
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CFA-7: an interpenetrated metal–organic framework of the MFU-4 family†
Phillip
Schmieder
,
Maciej
Grzywa
,
Dmytro
Denysenko
,
Manuel
Hambach
and
Dirk
Volkmer
*
Institute of Physics, Chair of Solid State and Material Science, Augsburg University, Universitaetsstrasse 1, 86159 Augsburg, Germany. E-mail: dirk.volkmer@physik.uni-augsburg.de; Fax: +49(0)821 598 5955; Tel: +49(0)821 598 3006
First published on 16th June 2015
Abstract
The novel interpenetrated metal–organic framework CFA-7 (Coordination Framework Augsburg University-7), [Zn5Cl4(tqpt)3], has been synthesized containing the organic linker {H2-tqpt = 6,6,14,14-tetramethyl-6,14-dihydroquinoxalino[2,3-b]phenazinebistriazole}. Reaction of H2-tqpt and anhydrous ZnCl2 in N,N-dimethylformamide (DMF) yields CFA-7 as pseudo-cubic crystals. CFA-7 serves as precursor for the synthesis of isostructural frameworks with redox-active metal centers, which is demonstrated by postsynthetic metal exchange of Zn2+ by different M2+ (M = Co, Ni, Cu) ions. The novel framework is robust upon solvent removal and has been structurally characterized by single-crystal X-ray diffraction, TGA and IR spectroscopy, as well as gas sorption (Ar, CO2 and H2).
Introduction
The appearance of interpenetration in metal–organic frameworks (MOFs) has been described for a lot of MOFs in many articles.1 The phenomenon has a great impact on the framework structure, and consequently also on potential functional properties of the resulting three-dimensional porous materials.2Interpenetration, i.e. the occurrence of two or more structurally independent networks which are intergrown with each other, is a frequently observed phenomenon in MOF structural chemistry. The iconic MOF-5 was intensively investigated in terms of interpenetration in recent years.3 Depending on water content, reaction time and temperature, molar ratio of metal salt-to-organic linker or the addition of agents like melamine, non-interpenetrated, partially or completely interpenetrated network variants of MOF-5 were obtained. Interpenetration in MOFs often leads to a reduced specific surface area of the network. On the other hand, interpenetrated frameworks may also feature some additional advantages such as improved network stability, increased heat of adsorption for certain molecular probes or enhanced molecular size selectivity.3c,4
Aiming to extend the range of isoreticular MFU-4-type frameworks, we here report on the synthesis and the structure of CFA-7 (Coordination Framework Augsburg University-7) containing the largest straight linker molecule when compared to the organic bistriazolate linkers previously employed in the synthesis of porous MFU-4-type frameworks.5–10
The modular MFU-4 family is based on Kuratowski-type11 pentanuclear secondary building units (SBUs) of the general formula [MII5X4(L6)] (Fig. 1). In the pentanuclear coordination units, six triazolate ligands are monodentately coordinated to the central octahedrally coordinated Zn ion. The triazolate ligands are twisted around their coordinative bond to the central metal ion such that the nitrogen atoms are placed on the six edges of an imaginary tetrahedron, the four corners of which are occupied by four peripheral Zn ions. Accordingly, each of the latter are coordinated by three N donor atoms of the organic triazolate linkers. The coordination environment of each peripheral zinc ions is completed by an additional (anionic) ligand, i.e. a halide anion.
 | ||
| Fig. 1 Kuratowski-type SBU of MFU-4-type frameworks (central octahedrally coordinated metal ion: purple octahedron, peripheral Zn: yellow tetrahedra; N: blue; C: grey, Cl: green). | ||
A special feature of the Kuratowski-type secondary building unit is the possibility to exchange the peripheral zinc ions by other (transition) metal ions. Adjusting appropriate reaction conditions it is possible to obtain heterometallic derivatives of CFA-7via postsynthetic metal exchange, as described previously for MFU-4l7,9,10 and CFA-1.8 Heating up CFA-7 with solutions of the corresponding metal salts in N,N-dimethylacetamide (DMAc) leads to replacement of peripheral Zn2+ by M2+ (M = Co, Ni, Cu) ions. Depending on the metal source and the initial stoichiometric ratio of metal ions in the suspension, different numbers of zinc centers can be replaced by M2+ ions. CFA-7 includes the fourth bistriazolate linker used to build MFU-4-type frameworks and represents the first framework of this type which is interpenetrated. Employing very long organic linkers we were aiming at MFU-4XL, a hypothetical cubic framework structure with extraordinary high specific surface area. The non-interpenetrated 3D structure of CFA-7 is termed MFU-4extra large (MFU-4XL, calc. surface17 up to 4500 m2 g−1, Fig. 2 top right) due to its very large unit cell as compared to the previously published cubic networks of MFU-45 and MFU-4large6 (Fig. 2). However, all attempts to synthesize the non-interpenetrated framework have failed in our hands so far.
 | ||
Fig. 2 An overview of different bistriazolate frameworks of the MFU-4 family with Kuratowski-type secondary building units. MFU-4, MFU-4l and the DFT-calculated MFU-4XL are representatives of the MFU-4 family which crystallize in a cubic crystal system, whereas CFA-1 exhibits a trigonal crystal lattice. The interpenetrated CFA-7 framework described herein crystallizes in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 166). m (no. 166). | ||
Results and discussion
Syntheses and characterization
The H2-tqpt ligand was synthesized in three steps. The 2,2,5,5-tetramethylcyclohexane-1,3-dione and the 3,3,6,6-tetramethylcyclohexane-1,2,4,5-tetraone were synthesized by conventional procedures.12 The condensation reaction of the latter tetraone and two molecules of 1H-benzotriazole-5,6-diamine13 leads to the organic triazolate linker H2-tqpt (Scheme 1). The overall yield in these three steps is 21% based on dimedone as starting material. Metal–organic framework CFA-7 can be synthesized from H2-tqpt and ZnCl2 in a good yield by using N,N-dimethylformamide (DMF) as solvent at 130 °C (see Scheme 2). At these reaction conditions the network forms light yellow pseudo-cubic crystals (Fig. 3a). A second, different morphology is obtained by adjusting a lower reaction temperature, i.e. at 90 °C. Under these conditions CFA-7 crystals assume a hexagonal shape (Fig. 3b). Under reaction conditions identical to those in the synthesis of pseudo-cubic CFA-7-chloride (at 130 °C), employing ZnI2 instead of ZnCl2 however, pseudo-cubic yellow crystals of CFA-7-iodide were obtained. CFA-7 and its iodide derivative CFA-7-iodide are isomorphous. A Le Bail fit14 of the XRPD pattern of the compound CFA-7-iodide is presented in Fig. S1.† The refined unit cell parameters are: a = 28.6916(9) Å, c = 69.452(4) Å, V = 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 514(2) Å3.
514(2) Å3.
 | ||
| Scheme 1 Synthesis of the organic bistriazolate ligand H2-tqpt. | ||
 | ||
| Scheme 2 Different synthesis procedures to obtain CFA-7. | ||
 | ||
| Fig. 3 Optical microscopy images of CFA-7: (a) overview of the as synthesized pseudo-cubic crystals; (b) hexagonal CFA-7 crystals. | ||
It is also possible to synthesize CFA-7 in a microwave reactor at 250 W and 160 °C for 10 min. Morphology and surface details of the hexagonal crystals were investigated by DIC, SEM and AFM micrographs (Fig. 4–6). Differential interference contrast microscopy shows uniform hexagonal crystals with a size up to 30 μm (Fig. 4). Inspection of electron microscopic images reveals that the crystal faces of the hexagonal specimen are smooth and flat. An AFM measurement of a hexagonal single crystal of a size of about 30 × 30 μm was carried out in order to analyze the crystal's surface topography. In Fig. 6, it can be seen, that the surface is flat and exhibits no macroscopic height differences within the measured area.
 | ||
| Fig. 4 Differential interference contrast (DIC) microscopy of hexagonal CFA-7 crystals. | ||
 | ||
| Fig. 5 SEM micrographs of CFA-7; (a) hexagonal shape of a single- crystal of CFA-7; (b) partially intergrown crystals. | ||
 | ||
| Fig. 6 AFM micrographs; (a) 3 dimensional view of a hexagonal CFA-7 single crystal; (b) 3 dimensional view of CFA-7 crystal surface. | ||
Crystal structure analysis
Aiming to extend the range of isoreticular MFU-4-type frameworks, we calculated the hypothetical model for the cubic MFU-4extra large. The DFT calculated model (cell optimization employing plane-wave (norm-conserving) pseudo potentials, PBE functional, energy cut-off at 830 eV) of the structure is based on Kuratowski-type pentanuclear SBUs of the formula [Zn5Cl4(tqpt)3]. Instead of the previously published cubic networks of MFU-4 or MFU-4large, respectively, the novel CFA-7 network is twofold interpenetrated. Both subnetworks of CFA-7 represent the same bond connectivity scheme as predicted for the hypothetical compound MFU-4XL. The previously reported MFU-4l has two different kinds of pores depending on the position of the four Cl-atoms from different Kuratowski SBUs. The A-and B-cells in MFU-4large have a diameter of 11.97 Å and 18.56 Å, respectively. The DFT calculated model of MFU-4XL shows pores with a diameter of 20.60 Å and 22.46 Å, respectively. The pore aperture in the non-interpenetrated framework is 11.94 Å.A single-crystal X-ray diffraction study of a hexagonal crystal reveals that CFA-7 crystallizes in the trigonal space group Rm (no. 166). Pseudo-cubic crystal of a size ranging from about 25–40 μm in each direction were also investigated but the single crystal structural analyses lead to the same solutions (trigonal crystal system and Rm space group), but the quality of the obtained data sets was lower in terms of resolution at high diffraction angles if compared to hexagonal crystal. The asymmetric unit including atom labels is presented in Fig. S15.†CFA-7 is constructed from {Zn5Cl4−x((H2O)3)x}(6+x)+ secondary building units interconnected by the two-fold deprotonated organic linker tqpt2−. Due to the conformationally flexible cyclohexyl unit in the middle of each organic linker molecule, the tqpt2− ligand in the CFA-7 network is not completely planar. About 80% of the CFA-7 network is constructed from regular {Zn5Cl4(triazolate)6} Kuratowski-type SBUs. The remaining 20% SBUs consist of singly charged {Zn5Cl3(H2O)3(triazolate)6} units, in which a single chloro ligand is replaced by 3 aqua ligands, thus leading to a non-coordinated (positionally strongly disordered) chloride anion which is placed in the void volume of the crystal lattice in order to retain a net zero charge. The zinc ions in the centers of the SBUs (Zn1 or Zn5, placed at a site of 3m symmetry) are octahedrally coordinated by six nitrogen atoms of six different organic linker molecules. The four peripheral zinc ions (Zn2 or Zn4, placed at sites of m symmetry) are four-fold coordinated by three nitrogen atoms of the organic linker and by the chloride anion. A fraction of 20% of the fourth peripheral zinc ions (Zn3 or Zn6, positioned at a site of 3m symmetry) is octahedrally coordinated by three nitrogen atoms of the linker molecules and by three oxygen atoms from water molecules. The different coordination schemes for both kinds of SBUs are shown in the plots in Fig. 8. The Zn–O distances are between 2.07–2.09 Å. These values are in good agreement with those found in the literature for similar Zn(II) compounds in which the octahedrally coordinated Zn ion binds three nitrogen containing ligands and three water molecules.15 The Zn–N bond distances span a range of 2.150(10)–2.209(10) Å for the octahedrally coordinated Zn1 and Zn5 and 2.010(6)–2.135(13) Å for Zn2, Zn3, Zn4 and Zn6. These values are similar to those found in structurally related Zn triazolate compounds of the MFU-4 family.10CFA-7 features a 3-D two-fold interpenetrated structure constructed from Kuratowski-type SBUs. The packing diagram of CFA-7, depicted in Fig. 7, shows two subnetworks. The simplified networks, displayed in red and blue colors, are shifted by 4.51 Å with respect to each other in [001] direction, which represents the shortest distance between two adjacent Zn ions belonging to different subnetworks). Fig. 7 also indicates the pseudo-cubic nature of the CFA-7 network pointing at the close structural relation to the non-interpenetrated (hypothetical) MFU-4extra large network. Fig. 9 (left) shows a schematic representation of both subnetworks of CFA-7. The minimal divergence from right angles (Fig. 9 left) makes it impossible to solve the structure in a cubic space group.
 | ||
| Fig. 7 Interpenetrated structure of CFA-7. In yellow the apparent pore aperture of the channels of the network is given (calculated with the Poreblazer software17). | ||
 | ||
| Fig. 8 Coordination environments of Zn ions in the Kuratowski-type secondary building units of CFA-7; (a) staggered arrangement of six water ligands from two different SBUs; (b) peripheral Zn ions which are coordinated to three N-atoms and one Cl-atom or to three N-atoms and three O-atoms. | ||
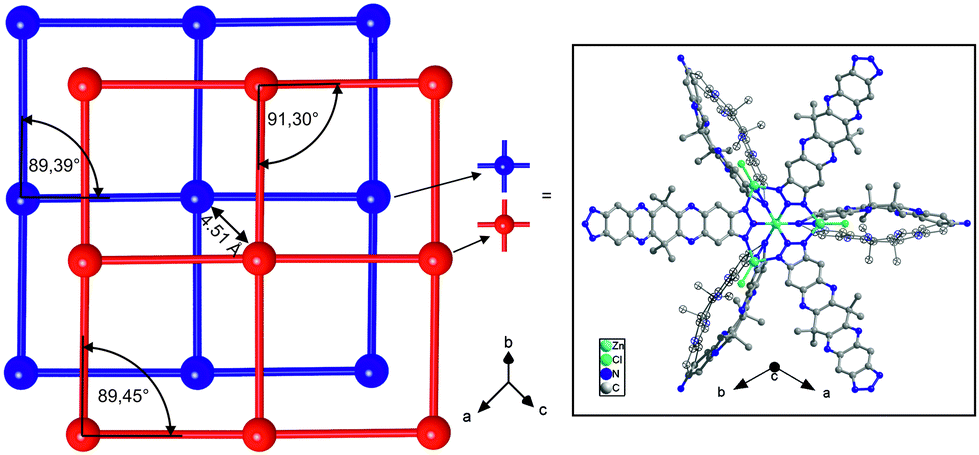 | ||
| Fig. 9 Schematic representation of the two-fold interpenetrated CFA-7 networks (left) and its SBU with the disordered organic linker in c-direction (right, positional disorder of two parts of the ligand with the occupancy of 0.5/0.5, one part shown in ellipses). | ||
Calculations with the program SQUEEZE16 reveal that the initial solvent accessible void volume is 28982 Å3, which is about 58.3% of the unit cell volume (49703 Å3) for a probe radius of 1.68 Å, corresponding to the approximate van der Waals radius of argon.17 In the crystal structure of CFA-7 the pores are occupied by disordered Cl− ions and DMF molecules. The positions of the molecules were impossible to resolve and to refine from the electron density distribution. According to the crystallographic data there is an electron count of 7108 per unit cell (rest of the electron density), which corresponds to approx. 5.34 Cl− and 175 DMF molecules in the unit cell of CFA-7. Thus, the composition of the framework is determined as [Zn5Cl3.11(H2O)2.65(tqpt)3](0.89+) (Cl−)0.89·29.25DMF. This means, 0.89 chloride ions per SBU are displaced in the pores in order to retain neutral charge.
The calculation of maximal pore diameter and pore limiting diameter was performed using the Poreblazer software.18 The limiting diameter in the CFA-7 network is 6.83 Å and the maximum pore diameter is 11.71 Å. The diameter of the channels in the interpenetrated CFA-7 network is 10.80 Å. The organic triazolate ligand of the network is disordered along the c-direction (Fig. 9 right). Three oxygen atoms of water ligands coordinating to Zn3 or Zn6 are placed in the interpenetrated frameworks in a way such that a staggered arrangement of the altogether six oxygen atoms of adjacent Kuratowski units is obtained (Fig. 8a). The non-bonding distances between two oxygen atoms of each SBU range from 2.87 Å to 4.16 Å. It is possible that hydrogen bridges are formed between the water ligands of the octahedrally coordinated zinc ions.
Up to now, it was not possible to synthesize the non-interpenetrated network of CFA-7 (MFU-4extra large, Fig. 2). It is necessary to develop strategies to suppress the interpenetration in order to construct highly porous MOFs with high surface area. In literature different approaches for controlling the interpenetration in MOFs have been described.3c,4 In our hands, varying the reaction temperature (in the range of 60–150 °C) and the concentration of the reaction mixture (in the range of 2–25 μmol ml−1) had no influence on the interpenetration in CFA-7. Moreover, selecting solvents of different molecular size (e.g. N,N-dibutylformamide instead of DMF) did not lead to the synthesis of non-interpenetrated frameworks as confirmed by BET surface area measurements.
Thermal analysis and VTXRPD studies
The structural and thermal stability of CFA-7 was investigated by TGA and VTXRPD measurements (Fig. 10 and 11). In the TGA curve of pre-dried (300 °C per vacuum, blue curve) CFA-7 no weight loss in the range of 20–400 °C is observed. The as-synthesized sample of CFA-7 shows a weight loss of 35%, which is due to the loss of DMF molecules. The decomposition of the framework under nitrogen flow starts at approx. 420 °C, which is in good agreement with VTXRPD data (Fig. 11).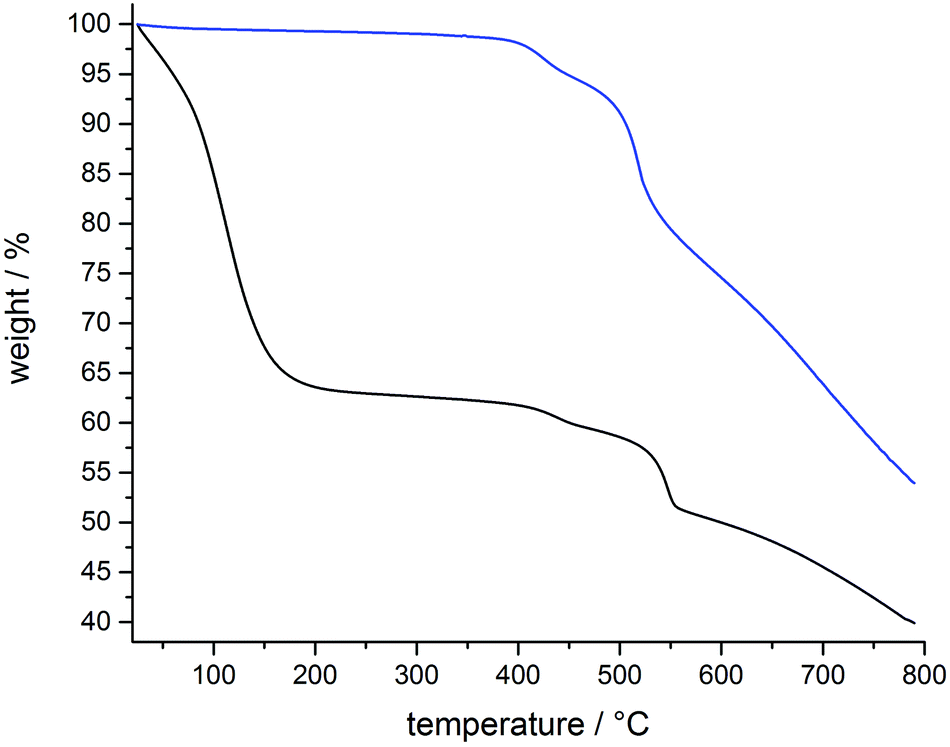 | ||
| Fig. 10 Thermogravimetric analysis of pre-dried (blue) and not-dried (black) CFA-7 under nitrogen atmosphere. | ||
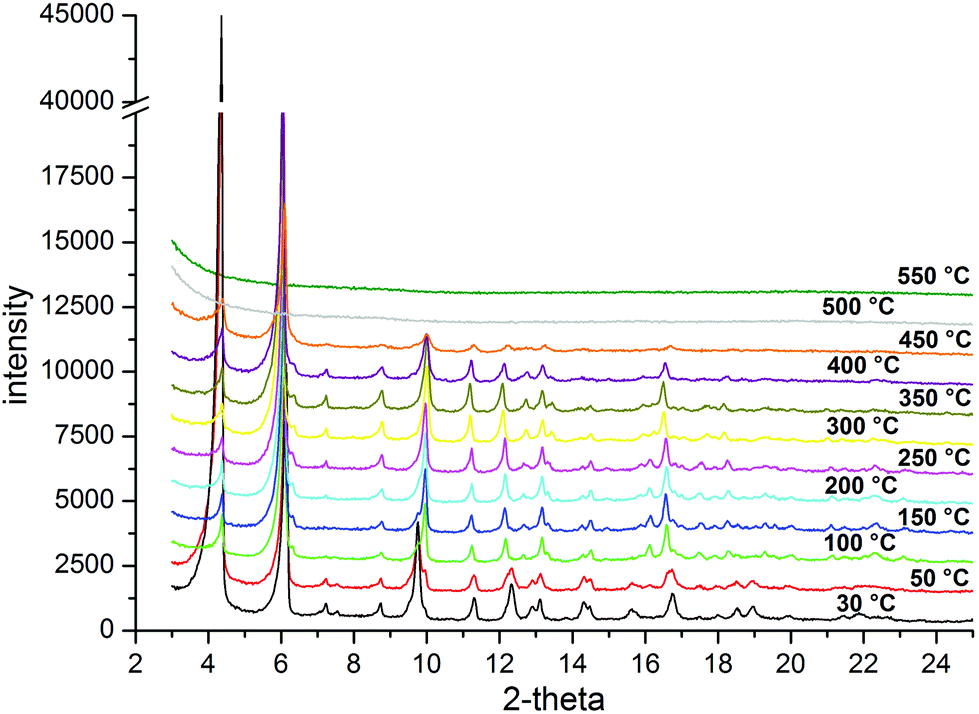 | ||
| Fig. 11 VTXRPD plot of CFA-7 in the range of 30–550 °C in air. | ||
Variable temperature X-ray powder diffraction studies (VTXRPD) show, similarly to the TGA data, that CFA-7 is stable up to 400 °C in air.
Metal-exchange reactions
Heating up the framework with solutions of the corresponding metal salts in DMAc leads to replacement of peripheral Zn2+ by M2+ ions (M = Co, Ni, Cu). Depending on the stoichiometric ratio between metal salt and framework in the suspension, different fractions of zinc ions can be replaced by M2+ ions (Table 1). The octahedrally coordinated zinc centers in the centre of the SBUs cannot be replaced under the given experimental conditions, similar to experimental findings previously reported for MFU-4l10 and CFA-1.8 UV/Vis spectra of metal-exchanged CFA-7 derivatives are shown in S 16. The spectroscopic measurements give no indication of octahedrally coordinated heterometallic ions. The diffuse reflectance UV/Vis spectra of Co-CFA-7 shows strong absorption bands at ca. 600 nm, typical for Co(II) centers in tetrahedral coordination environments and correspond to the spin-allowed 4A2(F)–4T1(P) transition. The Mn-CFA-7 shows no absorption bands in the UV/Vis spectrum relating to d–d metal-centered valence electron transitions. Copper(II)-exchanged derivatives show a weak band at ca. 950 nm. The position of the band maximum is in agreement with four coordinate tetrahedral Cu(II) complexes described in literature.19,20 For Co-CFA-7 the Le Bail fit of its X-ray powder pattern was performed in order to confirm that the structure of the framework remains intact. The refined unit cell parameters of Co-CFA-7 are: a = 28.848(2) Å, c = 68.585(9) Å, V = 49430(6) Å3. The resulting plots are presented in Fig. S2.† The XRPD patterns of the other metal exchanged CFA-7 samples, shown in Fig. S4,† also confirm the stability of the framework upon metal exchange. In the case of manganese the Mn/Zn ratio obtained upon metal exchange is quite high, which is probably due to the precipitation of amorphous manganese impurities on the surface of the crystals (Fig. 12c). All exchange reactions were performed at 60 °C for 16 hours. The exchange reaction, when performed with copper(II) perchlorate gives a higher exchange ratio of peripheral zinc ions if compared to copper chloride employed as the source of metal ions. These transition metal containing frameworks are of potential interest for catalytic applications. The XRPD and IR data (Fig. S4 and S17†) of dry samples (300 °C per vacuum) indicate phase purity and thermal stability of all derivatives thus obtained.
 | ||
| Fig. 12 SEM micrographs of CFA-7: (a) cubic CFA-7 crystals after the postsynthetic metal exchange with CoCl2; (b) exchange with NiCl2; (c) metal exchange with MnCl2; (d) crystals after exchange with Cu(ClO4)2. | ||
| Metal salt | EDX [At%] | Molar ratioa Zn2+:M2+ |
Morphology | Color |
|---|---|---|---|---|
|
a Theoretical Zn2+:M2+ ratio for total exchange of the peripheral metal atoms is 1:4.
|
||||
| CuCl2·2H2O | Zn: 35.91 | 3.0:2.0 |
Cubes | Light brown |
| Cu: 23.72 | ||||
| Cl: 40.37 | ||||
| Cu(ClO4)2·6H2O | Zn: 19.70 | 1.6:3.4 |
Cubes | Brown |
| Cu: 40.80 | ||||
| Cl: 39.50 | ||||
| CoCl2·6H2O | Zn: 38.35 | 3.3:1.7 |
Cubes | Green |
| Co: 20.55 | ||||
| Cl: 41.10 | ||||
| NiCl2 (wf) | Zn: 36.16 | 3.0:2.0 |
Cubes | Dark yellow |
| Ni: 23.64 | ||||
| Cl: 40.20 | ||||
| MnCl2·2H2O | Zn: 2.04 | 0.1:4.9 |
Cubes with contaminated surface | Dark brown |
| Mn: 94.01 | ||||
| Cl: 3.95 | ||||
Gas sorption measurements
The argon adsorption/desorption isotherm for CFA-7 measured at 77.3 K follows type I behavior typical for microporous materials, no hysteresis is observed (Fig. S6†). Data analysis reveals the BET surface area of 1718 m2 g−1 and Langmuir surface area of 1939 m2 g−1 (calculated value with Poreblazer software: 1651 m2 g−1). The total pore volume determined at p/p0 = 0.99 is 0.70 cm3 g−1, the micropore volume determined using an NLDFT method (pores up to 20 Å) is 0.61 cm3 g−1 (calc. 0.66 cm3 g−1). Porosity data for metal-exchanged CFA-7 derivatives are given in ESI (Table S1 and Fig. S6†). Analysis using the non-local density functional theory (NLDFT) implementing a carbon equilibrium transition kernel for argon adsorption at 77 K based on a slit–pore mode21 reveals quite broad distribution of micropores in the range of 5–16 Å with a maximum at 13.5 Å (Fig. 13). These values are in good agreement with the calculated values between 6.83–11.71 Å. | ||
| Fig. 13 Pore size distribution for CFA-7 calculated by fitting NLDFT model to the Ar adsorption/desorption data. | ||
High-resolution CO2 adsorption/desorption isotherms measured at 194.7 K show no hysteresis over the whole pressure range and thus confirm the structural rigidity of the interpenetrated CFA-7 framework (Fig. 14).
 | ||
| Fig. 14 CO2 adsorption/desorption isotherm for CFA-7 at 194.7 K. | ||
The isosteric heat of CO2 adsorption in CFA-7 determined from the adsorption isotherms in the temperature range 273–293 K, as described in the ESI,† is nearly constant at 22.5 kJ mol−1 (Fig. 15). This value is considerably higher as the one determined previously for MFU-4l (16.5 kJ mol−1),22 which is most likely due to the presence of basic amino functions in the tqpt2− ligand. Alternatively, the increased heat of CO2 adsorption could also be a special feature of the interpenetrated framework. However, the second explanation seems to be less likely since the heat of H2 adsorption is very similar to MFU-4l and decreases constantly from 7.4 kJ mol−1 at 0.18 mmol g−1 loading to 6.2 kJ mol−1 at 2.2 mmol g−1 loading (Fig. 16).
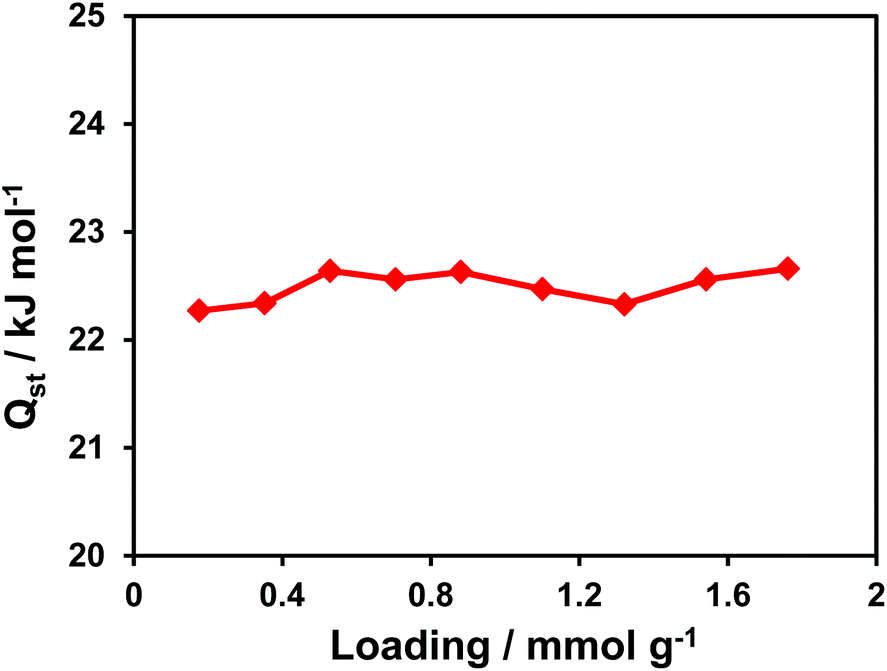 | ||
| Fig. 15 Loading-dependent isosteric heat of CO2 adsorption in CFA-7. | ||
 | ||
| Fig. 16 Loading-dependent isosteric heats of H2 adsorption in CFA-7 and MFU-4l. | ||
Interpenetrated MOFs sometimes show flexible character due to displacement of individual sub-frameworks against each other. Dramatic steps in the adsorption and hysteresis in the desorption of CO2 have been observed in a twofold interpenetrated MOF, Zn2(bttb)(dpni) (bttb = 4,4,4,4-benzene-1,2,4,5-tetrayltetrabenzolate, dpni = N,N-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) reported by Hupp and coworkers.23 Characterization of the structure by XRPD and pair distribution function (PDF) analysis indicated that structural changes upon CO2 sorption most likely involve the moving of the interpenetrated frameworks.24
In the interpenetrated CFA-7 it seems as though the interpenetration has no direct influence on the sorption properties. The much higher heat of absorption from CFA-7 for CO2 compared to MFU-4l is mostly due to the interaction between the carbon dioxide and the additional nitrogen atoms with pyrazine character in the organic linker molecule. Based on theoretical considerations, the incorporation of accessible nitrogen-donor functional groups, such as pyridine, pyrazine, imidazole, and tetrazole, into the pore walls of porous materials can dramatically affect the gas uptake capacity and selectivity of the materials, especially for CO2 capture, which most likely accounts for dipole–quadrupole interactions between the polarizable CO2 molecule and the accessible nitrogen site.25–31
Conclusion
In conclusion, we have synthesized and structurally characterized a novel metal–organic framework (CFA-7) constructed from tqpt2− ligands and {Zn5Cl4}6+ coordination units. This novel MOF contains Kuratowski-type pentanuclear SBUs, a characteristic feature of MFU-4-type frameworks. The CFA-7 network is two-fold interpenetrated and thus the first example of an interpenetrated network of the MFU-4 family. The isolated subnetworks of the interpenetrated CFA-7 show clearly the similarity with the cubic MFU-4, MFU-4large and the DFT-calculated hypothetical MFU-4extra large networks. Compared to the DFT-calculated MFU-4extra large model (calc. surface is 4460 m2 g−1, limiting diameter is 11.91 Å and the maximum pore diameter is 22.46 Å) the interpenetrated CFA-7 network shows much smaller values (BET surface area is 1718 m2 g−1, limiting pore diameter is 6.83 Å and the maximum pore diameter is 11.71 Å). To compare the thermal stability of the network and the results from gas sorption measurements it is needful to synthesize the non-interpenetrated CFA-7.A special feature of the system is the extended organic linker molecule with four additional (pyrazine) nitrogen atoms per linker molecule, which do not coordinate any metal ions under the reaction conditions described in this article. However, it might be possible to functionalize these N-donor sites under suitable reaction conditions, employing various organometallic reagents, for instance.
Similarly to MFU-4large and CFA-1 frameworks it is possible to exchange the peripheral Zn ions of each Kuratowski unit by Co(II), Cu(II) or Ni(II) to form open-shell 3d transition metal CFA-7 derivatives as a versatile approach towards redox-active MOFs. Further attempts to synthesize the non-interpenetrated CFA-7 framework (“MFU-4XL”) are currently underway. Studying this framework would help to understand the nature of the increased isosteric heat of CO2 adsorption which could also be a special feature of the interpenetrated framework.
Experimental section
Materials and methods
All starting materials were of reagent grade and used as received from the commercial supplier. Thermogravimetric analysis (TGA) was performed with a TGA Q500 analyzer in the temperature range of 25–800 °C in flowing nitrogen at a heating rate of 10 K min−1. Argon gas sorption isotherms at 77.3 K in the range of 1.0 × 10−5 ≤ p/p0 ≤ 0.99 were measured with a Quantachrome Autosorb-I ASI-CP-8 instrument. CO2 and H2 sorption isotherms were measured with a BELSORP-max instrument combined with a BELCryo system. Adsorbed gas amounts are given in cm3 g−1 [STP], where STP = 101.3 kPa and 273.15 K. Prior to measurements, the sample was heated at 260 °C for 20 h in high vacuum in order to remove occluded solvent molecules. Fourier transform infrared (FTIR) spectra were recorded with ATR in the range 4000–180 cm−1 on a Bruker Equinox 55 FT-IR spectrometer. Energy-dispersive X-ray spectroscopy (EDX) was performed with a Philips XL 30 FEG scanning electron microscope equipped with an EDAX SiLi detector. Atomic force micrographs were recorded on an Agilent 5500 AFM with MAC III controller operating in tapping mode. Olympus AC160TS cantilevers with a force constant of 26 N m−1 (tip radius 9 ± 2 nm) were used at a resonance frequency of about 300 kHz.The H2-tqpt ligand was synthesized in three steps from commercially available 5,5-dimethylcyclohexane-1,3-dione.11 The following methylation and oxidation reactions are documented in literature.11 The 1H-benzotriazole-5,6-diamine was synthesized as described in literature.12
Synthesis of 6,6,14,14-tetramethyl-6,14-dihydroquinoxalino-[2,3-b]phenazinebistriazole, (H2-tqpt)
To a suspension of 3,3,6,6-tetramethylcyclohexane-1,2,4,5-tetraone (1.00 g, 5.10 mmol) in CH3COOH (200 mL), 1H-benzotriazole-5,6-diamine (1.60 g, 10.70 mmol) was added under good stirring. The suspension was heated to reflux for 24 hours. The dark red solution was cooled with an ice bath and diluted with 400 mL of water to precipitate a light yellow solid. The formed precipitate was filtered off and washed with H2O and a small amount of MeOH.Yield: 2.15 g (73%). 1H NMR (400 MHz, DMSO-d6/TFA-d, δ (ppm)): 11.67 (s, 4H, NH), 8.64 (s, 4H, ArH), 2.01 (s, 12H, –CH3). 13C NMR (100 MHz, DMSO-d6/TFA-d, δ (ppm)): 156.65, 139.88, 138.47, 112.52, 45.83, 31.23. IR (ν (cm−1)): 1459.53, 1225.48, 1097.79, 850.99, 611.90, 412.05. Elemental analysis calcd for C22H18N10 (%): C 62.55, H 4.29, N 33.16; found: C 62.58, H 4.20, N 33.22.
Synthesis of CFA-7
Yield: 17.65 mg (51%). IR (ν (cm−1)): 1456, 1271, 1211, 1146, 1105, 1070, 850, 613, 310. Elemental analysis calcd (%) for C66H48Cl4N30Zn5: C 45.82, H 2.80, N 24.29; found: C 45.89, H 2.71, N 24.30.
Synthesis of CFA-7-iodide derivative
A mixture of zinc iodide (25.6 mg, 0.08 mmol) and H2-tqpt (10 mg, 0.02 mmol) was dissolved in 4 mL of DMF and the solution was placed in a glass tube (10 mL). The tube was sealed and heated at 140 °C for 3 days and then cooled to room temperature. The precipitate was filtered, washed with DMF (3 × 1 mL) and MeOH (3 × 3 mL) and dried at 300 °C under vacuum.Yield: 20.11 mg (48%). IR (ν (cm−1)): 1410, 1311, 1196, 866, 422, 230. Elemental analysis calcd (%) for C66H48I4N30Zn5: C 37.82, H 2.31, N 20.05; found: C 37.89, H 2.41, N 20.30.
General procedure for metal exchange reactions
The corresponding metal(II) salt (0.80 mmol) was dissolved in N,N-dimethylacetamide (DMAc) (4 mL) and activated CFA-7 (5 mg, 0.003 mmol) was added to the solution. The suspension was heated for 16 hours at 60 °C in a sealed tube.The particles were filtrated and washed several times with DMAc, methanol and dichloromethane. The sample was dried at 300 °C under vacuum.
Yield: 4.23–4.90 mg (90–97%); the number of M2+ ions in the formula unit calculated from the M2+/Zn ratio was determined by EDX. All metal exchange reactions can be carried out in the same procedure with the corresponding metal salt. UV/vis and IR spectra are displayed in the ESI.†
Single-crystal X-ray diffraction
Several crystals of CFA-7 were taken from mother liquids, mounted on a MiTeGen MicroMounts and tested on a diffractometer. Unfortunately, most of the crystals scattered only up to 40–42° θ (1.05–1.0 Å resolution). The best recorded data were obtained for a hexagonal single crystal of CFA-7 of approx. dimensions 44 × 39 × 36 μm3. The X-ray data for the single crystal structure determination of CFA-7 were collected on a Bruker D8 Venture diffractometer. Intensity measurements were performed using monochromated (doubly curved silicon crystal) MoKα radiation (0.71073 Å) from a sealed microfocus tube. Generator settings were 50 kV, 1 mA. Data collection temperature was −173 °C. APEX2 software was used for preliminary determination of the unit cell.32 Determination of integrated intensities and unit cell refinement were performed using SAINT.33 The structure was solved and refined using the Bruker SHELXTL Software Package.34 Selected crystal data and details of structure refinements for CFA-7 are provided in Table 2. Complete crystallographic data for the structure [Zn5Cl3.11(H2O)2.65(tqpt)3](0.89+) (Cl−)0.89·29.25DMF reported in this paper have been deposited in the CIF format with the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB21EZ, UK as supplementary publication no. CCDC 1062243.| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]1/2. | |
|---|---|
| Compound | CFA-7; [Zn5Cl3.11(H2O)2.65(tqpt)3](0.89+) (Cl−)0.89·29.25DMF |
| Empirical formula | C307.5H516.1 Cl8N118.5O63.8Zn10 |
| Formula | C66H53.3Cl3.11N30O2.65Zn5, Cl0.8929.25 (C3H7NO) |
| M r/g mol−1 | 7831.59 |
| T/K | 100(2) |
| Wavelength/Å | 0.71073 |
| Crystal system | Trigonal |
| Space group |
Rm (no. 166) |
| a/Å | 28.764(4) |
| c/Å | 69.369(9) |
| V/Å3 | 49703(12) |
| Z | 6 |
| D c/g cm−3 | 1.570 |
| μ/mm−1 | 0.873 |
| F(000) | 24822 |
| θ Range/° | 2.18 to 25.06 |
| Refls. collected | 200917 |
| Refls. unique | 10406 |
| Data/restraints/parameters | 10406/264/453 |
| R(int) | 0.1992 |
| GooF | 1.129 |
| R 1 (I > 2σ(I)]a | 0.1299 |
| wR2 (all data)b | 0.3676 |
| Largest diff. peak and hole/Å−3 | 1.855 and −1.798 |
(VT-)X-ray powder diffraction
VT-XRPD measurements were collected using a Bruker D8 Advance θ–2θ diffractometer in transmittance Bragg–Brentano geometry employing a Göbel mirror, Cu-radiation and equipped with a LYNXEYE 1-D detector. The samples were ground in an agate mortar and loaded into quartz capillaries (Hilgenberg) with 0.5 mm diameter and 0.01 mm wall thickness. The patterns were recorded in a temperature range of 30 to 550 °C, in the 3–60° 2θ range, with a step time of 1 s and a step width of 0.02° 2θ. Temperature program between measurements: 0.5 °C s−1 heating rate, followed by 10 min isothermal steps required for recording diffraction data sets. The Le Bail fits were performed using the Jana2006 program.35Acknowledgements
Financial support by the DFG (Priority Program SPP 1362 “Porous Metal–organic Frameworks”) is gratefully acknowledged.Notes and references
- (a) T. Reineke, M. Eddaoudi, D. Moler, M. O'Keeffe and O. Yaghi, J. Am. Chem. Soc., 2000, 122, 4843 CrossRef CAS; (b) H.-L. Jiang, T. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232 CrossRef CAS PubMed; (c) B. Chen, X. Wang, Q. Zhang, X. Xi, J. Cai, H. Qi, S. Shi, J. Wang, D. Yuan and M. Fang, J. Mater. Chem., 2010, 20, 3758 RSC; (d) H. Xu, W. Bao, Y. Xu, X. Liu, X. Shen and D. Zhu, CrystEngComm, 2012, 14, 5720 RSC; (e) E. Procopio, N. Padial, N. Masciocchi, S. Galli, J. Oltra, E. Barea and J. A. R. Navarro, CrystEngComm, 2013, 15, 9352 RSC.
- (a) K. Mulfort, O. Farha, C. Malliakas, M. Kanatzidis and J. Hupp, Chem. – Eur. J., 2010, 16, 276 CrossRef CAS PubMed; (b) K. Mulfort and J. Hupp, J. Am. Chem. Soc., 2007, 129, 9604 CrossRef CAS PubMed; (c) S. Bureekaew, H. Sato, R. Matsuda, Y. Kubota, R. Hirose, J. Kim, K. Kato, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 7660 CrossRef CAS PubMed.
- (a) J. Hafizovis, M. Bjoergen, U. Olsbye, P. Dietzel, S. Borgida, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612 CrossRef PubMed; (b) B. Chen, X. Wang, Q. Zhang, X. Xi, J. Cai, H. Qi, S. Shi, J. Wang, D. Yuan and M. Fang, J. Mater. Chem., 2010, 20, 3758 RSC; (c) H. Kim, S. Das, M. G. Kim, D. N. Dybtsev, Y. Kim and K. Kim, Inorg. Chem., 2011, 50, 3691 CrossRef CAS PubMed.
- (a) M. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142 CrossRef PubMed; (b) S. Ma, X.-S. Wang, D. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130 CrossRef CAS PubMed; (c) B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72 CrossRef CAS PubMed.
- S. Biswas, M. Grzywa, H. P. Nayek, S. Dehnen, I. Senkovska, S. Kaskel and D. Volkmer, Dalton Trans., 2009, 6487 RSC.
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem. – Eur. J., 2011, 17, 1837 CrossRef CAS PubMed.
- D. Denysenko, T. Werner, M. Grzywa, A. Puls, V. Hagen, G. Eickerling, J. Jelic, K. Reuter and D. Volkmer, Chem. Commun., 2012, 48, 1236 RSC.
- P. Schmieder, D. Denysenko, M. Grzywa, B. Baumgärtner, I. Senkovska, S. Kaskel, G. Sastre, L. v. Wüllen and D. Volkmer, Dalton Trans., 2013, 42, 10786 RSC.
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832 CrossRef CAS PubMed.
- D. Denysenko, J. Jelic, K. Reuter and D. Volkmer, Chem. – Eur. J., 2015, 21, 8188 CrossRef CAS PubMed.
- (a) S. Biswas, M. Tonigold and D. Volkmer, Z. Anorg. Allg. Chem., 2008, 634, 2532 CrossRef CAS PubMed; (b) S. Biswas, M. Tonigold, M. Speldrich, P. Kögerler, M. Weil and D. Volkmer, Inorg. Chem., 2010, 49, 7424 CrossRef CAS PubMed; (c) Y.-Y. Liu, M. Grzywa, M. Tonigold, G. Sastre, T. Schüttrigkeit, N. S. Leeson and D. Volkmer, Dalton Trans., 2011, 40, 5926 RSC.
- (a) Y. Gaoni and E. Wenkert, J. Org. Chem., 1966, 31, 3809 CrossRef CAS; (b) G. M. Whitesides and W. J. Ehmann, J. Org. Chem., 1970, 35, 3565 CrossRef CAS; (c) R. Gleiter, T. Doerner and H. Irngartinger, Liebigs Ann., 1996, 381 CAS.
- J. Liu, J. M. Balkovec, A. D. Krikorian, D. Guiadeen, G. Yang, T. Jian, Z. Wu, Y. Yu, R. P. Nargund and P. Vachal, PCT Int. Appl, WO 2012044567, 2012 Search PubMed.
- (a) A. Le Bail, Mater. Res. Bull., 1988, 23, 447 CrossRef CAS; (b) A. Le Bail, Powder Diffr., 2005, 20, 316 CrossRef CAS.
- (a) V. Gomez, J. Benet-Buchholz, E. Martin and J. R. Galan-Mascaros, Chem. – Eur. J., 2014, 20, 5369 CrossRef CAS PubMed; (b) D. Hutchinson, M. James and S. Moratti, Inorg. Chem., 2014, 53, 2122 CrossRef CAS PubMed; (c) Z.-Q. Liu, W. Dong and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, 2943 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- Quantachrome Autosorb, Version 1.56, Quantachrome GmbH & Co. KG, Odelzhausen, Germany, 2009 Search PubMed.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37(14), 1248 CrossRef CAS PubMed.
- A. B. P. Lever, in Inorganic Electronic Spectroscopy, 1984, p. 570ff Search PubMed.
- O. R. Rodig, T. Brueckner, B. K. Hurlburt, R. K. Schlatzer, T. L. Venable and E. Sinn, J. Chem. Soc., Dalton Trans., 1981, 196 RSC.
- J. Jagiello and M. Thommes, Carbon, 2004, 42, 1227 CrossRef CAS PubMed.
- G. Sastre, J. van den Bergh, F. Kapteijn, D. Denysenko and D. Volkmer, Dalton Trans., 2014, 43, 9612–9619 RSC.
- K. Mulfort, O. Farha, C. Malliakas, M. Kanatzidis and T. Hupp, Chem. – Eur. J., 2010, 16, 276 CrossRef CAS PubMed.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS PubMed.
- X.-J. Wang, P.-Z. Li, Y. Chen, Q. Zhang, X. Chan, R. Ganguly, Y. Li, J. Jiang and Y. Zhao, Sci. Rep., 2013, 3, 1149 Search PubMed.
- R. Vaidhyanathan, S. Iremonger, G. Shimizu, P. Boyd, S. Alavi and T. Woo, Science, 2010, 330, 650 CrossRef CAS PubMed.
- W. Li, H. Shi and J. Zhang, ChemPhysChem, 2014, 15, 1772 CrossRef CAS PubMed.
- S. Couck, J. Denayer, G. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS PubMed.
- N. Du, H. Park, G. Robertson, M. Dal-Cin, T. Visser, L. Scoles and M. Guiver, Nat. Mater., 2011, 10, 372 CrossRef CAS PubMed.
- F. Wang, Y.-X. Tan, H. Yang, Y. Kang and J. Zhang, Chem. Commun., 2012, 48, 4842 RSC.
- R. Vaidhyanathan, S. Iremonger, K. Dawson and G. Shimizu, Chem. Commun., 2009, 5230 RSC.
- APEX2 Version 2011.6, Bruker AXS Inc Search PubMed.
- SAINT Version 8.32B, Bruker AXS Inc., 2013 Search PubMed.
- XL Version 2013/3 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- V. Petricek, M. Dusek and L. Palatinus, Jana2006, Structure Determination Software Programs, Institute of Physics, Praha, Czech Republic, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRPDs from CFA-7 and its derivatives, NMR measurements of the organic linker, IR spectras and crystallographic data. CCDC 1062243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01673h |
| This journal is © The Royal Society of Chemistry 2015 |