2), pincer based moieties have been applied.
The oxidative addition of the Sn–Cl bond in 2,6-Mes2-C6H3SnCl to trans-[dppe]2W(N2)2, followed by salt elimination afforded the cation, 72 (Scheme 32). Replacing the 2,6-Mes2-C6H3 substituent by a Cp* moiety, the same group isolated the germanium variant, 73.88 Both Sn and Ge atoms adopt a nearly linear geometry with WE triple bond distances of 2.4641(7) and 2.303(1) Å, respectively. Subsequent theoretical calculations suggested that the π-back donation from W to germanium/tin may indeed contribute to the stability of these cations. One may raise the question here that the formal oxidation state of Sn in 72 and 73 is not +2, so they are not “true” germylium and stannylium ylidenes. Although we do not completely disagree with this tenet, but such a classification for transition metal supported p-block cations is always questionable taking into account other resonance contributors manifesting a germylium or stannylium character. Therefore, for the sake of completion we incorporated these cations in the review.
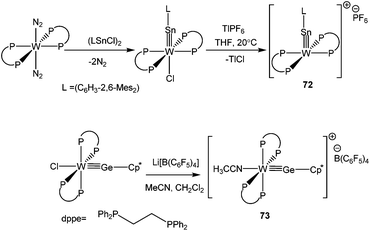 |
| | Scheme 32 Tungsten supported Sn and Ge cations. | |
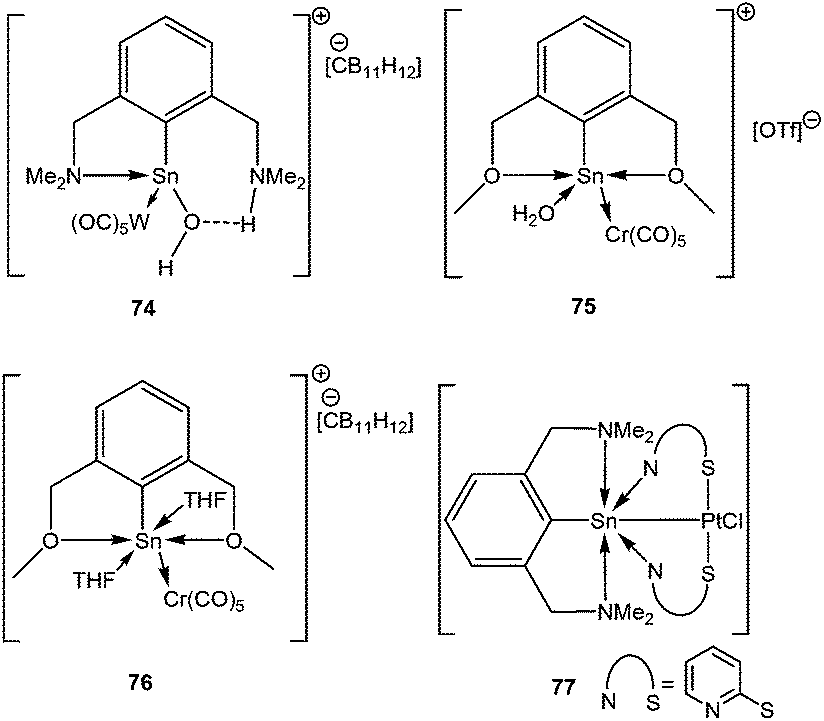
The metathetical reaction between bulky pincer based ligands with (THF)W(CO)5·SnCl2 or (THF)Cr(CO)5·SnCl2, followed by the abstraction of the halides resulted in several Sn(II) cations, such as [2,6-(Me2-NCH2)2C6H3(H2O)Sn{W(CO)5}]+ (74+), [{(2,6-MeOCH2)2C6H3}Sn(OH2){Cr(CO)5}]+ (75+) and [{(2,6-MeOCH2)2C6H3}Sn(THF)2{Cr(CO)5}]+ (76+) with various counter anions (Scheme 33).89 Another pincer based stannylene, 77 has recently been reported by Jambor et al. featuring a Sn–Pt bond.90 Although one might be tempted to assign the tin atom the +2 oxidation state, NBO analysis revealed that there was no lone pair at the tin atom. A plausible explanation is that the lone pair was shared by both Sn and Pt atoms and used up for the formation of a Sn–Pt bond, leading to some, by no means complete, stannylium ion character. Low-field 119Sn NMR (70.6 ppm) resonance indicated accumulation of negative charge on the Pt atom. DFT calculations on this complex indicated that there is significant charge donation from the [N→Sn]+ fragment to the [Pt(pyt)2Cl]− moiety. This finding was in accord with the NPA analysis which disclosed a significant fraction of positive charge at the Sn atom (+1.709) and negative charge on the Pt (−0.486). The decrease of the Sn–Pt bond length (2.466(1) Å) with respect to the other reported Sn–Pt bond lengths (vide infra) reflects an increased contribution of backbonding from the metal.
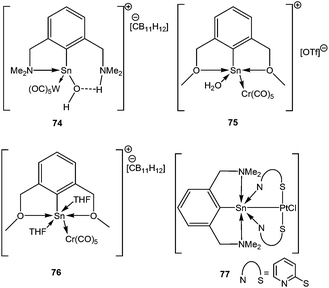 |
| | Scheme 33 Various transition metal supported Sn(II) cations. | |
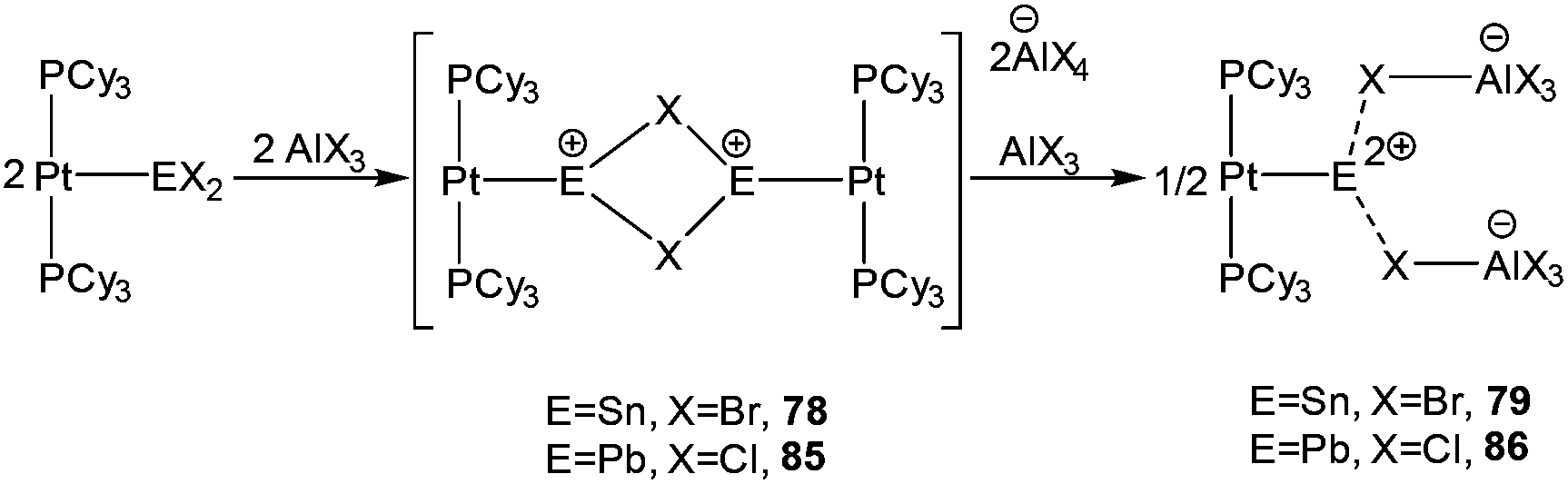
Very recently, Braunschweig et al. showed the latent σ-donor ability of the Pt(PCy3)2 to coordinate with Ge(II), Sn(II), and Pb(II) dihalides, which resulted in the formation of the respective Metal Only Lewis Pairs (MOLPs).91 The mono stannylene complex, [(Cy3P)2Pt–SnBr2] was found to be an excellent starting material for realizing the low-coordinate Sn(II) mono- and dications via halide abstraction reaction, as manifested by the formation of [(Cy3P)2Pt–SnBr]22+ (78+) and [(Cy3P)2Pt(Sn)] (792+).92 The Sn–Pt bond lengths in these cations [78+: 2.524(1) and 792+: 2.502(1) Å] are significantly shorter than the Sn–Pt bond present in their precursor complex [(Cy3P)2Pt–SnBr2] (2.605(2) Å) (Scheme 34). Although exhibiting no interaction with the solvent molecule, these cations were not totally free, being weakly bound to the counter anion through their bromide atoms. No 119Sn NMR signals were detected presumably due to the broadening of resonance to an undetectable level caused by the increased electric field gradient and large chemical shift anisotropies induced by the tin environment.
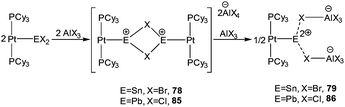 |
| | Scheme 34 Formation of Sn(II) & Pb(II) mono and pseudo dications in the coordination sphere of platinum. | |
4.2. Application of Sn(II) cations in alkene polymerization
Rhodes, Chien and Rausch prepared a [(η5-C5Me5)Sn]+ (17+) derivative as its (B(C6F5)4)− salt93 which was earlier reported by Jutzi et al. with a BF4 counter-anion (vide supra). The compound was found to be an effective co-catalyst in the Ziegler–Natta polymerization of ethylene and propylene. The co-catalyst had an important role in the initiation step as well as in the propagation step. Alkene polymerization using various Zr complexes such as Cp2ZrMe2, rac-Et(Ind)2ZrMe2, rac-Et(Ind)2ZrCl2, and Cp2ZrCl2 as catalyst precursors was carried out at variable temperatures (−20 °C to 70 °C). It was found that as the polymerization temperature was increased, the activity of the alkene polymerization was also increased. However, a direct comparison of 17+·B(C6F5)4 with Ph3C+·B(C6F5)4 as the co-catalyst for ethylene and propylene polymerization indicated that the activities were substantially higher when Ph3C+ was used as a co-catalyst. For example, the activity dropped from 1.2 × 106 to 4.2 × 105 for ethylene polymerization when Ph3C+·B(C6F5)4 was replaced with 17+·B(C6F5)4 as the co-catalyst.
5. Pb(II) cations and dications
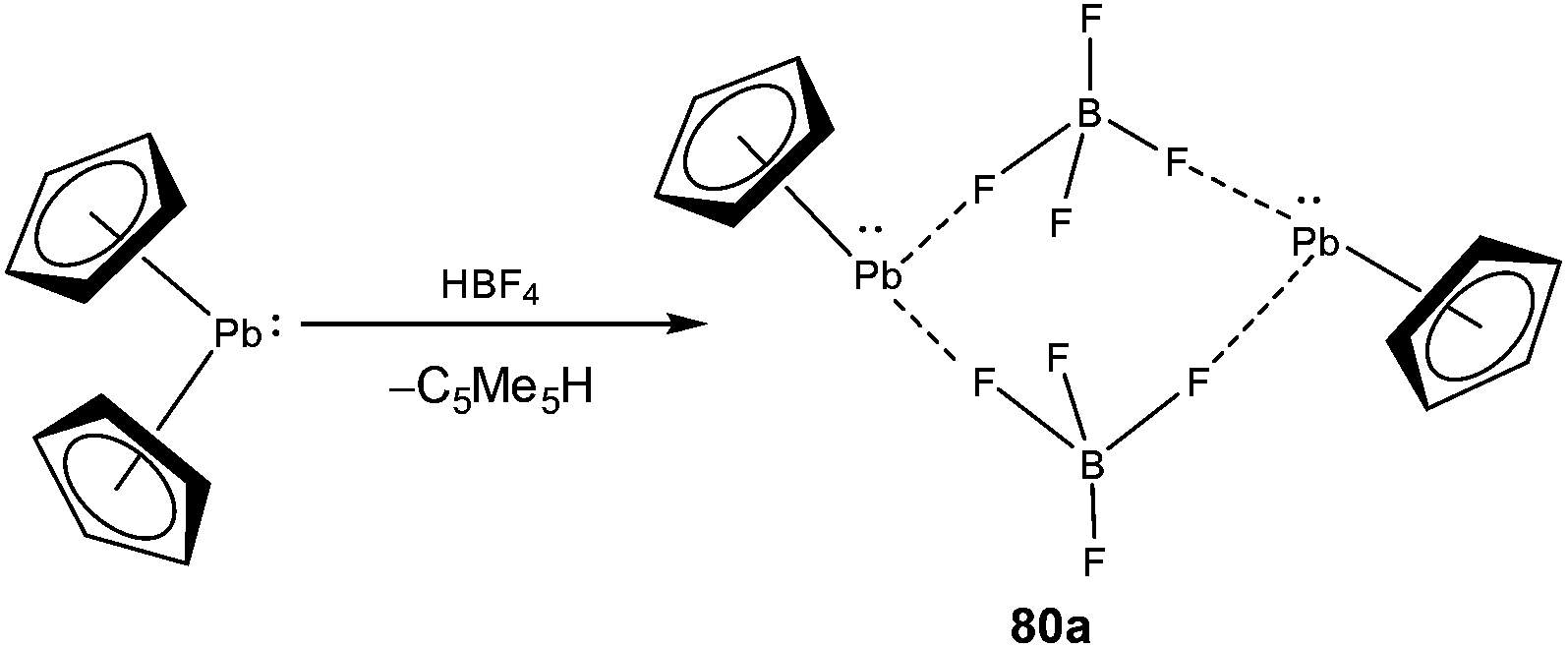
Low coordinate Pb(II) cations are by far the least ubiquitous among the classes of tetrel(II) cations. The isolation of low coordinate Pb(II) cations under ambient conditions was plagued by the highly electrophilic nature of these species. Like in all previous cases, the first isolation of a low coordinate Pb(II) cation came from the lab of Jutzi, who obtained half-sandwich complexes [Me5C5Pb][BF4] (80a) and [Me5C5Pb][OSO2CF3] (80b) as dimers from the reaction of [Me5C5]2Pb with tetrafluoroboric acid and trifluoromethanesulfonic acid, respectively (Scheme 35).21b The high field resonance of the cationic Pb center in 80a (−5041 ppm) and 80b (−4961 ppm) was observed presumably due to η5 coordination of the cyclopentadienyl ligand to the metal center. 80a was structurally characterized which revealed a pentagonal–pyramidal geometry around the Pb atom. The shortest Pb–F bond length in 80a was of 2.831(9) Å, which was relatively long and can be classified as a weak interaction, but was still sufficient for the preferred formation of the dimeric units. The Lewis acidic nature of 80a was confirmed through its reactions with 2,2′-bipyridine and 1,8-naphthyridine, which formed 1:1 adducts.21b
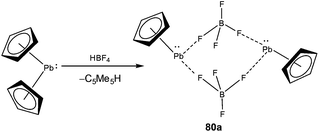 |
| | Scheme 35 Pentamethylcyclopentadienyl Pb(II) cations 80a. | |
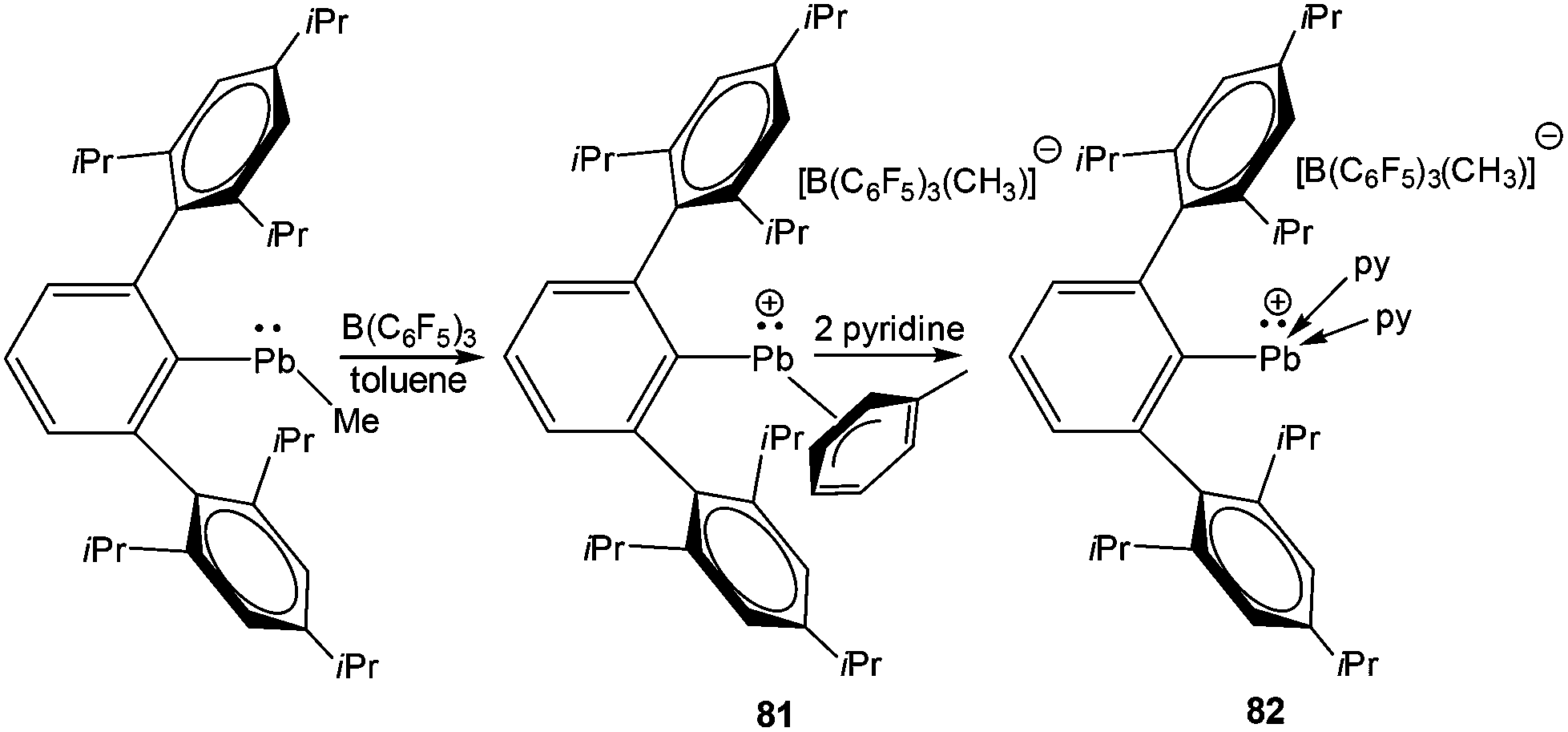
The next milestone contribution to the low coordinate Pb(II) cations was achieved by Power et al., who synthesized [Ar*Pb·η2-toluene][MeB(C6F5)3] (Ar* = 2,6-(2,4,6-iPr3C6H2)2-C6H3) (81) (Scheme 36) from the reaction of Ar*PbMe with B(C6F5)3 in toluene.94 The low coordination of the Pb center in 81 was manifested in its 207Pb NMR resonances observed at δ = 8974 ppm, shifted 13500 ppm downfield of the [(η5-C5Me5)Pb]+ salts. This very large difference is consistent with a lower effective coordination number of Pb in 81+ and weak interactions with toluene. The solid-state structure of 81 was determined by single crystal X-ray analysis (Fig. 7). The analysis revealed that there are no close interactions (3.963(6) Å) between the lead atom and the anion in 81. However, the Pb center interacted with the solvent toluene and the Pb–toluene interaction can be best described as being of η2 type. The weakly coordinating toluene molecule can be easily displaced by two pyridine molecules to yield [Ar*Pb(py)2][MeB(C6F5)3] (82). The upfield shift of the 207Pb NMR resonance (δ = 4764 ppm) of 82 with respect to that of 81 (δ = 8974 ppm) and the pyramidalization at the Pb(II) cationic center clearly reflected the decrease of the cationic character in line with the increase of the coordination number of the Pb atom.
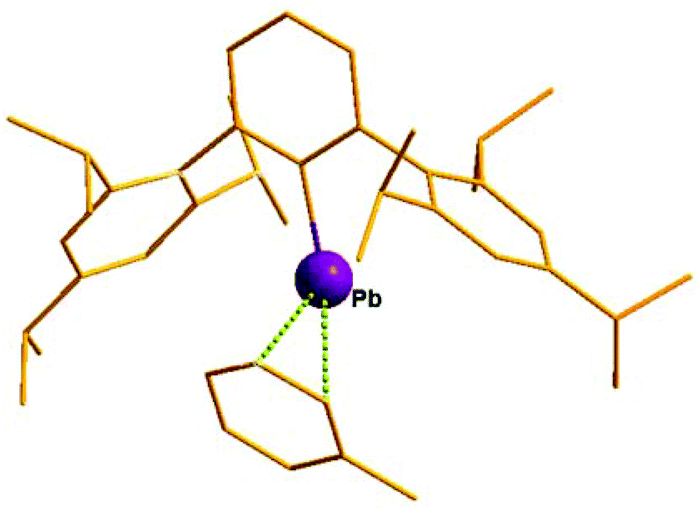
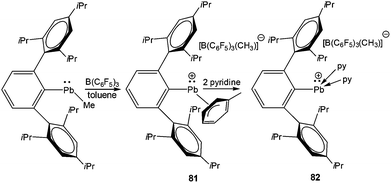 |
| | Scheme 36 Quasi-monocoordinate Pb(II) monocation, 81 and donor supported monocation, 82. | |
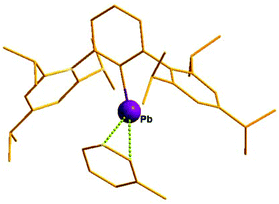 |
| | Fig. 7 Molecular structure of 81. | |
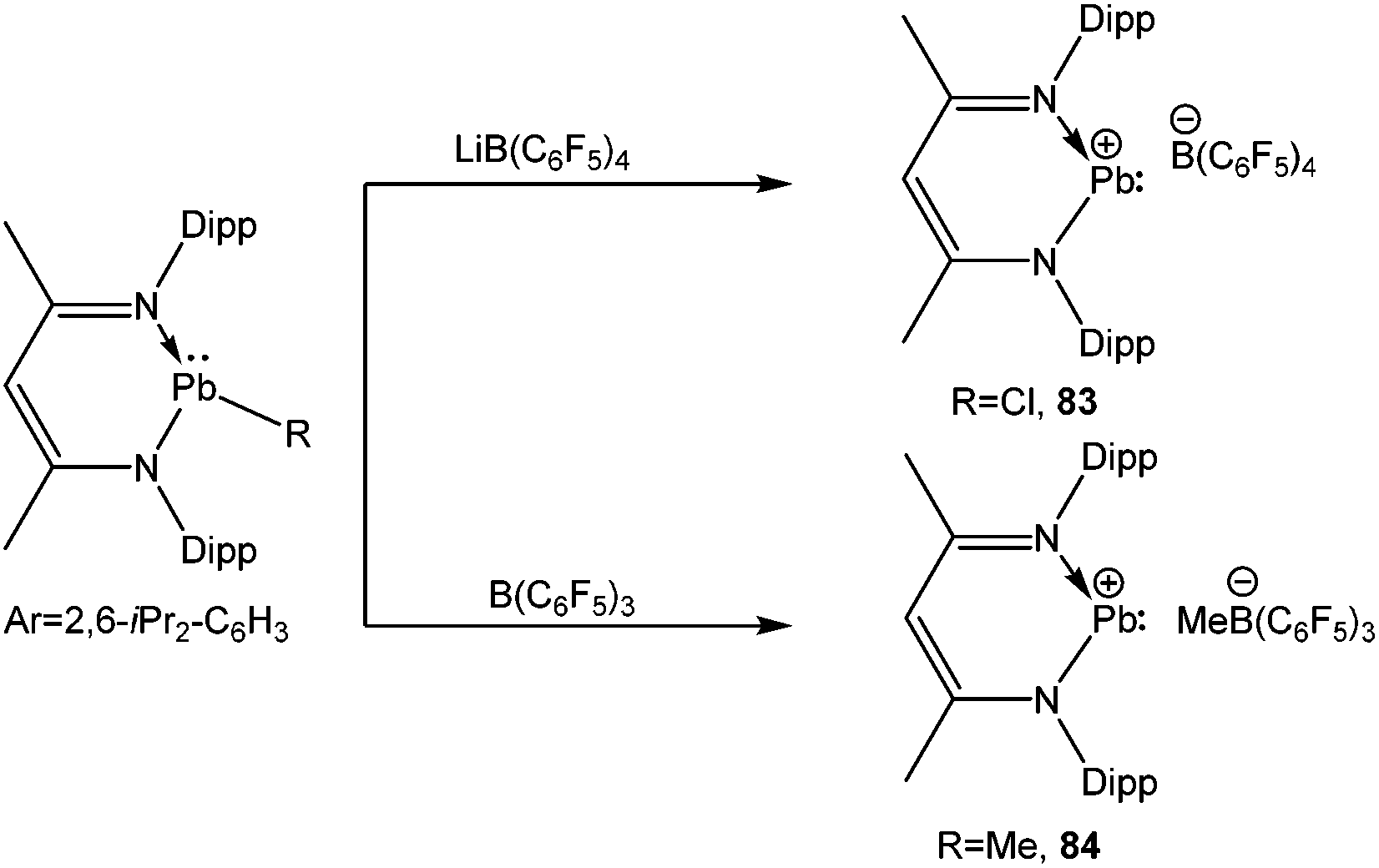
A series of tetrel(II) cations supported by the β-diketiminato ligand has recently been completed by Fulton et al. with the isolation of lead(II) cations (83+ and 84+) with the B(C6F5)4 and MeB(C6F5)3 counter-anions (Scheme 37). The Pb center of these compounds was well separated from its anion (the closest approach from Pb to the nearest F atom was 3.319 (4) Å), reminiscing the structural properties of their tin homologue. However, in the solid-state structure of 83+ a dichloromethane molecule was found with 64% occupancy to give a long-range lead–chlorine interaction (Pb–Cl(1) = 3.213(4) Å). Calculation points to the increased stability of the solvated complexes LPb+·CH2Cl2 over LPb+ by 3 kcal mol−1 (L = CH(CMeNDipp)2). No 207Pb NMR resonances for 83 and 84 were observed, which according to the authors, may be attributed to the fast relaxation of the lead nucleus.
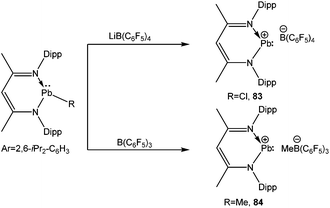 |
| | Scheme 37
β-Diketiminato ligand supported Pb(II) cations, 83 and 84. | |
The success of stabilizing Sn(II) mono- and dications in the coordination sphere of [Pt(PCy3)2] led Braunschweig and co-workers to isolate the lead variants. Adopting the same synthetic protocol that was employed for 78+ and 792+, they reported two Pb(II) mono-cations with AlCl4 (85a) and [BArCl4] [ArCl = 3,5-Cl2-C6H3] (85b) and a rare example of Pb(II) dication [{(Cy3P)2Pt(Pb)}{AlCl4}2 (86)] (Scheme 34, vide supra).92 The Pb(II) cations and dications were structurally similar to the analogous Sn(II) cations upon comparison of bonding motifs and metrical parameters. The Pb centers in 85a and 85b revealed weak bonding interactions (3.420(1) Å) to the chloride ion in the counter-anions. In the solid-state form, 86 also exhibited a weak interaction of the Pb atom with the two chloride atoms of two aluminate counter-anions. Detailed theoretical investigations were additionally carried out to further characterize the bonding situation in these complexes. It was proposed before that the Pt(0) and Pb(II) fragments in the starting material (Pt(PCy3)2→PbCl2) donate σ-electron density to each other, leading to the concept of “synergic σ-donation”,91b which was further proposed by Su et al. in the transition metal complexes featuring multiple bonds between group 10 and group 14 elements.95 A recent EDA-NOCV study by Braunschweig et al. revealed that the Pt–Pb bond in (Pt(PCy3)2→PbCl2) is a Pt→Pb dative bond whereas in 85 and 86, it is an electron sharing bond. So, the real bonding phenomena of these complexes are not very well defined and clearly require further theoretical investigation.
6. Conclusions
It is clear from the foregoing discussion that despite being a relatively new field, the chemistry of cations featuring heavier group 14 elements in low oxidation states is rich and diverse. The synthesis of [(C5Me5)E]+ (E = Si–Pb) triggered the research in this field, which has resulted in the isolation of [Si–Cl]+, Si(II), Ge(II), Sn(II) dications which were previously known only by theoretical calculation and/or in gas phase studies. A number of synthetic methods provide access to a variety of coordination environments for the cations of heavier group 14 elements. Most of the compounds were structurally characterized and understanding their bonding phenomena has become one of the most fundamental objectives in modern day main group chemistry. For instance, most of the cations were stabilized by N- or C-donor ligands and it remains a question whether the charge is localized on the central atom or delocalized over the ligand/ligands. None of the charge calculations clearly conclude about the ionicity of these cations and given their high-field shifts especially in 29Si and 119Sn NMR, one would expect that the charges do not completely reside on the central atom. Recently, the group of Stalke extensively studied the charge density of various low valent silicon compounds96 like 1,4-disilabenzene,97 hexasilabenzene98etc. Perhaps experimental charge density studies of a few of these cations would bring more clear pictures of their bonding situation.
The selection of ligands has turned out to be often decisive in the synthesis of these compounds because the stabilization requires the usage of sterically encumbering ligands with donor substituents. Therefore, the design of many more new ligands for stabilizing low oxidation species continues to be a central theme in this area and will remain so for many more years. For example, the emergence of carbodiphosphorane, which is a simultaneous σ- and π-donor and its utilization for realizing two coordinate [Ge–Cl]+ (36+) will rival NHC for the stabilization of low coordinate group 14 cations in the coming years. In a similar way, bis-carbene has already obtained a foothold in silicon (13+) and germanium (48+ and 53) chemistry and many more such fascinating compounds supported by bis-carbene ligands are expected. The use of macrocycles to protect and stabilize germanium(II) and tin(II) cations and dications rendered an alternative route to the stabilization of low valent species.
Bearing in mind the infancy of cations of heavier group 14 elements in low oxidation states, there are plenty of objectives to achieve and problems to solve to develop the fundamental aspects of this field. For example, a monocoordinate silicon(II) cation, which is truly a higher homologue of HSi+ is elusive. The scenario is the same for other heavier group 14 elements. Similarly, a Si(II) dication encapsulated in crown ethers or cryptands is yet to be realized and remains as a sought after goal. One would also notice that unlike silicon and germanium, no monomeric tin(II) cation was reported with neutral ligands like NHC or carbodiphosphorane which is due to the poor overlap between 2p (C) and 5p (Sn). The examples of Pb(II) cations and dications are still very scant and isolation of many more Pb(II) cations is highly desirable.
The reactivity of cations of low valent heavier group 14 elements has not been very well studied but the seminal [Cp*Si]+ (1) was found to be a potential workhorse for a wider range of transformations. In fact, 1 led to novel neutral silicon(II) compounds as well as a cyclotrisilene simply by the addition of appropriate anionic nucleophiles. Most of the other cations were only reacted with some Lewis bases like 4-DMAP to establish the Lewis acidic nature of the cations. However, Driess et al. recently demonstrated that bis-NHC stabilized Si(II) cation, 13 and Ge(II) cations, 48 are excellent precursors to give rise to the hitherto elusive cyclic silylone55 and germylone,77b (heavier analogues of carbodiphosphorane) respectively (Scheme 38). These results ensure that the search for new chemistry involving heavier group 14 elements will not abate anytime soon. However, more systematic studies of the reactivities of such cations are highly desirable. The main target in this chemistry is metal-free catalysis which is yet to be realized. Nevertheless, catalytic conversion of 1,2-dimethoxyethane to 1,4-dioxane by 1 is a very promising step towards metal free catalysis and it is only a matter of time before this goal will be accomplished. Besides, Rausch et al. reported [(η5-Me5C5)Sn]+[B(C6F5)4]− to be an effective co-catalyst for Ziegler–Natta olefin polymerization, which manifests that low-valent group 14 cations are not only of academic interest but also have potential for applications in industrial processes.
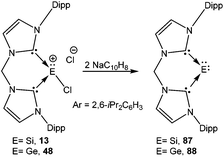 |
| | Scheme 38 Synthesis of silylone and germylone from silicon(II) and germanium(II) cations. | |
Acknowledgements
SK and SSS thank SERB (India) for financial support. SK also acknowledges the support from IISER. SSS is indebted to CSIR-NCl for the start-up grant. VSVSN thanks CSIR, India and SP thanks UGC, India for the research fellowships.
Notes and references
-
(a) J. F. Norris and W. W. Sanders, Am. Chem. J., 1901, 25, 54 CAS;
(b) J. F. Norris, Am. Chem. J., 1901, 25, 117 CAS;
(c) F. Kehrmann and F. Wentzel, Chem. Ber., 1901, 34, 3815 CrossRef CAS PubMed.
- A. v. Baeyer and V. Villiger, Chem. Ber., 1901, 34, 3815 CrossRef PubMed.
- G. A. Olah, J. Am. Chem. Soc., 1972, 94, 808 CrossRef CAS.
-
(a) J. B. Lambert, S. Zhang, C. L. Stern and J. C. Huffmann, Science, 1993, 260, 1917 CAS;
(b) C. A. Reed, Z. Xie, R. Bau and A. Benesi, Science, 1993, 262, 402 CAS.
- G. A. Olah, G. Rasul, X. Li, H. A. Buchholz, G. Sanford and G. K. Surya Prakash, Science, 1994, 263, 983 CAS.
-
(a) C. A. Reed and Z. Xie, Science, 1994, 263, 985 CAS;
(b) J. B. Lambert, S. Zhang and S. M. Ciro, Organometallics, 1994, 13, 2430 CrossRef CAS.
-
(a) J. B. Lambert and B. Kuhlmann, J. Chem. Soc., Chem. Commun., 1992, 931 RSC;
(b) M. Kira, T. Oyamada and H. Sakurai, J. Organomet. Chem., 1994, 471, C4 CrossRef CAS.
- M. Arshadi, D. Johnels and U. Edlund, Chem. Commun., 1996, 1279 RSC.
- K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Müller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825 CrossRef CAS PubMed.
- J. B. Lambert, Y. Zhao, H. Wu, W. C. Tse and B. Kuhlmann, J. Am. Chem. Soc., 1999, 121, 5001 CrossRef CAS.
- J. B. Lambert, L. Lin, S. Keinan and T. Müller, J. Am. Chem. Soc., 2003, 125, 6022 CrossRef CAS PubMed.
-
(a) J. B. Lambert and Y. Zhao, Angew. Chem., Int. Ed. Engl., 1997, 36, 400 CrossRef CAS PubMed;
(b) C. Chuit, R. J. P. Corriu, A. Mehdi and C. Reyé, Angew. Chem. Int., Ed. Engl., 1993, 32, 1311 CrossRef PubMed;
(c) R. Carré, C. Chuit, R. J. P. Corriu, A. Mehdi and C. Reyé, Angew. Chem., Int. Ed. Engl., 1994, 33, 1097–1099 CrossRef PubMed;
(d) T. Müller, Adv. Organomet. Chem., 2005, 53, 155 Search PubMed;
(e) T. Müller, Y. Zhao and J. B. Lambert, Organometallics, 1998, 17, 278 CrossRef;
(f) J. Belzner, D. Schär, B. O. Kneisel and R. Herbst-Irmer, Organometallics, 1995, 14, 1840 CrossRef CAS;
(g) H.-U. Steinberger, C. Bauch, T. Müller and N. Auner, Can. J. Chem., 2003, 81, 1223 CrossRef CAS;
(h) Y. Zhang, K. Huynh, I. Manners and C. A. Reed, Chem. Commun., 2008, 494 RSC;
(i) H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176 RSC;
(j) K. Müther, R. Fröhlich, C. Mück-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2011, 133, 12442 CrossRef PubMed;
(k) S. S. Sen, J. Hey, M. Eckhardt, R. Herbst-Irmer, R. Mata, E. Maedl, H. W. Roesky, M. Scheer and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 12510 CrossRef CAS PubMed.
-
(a) H. F. T. Klare, K. Bergander and M. Oestreich, Angew. Chem., Int. Ed., 2009, 48, 9077 CrossRef CAS PubMed;
(b) R. K. Schmidt, K. Müther, C. Muck-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2012, 134, 4421 CrossRef CAS PubMed.
-
(a) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldrige and J. S. Siegel, Science, 2011, 332, 574 CrossRef CAS PubMed;
(b) S. Duttwyler, C. Douvris, C. D. Nathanael, L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldrige and J. S. Siegel, Angew. Chem., Int. Ed., 2010, 49, 7519 CrossRef CAS PubMed;
(c) N. Lühmann, R. Panisch and T. Müller, Appl. Organomet. Chem., 2010, 24, 533 CrossRef PubMed.
-
(a) C. Douvris and O. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed;
(b) R. Panisch, M. Boldte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676 CrossRef CAS PubMed.
- A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636 CrossRef PubMed.
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849 CrossRef CAS PubMed.
-
(a) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683 CrossRef CAS PubMed;
(b) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687 CrossRef CAS PubMed.
-
(a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed;
(b) S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311 CrossRef CAS PubMed;
(c) S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 2, 659 RSC;
(d) S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578 CrossRef CAS PubMed.
- M. Driess, S. Yao, M. Brym and C. van Wüllen, Angew. Chem., Int. Ed., 2006, 45, 6730 CrossRef CAS PubMed.
-
(a) P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y. Tsay, Chem. Ber., 1980, 113, 757 CrossRef CAS PubMed;
(b) P. Jutzi, R. Dickbreder and H. Nöth, Chem. Ber., 1989, 122, 865 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef CAS PubMed.
-
V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-coordinate Si, Ge, Sn and Pb: From Phantom atom to Stable Compounds, Wiley, 2010, ch. 1 Search PubMed.
-
(a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500 CrossRef CAS PubMed;
(b) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568 CrossRef CAS PubMed;
(c) N. Kuriakose and K. Vanka, Dalton Trans., 2013, 43, 2194 RSC;
(d) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed.
- P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138 CrossRef CAS PubMed.
-
(a) E. Rivard, Dalton Trans., 2014, 43, 8577 RSC;
(b) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337 RSC;
(c) G. Prabusankar, A. Sathyanarayana, P. Suresh, C. N. Babu, K. Srinivas and B. P. R. Metla, Coord. Chem. Rev., 2014, 269, 96 CrossRef CAS PubMed.
-
(a) W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS PubMed;
(b) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841 CrossRef CAS PubMed.
-
(a) B. Inés, M. Patil, J. Carreras, R. Goddard, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 8400 CrossRef PubMed;
(b) S. Khan, G. Gopakumar, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2013, 52, 5644 CrossRef CAS PubMed.
- M. Q. Y. Tay, Y. Lu, R. Ganguly and D. Vidović, Angew. Chem., Int. Ed., 2013, 52, 3132 CrossRef CAS PubMed.
- R. Toner and G. Frenking, Chem. Commun., 2008, 1584 RSC.
- J. M. Lehn, Acc. Chem. Res., 1978, 11, 49 CrossRef CAS.
- T. Probst, O. Steigelmann, J. Riede and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1990, 29, 1397 CrossRef PubMed.
- P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360 CrossRef CAS PubMed.
-
(a) H. Braunschweig, K. Radacki, D. Rais and K. Uttinger, Angew. Chem., Int. Ed., 2006, 45, 162 CrossRef CAS PubMed;
(b) H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345 CrossRef CAS PubMed;
(c) J. Brand, H. Braunschweig and S. S. Sen, Acc. Chem. Res., 2014, 47, 180 CrossRef CAS PubMed.
- J. Brand, H. Braunschweig, F. Hupp, A. K. Phukan, K. Radacki and S. S. Sen, Angew. Chem., Int. Ed., 2014, 53, 2240 CrossRef CAS PubMed.
- A. C. Filippou, O. Chernov, K. W. Stumpf and G. Schnakenburg, Angew. Chem., Int. Ed., 2010, 49, 3296 CrossRef CAS PubMed.
- A. C. Filippou, U. Chakraborty and G. Schnakenburg, Chem. – Eur. J., 2013, 19, 5676 CrossRef CAS PubMed.
-
(a) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565 CrossRef CAS PubMed;
(b) S. S. Sen, Angew. Chem., Int. Ed., 2014, 53, 8820 CrossRef CAS PubMed.
- A. E. Douglas and B. L. Lutz, Can. J. Phys., 1970, 48, 247 CrossRef CAS PubMed.
- N. Grevesse and A. Sauval, Astron. Astrophys., 1970, 9, 232 CAS.
-
D. Rehder, Chemistry in Space, Wiley-VCH, Verlag and Co., 2010, ch. 3 Search PubMed.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164 CrossRef PubMed.
- E. D. Jemmis and P. v. R. Schleyer, J. Am. Chem. Soc., 1982, 104, 4780 Search PubMed.
- P. Jutzi, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller, H.-G. Stammler and A. B. Rozhenko, J. Am. Chem. Soc., 2009, 131, 12137 CrossRef CAS PubMed.
- S. Khan, S. S. Sen, H. W. Roesky, D. Kratzert, R. Michel and D. Stalke, Inorg. Chem., 2010, 49, 9689 CrossRef CAS PubMed.
- P. Jutzi, K. Leszczyńska, B. Neumann, W. W. Schoeller and H.-G. Stammler, Angew. Chem., Int. Ed., 2009, 48, 2596 CrossRef CAS PubMed.
- P. Jutzi, K. Leszczyńska, A. Mix, B. Neumann, W. W. Schoeller and H.-G. Stammler, Organometallics, 2009, 28, 1985 CrossRef CAS.
- P. Jutzi, K. Leszczyńska, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller and H.-G. Stammler, Organometallics, 2010, 29, 4759 CrossRef CAS.
- K. Leszczyńska, K. Abersfelder, A. Mix, B. Neumann, H.-G. Stammler, M. J. Cowley, P. Jutzi and D. Scheschkewitz, Angew. Chem., Int. Ed., 2012, 51, 6785 CrossRef PubMed.
- K. Leszczyńska, A. Mix, R. J. F. Berger, B. Rummel, B. Neumann, H.-G. Stammler and P. Jutzi, Angew. Chem., Int. Ed., 2011, 50, 6843 CrossRef PubMed.
- M. Driess, S. Yao, M. Brym, C. van Wüllen and D. Lentz, J. Am. Chem. Soc., 2006, 128, 9628 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 10074 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147 CrossRef CAS PubMed.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974 CrossRef CAS PubMed.
- S. Inoue, J. D. Epping, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 8514 CrossRef CAS PubMed.
- H.-X. Young, H.-W. Xi, Y. LI, K. H. Kim and C.-W. So, Chem. – Eur. J., 2013, 19, 11786 CrossRef PubMed.
- N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2008, 27, 2361 CrossRef CAS.
- D. L. Reger and P. S. Coan, Inorg. Chem., 1996, 35, 258 CrossRef CAS.
-
(a) H. V. R. Dias and Z. Wang, J. Am. Chem. Soc., 1997, 119, 4650 CrossRef CAS;
(b) H. V. R. Dias and W. Jin, J. Am. Chem. Soc., 1996, 118, 9123 CrossRef CAS.
- Y. Ding, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2001, 20, 1190 CrossRef CAS.
- M. Stender, A. D. Philips and P. P. Power, Inorg. Chem., 2001, 40, 5314 CrossRef CAS.
- M. Driess, S. Yao, M. Brym and C. van Wüllen, Angew. Chem., Int. Ed., 2006, 45, 4349 CrossRef CAS PubMed.
- M. J. Taylor, A. J. Saunders, M. P. Coles and J. R. Fulton, Organometallics, 2011, 30, 1334 CrossRef CAS.
- H. Arii, F. Nakadate, K. Mochida and T. Kawashima, Organometallics, 2011, 30, 4471 CrossRef CAS.
-
(a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed;
(b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed;
(c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed.
- A. Schäfer, W. Saak, D. Haase and T. Müller, Chem. – Eur. J., 2009, 15, 3945 CrossRef PubMed.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557 CrossRef CAS PubMed.
- P. E. Lippens, Phys. Rev. B: Condens. Matter, 1999, 60, 4576 CrossRef CAS.
-
(a) A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701 CrossRef CAS PubMed;
(b) K. C. Thimer, S. M. I. Al-Rafia, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2009, 7119 RSC.
- G. W. Parshall, J. Am. Chem. Soc., 1972, 94, 8716 CrossRef CAS.
- F. Cheng, J. M. Dyke, F. Ferrante, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2010, 39, 847 RSC.
- A. P. Singh, H. W. Roesky, E. Carl, D. Stalke, J.-P. Demers and A. Lange, J. Am. Chem. Soc., 2012, 134, 4998 CrossRef CAS PubMed.
- M. Bouska, L. Dostal, A. Ruzicka and R. Jambor, Organometallics, 2013, 32, 1995 CrossRef CAS.
-
(a) Y. Xiong, S. Yao, S. Inoue, A. Berkefeld and M. Driess, Chem. Commun., 2012, 48, 12198 RSC;
(b) Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004 CrossRef CAS PubMed.
- M. J. Ward, P. A. Rupar, M. W. Murphy, Y.-M. Yiu, K. M. Baines and T. K. Sham, Chem. Commun., 2010, 46, 7016 RSC.
- Y. Xiong, T. Szilvási, S. Yao, G. Tan and M. Driess, J. Am. Chem. Soc., 2014, 136, 11300 CrossRef CAS PubMed.
- A. Schäfer, F. Winter, W. Saak, D. Haase, R. Pöttgen and T. Müller, Chem. – Eur. J., 2011, 17, 10979 CrossRef PubMed.
-
(a)
N. N. Greenwood and A. Earnshaw, Chemistry of the elements, Pergamon Press, 1989 Search PubMed;
(b) B. L. Haymore, J. D. Lamb, R. M. Izatt and J. J. Christensen, Inorg. Chem., 1982, 21, 1598 CrossRef CAS;
(c) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson and M. Nieuwenhuyzen, Chem. Commun., 1996, 1967 RSC;
(d) T. Gunnlaugsson, A. Harte, J. P. Leonard and M. Nieuwenhuyzen, Chem. Commun., 2002, 2134 RSC.
-
(a) J. B. Lambert, Science, 2008, 322, 1333 CrossRef CAS PubMed;
(b) T. Müller, Angew. Chem., Int. Ed., 2009, 48, 3740 CrossRef PubMed.
- P. A. Rupar, R. Bandyopadhyay, B. F. T. Cooper, M. R. Stinchcombe, P. J. Ragogna, C. L. B. Macdonald and K. M. Baines, Angew. Chem., Int. Ed., 2009, 48, 5155 CrossRef CAS PubMed.
-
(a) F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5152 CrossRef CAS PubMed;
(b) W. Levason, G. Reid and W. Zhang, Coord. Chem. Rev., 2011, 255, 1319 CrossRef CAS PubMed.
- R. Bandyopadhyay, J. H. Nguyen, A. Swidan and C. L. B. Macdonald, Angew. Chem., Int. Ed., 2013, 52, 3469 CrossRef CAS PubMed.
-
(a) C. L. B. Macdonald, R. Bandyopadhyay, B. F. T. Cooper, W. W. Friedl, A. J. Rossini, R. W. Schurko, S. H. Eichhorn and R. H. Herber, J. Am. Chem. Soc., 2012, 134, 4332 CrossRef CAS PubMed;
(b) J. C. Avery, M. A. Hanson, R. H. Herber, K. J. Bladek, P. A. Rupar, I. Nowik, Y. Huang and K. M. Baines, Inorg. Chem., 2012, 51, 7306 CrossRef CAS PubMed.
- A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339 CrossRef CAS.
- A. C. Filippou, A. I. Philippopoulos, P. Portius and G. Schnakenburg, Organometallics, 2004, 23, 4503 CrossRef CAS.
-
(a) R. Jambor, B. Kašná, S. G. Koller, C. Strohmann, M. Schürmann and K. Jurkschat, Eur. J. Inorg. Chem., 2010, 902 CrossRef CAS PubMed;
(b) R. Dostalova, L. Dostal, A. Ruzicka and R. Jambor, Organometallics, 2011, 30, 2405 CrossRef CAS.
- J. Martincov, L. Dostal, S. Herres-Pawlis, A. Ruzcka and R. Jambor, Chem. – Eur. J., 2011, 17, 7423 CrossRef PubMed.
-
(a) J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed;
(b) H. Braunschweig, A. Damme, R. D. Dewhurst, F. Hupp, J. O. C. Jimenez-Halla and K. Radacki, Chem. Commun., 2012, 48, 10410 RSC.
- H. Braunschweig, M. A. Celik, R. D. Dewhurst, M. Heid, F. Hupp and S. S. Sen, Chem. Sci., 2015, 6, 425 RSC.
- B. Rhodes, J. C. W. Chien and M. D. Rausch, Organometallics, 1998, 17, 1931 CrossRef CAS.
- S. Hino, M. Brynda, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2004, 43, 2655 CrossRef CAS PubMed.
- W. H. Liao, P. Y. Ho and M. D. Su, Inorg. Chem., 2013, 52, 1338 CrossRef CAS PubMed.
-
(a) D. Kratzert, D. Leusser, J. J. Holstein, B. Dittrich, K. Abersfelder, D. Scheschkewitz and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 4478 CrossRef CAS PubMed;
(b) J. Hey, D. Leusser, D. Kratzert, H. Fliegl, J. M. Dieterich, R. A. Mata and D. Stalke, Phys. Chem. Chem. Phys., 2013, 15, 20600 RSC and references therein.
- S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873 RSC.
- K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564 CrossRef CAS PubMed.
Footnote |
| † This review is dedicated to Prof. Herbert W. Roesky on the occasion of his 80th birthday. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a