
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uranium-mediated oxidative addition and reductive elimination
Erli
Lu
and
Stephen T.
Liddle
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: stephen.liddle@nottingham.ac.uk
First published on 23rd June 2015
Abstract
Oxidative addition, and its reverse reaction reductive elimination, constitute two key reactions that underpin organometallic chemistry and catalysis. Although these reactions have been known for decades in main group and transition metal systems, they are exceptionally rare or unknown for the f-block. However, in recent years much progress has been made. In this Perspective article, advances in uranium-mediated oxidative addition/reductive elimination, since the point that this research area was initiated in the early-1980s, are summarised. We principally divide the Perspective into two parts of oxidative addition and reductive elimination, along with a separate section concerning reactions where there is no change of uranium oxidation state in reactant and product but the reaction has the formal appearance of a ‘concerted’ reductive elimination/oxidative addition from the perspective of the net result. This body of work highlights that whilst uranium is capable of performing reactions that to some extent conform to traditional reactivity types, novel reactivity that has no counterpart anywhere else can be performed, thus adding to the rich palate of redox chemistry that uranium can mediate.
 Erli Lu | Erli Lu received his PhD degree at the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS), in 2012, under the supervision of Yaofeng Chen, where he studied rare-earth organometallic chemistry. Later in 2012, he joined the research group of Steve Liddle at the University of Nottingham as a recipient of a Marie Curie International Incoming Fellowship to investigate actinide organometallic chemistry. He has 14 journal publications, two book chapters, and a patent to date. |
 Steve Liddle | Steve Liddle received his BSc (Hons) in 1997 and PhD in 2000, both from Newcastle University. After postdoctoral fellowships at the Universities of Edinburgh, Newcastle, and Nottingham he took up a Royal Society University Research Fellowship and Lectureship in 2007. He was promoted to Associate Professor and Reader in 2010 and Professor of Inorganic Chemistry in 2013. He was elected FRSC in 2011 and has been awarded a number of prizes for his independent work on uranium chemistry, including most recently the RSC Corday-Morgan prize in 2015. He has published over 140 research articles, reviews, and book chapters. |
1. Introduction
Oxidative addition, and its reverse reaction reductive elimination, constitute two indispensable and elementary cornerstones of organometallic reactivity. The importance of these two reactions cannot be overstated since together they have underpinned key processes in organometallic chemistry and catalysis for over five decades.1 Oxidative addition/reductive elimination are, overall, 2-electron processes that accompany a change in the oxidation state of a metal and cleavage/formation of chemical bonds but, overall, do not have any mechanistic implications. Classically, there are two kinds of oxidative addition: type (a) a two-electron redox process at one metal centre where an X–Y bond is cleaved, M–X and M–Y bonds are formed at the same metal, and the metal is formally oxidised by two units (Scheme 1a); type (b) two one-electron redox processes at two metal centres, which may be independent monomers or a dinuclear complex, where an X–Y bond is cleaved, M–X and M–Y bonds are formed at different metals, and each metal is formally oxidised by one unit (Scheme 1b).2 In the former process the metal oxidation state, coordination number, and valence electron count all increase by two whereas in the latter they all increase by one. In each case reductive elimination is simply the reverse reaction. The oxidative addition/reductive elimination couple can be considered to be an equilibrium process whose precise position is controlled by the relative strengths of the X–Y, M–X, and M–Y bonds, the size and electron richness of the metal, and how coordinatively saturated the reactant metal centre is. Of the two types of oxidative addition/reductive elimination couple, the type (a) two-electron single metal variant is by far the most important with respect to applications in catalysis.1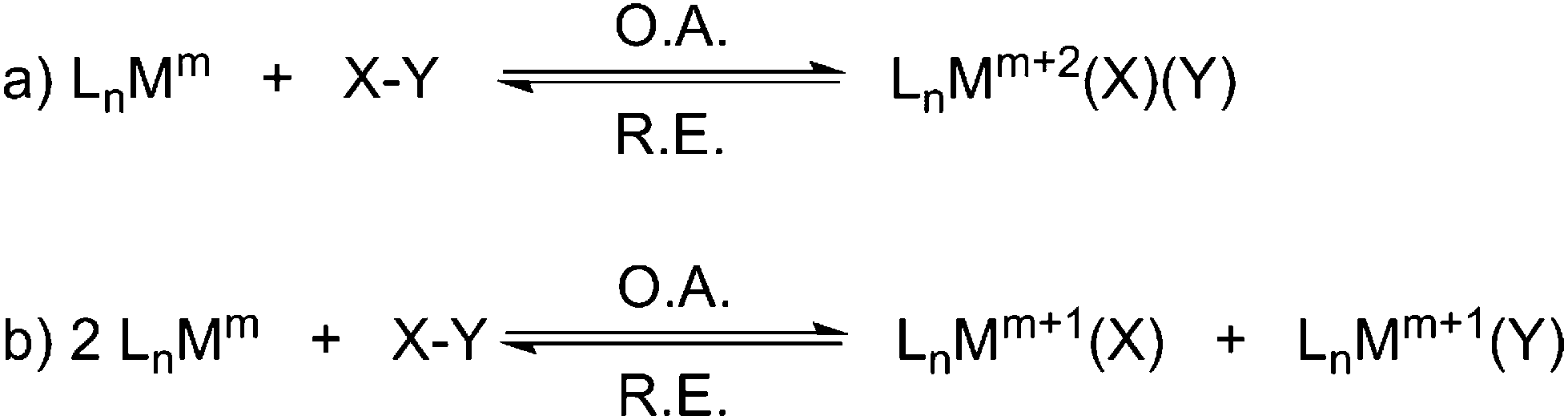 | ||
| Scheme 1 The two classical 2-electron oxidative addition (O.A.)/reductive elimination (R.E.) couples – Ln = generic supporting ligands; M = metal; m = oxidation state; X–Y = substrate. | ||
The vast majority of classical oxidative addition/reductive elimination chemistry is dominated by the d-block, especially late transition-metals. Their electron-richness, diversity of easily accessible oxidation states and coordination sites, in combination with a richness of non-bonding d-electrons, render these metals ideal for supporting classical oxidative addition and reductive elimination. The most prevalent late transition-metals for application in catalysis are group 9 and 10 metals, especial Pd, Pt, Rh, and Ir.1 The ability of these metals to form strong M–L bonds and the large energy splitting of d-orbitals renders them capable of activating strong chemical bonds e.g. C–H, C–C etc., and their electron richness favours 2-electron type (a) oxidative additions/reductive eliminations. Apart from late transition-metals, although much less prevalent, oxidative addition/reductive elimination has been studied in main group chemistry resulting in fundamental contributions to organic/organometallic chemistry.1 For example, oxidative addition of chlorine to phosphorus trichloride to give PCl5, oxidative addition of RX (R = alkyl, aryl; X = halide) to magnesium to prepare Grignard reagents,3 oxidative addition at tin(II) to give tin(IV) derivatives,4 and more recently oxidative addition at low valent group 13 centres are all known and established.5
In comparison to d-block metals, f-block elements are traditionally recognised as being unable to mediate classical 2-electron oxidative addition/reductive elimination, due to their propensity to perform 1-electron redox couples, and highly ionic and thus weak M–E bonds (M = f-element; E = group 14–17 element). Instead, their organometallic chemistry is dominated by salt metathesis, insertion reactions of unsaturated bonds, 1-electron redox processes, and σ-bond metathesis from their highly polarising nature, and these reactivities have played a pivotal role in many important catalytic processes.6 One exception, however, is uranium, which is known to have one of the most diverse range of oxidation states amongst all f-elements in an organometallic context (+2,7 +3, +4, +5, +6). The range of accessible and variable oxidation states, along with the ability to form reasonably covalent and relatively strong U–E bonds, which is a result of availability of 5f- and 6d-orbitals to interact with ligand frontier orbitals, renders uranium the most promising f-element for conducting oxidative addition/reductive elimination reactions.
The history of well-defined, homogeneous uranium-mediated oxidative addition/reductive elimination dates back to the early 1980s. But until very recently it was a relatively obscure research topic and only a handful of discrete examples were reported. In the 21st century, mainly boosted by the introduction of redox non-innocent ligands and the concept of sterically induced reduction (SIR),8 the field has flourished and is gathering momentum. Both redox non-innocent ligands and SIR provide a viable route to manipulate f-elements, which inherently prefer 1-electron process, to participate in these overall n-electron (n ≥ 2) oxidative addition/reductive elimination processes. Together with the widely accessible oxidation states of uranium, the oxidative addition/reductive elimination chemistry mediated by uranium is often distinct to late transition-metal counterparts. Due to the importance of the oxidative addition/reductive elimination couple, and the unique role of uranium in the periodic table,9 this research area has great potential to extend the boundaries of organometallic chemistry as well as catalysis; thus, an up-to-date summary is warranted.
In this Perspective article, advances in uranium-mediated oxidative addition/reductive elimination, since the point that this research area was initiated in the early-1980s,10 are summarised. We principally divide the perspective into two parts of oxidative addition and reductive elimination, along with a separate section concerning reactions where there is no change of uranium oxidation state in reactant and product but the reaction has the formal appearance of a ‘concerted’ reductive elimination/oxidative addition from the perspective of the net result.11 This overview serves to highlight some similarities to transition metal chemistry, but also that uranium is capable of effecting some unique reactivity of its own.
2. Oxidative addition
According to Scheme 1, there are three essential criteria that must be met to classify a reaction to be oxidative addition, rather than simply an oxidation: (i) increment of oxidation state of metal; (ii) cleavage of an X–Y bond; (iii) formation of both M–X and M–Y bonds. Thus, oxidations of low-valent uranium centres that are accompanied by extrusion of a small molecule, such as oxidation of U(III) precursors to U(V)-imides12 or -nitrides13 by azides with extrusion of N2, do not meet the IUPAC criteria and are not included due to the fact that the extruded small molecule (e.g. N2) does not remain bonded to the metal centre. The U(III)-mediated reductive coupling of small molecules, e.g. CO,14 is not included either, because of the absence of formal bond cleavage in these reactions.2.1. Oxidative addition with uranium as the only electron donor
In the following section, oxidative additions where uranium acts as the only electron donor are discussed (in comparison with reactions involving redox non-innocent ligands, vide infra). Generally, a low-valent (usually U(III)) and thus reducing uranium precursor is oxidised to form a high-valent uranium compound, along with the cleavage of E–E′ or E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E′ bonds (E,E′ = group 14–17 elements) and formation of U–E(E′) or UE(E′) bonds. Due to the unique chemical properties of uranium, the 2-electron mono-metal type (a) oxidative addition, which is prevalent in late transition-metal chemistry, is still unknown for uranium. The number of strictly defined 1-electron bis-metal type (b) oxidative additions remains relatively few. In this section, instances of clear-cut type (b) oxidative additions are summarised first. The rest of this section then deals with uranium-based oxidative additions with wider and less clear-cut definitions, which are cataloged according to substrate scopes.
E′ bonds (E,E′ = group 14–17 elements) and formation of U–E(E′) or UE(E′) bonds. Due to the unique chemical properties of uranium, the 2-electron mono-metal type (a) oxidative addition, which is prevalent in late transition-metal chemistry, is still unknown for uranium. The number of strictly defined 1-electron bis-metal type (b) oxidative additions remains relatively few. In this section, instances of clear-cut type (b) oxidative additions are summarised first. The rest of this section then deals with uranium-based oxidative additions with wider and less clear-cut definitions, which are cataloged according to substrate scopes.
In 1981, Finke and co-workers reported reactions between the U(III) complex [Cp*2U(Cl)(thf)] (1) and a series of haloalkanes.10a For chloroalkanes, these reactions yielded U(IV) bis-chloride (2) and U(IV) chloride alkyl (3) products, with cleavage of the C–Cl bond (Scheme 2). The oxidation state of uranium increases by one, whilst the R- and X-groups attach to two different U(IV) centres. Thus, the overall reaction can be described as type (b) oxidative addition. The free-radical nature of these reactions was indicated by the observation of R–R coupling products along with other radical decay products. If X is not chloride, ligand-scrambling processes were observed. Kinetic and mechanistic studies revealed that the active U(III) species ‘[Cp*2U(Cl)]’ is slowly produced by dissociation of the thf molecule from 1, whilst the subsequent atom-abstraction step from R–X is very fast.10b
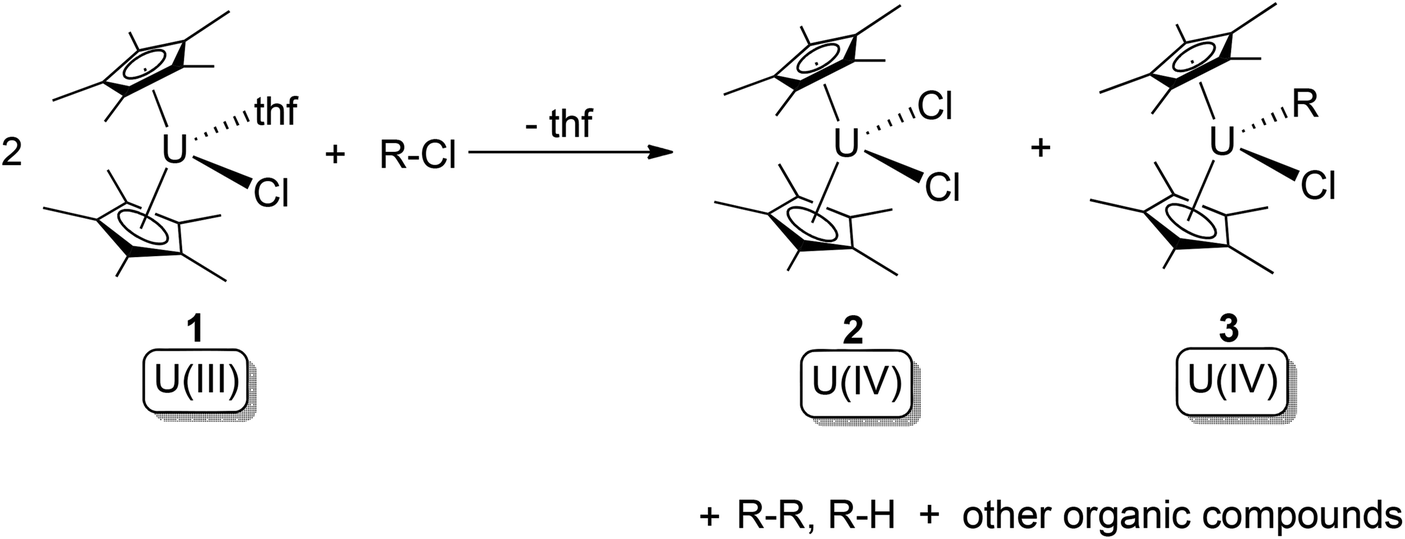 | ||
| Scheme 2 Oxidative addition of [Cp*2U(Cl)(thf)] (1) by haloalkanes.10a,b | ||
In related work, as a part of their systematic study of U(III) compounds, Marks and co-workers observed similar oxidative addition towards haloalkanes, but used the solvent-free U(III) trimer complex [Cp*2U(μ-Cl)]3 (4) (Scheme 3).10d The outcome of the reactions was reported to be dependent on the haloalkane substrate employed: with chloromethane the reaction produced two U(IV) products, [Cp*2UCl2] (2) and [Cp*2U(Cl)(CH3)] (3-Me), as expected as a clear-cut type (b) oxidative addition. However, with chlorobenzene [Cp*2UCl2] (2) was the only U-containing product along with other free-radical decay products. These results support the hypothesis that these uranium-mediated oxidative additions towards C–X bonds are free-radical processes.
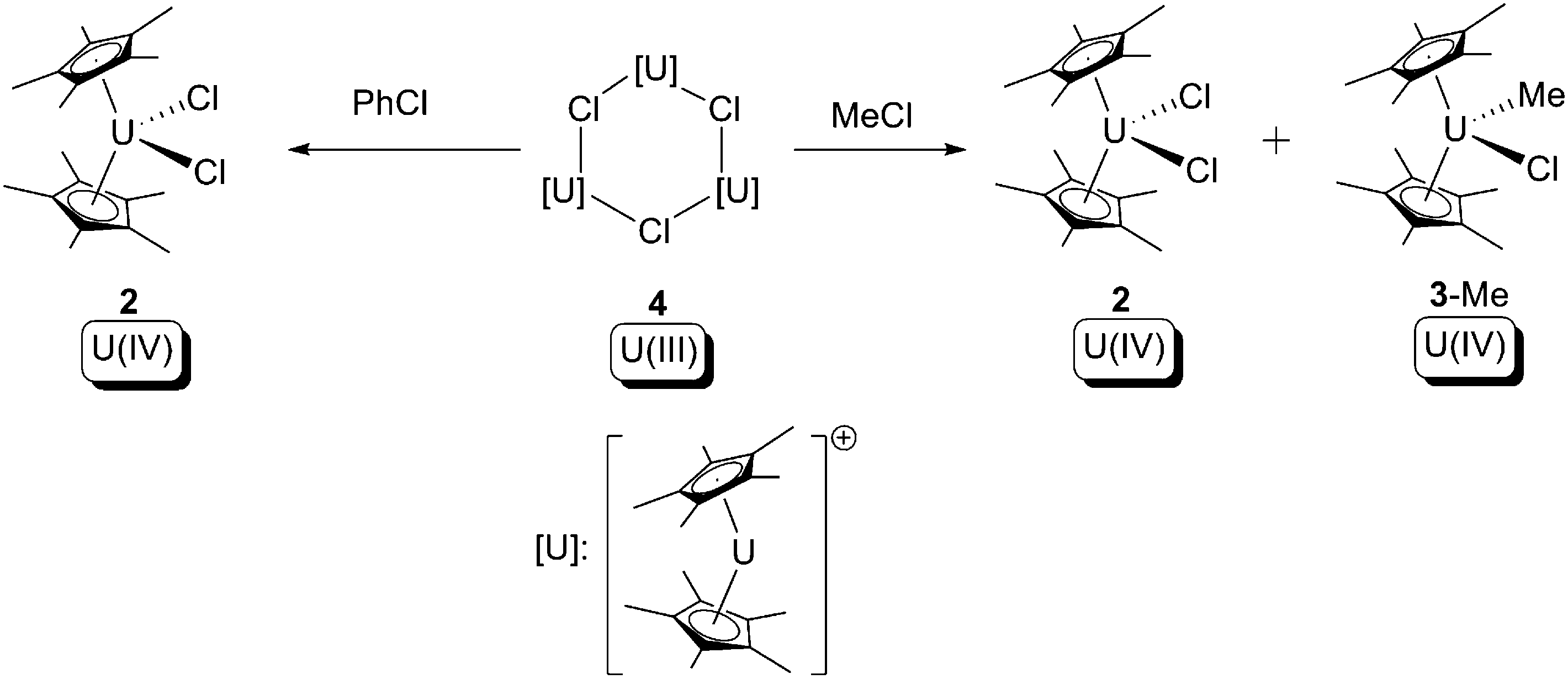 | ||
| Scheme 3 Oxidative addition of [Cp*2U(μ-Cl)]3 (4) by haloalkanes.10d | ||
In 2002, Scott and co-workers reported reactions between two equivalents of the U(III) complex [U(NN′3)] (5) (NN′3 = [N(CH2CH2NSiMe2tBu)3]3−) and halogens (Cl2, Br2, I2) to produce the corresponding U(IV) halide complexes [(NN′3)UX] (6) (X = Cl, Br, I) (Scheme 4).15 The reactions are formally type (b) oxidative additions, whilst the NN′3 triamidoamine ligand acts as an inert supporting ligand.
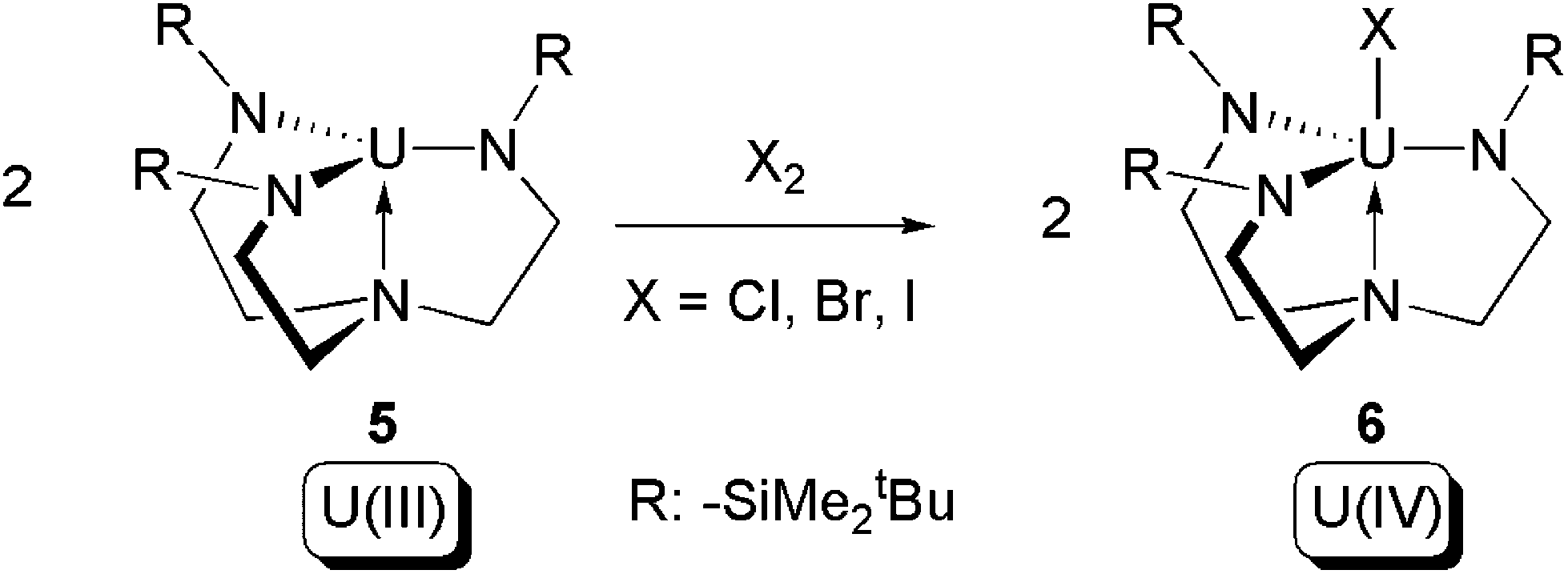 | ||
| Scheme 4 Oxidative addition of [U(NN′3)] (5) by halogens.15 | ||
In 2009, Boncella and co-workers reported reactions between a U(V) bridging imido dimer (7) and Ph–E–E–Ph (E = S, Se, Te), which produced a series of U(VI) trans-bis-imide products (Scheme 5).16 Amongst the products, 9 and 10 can be considered to result from ligand redistribution reactions of the initial products 8. The reactions result in a +1 increase of the oxidation state for each uranium centre, cleavage of an E–E bond, and formation of U–E bonds, and thus can be classified as type (b) oxidative addition.
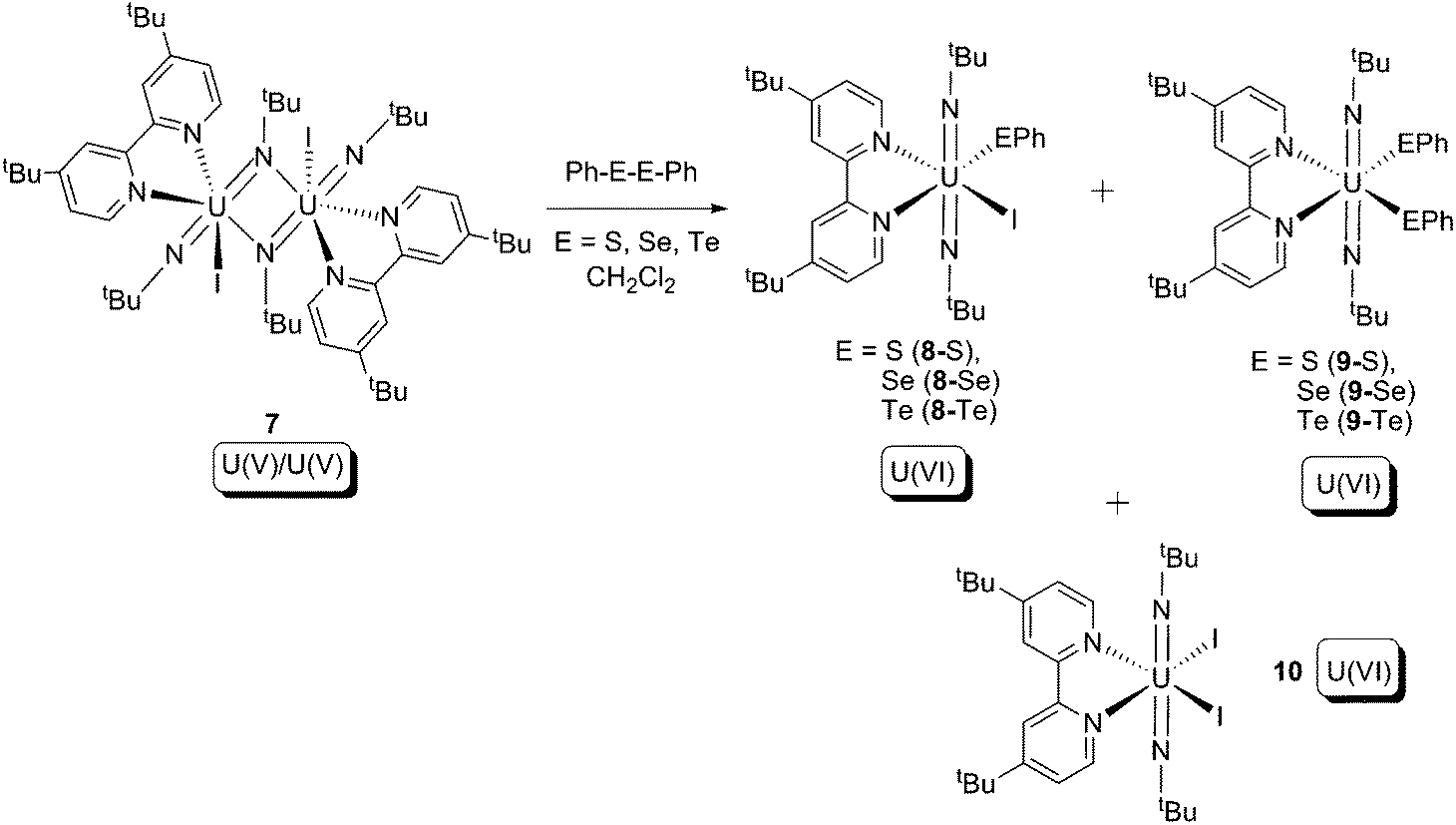 | ||
| Scheme 5 Oxidative addition of U(V) bridging imide dimer (7) by PhE–EPh (E = S, Se, Te).16 | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N bonds.
The 4-electron non-classical oxidative addition of EE′ bond (E,E′: group 14–16 element) on a single metal centre (Scheme 6), which leads to cleavage of the EE′ double bond, formation of ME(E′) double bonds, along with increase of +4 of the metal oxidation state, has been observed for transition-metals.17 Cleavage of the strong EE′ bond and formation of the generally highly reactive ME(E′) bond renders this type of oxidative addition to be attractive for the purpose of functionalisation and utilisation of the EE′ species, e.g. carbonyl and azo compounds.
N bonds.
The 4-electron non-classical oxidative addition of EE′ bond (E,E′: group 14–16 element) on a single metal centre (Scheme 6), which leads to cleavage of the EE′ double bond, formation of ME(E′) double bonds, along with increase of +4 of the metal oxidation state, has been observed for transition-metals.17 Cleavage of the strong EE′ bond and formation of the generally highly reactive ME(E′) bond renders this type of oxidative addition to be attractive for the purpose of functionalisation and utilisation of the EE′ species, e.g. carbonyl and azo compounds.
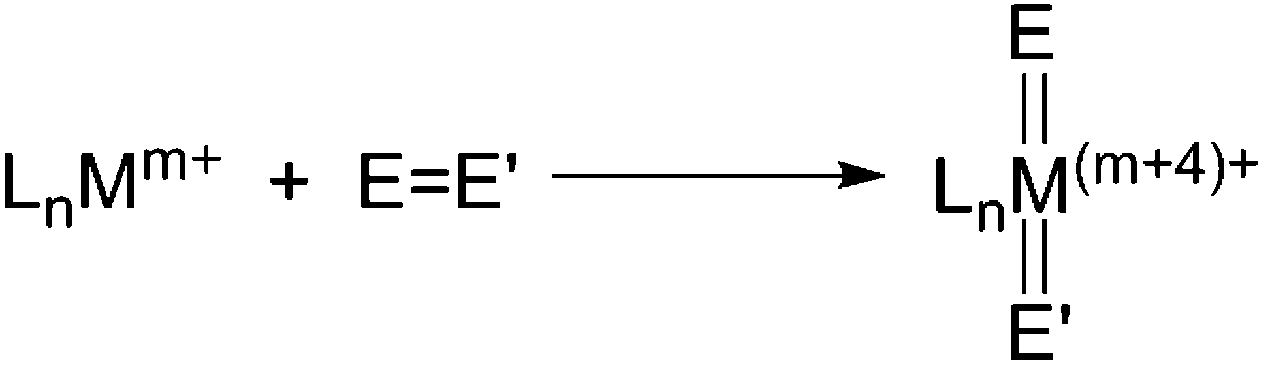 | ||
| Scheme 6 Generic 4-electron oxidative addition of a EE′ bond (E, E′: group 14–16 element). | ||
In 1998, Burns and co-workers reported that a U(III) halide ‘ate’ compound, [Cp*2UCl(NaCl)(thf)2] (11), undergoes a 4-electron oxidative addition reaction overall towards azobenzene, to produce a U(VI) bis-imide (12) and the known U(IV) bis-chloride (2) as products (Scheme 7a).18 The reaction was postulated to proceed firstly via a 2-electron oxidative cyclometallation between 11 and azobenzene, yielding a chlorohydrazineuranium(V) intermediate; this intermediate is reduced by the U(III) starting material 11 yielding an azouranium(IV) intermediate, then a 2-electron oxidative ring-opening of the U–N–N three membered ring occurs to form the final product 12. It should be noted that 2 can be treated by sodium amalgam to reform 11. This is the first instance of azo-to-imide conversion in actinide organometallic chemistry.
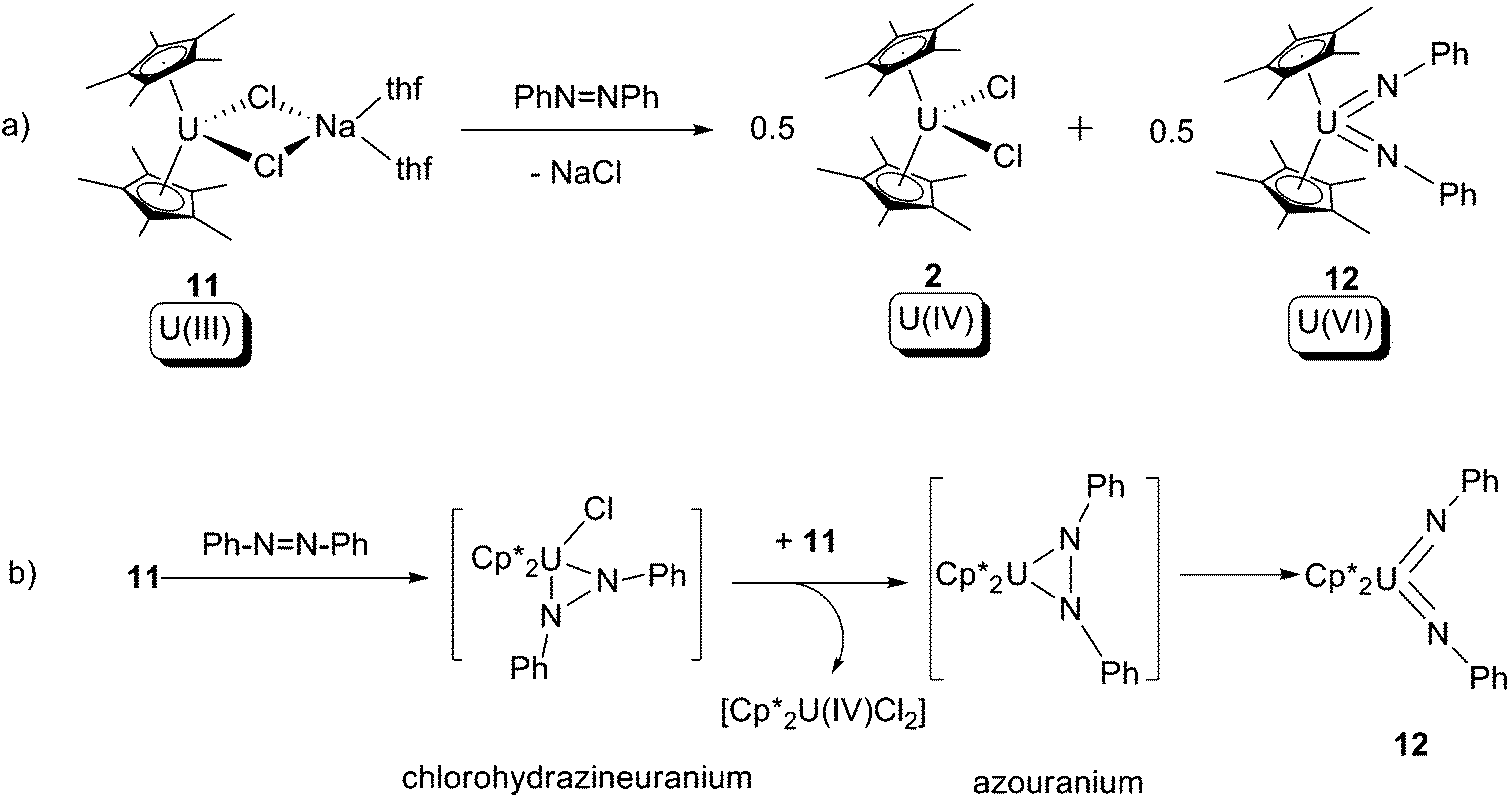 | ||
| Scheme 7 (a) 4-Electron oxidative addition of a U(III) compound by azobenzene to form U(VI) bis-imide 12; (b) proposed mechanism for the reaction.18 | ||
Another example of this type of reaction was reported by Evans and co-workers in 2013.19 In this case the U(III) allyl compound 13 was treated with 1 equivalent of azobenzene, to produce U(IV) bis-allyl (14) as well as the known U(VI) bis-imide (12) (Scheme 8).
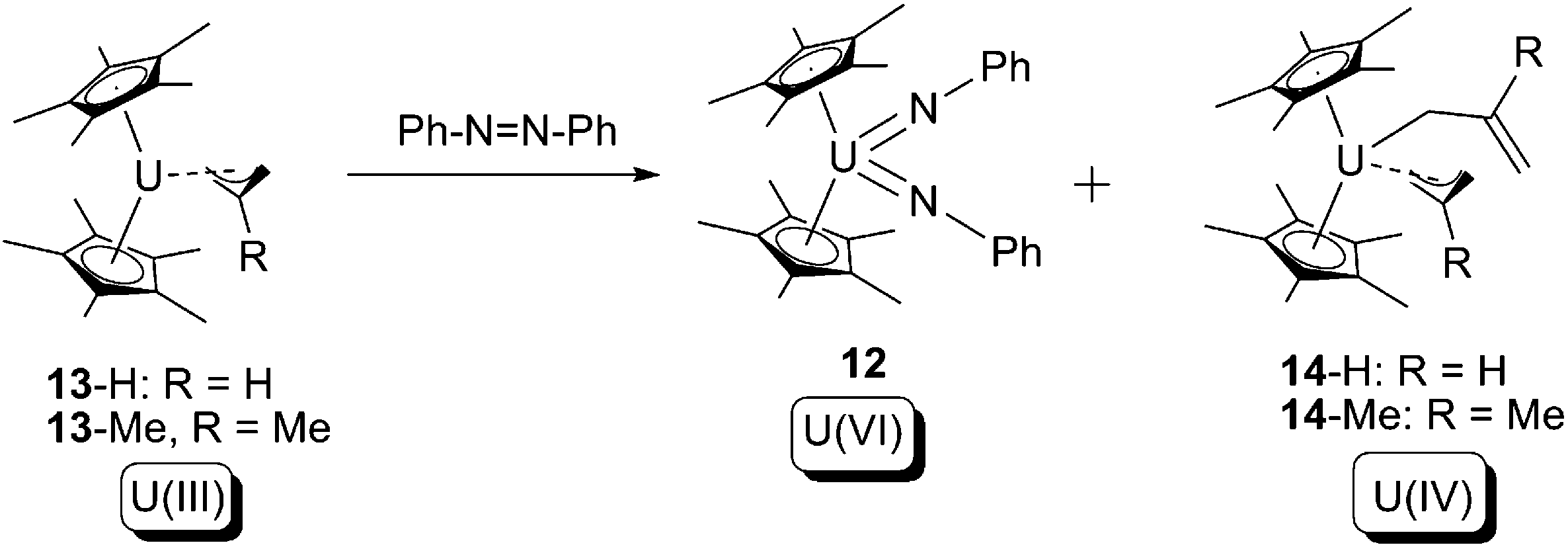 | ||
| Scheme 8 4-Electron oxidative addition of a U(III) allyl by azobenzene.19 | ||
N–Ph in Scheme 7. The intramolecular nature of the oxidative ring-opening step was supported by crossover experiments using asymmetrically substituted hydrazine as the substrate.
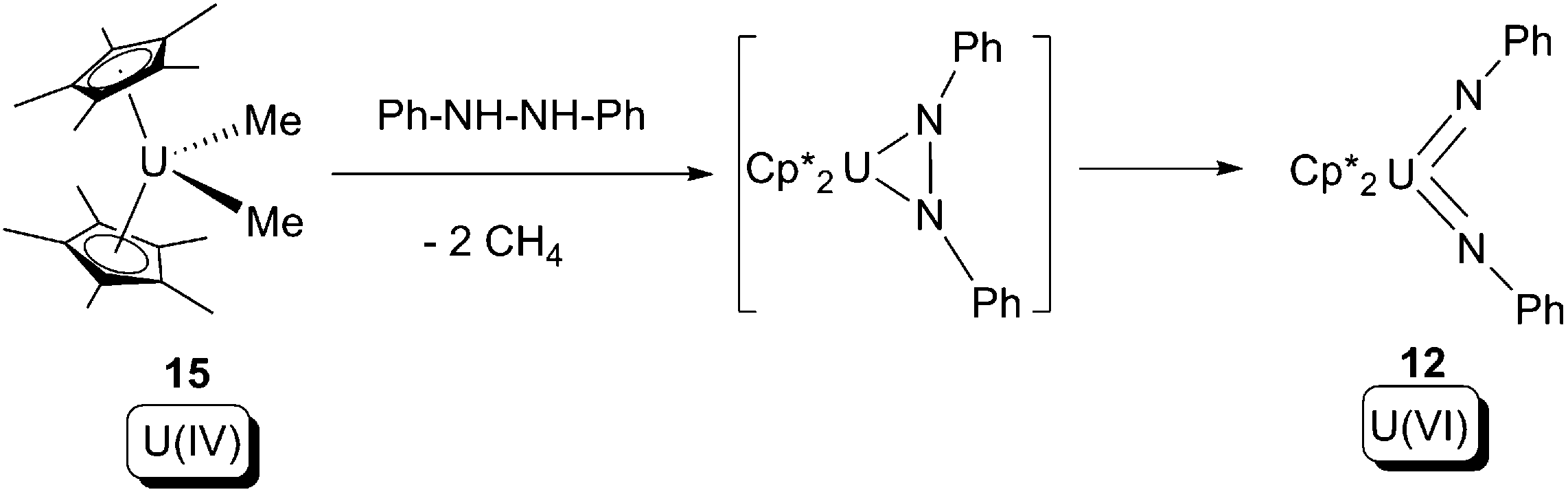 | ||
| Scheme 9 2-Electron oxidative addition of 15 by a N–N bond substrate.20 | ||
Based on the aforementioned oxidative additions towards NN and N–N bonds, U-mediated catalytic conversion of hydrazine to aniline and azobenzene was reported (Scheme 10).21 The presence of aniline as a product suggested the formation of U(IV) bis-anilide [Cp*2U(NHPh)2] during the reaction, however this species could not be detected.
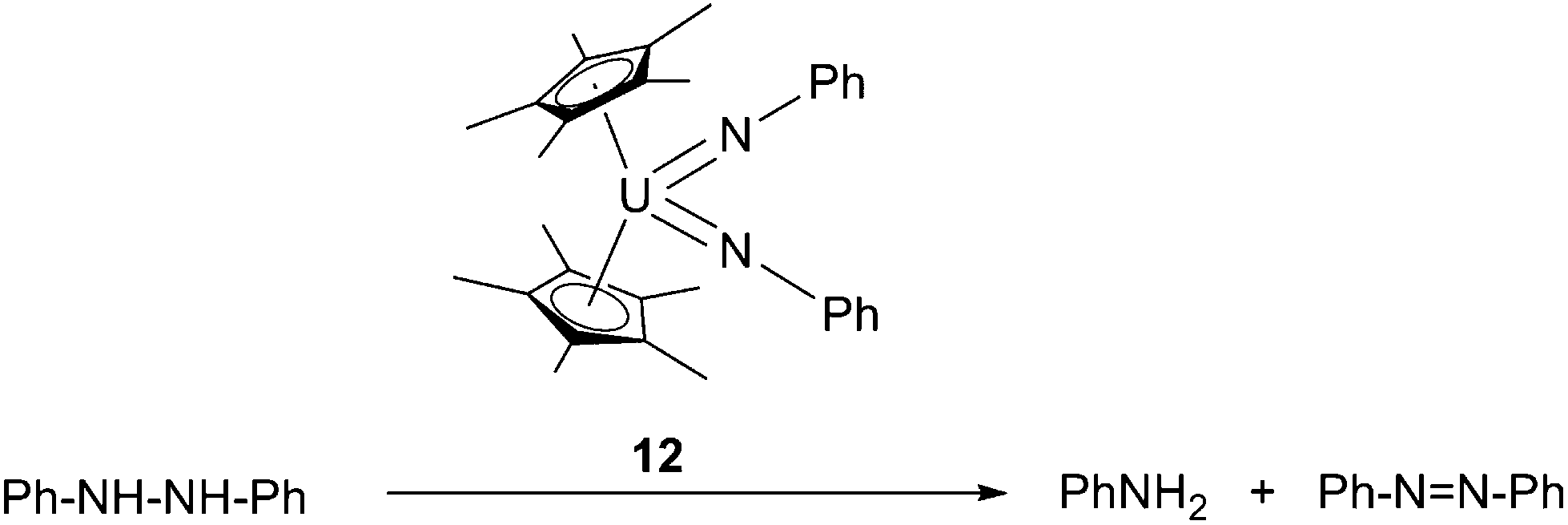 | ||
| Scheme 10 Catalytic conversion of hydrazine into aniline and azobenzene.21 | ||
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond have been reported.22 One reason for this scarcity, in addition to the strength of NN bond, is the fact that the conversion of N2 to two nitrides (N3−) requires a 6-electron redox process overall. Uranium compounds have exhibited significant potential to activate N2. The general strategy utilises low-valent and thus reducing uranium centres to reduce N2 by populating the anti-bonding orbitals of N2 thus weakening the NN bond.23 The versatile range of oxidation states of uranium renders it a most promising f-element to mediate N2 cleavage reaction.
N bond have been reported.22 One reason for this scarcity, in addition to the strength of NN bond, is the fact that the conversion of N2 to two nitrides (N3−) requires a 6-electron redox process overall. Uranium compounds have exhibited significant potential to activate N2. The general strategy utilises low-valent and thus reducing uranium centres to reduce N2 by populating the anti-bonding orbitals of N2 thus weakening the NN bond.23 The versatile range of oxidation states of uranium renders it a most promising f-element to mediate N2 cleavage reaction.
One of only two examples of uranium-mediated complete cleavage of the NN bond was reported by Gambarotta and co-workers in 2002.24 The U(III) calix-tetrapyrrole compound 16 reacted with N2, with the assistance of [K(naphthalenide)], to produce a U(V)/U(IV) dinuclear mixed-valent compound 17, which has two anionic μ-nitrido (N3−) ligands (Scheme 11a). X-ray crystallographic characterisation of 17 revealed that the anion is centro-symmetric, and the two uranium centres are equivalent to each other. The N⋯N distance between the two nitrido centres is too long for there to be any N–N interaction. The overall formal redox couple of the reaction can be found in Scheme 11b. It is noteworthy that both the U(III) compound 16 and [K(naphthalenide)] on their own cannot reduce N2, thus a U(II) intermediate and/or a kind of cooperation between the alkali metal centre and the uranium centre is plausible for the unique reactivity.
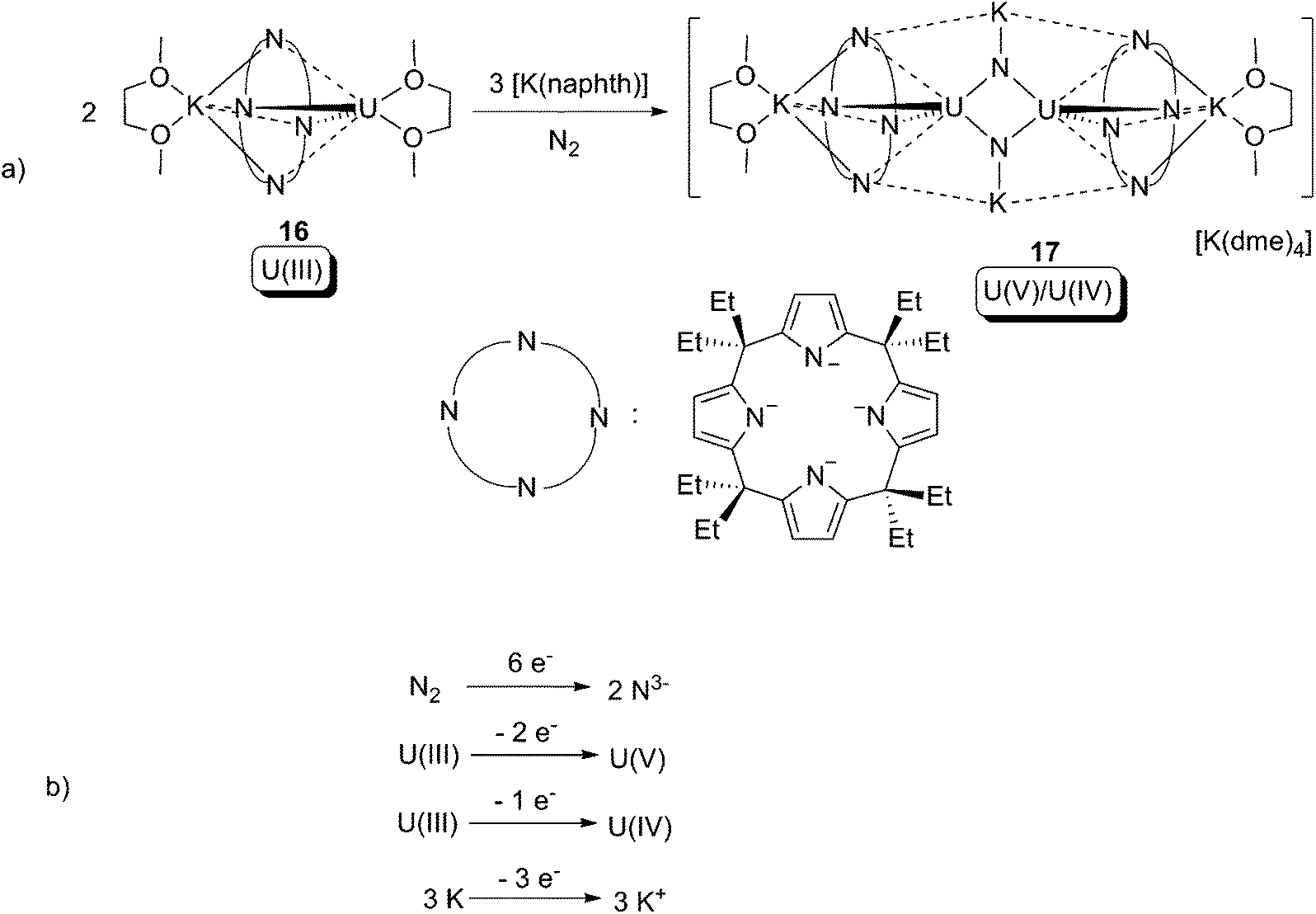 | ||
| Scheme 11 (a) Oxidative addition of the NN bond in N2 to uranium, and (b) the overall formal redox couple.24 | ||
The other instance of oxidative addition of low-valent uranium towards N2 can be found in Scheme 12. The U(III) compound [Cp*2U(BPh4)] (18) was reduced by KC8 in thf under an N2 atmosphere, affording a single crystal which was proven to be a U(IV) nitride 19 by a combination of DFT and X-ray crystallography. Unfortunately the reaction was not reproducible so neither spectroscopic nor elemental analysis could be provided for 19.25
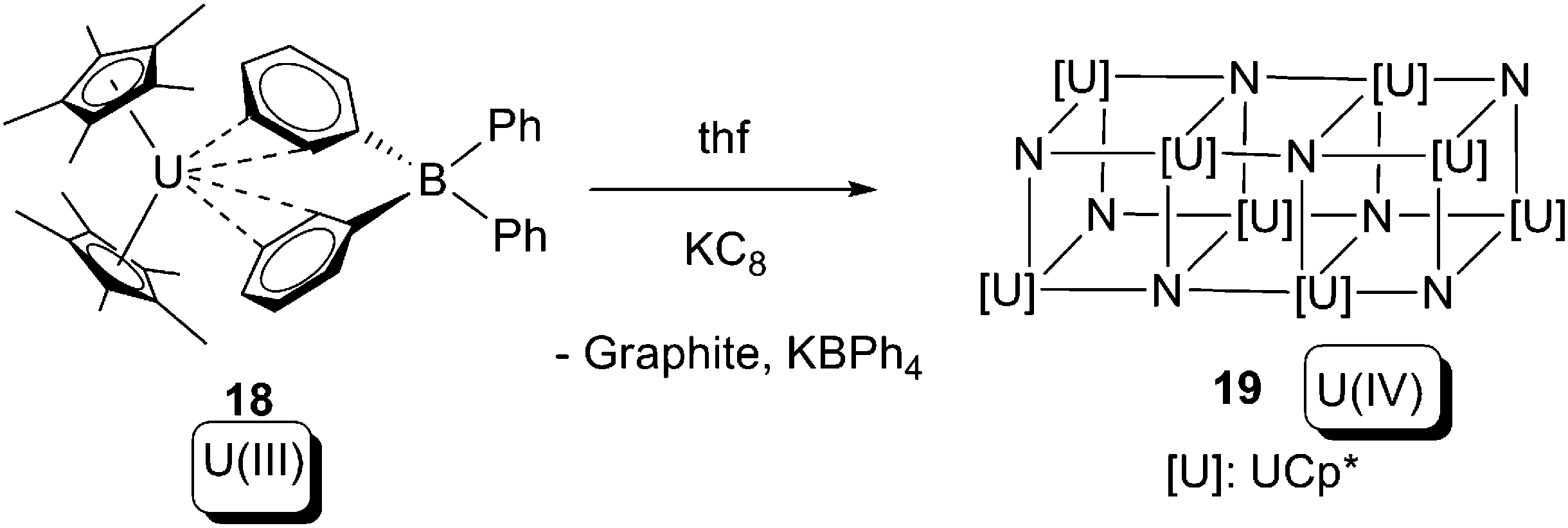 | ||
| Scheme 12 Oxidative addition of N2 to form a U(IV) nitride cluster.25 | ||
E multiple bonds, as well as 1-electron free-radical H-atom abstraction reactions.6c Classical oxidative addition of a low-valent f-block metal centre towards a C–H bond is still elusive, although a U(III)⋯H–C σ-complex has been reported.27
In 2008, Evans and co-workers reported a formal oxidative addition of a C–H bond at a U(III) centre.28 The U(III) hydride dimer [Cp*2U(μ-H)]2 (20), which was produced from a reversible bimetallic reductive elimination of H2 from a U(IV) hydride (vide infra),10c was reported to be able to activate the C–H bond of a methyl group on the Cp* ligand (Scheme 13). The formation of the U(IV) dinuclear tuck-in tuck-over compound (21) was confirmed by X-ray crystallography, and the H2 was probed by measuring the gas evolution by Toepler pump. Isotopic labelling experiments using [Cp*2U(μ-D)]2 (20-D) were hampered by H-D exchanging between protons of –CH3 and U–D.10c As a net result, the reaction leads to: (i) increase of uranium oxidation state by +1 for each of the two U centres; (ii) cleavage of two C–H bonds; and (iii) formation of two covalent U–CH2 bonds and two covalent U–H bonds. Thus, this reaction can formally be classified as an oxidative addition. The mechanism of the reaction is still ambiguous, but a U(II) intermediate is plausible, which would be produced by a U(III)/U(II) reductive elimination of H2 from 20, followed by U(II)/U(IV) oxidative addition towards C–H bonds to yield 21 (Scheme 14).
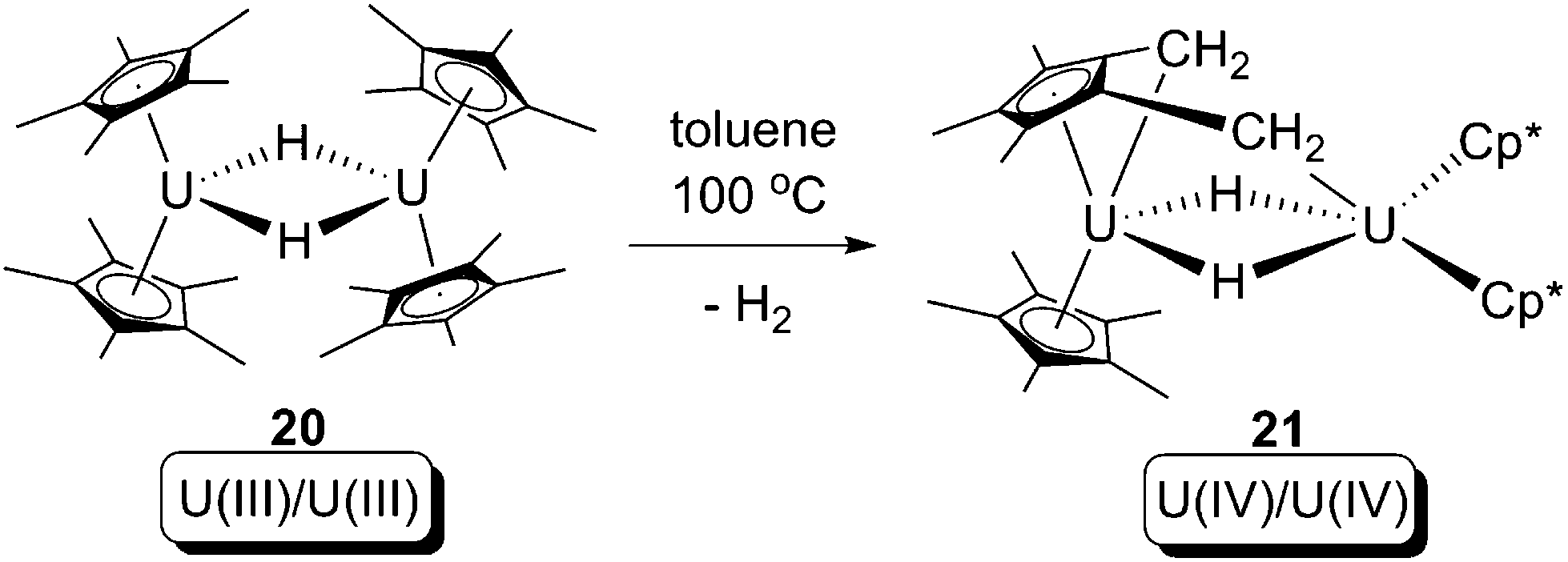 | ||
| Scheme 13 Oxidative addition of a U(III) hydride by a Cp* C–H bond.28 | ||
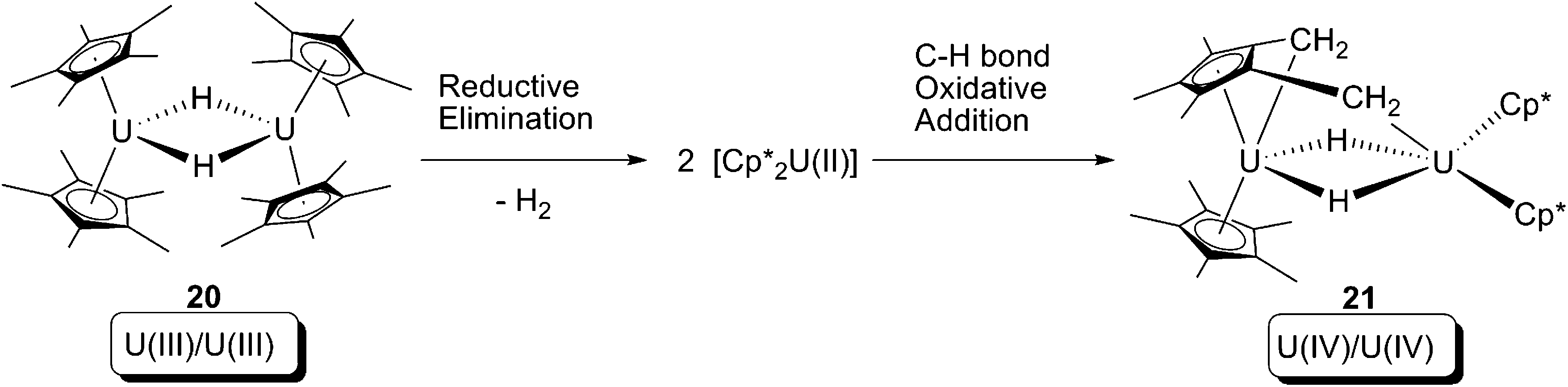 | ||
| Scheme 14 Possible mechanism of producing of 21, via a U(III)/U(II) reductive elimination and subsequently U(II)/U(IV) oxidative addition by a Cp* C–H bond. | ||
Although it does not fit the definition of oxidative addition, an example of ‘oxidative elimination’ of H2 from a U(III) hydroxide [Cp′′2U(μ-OH)]2 (Cp′′ = 1,3-(Me3Si)2C5H3) to yield U(IV) oxo [Cp′′2U(μ-O)]2 merits a mention here.29 The reaction was postulated to proceed via slow formation of a U(III)/U(IV) mixed-valent hydroxide oxo hydride [Cp′′2U(μ-O)(μ-OH)U(H)Cp′′2] species, which rapidly decays to the final product. The proposed mechanism was supported by kinetic data and isotopic labeling experiments.
An example of a more clear-cut uranium-mediated P4 activation that can be conclusively classified as oxidative addition was reported in 2011. Cloke, Green, and co-workers observed that the U(III) pentamethylcyclopentadienyl cyclooctatetraenyl complex [U(Cp*)(η8-C8H6-1,4-(SiiPr3)2)(thf)] (22) reacts with 0.5 equivalents of P4, producing a single product 23 (Scheme 15).31 The structure of 23 was comprehensively studied by X-ray crystallography as well as DFT computational methods: the planar, square P4 moiety was proven to be a dianion (P4)2−, and the oxidation states of both of the two uranium centres are +4. The reaction fulfils criteria for oxidative addition by: (i) cleavage of two P–P bonds in P4; (ii) increase of each uranium oxidation state by +1; (iii) formation of two new U–P covalent bonds per uranium ion.
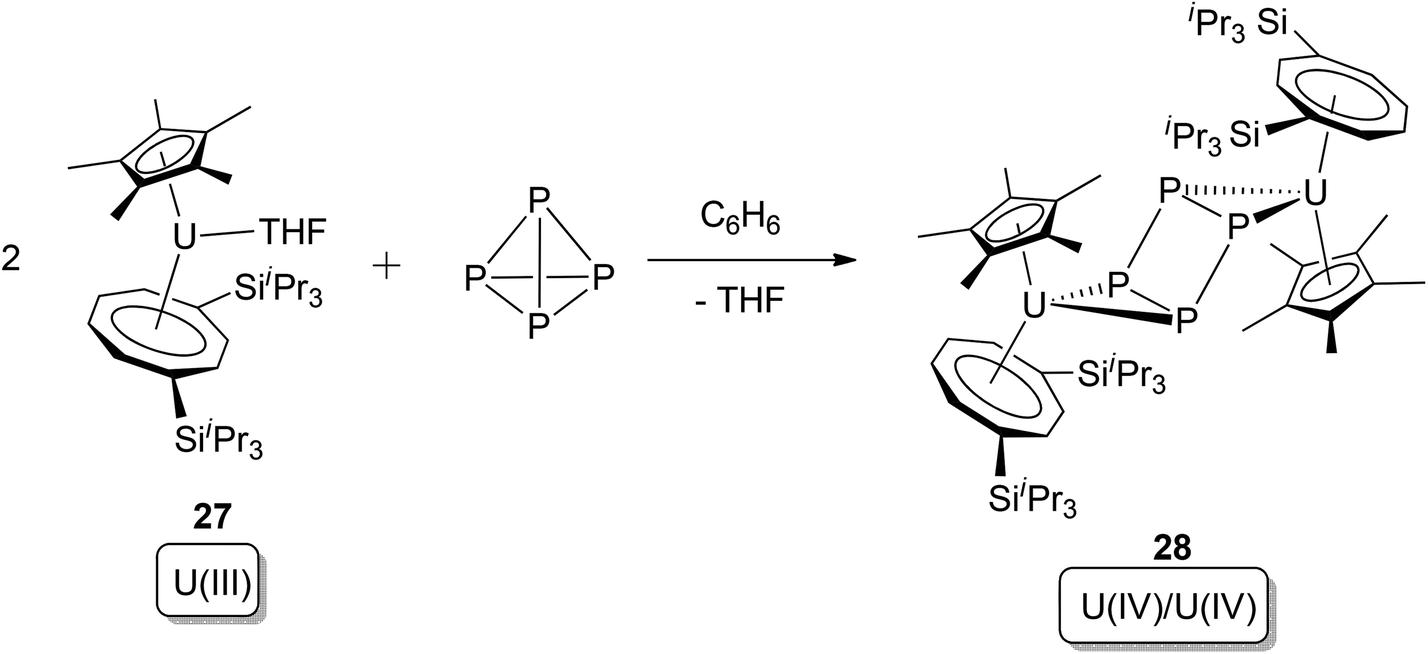 | ||
| Scheme 15 Oxidative addition of the U(III) complex 22 by P4.31 | ||
Some other oxidations of low-valent uranium compounds by group 16 elements (S, Se, Te) or equivalent reagents ({[K(18-crown-6)]2[Te2]}) have been reported, with the formation of high-valent uranium species with UE/U–E bonds.32 These reactions are oxidations, because they feature an increase of uranium oxidation state (by +2 or +1) and formation(s) of UE or U–E bond(s), but E⋯E interactions usually remain in the products.
2.2. Oxidative addition involving redox non-innocent ligands or other oxidisable ligands
The aforementioned examples of oxidative addition share a common character: uranium, but not the ligand(s), acts as the only electron donor. As a result, only the oxidation state of the uranium increases. In the following section, we discuss uranium-mediated oxidative addition where ligands act as electron donors. The introduction of redox non-innocent ligands into actinide organometallic chemistry, as well as contributions concerning ‘sterically induced reduction’ (SIR),33 have enabled this type of reaction to be possible and flourish in recently years.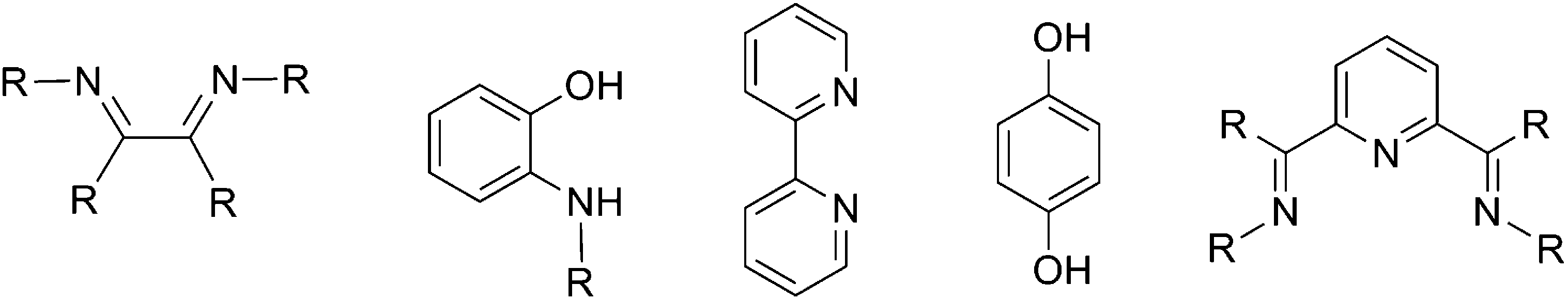 | ||
| Fig. 1 Representative redox non-innocent (precursor) ligands. | ||
In 2011, Bart and co-workers reported the reaction between a bis-(ene-α-diamide) U(IV) compound 24 and iodomethane (Scheme 16).36 In this reaction, the C–I bond in iodomethane is cleaved, whilst the uranium oxidation state does not change. One of the two redox non-innocent ene-α-diamide ligands in 24 is oxidised from its dianionic form (L2−) to a methylated monoanion form (MeL1−), with concomitant C–C bond formation. Although the reaction in Scheme 16 cannot be clearly defined as an oxidative addition, it does demonstrate the potential of a redox non-innocent ligand to take part in novel redox bond cleavage and formation reactions that are essential steps for a genuine oxidative addition.
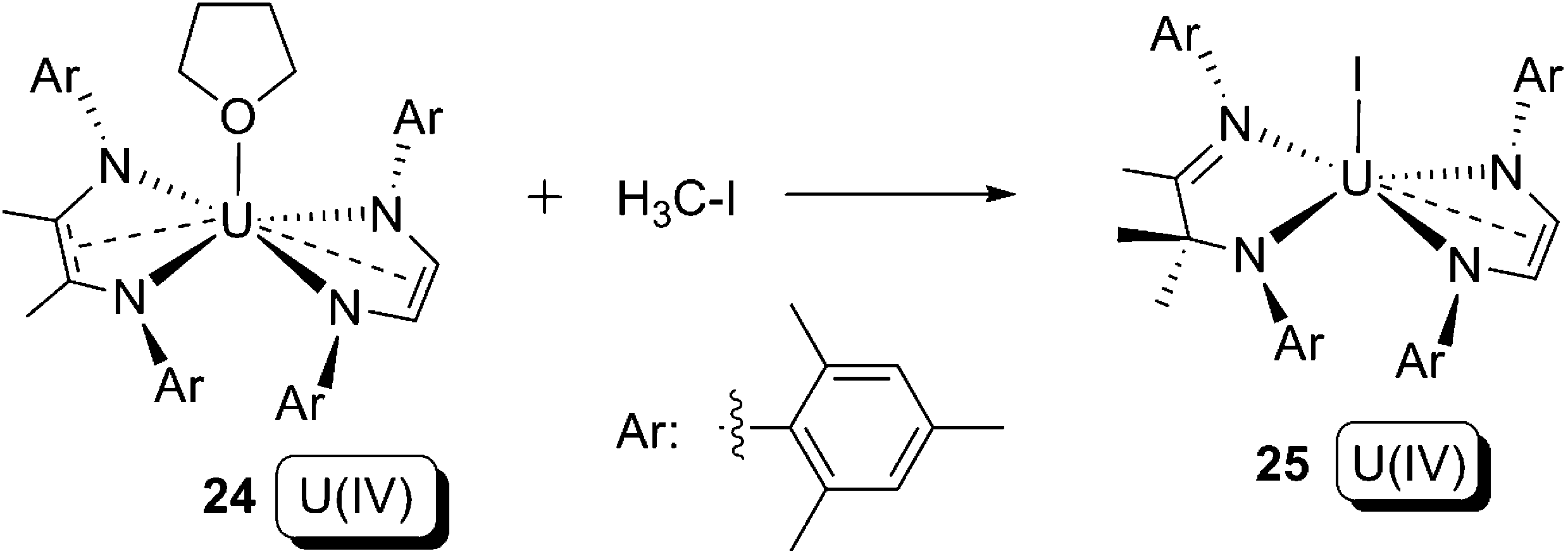 | ||
| Scheme 16 Reaction between bis-(α-diimine) U(IV) compound 24 and iodomethane.36 | ||
ortho-Iminoquinone is a relative of α-diimine, and three redox states are available for this ligand (Scheme 17). The U(IV) bis-amidophenolate compounds (series 26) were synthesised from salt elimination between UCl4 and the ligand bis-alkali metal salt.37 Compound series 26 were treated with PhICl2 or I2, resulting in the appearance of formal oxidative addition by the halogens (Scheme 18). However, the uranium oxidation states in the reactants and products are all the same (+4), while the ligands were oxidised from L2− (in series 26) to L1− (series 27 and 28) and so these reactions are not true type (a) oxidative additions. These reactions demonstrate that the L2−/L1− redox couple is more favourable than U(IV)/U(VI) or U(IV)/U(V) oxidations. The reaction to form 28 (Scheme 18b) is the closest to a clear-cut formal oxidative addition with the cleavage of an I–I bond and formation of two U–I bonds (but recall the uranium oxidation state does not change), whilst the reactions to form the 27 series (Scheme 18a) are more appropriately described as 1-electron chloride abstractions, since although two I–Cl bonds are broken there is no Cl–Cl bond in PhICl2 to be cleaved.38
 | ||
| Scheme 17 Different redox state structures of ortho-amidophenolate metal complexes. | ||
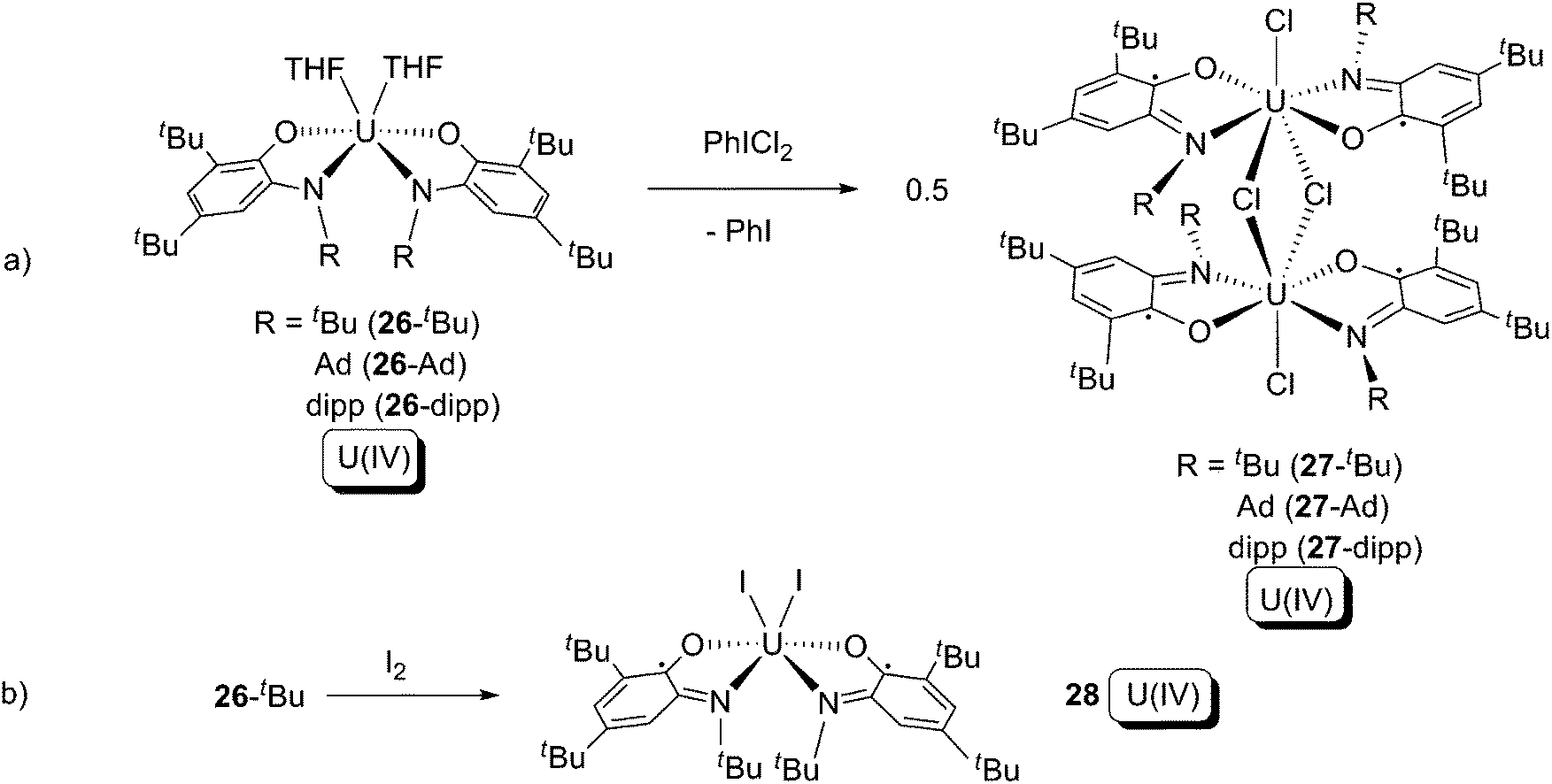 | ||
| Scheme 18 Formal oxidative addition of uranium by halogens in the presence of ortho-amidophenolate ligands.37 | ||
The pyridine(diimine) (PDI) ligand is another prevalent class of redox non-innocent ligand for transition-metal organometallic chemistry (Scheme 19). The capability of the PDI to host up to 4 electrons in combination with uranium has led to unique and fascinating chemistry.39
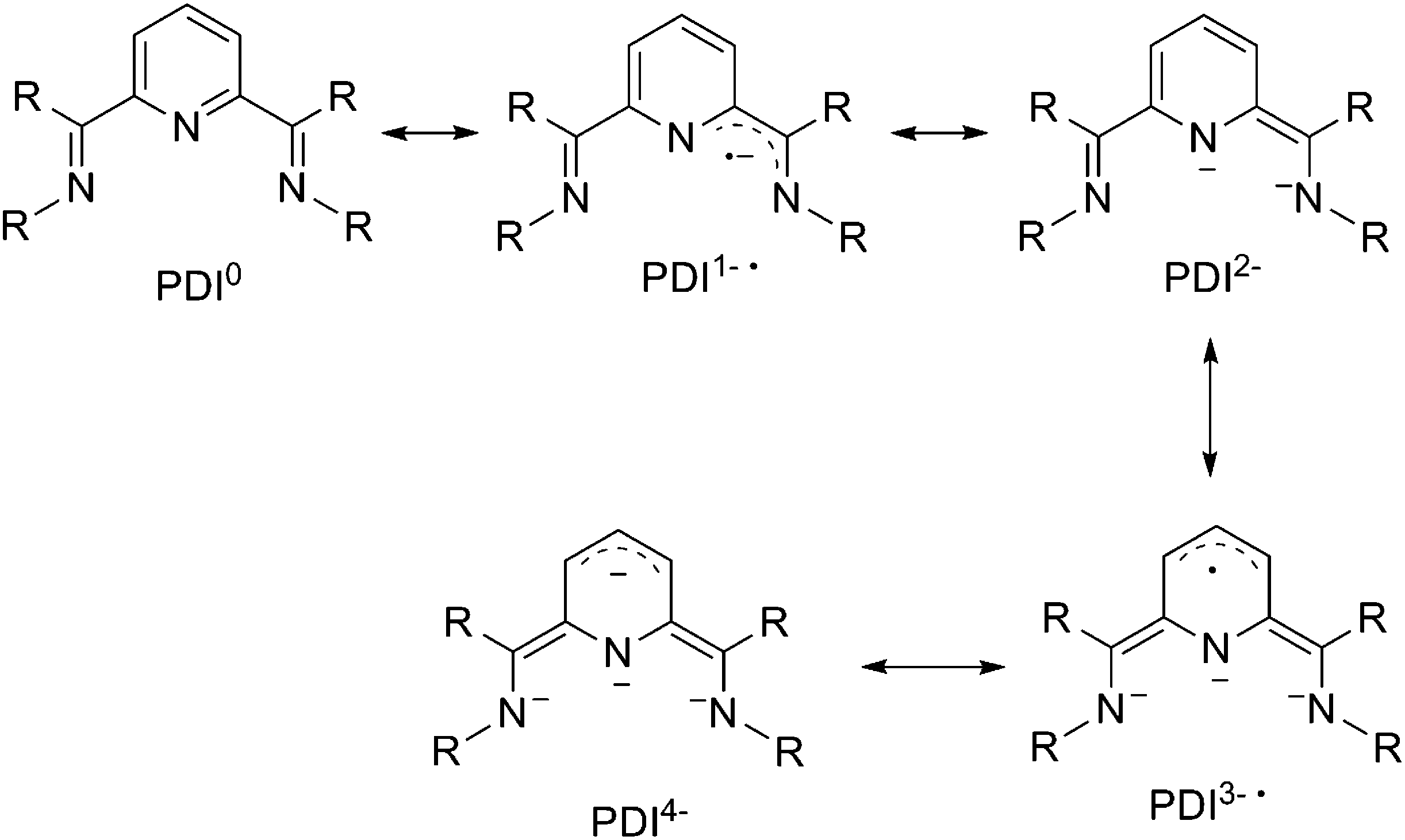 | ||
| Scheme 19 Different redox state structures of the PDI ligand. | ||
Very recently, the U(IV) compound 29 bearing a PDI3− ligand was synthesised by Bart and co-workers.40 Upon treatment with azobenzene, the U(V) bis-imide complex 30 containing a PDI0 ligand was produced (Scheme 20a). The overall 4-electron redox couple is summarised in Scheme 20b, where it can be seen that the uranium donates 1 out of the 4 requisite electrons, whilst the PDI ligand donates the other 3 electrons.
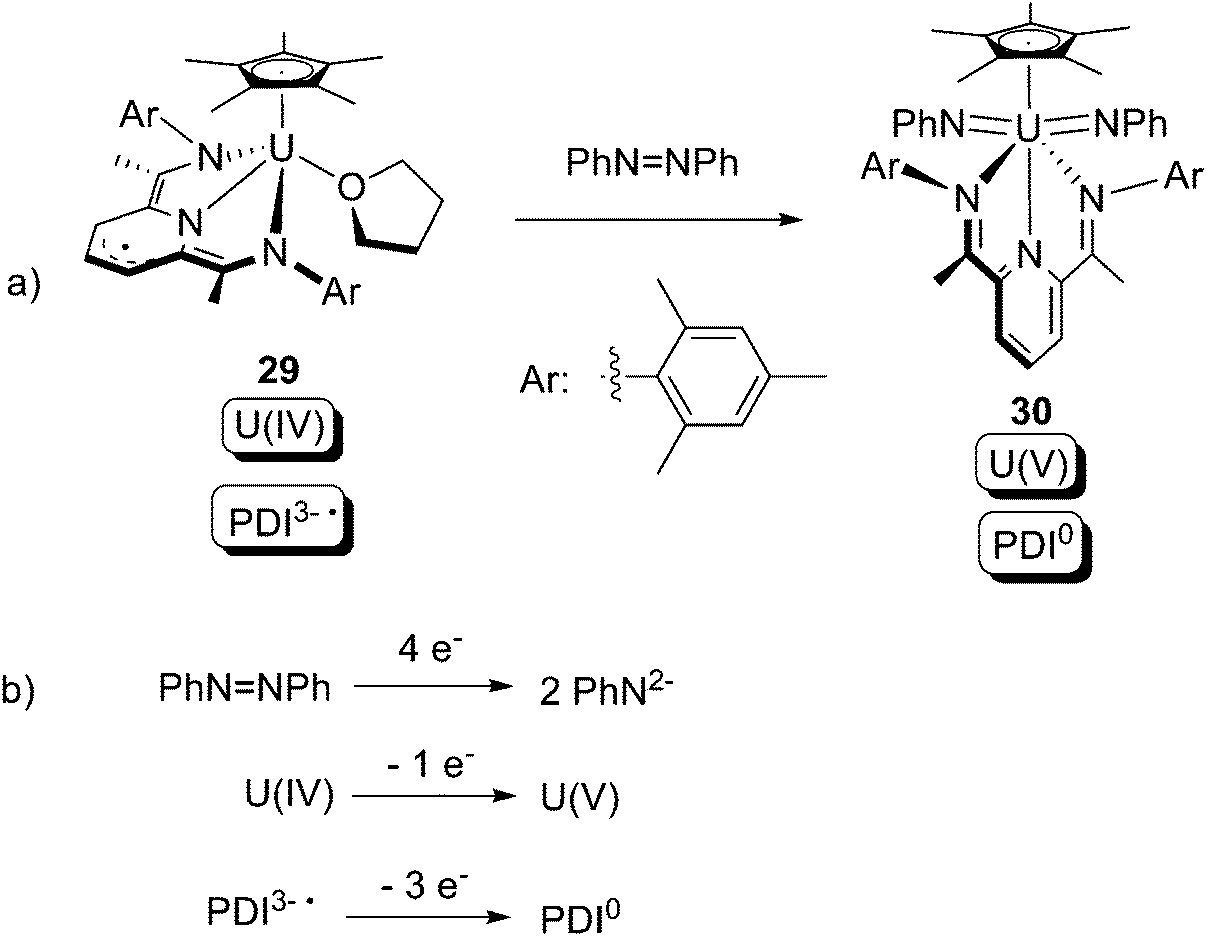 | ||
| Scheme 20 Oxidative addition of a U(IV) centre by PhNNPh, in cooperation with the redox non-innocent PDI ligand.40 | ||
Bart and co-workers subsequently reported that another U(IV) PDI1− compound, 31, which is akin to 29, is also capable of executing oxidative additions completely based on the non-innocent PDI ligand (Scheme 21).41 The uranium oxidation states (+4) do not change throughout the reactions, whilst the 2-electron donations are made by the PDI3− trianionic free-radical ligand in 31, which is converted to a PDI1− monoanionic free-radical ligand in the products 32–34. To elucidate the mechanism of the oxidative additions, a crossover reaction between 31 and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture PhS–SPh and PhSe–SePh was examined. A mixture of 34-S, 34-Se, and crossover product 34-S/Se were observed as products. Because ligand scrambling between 34-S and 34-Se was proven to be unlikely on the basis of control experiments, the formation of 34-S/Se is evidence of a free-radical mechanism.
:1 mixture PhS–SPh and PhSe–SePh was examined. A mixture of 34-S, 34-Se, and crossover product 34-S/Se were observed as products. Because ligand scrambling between 34-S and 34-Se was proven to be unlikely on the basis of control experiments, the formation of 34-S/Se is evidence of a free-radical mechanism.
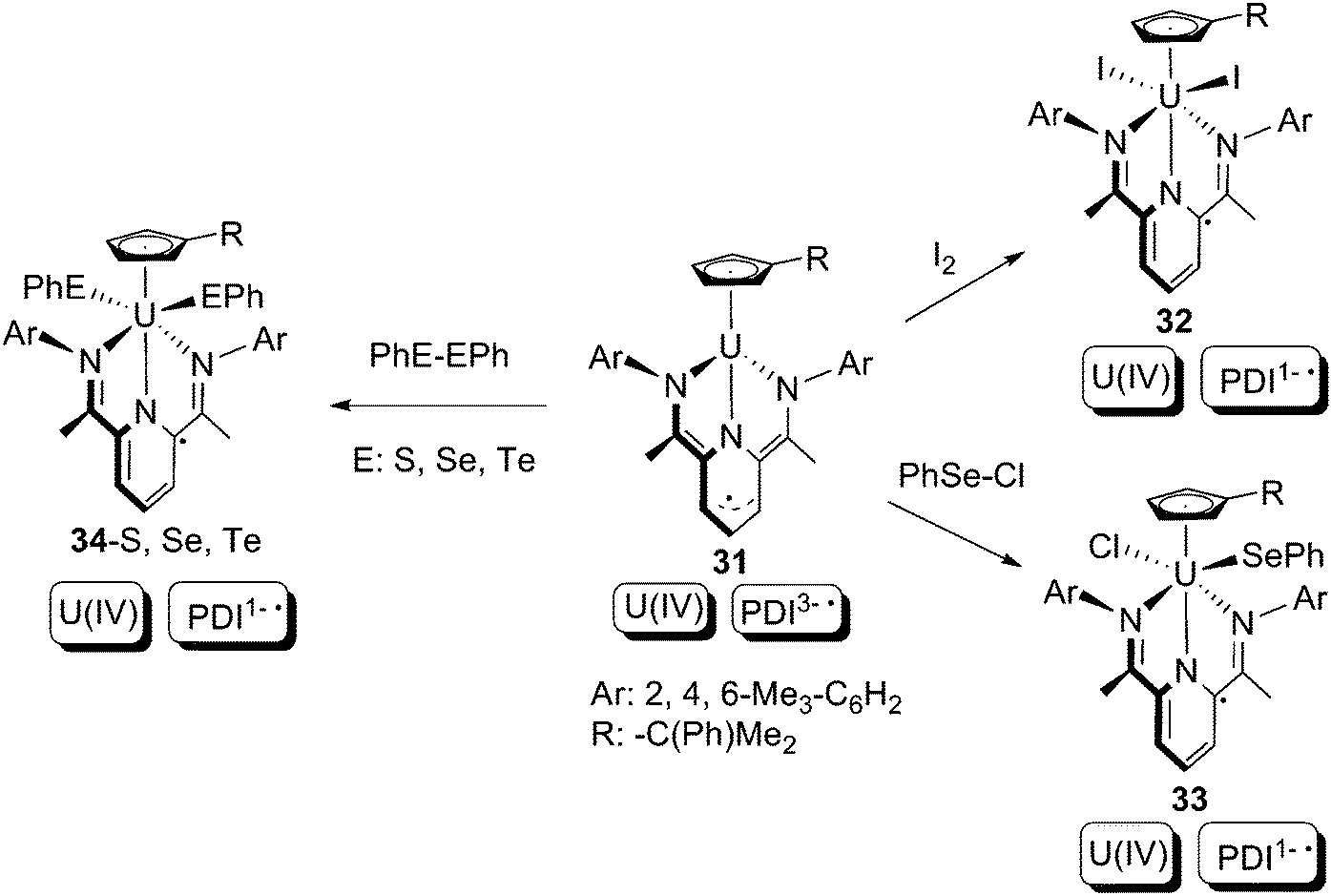 | ||
| Scheme 21 2-Electron oxidative addition based on redox non-innocent PDI ligands.41 | ||
The concept of SIR is also applicable to actinide compounds. In combination with the versatility of the range of oxidation states that uranium can adopt, multi-electron redox processes (>4-electron) are possible which can exceed the inherent 4-electron highest limit for the uranium-based redox couple (U(II)/U(VI), 4-electron).
The tris-pentamethylcyclopentadienyl U(III) compound [Cp*3U] (35) was first reported in 1997 by Evans and co-workers.46 The sterically-crowded 35 was found to be able to effect SIR towards the NN bond of azobenzene, to produce the known U(VI) bis-imide 12, along with Cp*2 (36) (Scheme 22).47 The overall 4-electron redox couple is listed in Scheme 22b. This oxidative addition led to cleavage of the NN bond, formation of two UN bonds, and an increase of uranium oxidation state from +3 to +6.
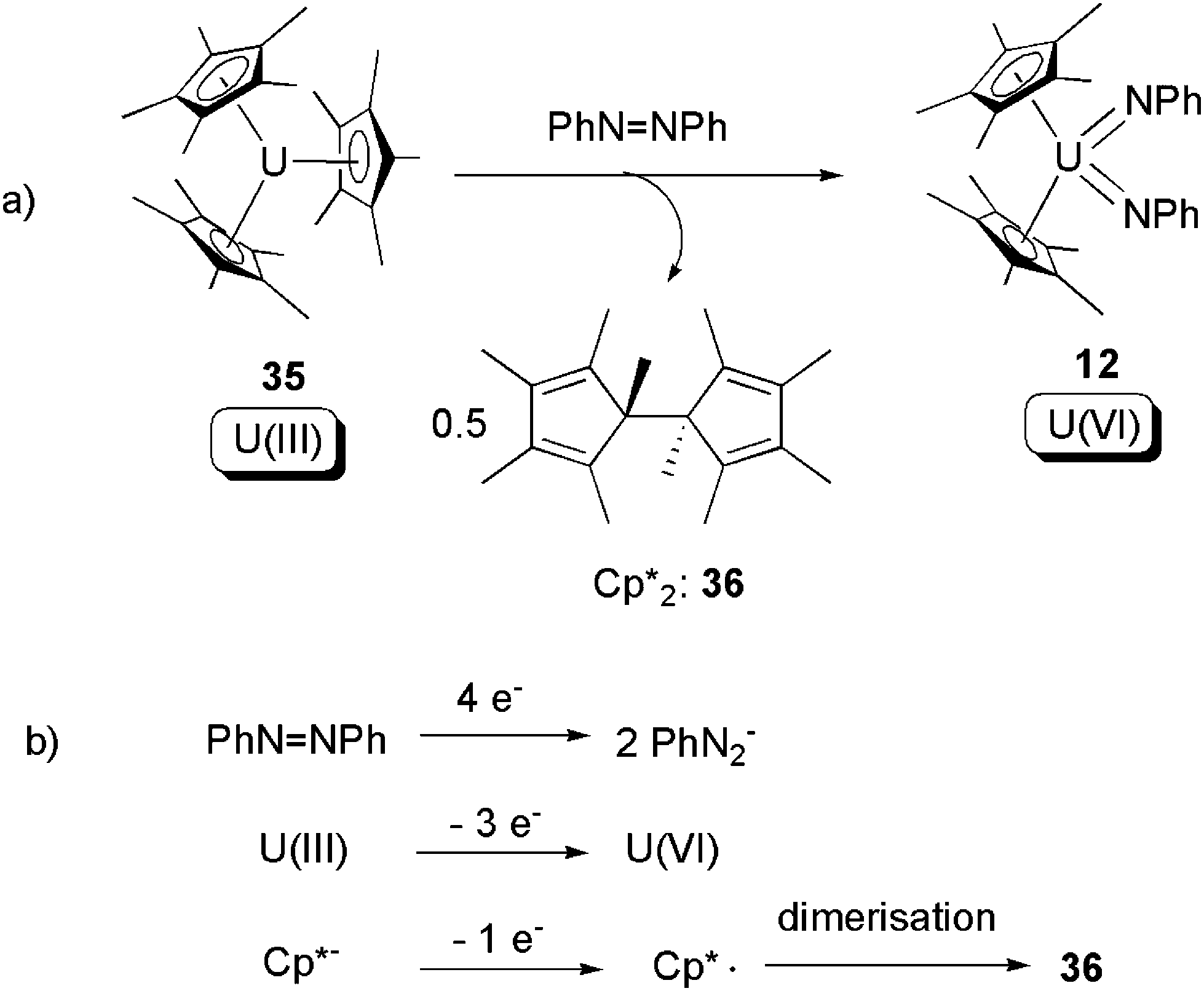 | ||
| Scheme 22 Oxidative addition of a U(III) compound by azobenzene, with the assistance of 1-electron donation from a Cp*− ligand.47 | ||
In the same paper, it was also found that along with Cp*−, both the dianionic arene ligand (C6H6)2− and even the conventionally inert (BPh4)− ligand can act as electron donors.47 These reactions and their overall redox couples can be found in Scheme 23. Complex 37 belongs to the uranium arene inverted-sandwich compound family, in which the arene anion (Arn−) was reported to act as multi-electron donor towards organic or organometallic substrates.48 But the tetra-phenyl borate anion (BPh4)− in 18 was generally regarded to be redox inert, although some instances of B–Ph bond cleavage have been reported.49
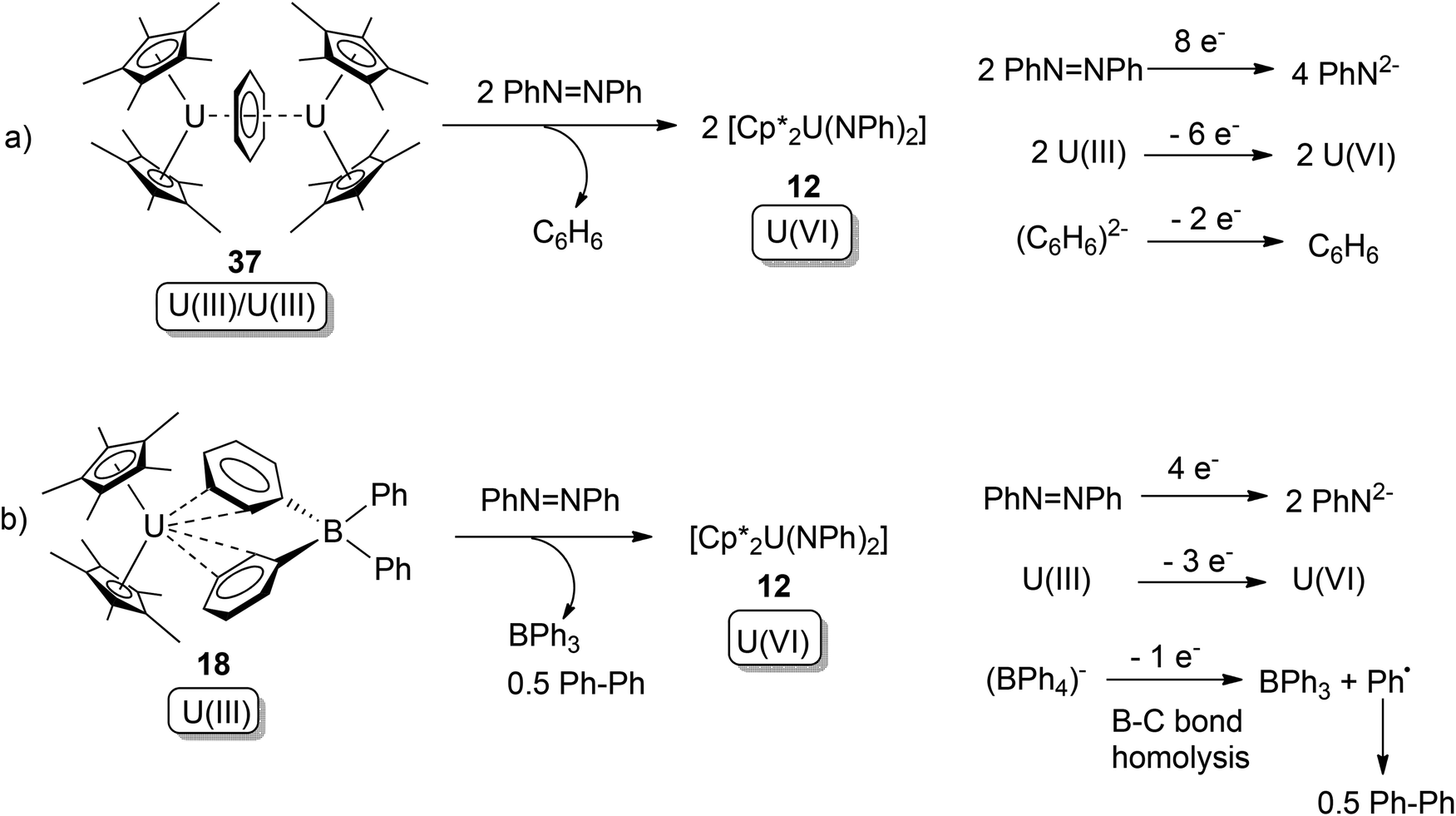 | ||
| Scheme 23 Oxidative additions of U(III) compounds by azobenzene, with (C6H6)2− and (BPh4)− acting as electron donors.47 | ||
The potential of (BPh4)− to act as a redox active ligand was further exploited shortly after the report of reactions in Scheme 23. In 2007, it was found that 38, which is closely related to 18, undergoes oxidative addition towards PhS–SPh. Here, both U(III) and (BPh4)− act as electron donors (Scheme 24).50 Further studies of lanthanide compounds consolidated the recognition of the (BPh4)− anion to act as a 1-electron donor via B–C bond cleavage to produce BPh3 and Ph as a free-radical.51
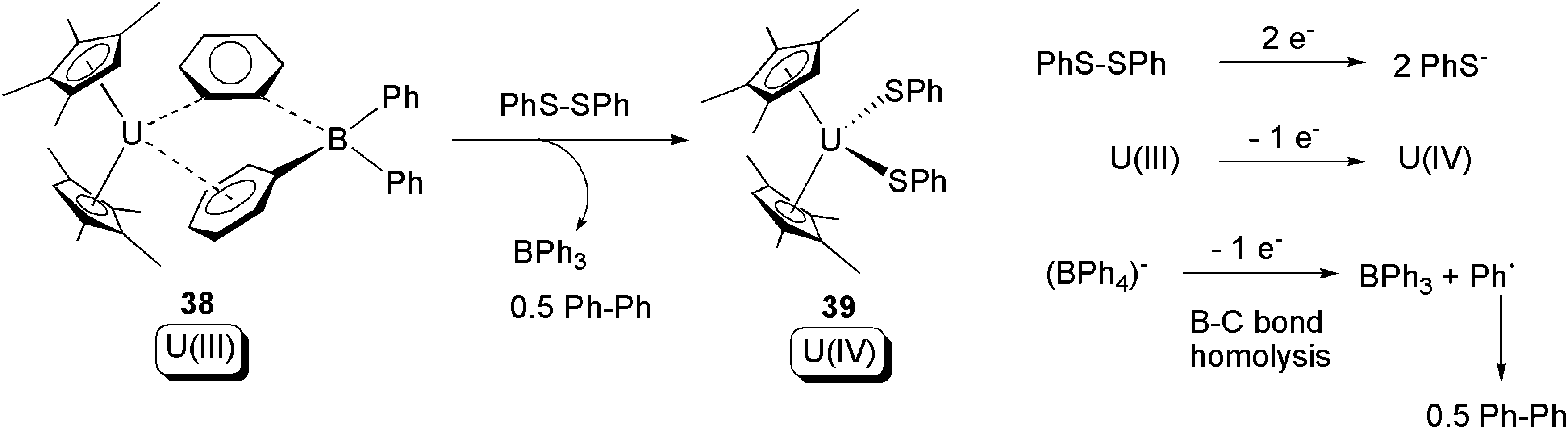 | ||
| Scheme 24 Oxidative addition of a U(III) compound by PhS–SPh, with the assistance of 1-electron donation from the (BPh4)− ligand.50 | ||
Hydride (H−) can also play a role as a 1-electron donor, to form free-radical H that couples to yield H2. In 2007, multi-electron (4-, 6- and 8-electron) redox reactions mediated by actinide hydrides were reported.52 In this work, the known U(III) hydride dimer [Cp*2U(μ-H)]2 (20) was found to be able to conduct overall 4- or 8-electron oxidative additions towards PhE–EPh (E: S, Se) or PhNNPh (Scheme 25).
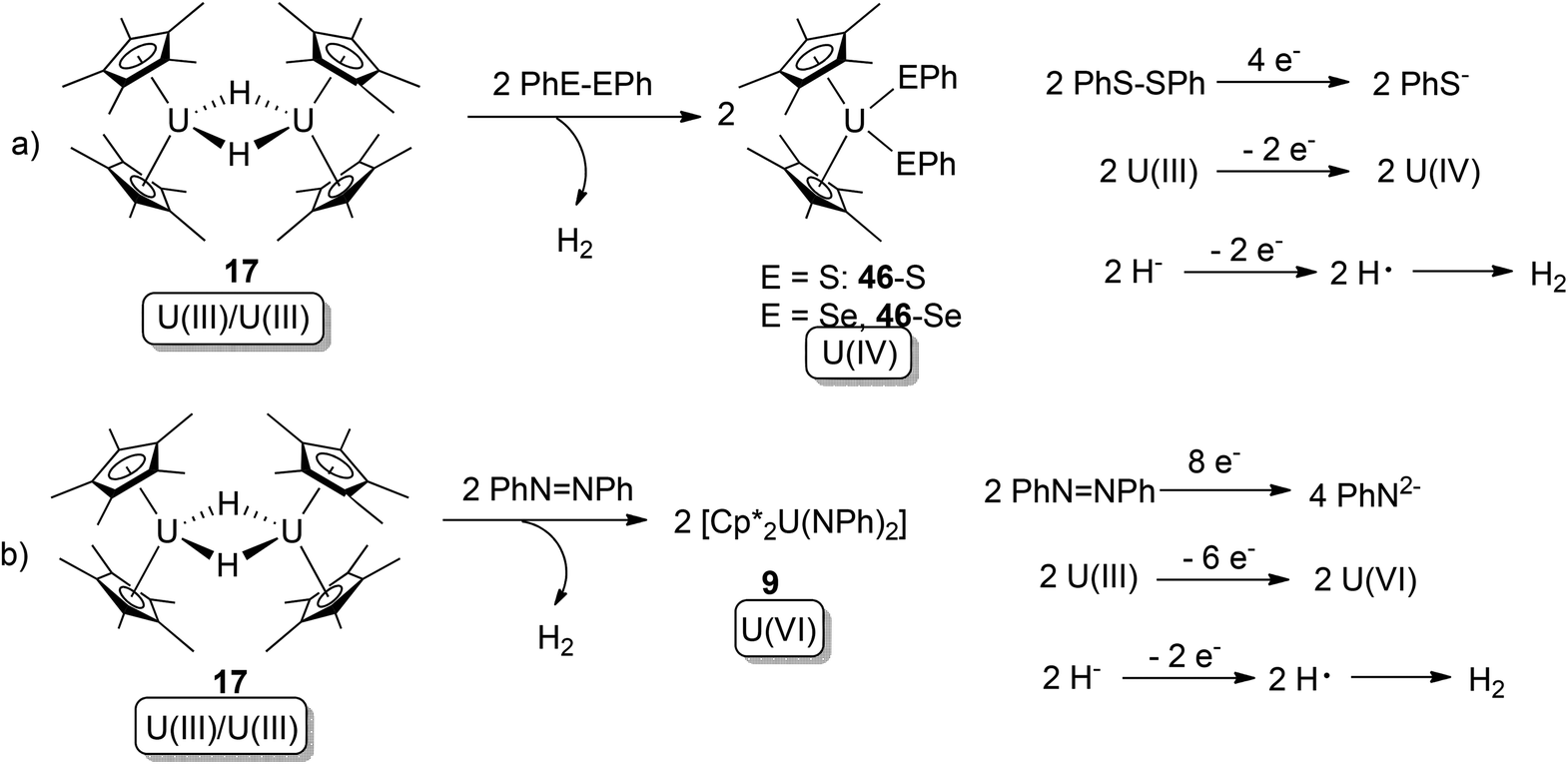 | ||
| Scheme 25 Oxidative addition of a U(III) hydride by PhE–EPh and PhNNPh, with the assistance of 1-electron donation from a H− ligand.52 | ||
3. Reductive elimination
According to Scheme 1, it follows that there are three criteria for a classical reductive elimination: (i) cleavage of M–X and M–Y bonds; (ii) formation of a X–Y bond; (iii) a decrease of the metal centre(s) oxidation state(s). For transition-metal organometallic chemistry, reductive elimination is widespread and usually exists as the reverse reaction of oxidative addition in a catalytic cycle. However, in the context of actinide organometallic chemistry, reductive elimination is quite elusive in comparison with oxidative addition. In the following section, instances of reductive elimination mediated by uranium compounds are summarised. A noteworthy trend in recent years is that redox non-innocent ligands are playing an increasingly important role in uranium-mediated reductive elimination.In 1981, as part of their work reporting the synthesis and properties of bis-pentamethylcyclopentadienyl actinide alkyls and hydrides, Marks and co-workers reported an equilibrium between [Cp*2U(H)(μ-H)]2 (41) and [Cp*2U(μ-H)]2 (20) (Scheme 26).10c Although the structures of these hydrides have not been characterised by X-ray/neutron diffraction until very recently,53 the reaction from left-hand-side to right-hand-side in Scheme 26 is the first example of reductive elimination for an actinide compound, which fits the definition of reductive elimination from all aspects: (i) cleavage of 2 U–H bonds; (ii) formation of an H–H bond; (iii) decrease of the oxidation state of each uranium from +4 to +3. This is a classical type (b) (bis-metallic) reductive elimination/oxidative addition couple according to Scheme 1.
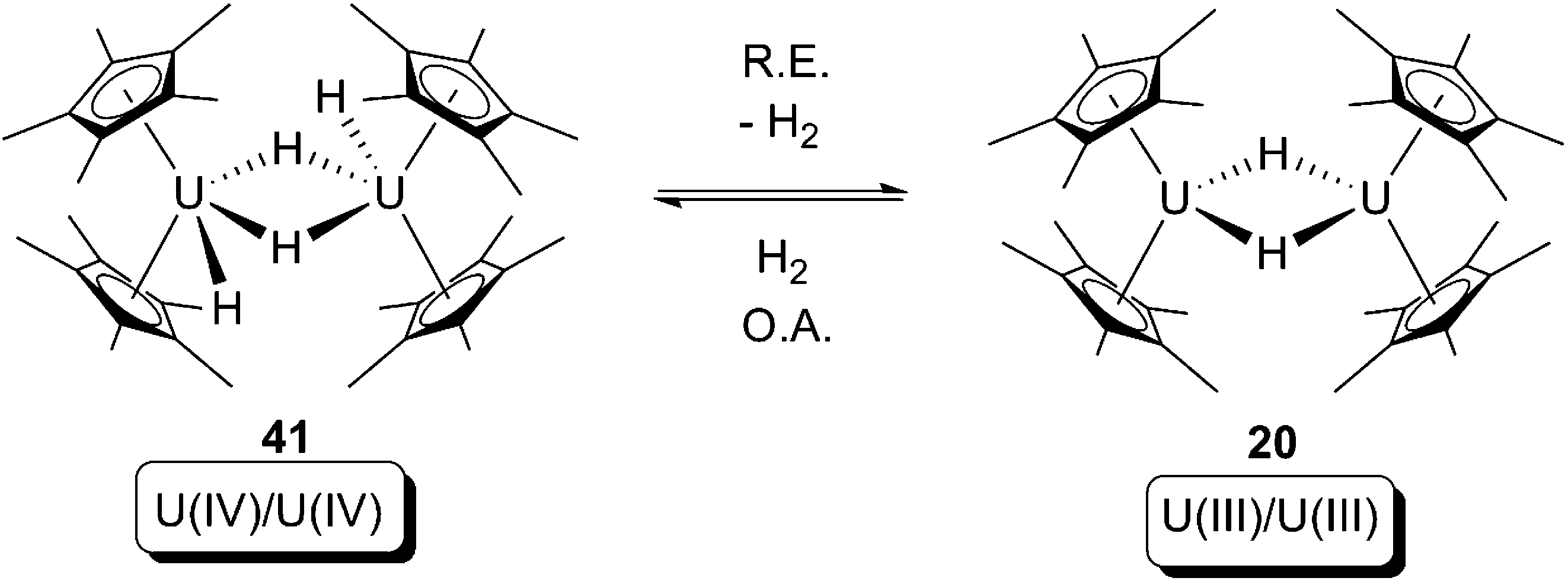 | ||
| Scheme 26 Bimetallic reductive elimination (R.E.)/oxidative addition (O.A.) equilibrium of U(IV) and U(III) hydrides.10c | ||
In 1982, Seyam and co-workers reported the reaction between uranyl dichloride [UO2Cl2] and 2 equivalents of phenyllithium at low temperature.10e The in situ generated UO2Ph2 species was allowed to warm to ambient temperature, yielding C–C bond-coupled biphenyl via a reductive elimination, along with UO2 (Scheme 27). However, neither the UO2Ph2 species nor the UO2 species were structurally authenticated. Nevertheless, the reaction fulfills all criteria of type (a) reductive elimination, and so far is the only genuine type (a) reductive elimination for any actinide compound. Other alkyllithium reagents (RLi, R = iPr, nBu, tBu, Me) were tested for the reaction, but instead of reductive eliminations, β-H elimination or free-radical H-abstraction were observed.
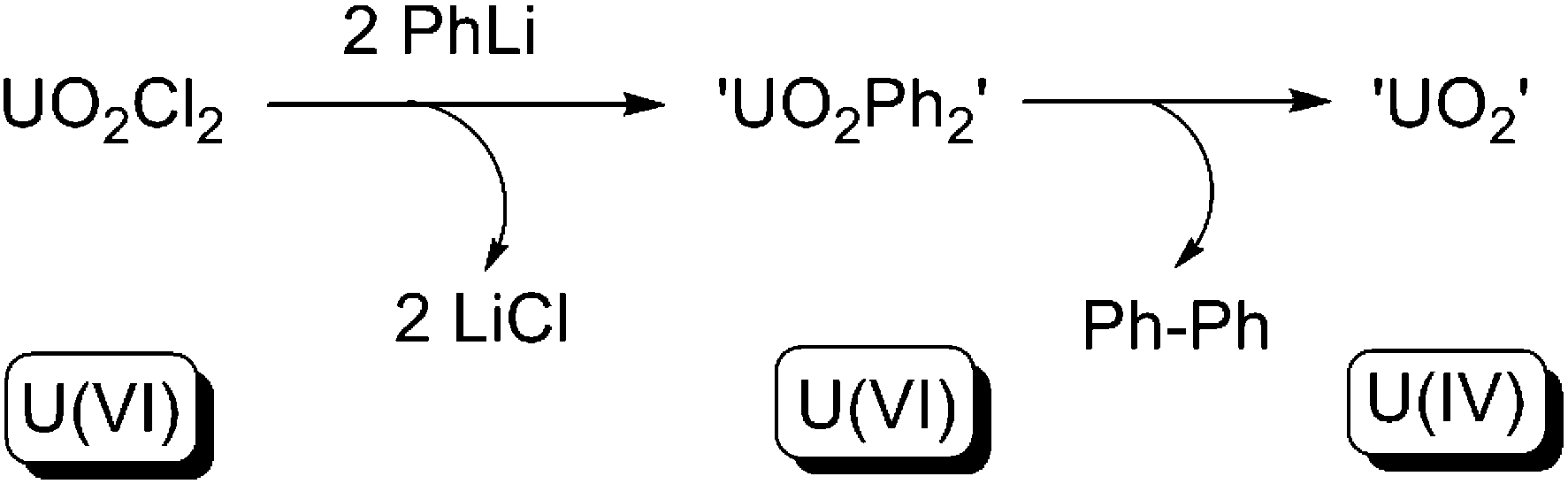 | ||
| Scheme 27 Reductive elimination from a U(VI) uranyl centre.10e | ||
After decades of dormancy, in 2012 Bart and co-workers reported a redox non-innocent ligand induced C–C bond forming reductive elimination from a U(IV) homoleptic alkyl (Scheme 28a).54 Reaction between the U(IV) tetra-benzyl compound 42 and α-diimine led to 43, which remains a U(IV) compound, along with oxidatively coupled PhCH2CH2Ph. The redox couple can be found in Scheme 28b. A noteworthy point here is that the U(IV) centre is not involved in the redox process. An isotope-labelling crossover experiment proved that the reductive elimination follows an intramolecular mechanism. Reactions between 42 and redox-inert ligands were also tested, but no reductive eliminations were observed.
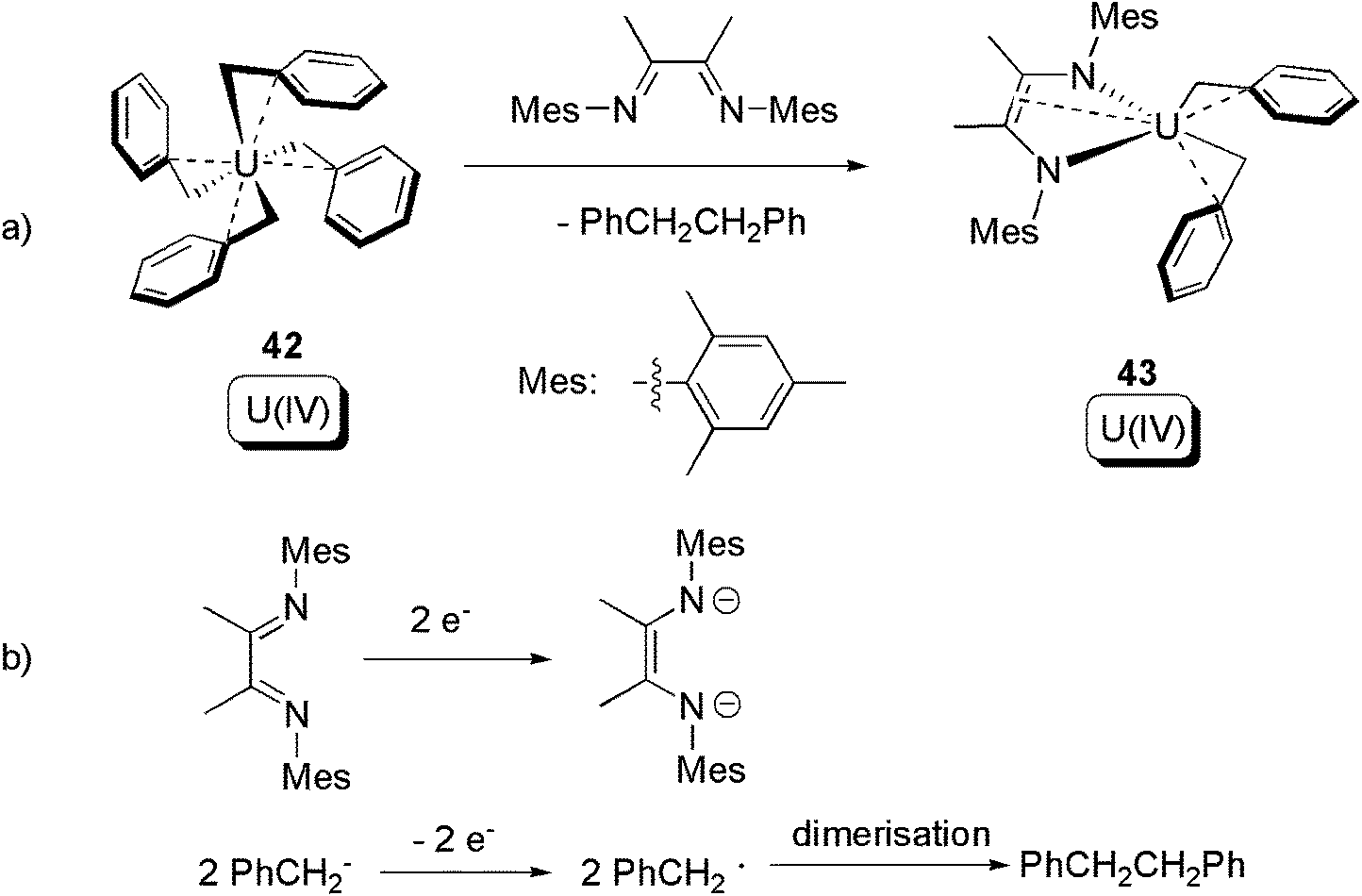 | ||
| Scheme 28 Reductive elimination from a U(IV) alkyl, with the assistance of a redox non-innocent α-diimine ligand.54 | ||
Beside the α-diimine ligand class, it was also found that iminoquinone (44) can induce similar reductive elimination of a C–C bond from 42 (Scheme 29) to give 45.55 An isotope labelling crossover experiment using [U(CD2C6D5)4] (42-D) revealed that the reaction occurs in distinct steps, and a U(IV) tris-benzyl with monoanionic free-radical ligand was postulated as the intermediate.
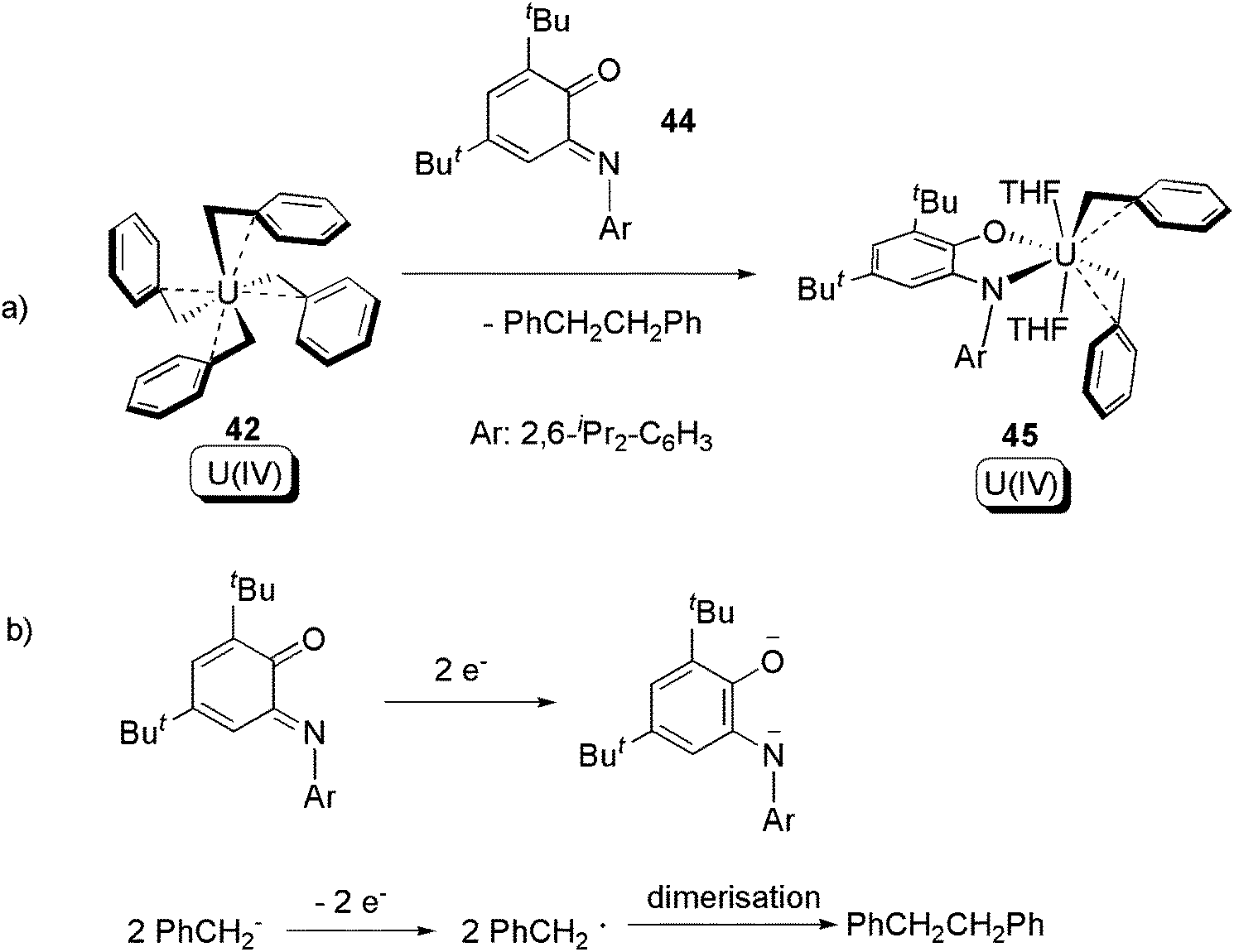 | ||
| Scheme 29 Reductive elimination from U(IV) tetra-benzyl, with the assistance of a redox non-innocent quinone ligand.55 | ||
A related reaction to the aforementioned non-innocent ligand-induced reductive eliminations was also reported by Bart and co-workers. In the presence of an organo-azide, the U(III) mono-alkyl complex 46, supported by two scorpionate hydrotris(pyrazolyl)borate ligands, was found to be able to yield bibenzyl as a result of intermolecular C–C coupling, as well as a U(IV) imide (47) (Scheme 30).56 The mechanism of these reactions was not discussed in the original work, but an initial intermolecular reductive elimination of 46 to produce PhCH2CH2Ph and a U(II) species is possible. The putative U(II) species could then be subsequently oxidised by azide to produce the U(IV) imide 47 and extruded N2. It should be noted, however, that this reaction defies any conventional classification because although oxidatively coupled bibenzyl is formed the uranium in fact undergoes a one-electron oxidation overall.
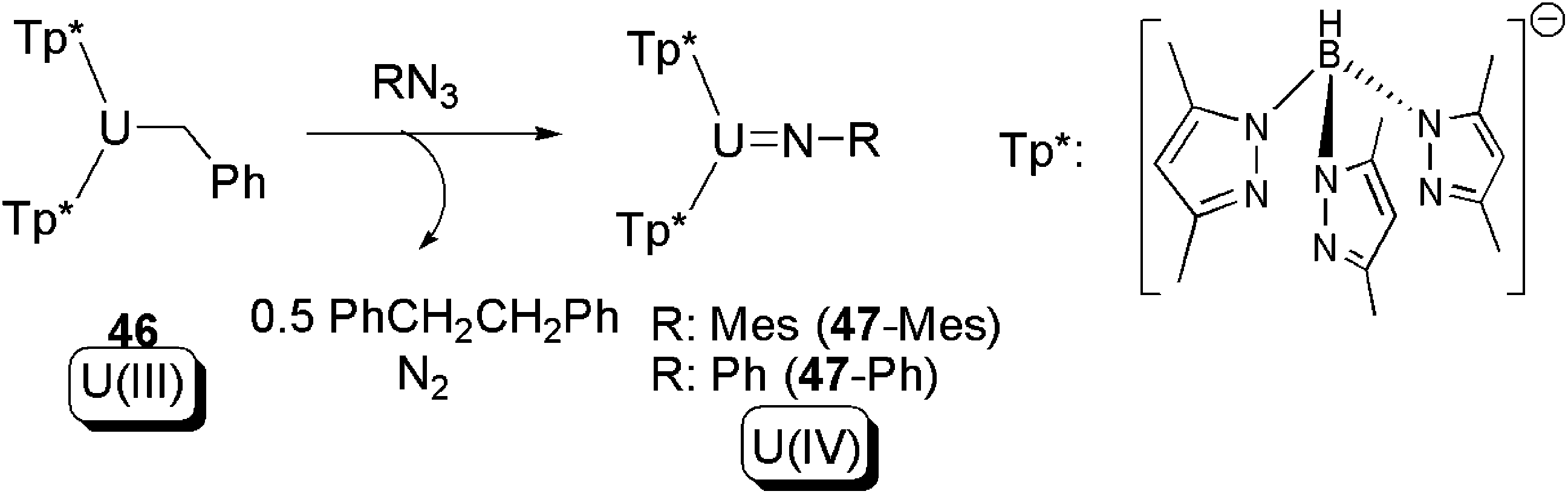 | ||
| Scheme 30 C–C bond coupling in a reaction between a U(III) alkyl and an organic azide.56 | ||
4. ‘Concerted’ reductive elimination/oxidative addition: bond formation and cleavage with no change of the uranium oxidation state
In many of the examples of oxidative addition/reductive elimination described above, the oxidation state of uranium does not change. Whilst most cases involve redox non-innocent ligands and the redox couples therein are not based on uranium at all, there are some instances where uranium is involved but its oxidation state is kept as a constant overall.Recent discoveries of the novel +2 oxidation state of actinide elements7,57 provides a new perspective from which to potentially view these redox reactions: an initial reductive elimination of a U(IV) centre to form a U(II) intermediate, which immediately undergoes an oxidative addition towards an incoming substrate (Scheme 31), thus can be rationalised as a concerted reductive elimination/oxidative addition. In this regard, the chemistry in Scheme 26 provides indirect evidence that such processes should be considered. However, it should be borne in mind that this hypothesis is currently limited to a theoretical construct and is open to debate since no supporting experimental evidence has been obtained.
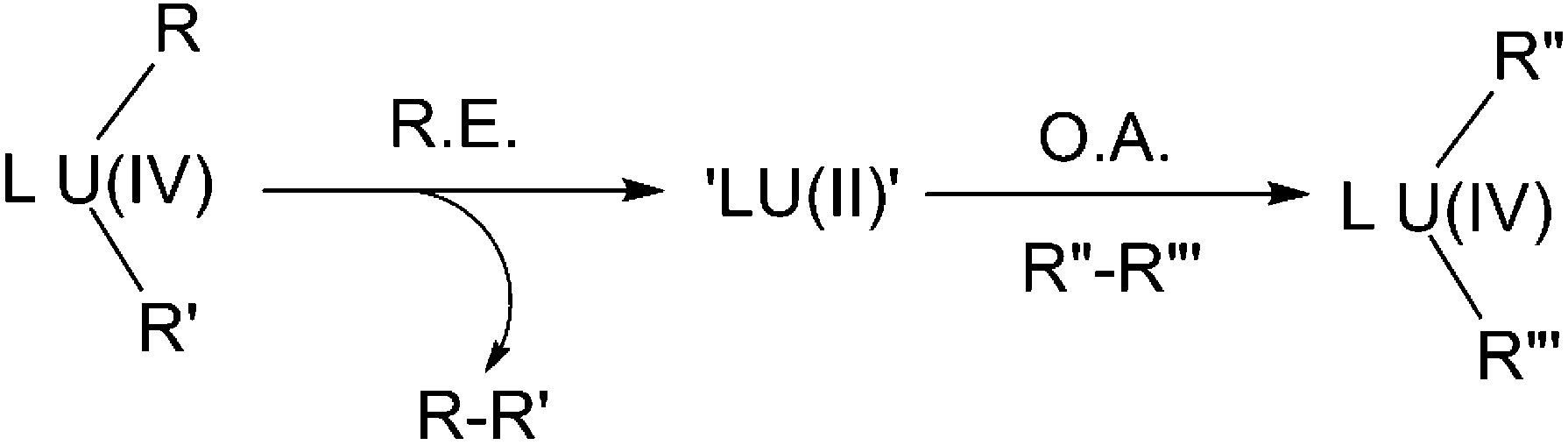 | ||
| Scheme 31 Concerted reductive elimination (R.E.)/oxidative addition (O.A.) via a hypothetical U(II) intermediate. | ||
In 2008, the U(IV) tuck-in tuck-over hydride 21 was found to be able to cleave the S–S bond of PhS–SPh, providing the U(IV) bis-phenylsulfide (40-S) (Scheme 32).28 The mechanism of the reaction was not clear according to the original paper, but concerted reductive elimination/oxidative addition is a plausible candidate: a reductive elimination to form C–H bond and a U(II) intermediate, which could then be followed by oxidative addition of a S–S bond.
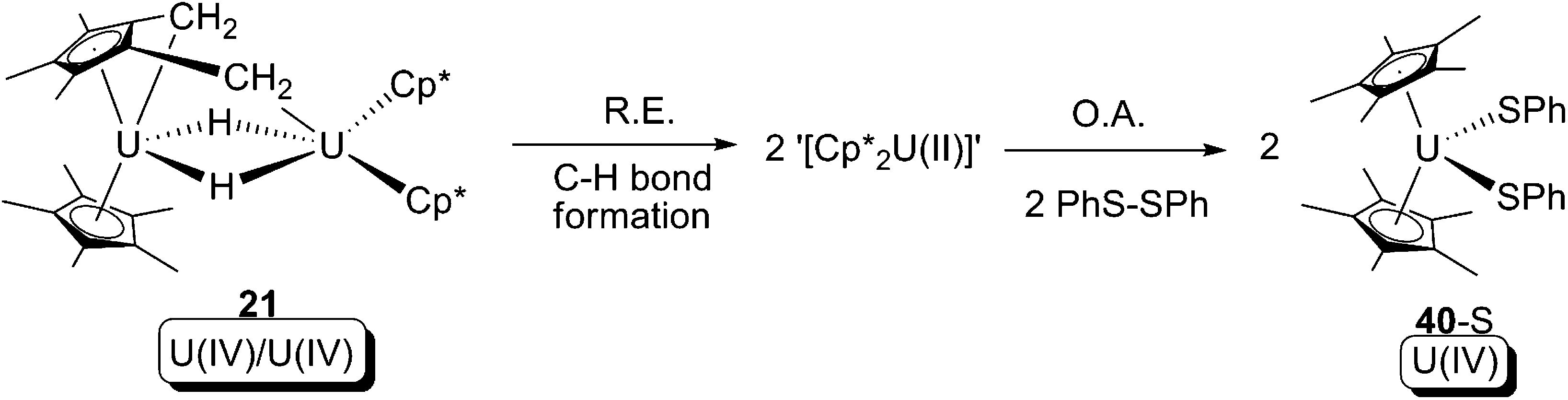 | ||
| Scheme 32 Concerted reductive elimination/oxidative elimination (R.E./O.A.) as a possible mechanism of formation of 40-S.28 | ||
5. Concluding remarks
After decades of limited progress, uranium-mediated oxidative additions/reductive eliminations are gathering increasing research interest, although the area is still in its infancy in comparison with transition-metal counterparts. The trend is significantly boosted by the introduction of sterically induced reduction (SIR), as well as the exploitation of redox non-innocent ligands.Among the pair of reactions, oxidative addition is better developed compared to reductive elimination. This fact is partially due to the significant reductive potential of low-valent uranium, e.g. U(III). On the other hand, reduction of high-valent uranium centres (in oxidation state of +6, +5, or +4) to lower oxidation state often requires harsh conditions (e.g. alkali metals), and is usually outside the scope of the conventional potential range of R−/R couples, which is essential for the occurrence of reductive elimination. Furthermore, stable high valent uranium polyalkyls, which would capable of performing reductive elimination, are sparse, and although lower valent polyalkyl uranium species are known reductive elimination from those complexes would give oxidation states of uranium that are inaccessible under normal conditions (e.g. I and II). Thus, there is a dearth of suitable complexes for such reactivity experimentally. As a result, the barriers to oxidative addition is much lower than that of the reductive elimination, and in most cases the reaction is irreversible. This is quite different from that in transition-metal chemistry, in which the reversible oxidative addition/reductive elimination is better balanced. To overcome the higher energy barrier for the reductive elimination, involving higher uranium oxidation states (in particular +6, which is quite oxidising, as proven by initial studies of uranyl bis-alkyls10e) redox non-innocent ligands deserve future research effort. A step further is uranium mediated reversible oxidative addition/reductive elimination, which is highly desirable and can only be possible if the energy barriers for each of the two reactions are comparable. In this regard, the reversible oxidative addition/reductive elimination of 41/20 are notable since this represents a reversible type (b) oxidative addition/reductive elimination couple. Despite the impressive array of oxidative addition/reductive elimination reactions that have emerged, it is important to note that a classical type (a) oxidative addition has not yet been observed, and thus a reversible type (a) oxidative addition/reductive elimination couple has not been realised. On the other hand, uranium often mediates transformations that have no precedent anywhere else.
Bond cleavage and formation is the very essence of chemistry. Thus, another point of concern to uranium-mediated oxidative addition/reductive elimination is the scope of substrates. So far the most prevalent substrates for oxidative addition are azobenzene and diphenyl disulfide (or, less commonly, diselenide or ditelluride): here the NN bond or E–E (E = S, Se, Te) bonds are either weak or bear energetically low-lying antibonding orbitals and are thus easy to reductively cleave. For reductive elimination, thermodynamically favourable C–C bond formation is the most prevalent, with the assistance of redox non-innocent ligands. Further endeavours must extend the substrate scope to more synthetically useful C–C/C–H/H–H/C–O/C–F bonds for both oxidative addition and reductive elimination.
Due to the distinctive chemical properties of uranium, the mechanism of uranium-mediated oxidative addition/reductive elimination is intriguing and may differ significantly from those for late transition-metals. Mechanistic study is not only important to understanding nature of these reactions, but also can be a practical guide for developing catalytic systems. However, for most of uranium-mediated oxidative addition/reductive elimination the mechanism is poorly understood. Thus, mechanism elucidation of these reactions can be a new horizon for both experimental and theoretical chemists. A noteworthy point here is incorporation of the novel U(II) oxidation state into mechanistic explanations: although molecular U(II) compounds under ambient conditions were unknown until very recently, and can only be synthesised under harsh reducing conditions, this oxidation state may play a much more important role as intermediate/synthon than was previously thought, as hinted at by the equilibrium between U(IV)/U(III) hydrides 41/20.10c
Acknowledgements
The Authors are grateful to the Royal Society, EPSRC, ERC, Marie Curie IIF Scheme, University of Nottingham, National Nuclear Laboratory, and COST for continued support.References
- (a) R. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, 2005, ISBN 0-471-66256-9 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, New York, 2010, ISBN 1-891389-53-X Search PubMed.
- IUPAC, Compendium of Chemical Terminology, 2nd edn (the “Gold Book”), compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997. XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat and B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. DOI:10.1351/goldbook.
- D. Seyferth, Organometallics, 2009, 28, 1598 CrossRef CAS.
- (a) C. Wills, K. Izod, W. Clegg and R. W. Harrington, Dalton Trans., 2010, 39, 2379 RSC; (b) L. Yun, H. Vazquez-Lima, H. Fang, Z. Zhao, G. Geisberger, C. Dietl, A. Ghosh, P. J. Brothers and X. Fu, Inorg. Chem., 2014, 53, 7047 CrossRef CAS PubMed; (c) T. Chlupatý, Z. Růžičková, M. Horáček, M. Alonso, F. De Proft, H. Kampová, J. Brus and A. Růžička, Organometallics, 2015, 34, 606 CrossRef.
- A. V. Protchenko, D. Dange, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 10902 CrossRef CAS PubMed.
- The fact is highlighted by seminal works concerning: SmI2 mediated organic reactions: (a) P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693 CrossRef CAS; (b) D. J. Procter, R. A. Flowers II and T. Skydtrup, Organic Synthesis Using Samarium Diiodide, Royal Society of Chemistry, 2009 Search PubMed, ISBN 978-1-84755-110-8; and C–H activation: (c) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82 CrossRef CAS PubMed.
- (a) H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158 CrossRef CAS PubMed; (b) M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310 CrossRef CAS PubMed.
- (a) W. J. Evans, Coord. Chem. Rev., 2000, 206–207, 263 CrossRef CAS; (b) W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119 CrossRef CAS PubMed.
- (a) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137 CrossRef CAS PubMed; (b) S. T. Liddle, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201412168.
- (a) R. G. Finke, Y. Hirose and G. Gaughan, J. Chem. Soc., Chem. Commun., 1981, 232 RSC; (b) R. G. Finke, D. A. Schiraldi and Y. Hirose, J. Am. Chem. Soc., 1981, 103, 1875 CrossRef CAS; (c) P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650 CrossRef CAS; (d) P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170 CrossRef CAS; (e) A. M. Seyam, Inorg. Chim. Acta, 1982, 58, 71 CrossRef CAS.
- W. J. Evans, E. Montalvo, S. A. Kozimor and K. A. Miller, J. Am. Chem. Soc., 2008, 130, 12258 CrossRef CAS PubMed.
- O. P. Lam, S. M. Franke, H. Nakai, F. W. Heinemann, W. Hieringer and K. Meyer, Inorg. Chem., 2012, 51, 6190 CrossRef CAS PubMed.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed.
- (a) I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS PubMed; (b) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef CAS PubMed; (c) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS PubMed; (d) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77 RSC; (e) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265 CrossRef CAS PubMed.
- P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423 RSC.
- L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615 CrossRef CAS PubMed.
- (a) J. C. Bryan and J. M. Mayer, J. Am. Chem. Soc., 1987, 109, 7213 CrossRef CAS; (b) J. C. Bryan and J. M. Mayer, J. Am. Chem. Soc., 1990, 112, 2298 CrossRef CAS; (c) M. A. Lockwood, P. E. Fanwick, O. Eisenstein and I. P. Rothwell, J. Am. Chem. Soc., 1996, 118, 2762 CrossRef CAS.
- B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959 CrossRef CAS.
- C. L. Webster, J. W. Ziller and W. J. Evans, Organometallics, 2013, 32, 4820 CrossRef CAS.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448 CrossRef CAS.
- R. G. Peters, B. P. Warner and C. J. Burns, J. Am. Chem. Soc., 1999, 121, 5585 CrossRef CAS.
- For selective examples of complete cleavage of NN bond in N2 mediated by transition-metal compounds, see:
(a) C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 862 Search PubMed;
(b) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76 CrossRef CAS PubMed;
(c) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, Nat. Chem., 2010, 2, 30 CrossRef CAS PubMed;
(d) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120 CrossRef CAS PubMed;
(e) K. C. Macleod and P. J. Holland, Nat. Chem., 2013, 5, 559 CrossRef CAS PubMed;
(f) H. Tanaka, K. Arashiba, S. Kuriyama, A. Sasada, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2014, 5, 3737 Search PubMed;
(g) T. Miyazaki, H. Tanaka, Y. Tanabe, M. Yuki, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2014, 53, 11488 CrossRef CAS PubMed;
(h) Y. Ishida and H. Kawaguchi, J. Am. Chem. Soc., 2014, 136, 16990 CrossRef CAS PubMed;
(i) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532 CAS.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS PubMed.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433 CrossRef CAS.
- T. K. Todorova, L. Gagliardi, J. R. Walensky, K. A. Miller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 12397 CrossRef CAS PubMed.
- (a) J. Chatt and J. M. Davidson, J. Chem. Soc., 1965, 843 RSC; (b) M. L. Green and P. J. Knowles, J. Chem. Soc. D, 1970, 24, 1677 RSC; (c) P. Foley and G. M. Whitesides, J. Am. Chem. Soc., 1979, 101, 2732 CrossRef CAS; (d) A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352 CrossRef CAS; (e) J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3723 CrossRef CAS.
- I. Castro-Rodriguez, H. Nakai, P. Gantzel, L. N. Zakharov, A. L. Rheingold and K. Meyer, J. Am. Chem. Soc., 2003, 125, 15734 CrossRef CAS PubMed.
- W. J. Evans, K. A. Miller, A. G. DiPasquale, A. L. Rheingold, T. J. Stewart and R. Bau, Angew. Chem., Int. Ed., 2008, 47, 5075 CrossRef CAS PubMed.
- W. W. Lukens, Jr., S. M. Beshouri, L. L. Blosch and R. A. Andersen, J. Am. Chem. Soc., 1996, 118, 901 CrossRef.
- For lanthanide metal complexes, see: (a) S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740 CrossRef CAS PubMed; (b) T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 9491 CrossRef CAS PubMed; (c) W. Huang and P. L. Diaconescu, Chem. Commun., 2012, 48, 2216 RSC; (d) W. Huang and P. L. Diaconescu, Eur. J. Inorg. Chem., 2013, 4090 CrossRef CAS PubMed. For actinide metal complexes, see: (e) O. J. Scherer, B. Werner, G. Heckmann and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1991, 30, 553 CrossRef PubMed; (f) D. Patel, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 13334 CrossRef CAS PubMed.
- A. S. P. Frey, F. G. N. Cloke, P. B. Hitchcock and J. C. Green, New J. Chem., 2011, 35, 2022 RSC.
- (a) J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468 CrossRef CAS PubMed; (b) J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193 CrossRef CAS; (c) J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352 CrossRef CAS PubMed; (d) D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 10240 CrossRef CAS PubMed; (e) C. Camp, M. A. Antunes, G. García, I. Ciofini, I. C. Santos, J. Pécaut, M. Almeida, J. Marçalo and M. Mazzanti, Chem. Sci., 2014, 5, 841 RSC.
- (a) W. J. Evans, J. Organomet. Chem., 2002, 647, 2 CrossRef CAS; (b) W. J. Evans, J. Alloys Compd., 2009, 488, 493 CrossRef CAS PubMed.
- C. K. Jørgensen, Coord. Chem. Rev., 1966, 1, 164 CrossRef.
- (a) I. Korobkov, S. Gorelsky and S. Gambarotta, J. Am. Chem. Soc., 2009, 131, 10406 CrossRef CAS PubMed; (b) E. J. Schelter, R. Wu, B. L. Scott, J. D. Thompson, T. Cantat, K. D. John, E. R. Batista, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2010, 49, 924 CrossRef CAS PubMed; (c) S. J. Kraft, P. E. Fanwick and S. C. Bart, Inorg. Chem., 2010, 49, 1103 CrossRef CAS PubMed; (d) E. J. Schelter, R. Wu, J. M. Veauthier, E. D. Bauer, C. H. Booth, R. K. Thomson, C. R. Graves, K. D. John, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2010, 49, 1995 CrossRef CAS PubMed; (e) G. L. Manni, J. R. Walensky, S. J. Kraft, W. P. Forrest, L. M. Pérez, M. B. Hall, L. Gagliardi and S. C. Bart, Inorg. Chem., 2012, 51, 2058 CrossRef PubMed; (f) J. J. Kiernicki, B. S. Newell, E. M. Matson, N. H. Anderson, P. E. Fanwick, M. P. Shores and S. C. Bart, Inorg. Chem., 2014, 53, 3730 CrossRef CAS PubMed; (g) E. M. Matson, J. J. Kiernicki, N. H. Anderson, P. E. Fanwick and S. C. Bart, Dalton Trans., 2014, 43, 17885 RSC.
- S. J. Kraft, U. J. Williams, S. R. Daly, E. J. Schelter, S. A. Kozimor, K. S. Boland, J. M. Kikkawa, W. P. Forrest, C. N. Christensen, D. E. Schwarz, P. E. Fanwick, D. L. Clark, S. D. Conradson and S. C. Bart, Inorg. Chem., 2011, 50, 9838 CrossRef CAS PubMed.
- E. M. Matson, S. R. Opperwall, P. E. Fanwick and S. C. Bart, Inorg. Chem., 2013, 52, 7295 CrossRef CAS PubMed.
- (a) E. M. Archer and T. G. D. van Schalkwyk, Acta Crystallogr., 1953, 6, 88 CrossRef CAS; (b) J. V. Carey, P. A. Chaloner, P. B. Hitchcock, T. Neugebauer and K. R. Seddon, J. Chem. Res., 1996, 358, 2031 Search PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919 CrossRef CAS PubMed.
- D. P. Cladis, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2013, 49, 4169 RSC.
- J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2014, 50, 8189 RSC.
- (a) W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 7423 CrossRef CAS; (b) W. J. Evans, K. J. Forrestal, J. T. Leman and J. W. Ziller, Organometallics, 1996, 15, 527 CrossRef CAS.
- W. J. Evans, K. J. Forrestal and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 9273 CrossRef CAS.
- W. J. Evans, J. M. Perotti, S. A. Kozimor, T. M. Champagne, B. L. Davis, G. W. Nyce, C. H. Fujimoto, R. D. Clark, M. A. Johnston and J. W. Ziller, Organometallics, 2005, 24, 3916 CrossRef CAS.
- W. J. Evans, B. M. Schmiege, S. E. Lorenz, K. A. Miller, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 8555 CrossRef CAS PubMed.
- W. J. Evans, K. J. Forrestal and J. W. Ziller, Angew. Chem., Int. Ed. Engl., 1997, 36, 774 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681 RSC.
- (a) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108 CrossRef CAS; (b) W. J. Evans, C. A. Traina and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 17473 CrossRef CAS PubMed; (c) D. P. Mills, F. Moro, J. McMaster, J. von Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454 CAS; (d) D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388 CrossRef CAS PubMed; (e) P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668 CrossRef CAS PubMed; (f) D. Patel, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Commun., 2013, 4, 2323 Search PubMed; (g) D. Patel, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224 RSC; (h) C. Camp, V. Mougel, J. Pécaut, L. Maron and M. Mazzanti, Chem. – Eur. J., 2013, 19, 17528 CrossRef CAS PubMed.
- (a) M. Aresta, E. Quaranta and I. Tommasi, Gazz. Chim. Ital., 1993, 123, 271 CAS; (b) M. Aresta, E. Quaranta and I. Tommasi, Organometallics, 1995, 14, 3349 CrossRef CAS.
- W. J. Evans, K. A. Miller, W. R. Hillman and J. W. Ziller, J. Organomet. Chem., 2007, 692, 3649 CrossRef CAS PubMed.
- (a) W. J. Evans, J. R. Walensky, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Organomet. Chem., 2009, 694, 1238 CrossRef CAS PubMed; (b) M. R. MacDonald, J. W. Ziller and W. J. Evans, Inorg. Chem., 2011, 50, 4092 CrossRef CAS PubMed.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568 CrossRef CAS.
- D. J. Grant, T. J. Stewart, R. Bau, K. A. Miller, S. A. Mason, M. Gutmann, G. J. McIntyre, L. Gagliardi and W. J. Evans, Inorg. Chem., 2012, 51, 3613 CrossRef CAS PubMed.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2012, 134, 6160 CrossRef CAS PubMed.
- E. M. Matson, S. M. Franke, N. H. Anderson, T. D. Cook, P. E. Fanwick and S. C. Bart, Organometallics, 2014, 33, 1964 CrossRef CAS.
- E. M. Matson, M. G. Crestani, P. E. Fanwick and S. C. Bart, Dalton Trans., 2012, 41, 7952 RSC.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Chem. Sci., 2015, 6, 517 RSC.
| This journal is © The Royal Society of Chemistry 2015 |