
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lead(II) nitrate and hexafluorosilicate complexes with neutral diphosphine coordination†
Jennifer
Burt
,
William
Grantham
,
William
Levason
* and
Gillian
Reid
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: wxl@soton.ac.uk
First published on 21st May 2015
Abstract
Rare examples of phosphine complexes of lead(II) are reported. The reaction of Pb(NO3)2 with Me2P(CH2)2PMe2, o-C6H4(PMe2)2 or Et2P(CH2)2PEt2 (L–L) in H2O/MeCN gave white [Pb(L–L)(NO3)2], irrespective of the ratio of reagents used. The X-ray structures of [Pb{Me2P(CH2)2PMe2}(NO3)2] and [Pb{o-C6H4(PMe2)2}(NO3)2] reveal chelating diphosphines and κ2-NO3− groups occupying one hemisphere about the lead centre with single oxygen bridges to two further nitrate groups from neighbouring molecules completing a distorted eight-coordinate geometry. Using Pb(SiF6)·2H2O produced [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O which has a chelating diphosphine, the water molecule and a coordinated SiF62− group (which could be described as either κ1- or asymmetric κ3-coordinated to the lead), with further Pb–F interactions to neighbouring molecules producing a chain polymer structure. The structure of [Pb{o-C6H4(PMe2)2}(DMF)2(SiF6)]·DMF was also determined and contains dimers with fluorosilicate bridges. Adventitious oxygen readily form diphosphine dioxide complexes, and the structures of [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2] and [Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2 produced in this way were determined. The former contains eight-coordinate lead with κ2-NO3− groups and bridging diphosphine dioxides, which results in an infinite polymer. In the latter the diphosphine is chelated but the diphosphine dioxide bridges between Pb(II) centres, with coordinated BF4− groups completing a very distorted ten-coordinate moiety. Attempts to isolate similar complexes with o-C6H4(PPh2)2 or o-C6H4(AsMe2)2 were unsuccessful.
Introduction
Classed as an intermediate Lewis acid by Pearson's hard and soft acid–base concept,1 lead(II) forms stable complexes with a broad range of neutral and charged donor ligands across Groups 14–17.2–4 Although the majority of complexes are with hard (oxygen and nitrogen) donor atom ligands,3 there is also considerable coordination chemistry with softer sulfur and selenium donor ligands.4 However, whilst phosphine complexes of most heavy main group metals (including Hg(II), In(III), Sn(II), Sn(IV) and Bi(III)) are well established,5 the coordination chemistry of lead(II) with neutral phosphines is extremely limited.5 While [Pb{Et2P(CH2)2PEt2}Br2] and [Pb{Et2P(CH2)2PEt2}2](ClO4)2 were claimed in 1960, characterisation was limited to partial microanalysis,6 and the only structurally characterised examples of neutral diphosphine complexes are the lead(II) thiolates [(2,6-Me2C6H3S)2Pb]2{μ-Ph2P(CH2)2PPh2} and [(2,6-Me2C6H3S)2Pb]3{Me2P(CH2)2PMe2}.7 The latter contains a chain of the three lead centres linked by thiolate bridges, with the Me2P(CH2)2PMe2 chelating to the central Pb.7 Several 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 polydentate phosphine complexes of Pb(SbF6)2 have also been studied by in situ31P{1H} and 207Pb NMR spectroscopy in MeNO2, although no complexes were isolated.8,9
:1 polydentate phosphine complexes of Pb(SbF6)2 have also been studied by in situ31P{1H} and 207Pb NMR spectroscopy in MeNO2, although no complexes were isolated.8,9
One of the major challenges in synthesising lead(II) complexes with neutral diphosphine ligands is in finding a suitable lead precursor. Main group phosphine coordination chemistry has mostly been performed with metal halides,5 but the insoluble and intractable nature of the lead dihalides precludes easy synthetic access to this area; instead lead(II) oxo-salts such as acetate, nitrate or perchlorate are often used.2,3 Previously we have investigated crown ether, oxathia- and oxaselena-macrocycle complexes of Pb(NO3)2, Pb(BF4)2 and Pb(PF6)2, finding that the weakly coordinating fluoroanions also readily enter the first coordination sphere of the lead.10 The very stable di- and tri-imine complexes of Pb(II) with Pb(NO3)2 or Pb(ClO4)2 exhibit high coordination numbers and irregular geometries,11 whilst complexes of these ligands with Pb(BF4)2 and Pb(SiF6) were shown to exhibit a variety of different fluoroanion coordination modes.12 Here we report the reactions of Pb(NO3)2, Pb(SiF6) and Pb(BF4)2 with the diphosphines o-C6H4(PMe2)2, Me2P(CH2)2PMe2 and Et2P(CH2)2PEt2, focussing on the coordination behaviour of both the diphosphine and counterion in the complexes formed. Lead is a large metal centre with a covalent radius of 1.46 Å and hence regularly forms complexes with high coordination numbers where the geometry is governed by the number and steric demands of the ligands present, as well as by inter-ligand repulsions. The presence of a formal lone pair on lead(II) will also have an effect on the geometry observed if it is stereochemically active.2,3
Experimental section
All preparations were carried out under a dry dinitrogen atmosphere using standard Schlenk and glove-box techniques. Lead(II) nitrate and lead(II) tetrafluoroborate (the latter as 50% solutions in water) were obtained from Aldrich and used as received. Lead(II) hexafluorosilicate dihydrate was obtained from Alfa Aesar and used as received. Aqueous solutions of the lead salts were degassed before use, by bubbling with N2. The ligands were obtained commercially, (Strem) apart from o-C6H4(PMe2)2 and o-C6H4(AsMe2)2 which were made by the literature methods.13 Solvents were dried by distillation from CaH2 (CH2Cl2, CH3CN) or sodium benzophenone ketyl (hexane, Et2O). IR spectra were recorded as Nujol mulls between CsI plates using a Perkin Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H and 31P{1H} NMR spectra were recorded using a Bruker AV300 or DPX400 spectrometer and referenced to the residual solvent resonance and external 85% H3PO4 respectively. Microanalytical measurements were performed by London Metropolitan University.[Pb{o-C6H4(PMe2)2}(NO3)2]
To o-C6H4(PMe2)2 (0.090 g, 0.45 mmol) in CH3CN (5 mL) was added a degassed solution of Pb(NO3)2 (0.150 g, 0.45 mmol) in deionised water (3 mL). A white precipitate formed rapidly on stirring; the reaction was stirred for 2 h, then the white powder was isolated by filtration and dried in vacuo. Yield: 0.056 g, 23%. Anal. Calc. for C10H16N2O6P2Pb: (529.4) C, 22.7; H, 3.1; N, 5.3. Found: C, 22.6; H, 3.1; N, 5.2%. IR (Nujol): ν = 1314 (s), 1295 (s), 1033 (s), 837 (sh), 818 (m) (NO3) cm−1.[Pb{Me2P(CH2)2PMe2}(NO3)2]
To Me2P(CH2)2PMe2 (0.091 g, 0.60 mmol) in CH3CN (5 mL) was added a degassed solution of Pb(NO3)2 (0.101 g, 0.30 mmol) in deionised water (5 mL), giving a colourless solution. The reaction was stirred for 1 h, then the solvent was concentrated in vacuo and the resulting white powder was filtered and dried in vacuo. Yield: 0.071 g, 48% based on Pb. Anal. Calc. for C6H16N2O6P2Pb: (481.4) C, 15.0; H, 3.4; N, 5.8. Found: C, 15.1; H, 3.3; N, 5.8%. IR (Nujol): ν = 1310 (vs, br), 1038 (s), 840 (w), 819 (m) (NO3) cm−1.[Pb{Et2P(CH2)2PEt2}(NO3)2]
To a degassed solution of Pb(NO3)2 (0.162 g, 0.49 mmol) in deionised water (3 mL) was added Et2P(CH2)2PEt2 (0.101 g, 0.49 mmol) in CH3CN (5 mL) to give a colourless solution. The reaction was stirred for 2 h, then the solvent volume was reduced to about 3 mL in vacuo. The resulting white precipitate was isolated by filtration and dried in vacuo to give a white powder. Yield: 0.092 g, 35%. Anal. Calc. for C10H24N2O6P2Pb: (537.5) C, 22.4; H, 4.5; N, 5.2. Found: C, 22.2; H, 4.6; N, 5.2%. 1H NMR (CD2Cl2, 295 K): δ = 1.19–1.27 (m, [12H], CH3), 1.92–2.02 (m, [4H], CH2), 2.20–2.35 (m, [4H], CH2), 2.40 (d, [4H], CH2). 31P{1H} NMR (CH2Cl2/CD2Cl2, 295 K): δ = 89.3 (s); (223 K): 89.5 (s, 1JPPb = 2465 Hz); (193 K): 89.6 (s, 1JPPb = 2532 Hz). IR (Nujol): ν = 1300 (s, br), 1029 (s), 816 (m) (NO3) cm−1. Small colourless single crystals of the phosphine oxide complex, [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2], were grown from layering hexane onto a solution of the isolated powder dissolved in the minimal amount of CH2Cl2.[Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O
To a degassed solution of Pb(SiF6)·2H2O (0.151 g, 0.39 mmol) in deionised water (10 mL) was added o-C6H4(PMe2)2 (0.077 g, 0.39 mmol) in CH3CN (5 mL) which caused the precipitation of a small amount of white solid. The reaction was stirred for 1.5 h, then the solid was removed by filtration to leave a colourless solution. The solvent was removed in vacuo and the resulting white powder was dried in vacuo. Yield: 0.110 g, 48%. IR (Nujol): ν = 3500 (br), 1628 (m, br) (H2O), 721 (vs, br), 473 (s), 446 (s) (SiF6) cm−1. Small colourless single crystals of [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O were grown by layering Et2O onto a solution of the isolated product dissolved in the minimal amount of DMF.In a similar reaction where the o-C6H4(PMe2)2 to Pb(SiF6)·2H2O ratio used was 2:1.
[Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2
To a degassed solution of Me2P(CH2)2PMe2 (0.087 g, 0.58 mmol) in MeNO2 (6 mL) was added Pb(BF4)2 as a 50% aqueous solution (0.229 g, 0.30 mmol) that had been degassed (bubbling with N2) beforehand. A small amount of white precipitate rapidly formed, which was removed by filtration, then Et2O (10 mL) was layered onto the colourless filtrate, giving a few small colourless crystals.X-Ray experimental
Details of the crystallographic data collection and refinement parameters are given in Table 1. Crystals suitable for single crystal X-ray analysis were obtained as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinements were performed with SHELX(S/L)9714 and were straightforward, except where detailed below. H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. Attempts to improve the absorption correction for both [Pb{o-C6H4(PMe2)2}(NO3)2] and [Pb{Me2P(CH2)2PMe2}(NO3)2] failed to eliminate the large residual density electron peaks <1 Å from Pb1. DFIX restraints were applied to the O–H distances of the lattice H2O in [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O. For [Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2 the BF4− anion was modelled as two distinct orientations (A and B) and DFIX restraints were applied to the B–F distances. The proportions of A and B were allowed to vary through FVAR2. The disorder in the methyl groups around P2 was satisfactorily modelled by splitting C5 and C6 into A and B sites, then applying DFIX restraints to the P–C distances. The proportions of A and B were allowed to vary through FVAR3 while isotropic restraints were used on C5A, C6A and C6B to maintain reasonable ellipsoids. The N1 of the fractionally occupied lattice MeNO2 is located on a site with 222 symmetry giving a very disordered molecule. DFIX restraints were applied to the N–C and N–O distances.
| a Common items: T = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑‖Fσ| − |Fc‖/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo2]1/2. | ||||||
|---|---|---|---|---|---|---|
| Compound | [Pb{o-C6H4(PMe2)2}(NO3)2] | [Pb{Me2P(CH2)2PMe2}(NO3)2] | [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2] | [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O | Pb{o-C6H4(PMe2)2}(DMF)2(SiF6)]·DMF | [Pb(Me2P(CH2)2PMe2)(Me2(O)P(CH2)2- P(O)Me2)(BF4)2]·½MeNO2 |
| Formula | C10H16N2O6P2Pb | C6H16N2O6P2Pb | C20 H48N2O10P4Pb | C10H20F6O2P2PbSi | C19H37F6N3O3P2PbSi | C12.50H33.50B2F8N0.50O3P4Pb |
| M | 529.38 | 481.34 | 807.67 | 583.48 | 766.74 | 743.59 |
| Crystal system | Monoclinic | Tetragonal | Monoclinic | Triclinic | Triclinic | Orthorhombic |
| Space group (no.) | P21/n (no. 14) | P41212 (no. 92) | P21/c (no. 14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
P (no. 2) |
Fddd (no. 70) |
| a/Å | 10.182(4) | 8.2128(18) | 15.1483(5) | 6.6164(17) | 8.404(2) | 16.938(6) |
| b/Å | 10.863(3) | 8.2128(18) | 13.8900(4) | 10.637(3) | 10.352(3) | 24.212(9) |
| c/Å | 14.771(5) | 20.989(7) | 16.2854(5) | 12.354(3) | 15.994(5) | 25.686(10) |
| α/° | 90 | 90 | 90 | 93.580(6) | 87.522(7) | 90 |
| β/° | 91.614(5) | 90 | 112.383(3) | 93.952(3) | 89.782(6) | 90 |
| γ/° | 90 | 90 | 90 | 100.854(6) | 86.063(7) | 90 |
| U/Å3 | 1633.0(9) | 1415.7(6) | 3168.44(17) | 849.4(4) | 1386.9(7) | 10534(7) |
| Z | 4 | 4 | 4 | 2 | 2 | 16 |
| μ(Mo-Kα)/mm–1 | 10.552 | 12.159 | 5.576 | 10.250 | 6.307 | 6.719 |
| F(000) | 1000 | 904 | 1616 | 552 | 752 | 5760 |
| Total no. reflns | 7115 | 6585 | 26042 |
7661 | 13101 |
11501 |
| R int | 0.050 | 0.034 | 0.040 | 0.035 | 0.069 | 0.028 |
| Unique reflns | 3709 | 1628 | 7234 | 3848 | 6322 | 3021 |
| No. of params, restraints | 188, 0 | 80, 0 | 342, 0 | 219, 2 | 326, 0 | 212, 223 |
| R 1, wR2 [I > 2σ(I)]b | 0.043, 0.101 | 0.026, 0.055 | 0.031, 0.067 | 0.026, 0.064 | 0.043, 0.087 | 0.053, 0.103 |
| R 1, wR2 (all data) | 0.052, 0.104 | 0.029, 0.056 | 0.045, 0.073 | 0.028, 0.065 | 0.048, 0.088 | 0.061, 0.108 |
Crystallographic data in cif format have been deposited with the Cambridge Crystallographic Data Centre (CCDC) and given numbers 1060048–1060053.
Results and discussion
The lead(II) salts used for the syntheses were Pb(NO3)2 and Pb(SiF6)·2H2O, both of which have negligible solubility in organic solvents that are commonly used for phosphine coordination chemistry, such as CH2Cl2 or toluene. Reactions were therefore performed by dissolving Pb(NO3)2 or Pb(SiF6)·2H2O in the minimum amount of water that had been purged with N2 beforehand, to which a solution of the relevant diphosphine in MeCN was added, affording white powders. Owing to the insolubility of most of the complexes in common NMR solvents (CD2Cl2, CD3CN, D2O and CD3OD), and the labile nature of Pb(II) in solution, the investigations have been driven by solid state characterisation, the key technique being single crystal X-ray diffraction, supported by identification of the anion using infrared spectroscopy, with elemental analysis to confirm the stoichiometries of the bulk samples. Crystals were typically grown by carefully layering a miscible solvent onto a solution of the pre-isolated complex as described in the Experimental. Of the isolated complexes only [Pb{Et2P(CH2)2PEt2}(NO3)2] was sufficiently soluble in a weakly coordinating solvent (CD2Cl2) to allow multinuclear NMR studies. In donor solvents, the diphosphines were partially displaced and no useful data were obtained.Lead(II) nitrate complexes
The reaction of Me2P(CH2)2PMe2 with Pb(NO3)2 in MeCN/H2O formed only the 1:1 [Pb{Me2P(CH2)2PMe2}(NO3)2], despite a 2:1 molar ratio of reagents being used. Elemental analysis of the bulk powder isolated confirmed the 1:1 stoichiometry, while colourless crystals of [Pb{Me2P(CH2)2PMe2}(NO3)2] (Fig. 1a) were grown by layering Et2O onto a DMF solution containing the complex. The structure shows the Me2P(CH2)2PMe2 ligand chelated symmetrically to the Pb(II) centre with d(Pb–P) = 2.7918(15) Å while the nitrate groups are asymmetrically κ2-coordinated (d(Pb–O) = 2.621(4), 2.859(5) Å), all on one hemisphere of the lead. Two further Pb–O contacts, 2.995(4) Å, well within the Van der Waals radii sum of 3.54 Å for Pb⋯O15 from nitrates on neighbouring molecules link the units into an infinite polymer network (Fig. 1b) where the coordination number around the lead(II) centre is eight. The Pb–P bonds are ∼0.05 Å shorter than those in [(2,6-Me2C6H3S)2Pb]3{Me2P(CH2)2PMe2} (2.8389(10) Å).7
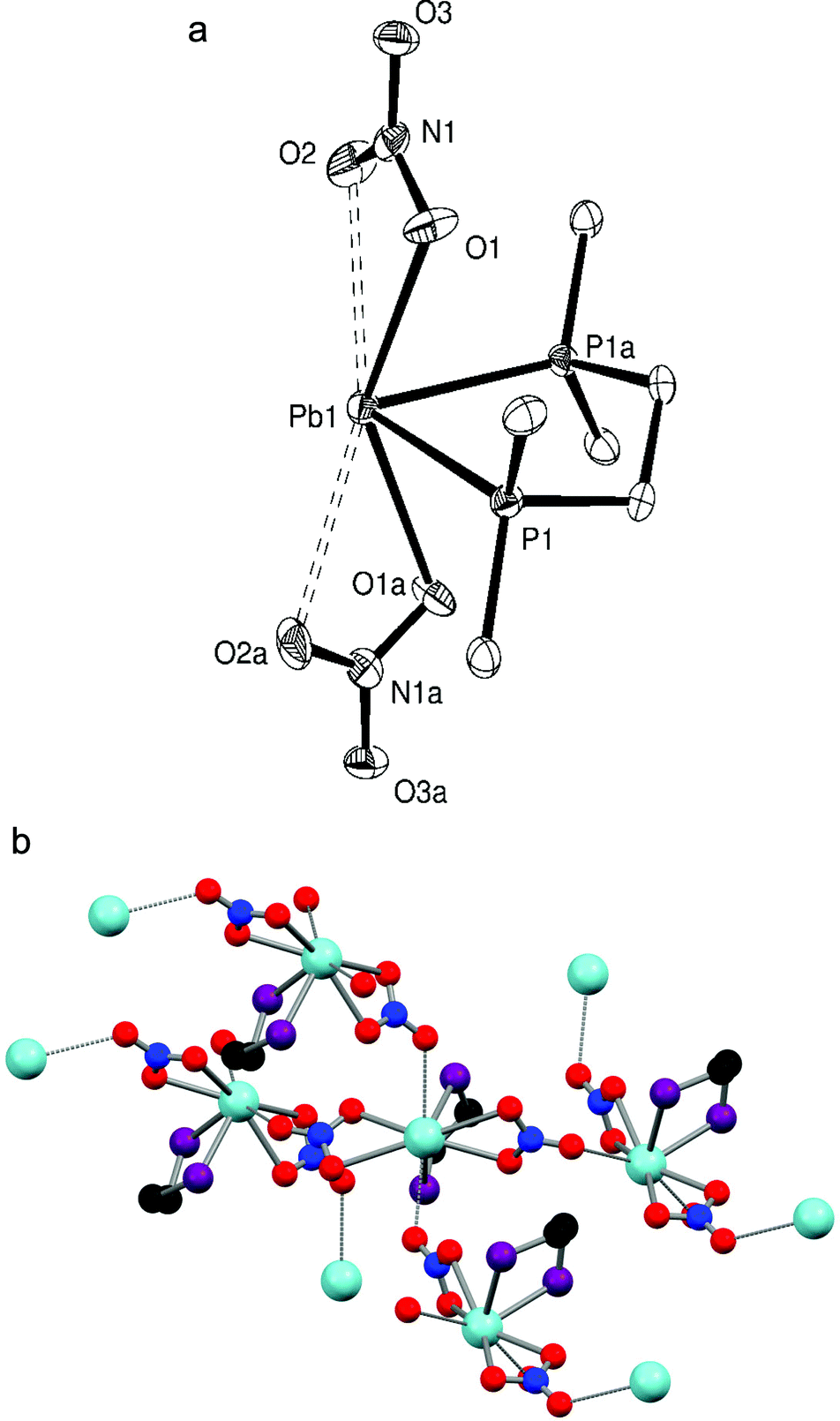 | ||
| Fig. 1 (a) Crystal structure of [Pb{Me2P(CH2)2PMe2}(NO3)2] showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, y, −z. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.7918(15), Pb1–O1 = 2.621(4), Pb1–O2 = 2.859(5), P1–Pb1–P1a = 74.19(6), O1–Pb1–O2 = 46.07(13), O1–Pb1–O1a = 139.98(19). (b) The extended structure of [Pb{Me2P(CH2)2PMe2}(NO3)2]. Methyl groups are omitted for clarity. | ||
Similarly, [Pb{o-C6H4(PMe2)2}(NO3)2] was isolated as a white powder from a 1:1 mol. ratio reaction of o-C6H4(PMe2)2 with Pb(NO3)2. Colourless crystals were obtained from a DMF solution layered with Et2O. A crystal structure determination revealed a similar molecular unit (Fig. 2a) to [Pb{Me2P(CH2)2PMe2}(NO3)2] with the lead(II) centre being eight coordinate. Though no longer symmetrical the Pb–P bond lengths are comparable, while the O–Pb–O angle formed by the nitrate oxygen atoms nearest to the diphosphine is larger in [Pb{o-C6H4(PMe2)2}(NO3)2] (147.36(16)° vs. 139.98(19)°). Again the third oxygen of the nitrate groups bridge (d(Pb–O) = 2.948(5), 2.959(5) Å) to neighbouring molecules to give an infinite chain structure (Fig. 2b).
 | ||
| Fig. 2 (a) Crystal structure of [Pb{o-C6H4(PMe2)2}(NO3)2] showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.806(2), Pb1–P2 = 2.8088(19), Pb1–O1 = 2.869(6), Pb1–O2 = 2.611(6), Pb1–O4 = 2.595(5), Pb1–O5 = 2.827(6), P1–Pb1–P2 = 71.95(6), O1–Pb1–O2 = 46.56(15), O2–Pb1–O4 = 147.37(16), O4–Pb1–O5 = 46.83(15). (b) The polymeric chain structure of [Pb{o-C6H4(PMe2)2}(NO3)2]. Methyl groups are omitted for clarity. | ||
The crystallographically identified κ2-coordinated nitrate groups can also be seen in the IR spectra of [Pb{Me2P(CH2)2PMe2}(NO3)2] and [Pb{o-C6H4(PMe2)2}(NO3)2]. The approximate C2v symmetry of κ2-NO3 groups means three IR active stretching modes (2A1 + B2) are expected;16 as seen with macrocyclic complexes of Pb(NO3)2 (e.g. [Pb(18-crown-6)(NO3)2]), which also exhibit high coordination numbers and κ2-NO3 groups, two stretches are observed at ∼1310 and 1030 cm−1 (the stretch anticipated at ∼1470 cm−1 is obscured by Nujol).10 A bending mode is also evident at ∼815 cm−1.16 Splitting of some of the bands was observed in the IR spectrum of [Pb{o-C6H4(PMe2)2}(NO3)2], which is less clearly resolved for [Pb{Me2P(CH2)2PMe2}(NO3)2].
Owing to the unpredictable geometries and the dissociative nature of complexes of Pb(II) in solution7,11 meaningful NMR spectra can generally only be obtained in non-competitive solvents such as CD2Cl2, in which both [Pb{Me2P(CH2)2PMe2}(NO3)2] and [Pb{o-C6H4(PMe2)2}(NO3)2] were found to be insoluble. By using Et2P(CH2)2PEt2 as the ligand with Pb(NO3)2 under analogous conditions a white powder, [Pb{Et2P(CH2)2PEt2}(NO3)2], was isolated which was found to be freely soluble in CD2Cl2. There is a significant high frequency shift in the 1H NMR spectrum of this complex confirming that the phosphine is coordinated, while a sharp singlet at δ = 89.3 is observed in the 31P{1H} NMR spectrum. This is a very large, high frequency shift from ‘free’ Et2P(CH2)2PEt2 (δ = −18) whose magnitude is mirrored in the shifts seen by Dean et al.8 in their in situ NMR studies of phosphine complexes of Pb(SbF6)2. Upon cooling the solution to 223 K, 207Pb satellites were observed, which sharpened upon further cooling to 193 K (1JPPb = 2532 Hz). The appearance of 207Pb satellites only at low temperatures has been reported before in a study of Pb(II) complexes of 2-phosphinobenzenethiolates,17 and was attributed to ligand lability. Similarly, no resonances were observed in the 207Pb NMR spectrum, even at low temperatures. To ensure that the resonances observed were indeed due to coordinated Et2P(CH2)2PEt2 (i.e. that no unwanted oxidation of the ligand had occurred), an NMR spectrum of the complex was taken in DMSO-d6, which displaced the diphosphine and showed the resonance of the uncoordinated Et2P(CH2)2PEt2. Despite numerous attempts, no crystals of [Pb{Et2P(CH2)2PEt2}(NO3)2] could be grown (instead a few crystals of the phosphine oxide complex [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2] were obtained, discussed below); the IR spectrum of [Pb{Et2P(CH2)2PEt2}(NO3)2] indicates that the nitrate groups are again κ2-coordinated (bands at 1300, 1029 and 816 cm−1), which when combined with the NMR spectroscopic evidence suggests that [Pb{Et2P(CH2)2PEt2}(NO3)2] has a similar structure to the other bidentate phosphine complexes.
Although [Pb{Me2P(CH2)2PMe2}(NO3)2] and [Pb{o-C6H4(PMe2)2}(NO3)2] were found to dissolve completely in DMF, a property exploited in order to grow crystals of these complexes, its relatively strong coordinating nature makes it problematic for NMR studies of these dynamic and labile systems. For both complexes a single resonance was observed in 31P{1H} NMR spectra run in DMF-d7, with chemical shifts intermediate between those expected by analogy with the complex [Pb{Et2P(CH2)2PEt2}(NO3)2] and the uncoordinated ligand shifts. The chemical shifts varied widely with concentration, indicating partial displacement of the diphosphine and rapidly exchanging systems. The values are not quoted. [Pb{Et2P(CH2)2PEt2}(NO3)2] was also found to be partially decomposed by DMF-d7.
Under the same reaction conditions the weaker σ-donor and sterically more crowded ligand o-C6H4(PPh2)2 did not form a complex with Pb(NO3)2. No complex could be isolated with o-C6H4(AsMe2)2, suggesting that the alkylphosphines are better donors in these systems than alkylarsines, an effect also observed with other Group 14 metal acceptors.5
Lead(II) hexafluorosilicate complexes
The reaction of Pb(SiF6)·2H2O with o-C6H4(PMe2)2 in a 1:1 ratio yielded a white powder which, when dissolved in DMF and layered with Et2O, gave small colourless crystals of [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O. The crystal structure (Fig. 3a) shows the diphosphine to be asymmetrically chelating, with a short Pb–F bond (d(Pb1–F5) = 2.573(3) Å) to a κ1-coordinated SiF62− group and a further bond to a water molecule, so that overall the core molecular geometry is very similar to that of [Pb{o-C6H4(PMe2)2}(NO3)2] (Fig. 2a), although the disparity in Pb–P bond lengths is greater (∼0.03 Å) and the F–Pb–O angle is large (154.76(11)°). The core geometry is therefore little affected by the anion present, with the primary contacts again covering one hemisphere of the lead. Although two more Pb–F distances (d(Pb1–F1) = 3.201(3), d(Pb1–F4) = 3.167(3) Å) lie well within the sum of the Van der Waal's radii of 3.49 Å for Pb⋯F,14 these long contacts are probably the result of packing within the crystal lattice causing these F atoms to lie close to the Pb(II) centre. Nevertheless, an alternative description for the molecular structure of [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O is that the SiF62− group is asymmetrically κ3-coordinated. These units are arranged into infinite strands by a Pb–F interaction from a neighbouring SiF62− group (d(Pb–F) = 2.815(3) Å); two further Pb–F interactions with a SiF62− group on a parallel strand (d(Pb–F) = 2.795(3), 3.083(3) Å) gives a polymeric chain structure overall (Fig. 3b), where the lead(II) centre is seven coordinate (or nine if the core SiF62− group is described as κ3-coordinated). The o-C6H4(PMe2)2 groups are arranged so that they point outwards from the chain. The coordinated water molecule is also hydrogen bonded to a lattice water molecule (HO–H⋯OH2 = 1.89(7) Å), and together these form a hydrogen-bonding network (Fig. 3b) with F atoms on nearby SiF62− groups (HO–H⋯F = 1.93(2), 2.05(6), 2.14(3) Å).
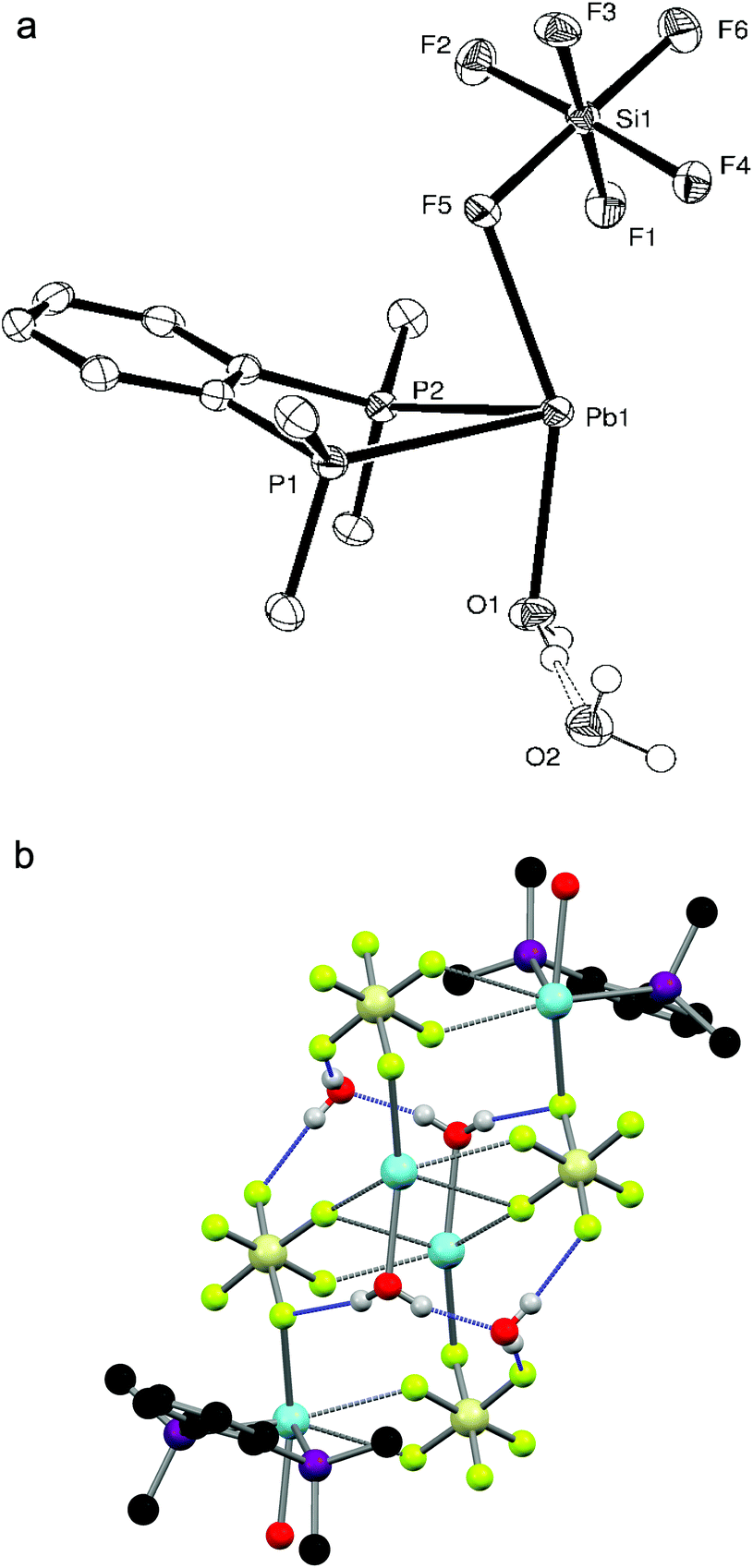 | ||
| Fig. 3 (a) Crystal structure of [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.8299(14), Pb1–P2 = 2.7976(13), Pb1–F5 = 2.573(3), Pb1–O1 = 2.496(3), P1–Pb1–P2 = 69.67(4), F5–Pb1–O1 = 154.76(11). (b) The polymeric chain structure of [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O. The intermolecular H-bonding network is shown in blue and the diphosphines on the two central lead atoms are omitted for clarity. | ||
The IR spectrum of the bulk solid confirms the presence of the SiF62− group, although the anion coordination mode cannot be reliably identified.16 The stretching mode is significantly broadened, indicating that the Pb–F interactions are too weak to lower the symmetry of the fluoroanion sufficiently to give resolved splittings.121 Water is also visible in the IR spectrum, indicating that the bulk solid is probably also [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·xH2O, though a satisfactory elemental analysis could not be obtained. Poor solubility precluded NMR analysis of this complex.
Repetition of the reaction with a 2:1 (diphosphine:Pb) ratio yielded a few crystals of [Pb{o-C6H4(PMe2)2}(DMF)2(SiF6)]·DMF, again demonstrating the preference of the Pb(II) centre to coordinate to just one diphosphine ligand. The asymmetric unit has the expected core bonding environment (P2FO) around the lead, with an O-bonded DMF molecule in place of H2O, giving a F–Pb–O angle of 140.82(12)°. The SiF62− group is now κ2- or κ3-coordinated depending on the viewpoint – the d(Pb1–F3) is ∼0.3 Å shorter than the comparable distance in [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O, while the d(Pb1–F2) remains long at 3.081(4) Å. The size of the DMF molecule and the lack of hydrogen bonding are presumably the main reasons the units now dimerise (Fig. 4) with the SiF62− groups bridging via one F atom per fluoroanion. The orientation of a second DMF molecule close to the Pb(II) centre suggests that is also interacting with the lead (d(Pb–O) = 3.263(6) Å), which gives an overall coordination number of seven or eight for Pb(II). There is also a DMF solvate molecule in the structure. Formation of this complex also shows that DMF will readily coordinate to lead(II), highlighting its unsuitability as a NMR solvent in these systems, but because of the lability of the complex in solution, the structure obtained is ultimately dependant on which complex crystallises out under the conditions employed.
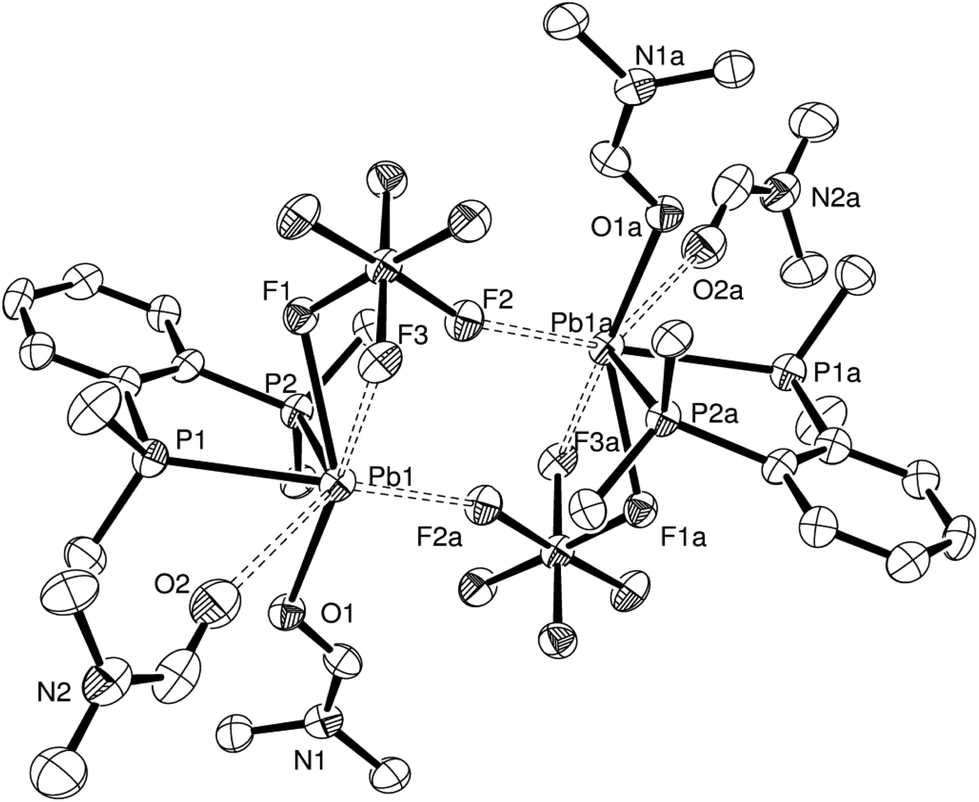 | ||
| Fig. 4 Crystal structure of the SiF6-bridged dimer present in [Pb{o-C6H4(PMe2)2}(DMF)2(SiF6)]·DMF showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level. H atoms and DMF solvate molecules are omitted for clarity. Symmetry operation: a = 2 − x, 2 − y, 1 − z. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.7671(19), Pb1–P2 = 2.7553(16), Pb1–F1 = 2.585(3), Pb1–F3 = 2.894(3), Pb1–F2a = 2.752(3), Pb1–O1 = 2.463(4), Pb1–O2 = 3.263(6), P1–Pb1–P2 = 72.30(5), F1–Pb1–F3 = 50.67(9), F1–Pb1–F2a = 110.58(10), F1–Pb1–O1 = 140.82(12), F1–Pb1–O2 = 109.10(12). | ||
Phosphine oxide complexes
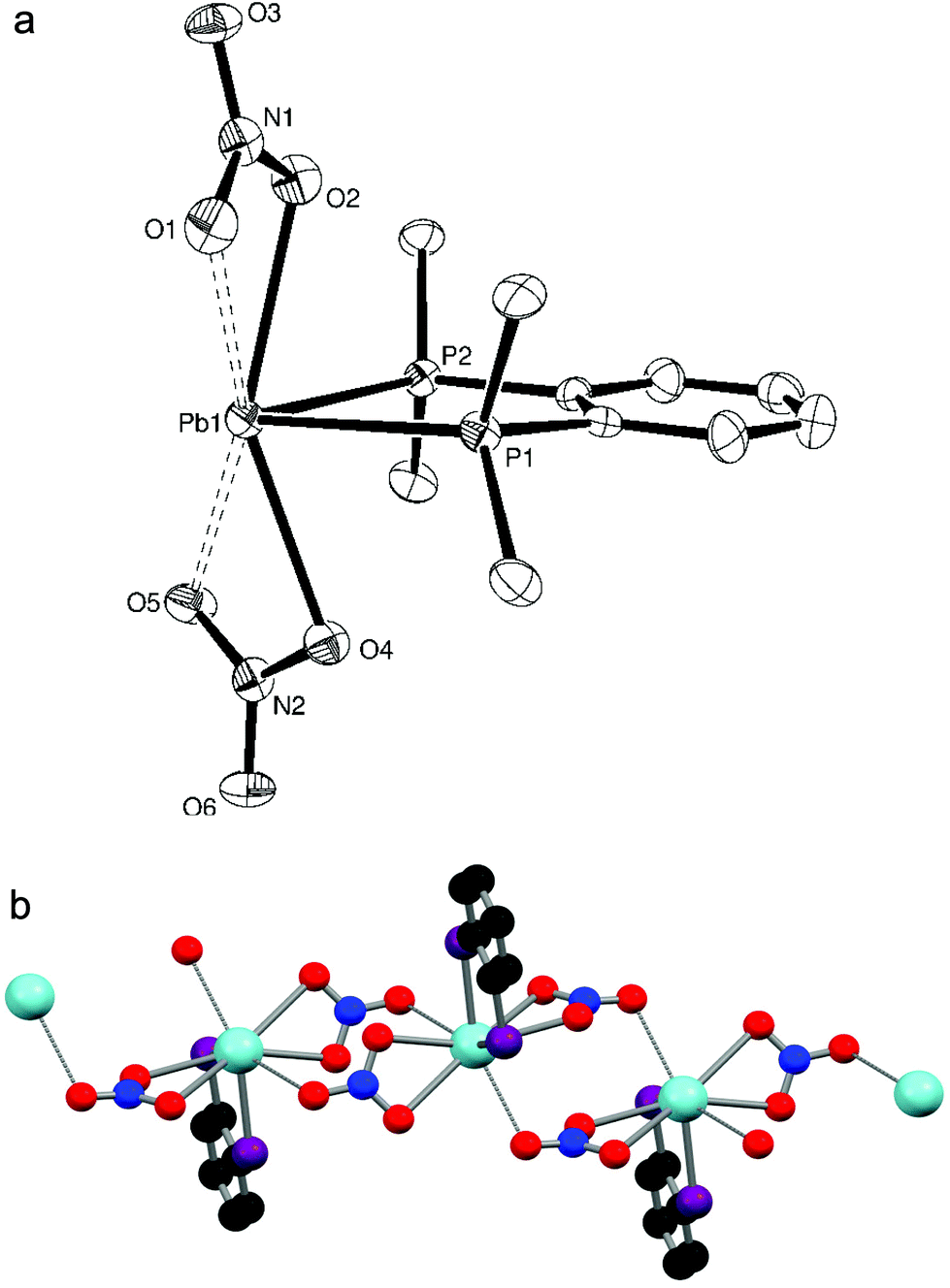
When trying to grow crystals of [Pb{Et2P(CH2)2PEt2}(NO3)2] by recrystallisation from CH2Cl2 and n-hexane, a few small colourless crystals of [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2] were instead isolated, indicating that in situ oxidation of the ligand has occurred, presumably due to adventitious O2. The X-ray structure determination reveals (Fig. 5a) that the Et2(O)P(CH2)2P(O)Et2 ligands each bridge to different Pb(II) atoms, and that there are two phosphine oxide ligands per metal centre, despite the parent phosphine complex having a 1:1 stoichiometry. This gives a core PbO4 geometry, with one Pb–O bond ∼0.1 Å longer than the other three and a O1–Pb1–O4 angle of 160.29(9)°. Interactions with the nitrate groups are weaker than in [Pb(L–L)(NO3)2] (L–L = o-C6H4(PMe2)2, Me2P(CH2)2PMe2) but they remain κ2-coordinated (the third oxygen is uncoordinated) and fill the remainder of the coordination sphere, to give eight coordinate lead. Due to the bridging nature of the phosphine oxide groups, the extended structure (Fig. 5b) is an infinite polymer network.
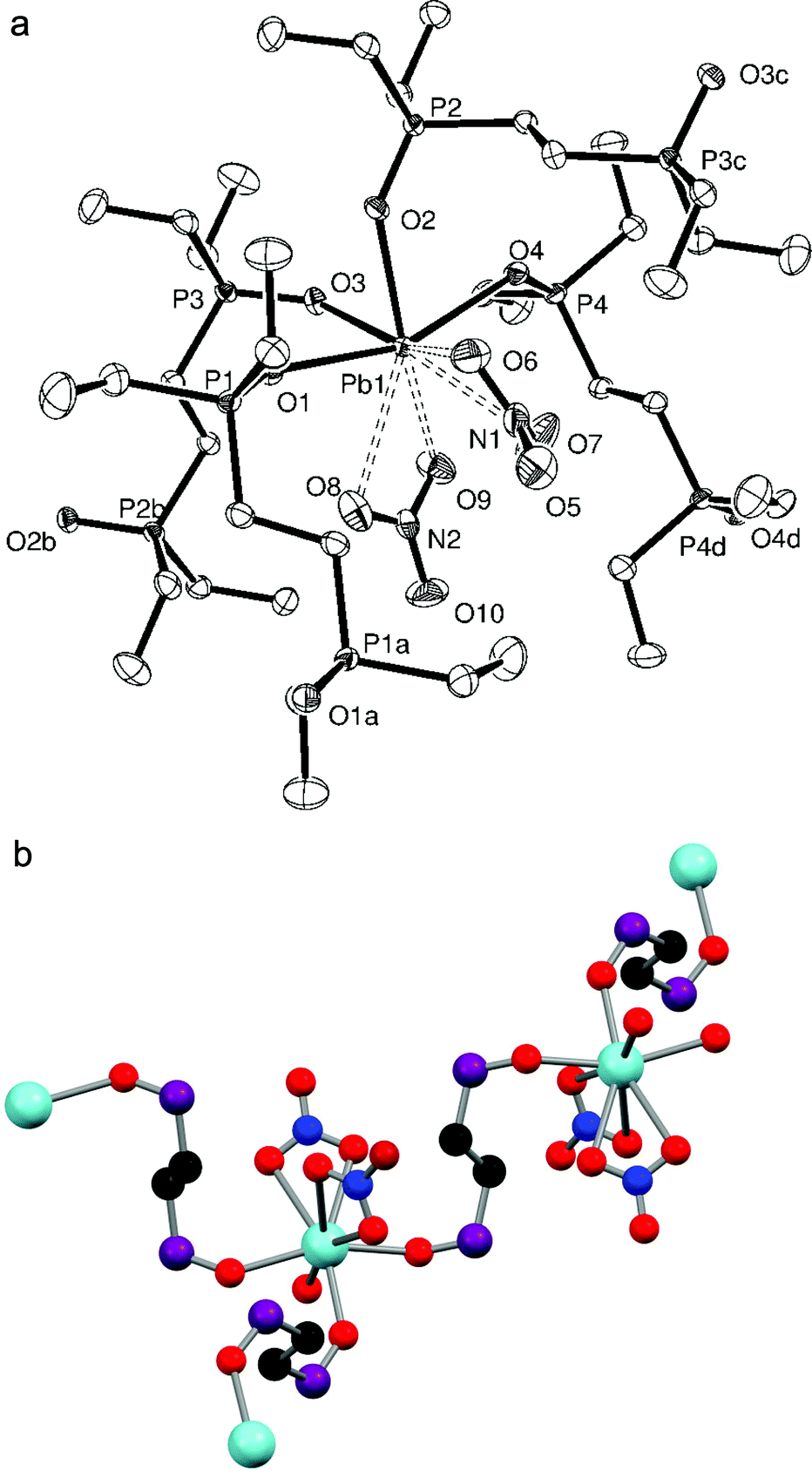 | ||
| Fig. 5 (a) Crystal structure of [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2] showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operations: a = −x, 1 − y, 1 − z; b = x, 1/2 − y, 1/2 + z; c = x, 1/2 − y, −1/2 + z; d = 1 − x, 1 − y, 1 − z. Selected bond lengths (Å) and angles (°): Pb1–O1 = 2.484(2), Pb1–O2 = 2.429(2), Pb1–O3 = 2.418(2), Pb1–O4 = 2.589(2), Pb1–O6 = 2.786(2), Pb1–O7 = 2.912(3), Pb1–O8 = 2.875(3), Pb1–O9 = 2.891(3), P1–O1 = 1.502(3), P2–O2 = 1.504(2), P3–O3 = 1.510(3), P4–O4 = 1.504(3), O1–Pb–O2 = 80.24(8), O1–Pb–O3 = 83.96(8), O1–Pb–O4 = 160.29(9), O2–Pb–O3 = 81.05(9), O2–Pb–O4 = 81.58(8), O3–Pb–O4 = 85.75(8), O6–Pb–O7 = 44.41(10), O8–Pb–O9 = 43.97(8), Pb1–O1–P1 = 144.23(15), Pb1–O2–P2 = 137.26(14), Pb1–O3–P3 = 132.28(15), Pb1–O4–P4 = 131.85(14). (b) The extended structure of [Pb{Et2(O)P(CH2)2P(O)Et2}2(NO3)2]. Ethyl groups are omitted for clarity. | ||
The in situ oxidation of diphosphine ligands in the presence of heavy p-block metals has been reported before;5 for example, the oxidation of the 1:1 adduct of BiCl3 and Ph2PCH2PPh2 to the crystallographically identified [BiCl3{Ph2P(O)CH2P(O)Ph2}]2 having occurred when trying to grow crystals of the phosphine complex.18 Interestingly, the in situ oxidation of diphosphine ligands has also been reported with Pb(II), when a few crystals of [{(Pb3(μ-I)6)(Ph2(O)P(CH2)2P(O)Ph2)2}]·EtOH were obtained from the solvothermal reaction of PbI2 with KI, I2 and Ph2P(CH2)2PPh2. Attempts to increase the yield of the phosphine oxide complex by introducing H2O2 into the reaction generated [{Pb2(μ-I)2(μ3-I)2}{Ph2(O)P(CH2)2P(O)Ph2}]. Both structures have octahedrally coordinated Pb(II) centres with the Ph2(O)P(CH2)2P(O)Ph2 groups bridging between metal centres.19
Attempts to form diphosphine complexes with Pb(BF4)2 (supplied as a 50% aqueous solution) were largely unsuccessful due to the acidity of the solution (which contains some excess acid to prevent hydrolysis), and led to protonation of the phosphine ligands. This tendency towards ligand protonation has been previously observed when crystals of [terpyH2][BF4]2 (terpy = 2,2′:6′,2′′-terpyridyl) were obtained from the reaction of aqueous Pb(BF4)2 and terpy.12 Despite this, on one occasion small colourless crystals of [Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2 were obtained from the reaction of aqueous Pb(BF4)2 (one mol. equiv.) with two mol. equiv. of Me2P(CH2)2PMe2 in MeNO2. The complexes crystallises in the unusual orthorhombic Fddd space group. The preferred coordination mode of both the diphosphine and diphosphine dioxide to lead(II) discussed above is also evident here, as the X-ray structure (Fig. 6a) reveals the Me2P(CH2)2PMe2 to be symmetrically chelating, while the Me2(O)P(CH2)2P(O)Me2 (from the in situ oxidation of the diphosphine ligand) bridges between Pb(II) centres, giving an infinite chain structure (Fig. 6b). The symmetry related BF4− groups are disordered, but seem to adopt two distinct and reasonably well-defined orientations. Although not discussed in detail because of the disorder, the Pb–F distances in the two orientations are similar at ∼3.1 and 3.3 Å, allowing the fluoroanion to be described as either κ1- or κ2-coordinated. Overall the Pb(II) exhibits the expected core (P2O2) geometry. There is also a disordered (the N atom is located on a site with 222 symmetry) fractionally occupied (50%) MeNO2 solvate molecule in the asymmetric unit.
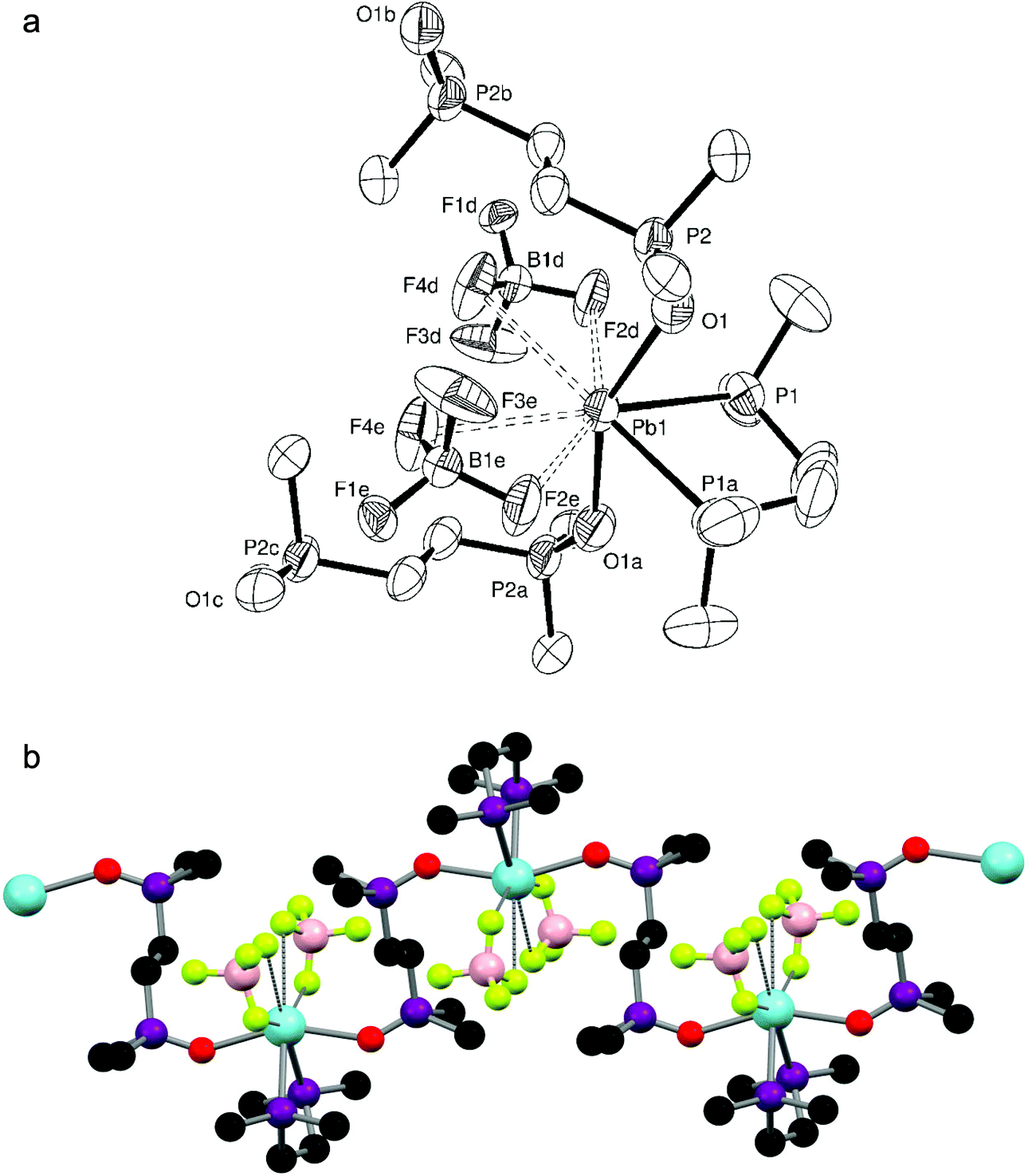 | ||
| Fig. 6 (a) Structure of [Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2 showing the atom labelling scheme. The displacement ellipsoids are drawn at the 50% probability level and H atoms and the fractionally occupied (50%) MeNO2 solvate molecule are omitted for clarity. Symmetry operations: a = 7/4 − x, 3/4 − y, z; b = 2 − x, 1 − y, −z; c = −1/4 + x, −1/4 + y, −z; d = 2 − x, −1/4 + y, −1/4 + z; e = −1/4 + x, 1 − y, −1/4 + z. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.789(2), Pb1–O1 = 2.454(6), O1–P2 = 1.478(6), P1–Pb1–P1a = 72.86(9), O1–Pb1–O1a = 154.5(3), Pb1–O1–P2 = 139.1(3). The BF4 is disordered with two distinct orientations (only one orientation is shown). (b) The extended structure of [Pb{Me2P(CH2)2PMe2}{Me2(O)P(CH2)2P(O)Me2}][BF4]2·½MeNO2. Only one orientation of the BF4 is shown. | ||
Conclusions
Several new and rare diphosphine complexes of the lead(II) salts Pb(NO3)2 and Pb(SiF6) have been isolated and structurally characterised, revealing a clear preference for a single bidentate phosphine ligand to chelate to lead(II). Unexpectedly, there is no evidence that a second diphosphine can be introduced to the Pb(II) coordination sphere, in marked contrast to the complexes of diimines where bis-ligand complexes predominate.11,12 This is unlikely to be due to steric constraints on the large metal centre, and presumably reflects rather limited affinity of the lead(II) for the phosphorus donors. Although the geometries around the Pb(II) are highly irregular, a core four-coordinate motif arranged on one hemisphere of the lead is identifiable in every complex, with longer (weaker) contacts to anions from neighbouring molecules occupying much of the remaining coordination sphere, leading to extended polymer chain and network structures. As with the di- and tri-imine complexes of Pb(SiF6),11 the little studied SiF62− dianion can bond to a single Pb(II) ion via one or more Pb–F interaction(s), or can bridge between metal centres. The SiF62− group is stable in these systems, with no evidence of hydrolysis or fragmentation of the fluoroanion having occurred. This contrasts with the degradation seen for some PF6− or BF4− salts with Pb(II) and Sn(II) macrocyclic species.10 In some cases adventitious oxygen leads to in situ oxidation of diphosphine ligands, yielding the corresponding diphosphine dioxide complexes, where the diphosphine dioxide bridges between lead(II) centres. This remains a challenging area, with poor solubility and the lability of Pb(II) in solution limiting the information that can be gathered from spectroscopic characterisation.Acknowledgements
We thank the EPSRC for support (EP/I033394/1). The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids. We also thank Dr M. Light and Dr M. Webster for help with the crystallographic analysis.References
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- J. Parr, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, p. 545 Search PubMed.
- E. S. Claudio, H. A. Godwin and J. S. Magyar, Prog. Inorg. Chem., 2003, 51, 1 CAS.
- R. L. Davidovich, V. Stavila and K. H. Whitmire, Coord. Chem. Rev., 2010, 254, 2193 CrossRef CAS.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65 CrossRef CAS.
- C. E. Wymore and J. C. Bailar, J. Inorg. Nucl. Chem., 1960, 14, 42 CrossRef CAS.
- A. J. Rossini, A. W. Macgregor, A. S. Smith, G. Schatte, R. W. Schurko and G. G. Briand, Dalton Trans., 2013, 42, 9533 RSC.
- P. A. W. Dean, D. D. Phillips and L. Polensek, Can. J. Chem., 1981, 59, 50 CrossRef.
- P. A. W. Dean, Can. J. Chem., 1983, 61, 1795 CrossRef CAS.
- P. Farina, T. Latter, W. Levason and G. Reid, Dalton Trans., 2013, 42, 4714 RSC; C. Beattie, P. Farina, W. Levason and G. Reid, Dalton Trans., 2013, 42, 15183 RSC.
- M. Kadarkaraisamy, D. P. Engelhart, P. M. Basa and A. G. Sykes, J. Coord. Chem., 2010, 63, 2261 CrossRef CAS; L. M. Engelhardt, J. M. Harrowfield, H. Miyamae, J. M. Patrick, B. W. Skelton, A. A. Soudi and A. H. White, Aust. J. Chem., 1996, 49, 1111 CrossRef; I. Bytheway, L. M. Engelhardt, J. M. Harrowfield, D. L. Kepert, H. Miyamae, J. M. Patrick, B. W. Skelton, A. A. Soudi and A. H. White, Aust. J. Chem., 1996, 49, 1099 CrossRef; L. M. Engelhardt, J. M. Harrowfield, H. Miyamae, J. M. Patrick, B. W. Skelton, A. A. Soudi and A. H. White, Aust. J. Chem., 1996, 49, 1135 CrossRef.
- J. Burt, W. Grantham, W. Levason, M. E. Light and G. Reid, Polyhedron, 2015, 85, 530 CrossRef CAS.
- E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877 CrossRef CAS; R. D. Feltham, A. Kasenally and R. S. Nyholm, J. Organomet. Chem., 1967, 7, 285 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlain, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, NY, 4th edn, 1986 Search PubMed.
- B. M. Barry, B. W. Stein, C. A. Larsen, M. N. Wirtz, W. E. Geiger, R. Waterman and R. A. Kemp, Inorg. Chem., 2013, 52, 9875 CrossRef CAS PubMed.
- G. R. Willey, M. D. Rudd, C. J. Samuel and M. G. B. Drew, J. Chem. Soc., Dalton Trans., 1995, 759 RSC.
- Z. J. Huang, H. J. Cheng, M. Dai, C. Y. Ni, H. X. Li, K. P. Hou, Z. G. Ren and J. P. Lang, Inorg. Chem. Commun., 2013, 31, 33 CrossRef CAS.
Footnote |
| † CCDC 1060048–1060053. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01448d |
| This journal is © The Royal Society of Chemistry 2015 |