
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyne substituted mononuclear photocatalysts based on [RuCl(bpy)(tpy)]+†
Ross J.
Davidson
a,
Lucy E.
Wilson
a,
Andrew R.
Duckworth
a,
Dmitry S.
Yufit
a,
Andrew
Beeby
a and
Paul J.
Low
*b
aDepartment of Chemistry, University of Durham, South Road, Durham DH1 3LE, England, UK
bSchool of Chemistry and Biochemistry, University of Western Australia, 35 Stirling Highway, Crawley, 6009 WA, Australia. E-mail: paul.low@uwa.edu.au
First published on 5th May 2015
Abstract
The ethynyl-phenylene substituted 2,2′:6′,2′′-terpyridine (tpy) derivatives, 4-(phenyl-ethynyl)-2,2′:6′,2′′-terpyridine (L1), 4-(methoxyphenyl-ethynyl)-2,2′:6′,2′′-terpyridine (L2), 4-(tolyl-ethynyl)-2,2′:6′,2′′-terpyridine (L3) and 4-(nitrophenyl-ethynyl)-2,2′:6′,2′′-terpyridine (L4) have been used to synthesize four new [RuCl(2,2′-bipyridine)(Ln)]PF6 based complexes. Electronic absorption, resonance Raman, cyclic voltammetry and spectroelectrochemistry aided by DFT calculations were used to explore the influence of the alkynyl substituents on the electronic structures, photochemical and redox properties of the complexes. Furthermore, it is shown that the addition of ethynyl phenyl moieties to the 4-position of the tpy ligand does not have a detrimental effect on these complexes, or the analogous aqua complexes, with respect to their ability to photocatalyse the oxidation of 4-methoxybenzyl alcohol to the corresponding benzaldehyde.
Introduction
With global energy demands rising at an alarming rate, and concerns over the supply, security and environmental impact of conventional energy resources, attention is being turned to alternative, non-carbon based forms of energy. In this regard, the use of solar power, either directly through photovoltaics or as a source of energy to produce fuels through artificial photosynthetic pathways is widely regarded as the most viable long term solution.1–4 Of the various solar fuel alternative to fossil-derived hydrocarbons, hydrogen is especially attractive, offering high energy density, yielding water as the only by-product of combustion and being available in almost limitless amounts, provided the challenges of coupling proton reduction to an efficient method of water oxidation can be overcome (eqn (1) and (2))| 4H+ + 4e− → 2H2 | (1) |
| 2H2O → O2 + 4H+ + 4e− | (2) |
The chemical challenges of water oxidation (eqn (2)), requiring the extraction of four protons and electrons from two water molecules and formation of an O–O bond, are the most demanding, as well as posing some significant fundamental questions concerning proton-coupled electron transfer reactions. Nature has overcome these various issues using an exquisitely balanced chain of light harvesting complexes, electron transfer centres and a catalytically active polynuclear {Mn4CaO5} cluster to achieve photosynthesis in green plants.5,6 Inspired in part by this biological template, metal complexes offering a range of stable oxidation states, photochemical properties and, at least in principle, a modular aspect to their construction, have been now widely explored as water oxidation catalysts,7–9 although recent rapid advances in solid-state catalysts must also be recognised.10–16
The seminal work of Meyer's group on the ‘blue dimer’ [{(bpy)2(H2O)Ru}2(μ-O)]4+, which turns over water in the presence of a sacrificial Ce(IV) oxidant17 focussed many studies of molecular catalysts for water oxidation on complexes of ruthenium.18 Refinement of the binuclear platform lead to the development of the Tanaka catalyst [{(3,6-tBu2Q)2(OH)Ru}2(μ-btpyan)]2+ (3,6-tBu2Q = 3,6-di-tert-butyl-1,2-quinone; btpyan = 2,2′:6′,2′′-terpyridyl)-anthracene) which displayed an impressive TON of 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 over 40 h of operation as an electrocatalyst immobilised on an ITO electrode,19 whilst later work lead largely by Meyer,20 Thummel21,22 and Sakai23 demonstrated the efficacy of mononuclear ruthenium complexes as water oxidation catalysts.24–26 Allied studies with mononuclear iridium complexes have demonstrated the broader scope of single site catalysts.27–29
500 over 40 h of operation as an electrocatalyst immobilised on an ITO electrode,19 whilst later work lead largely by Meyer,20 Thummel21,22 and Sakai23 demonstrated the efficacy of mononuclear ruthenium complexes as water oxidation catalysts.24–26 Allied studies with mononuclear iridium complexes have demonstrated the broader scope of single site catalysts.27–29
Within the ruthenium series of mononuclear complexes, a common design feature in the presence of five nitrogen donor ligands, typically as a tridentate and a bidentate or two monodentate ligands, and many catalysts featuring variations on this structural design are known.22,30–34 These systems are typically driven by a sacrificial chemical oxidant, such as Ce(IV), to regenerate the active form of the catalyst. Whilst this approach generates immense amounts of vital mechanistic information, complications can arise from the chemical non-innocence of the sacrificial reagent and counter ions,35,36 leading to interest in alternative strategies, such as immobilisation of these electrocatalysts on electrode surfaces.37–40
One alternative to the Ce(IV) based sacrificial oxidants, and which drives the area closer to the ultimate goal of a solar (light) driven process, utilises a photosensitizer such as [Ru(bpy)3]2+ or related derivatives which have reduction potentials in the excited state sufficiently low to oxidise many of the common Ru-based water oxidation catalysts, in conjugation with a chemically benign sacrificial oxidant such as [Co(NH3)5Cl]2+41 or Na2S2O8.42,43 The next logical step in the evolution of molecular water oxidation catalysts is the development of dyad systems in which the photosensitizer and catalyst complex are linked via a suitably positioned and constructed bridge.6–8,44–49
The design of sensitizer-catalyst dyads for water oxidation is an immense challenge, demanding a balance between the photochemical properties of the dyad, forward and reverse electron transfer rates, thermodynamic driving forces and chemical kinetics of an intricate PCET bimolecular reaction (eqn (2)), all within a synthetically achievable platform. As part of the effort to explore the different aspects of this overall reaction scheme, attention has been directed to models of the key transformation [{RuII}–OH2]2+ → [{RuIV}![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+,50–53 in which a [Ru(bpy)3]2+ style photosensitizer is used to photooxidise the catalyst centre, in conjunction with a mild sacrificial oxidant, [CoIII(NH3)5Cl]2+ to recycle the sensitizer. The photogenerated [{RuIV}O]2+ complexes can be used to oxidize benzyl alcohols, as a model for water, in a process that can be conveniently followed by 1H NMR spectroscopy, with concomitant 2-e/2H+ reduction of the catalyst back to the [{RuII}–OH2]2+ state. Both multi-component53 and covalently linked dyads50–52 have explored in this fashion.
O]2+,50–53 in which a [Ru(bpy)3]2+ style photosensitizer is used to photooxidise the catalyst centre, in conjunction with a mild sacrificial oxidant, [CoIII(NH3)5Cl]2+ to recycle the sensitizer. The photogenerated [{RuIV}O]2+ complexes can be used to oxidize benzyl alcohols, as a model for water, in a process that can be conveniently followed by 1H NMR spectroscopy, with concomitant 2-e/2H+ reduction of the catalyst back to the [{RuII}–OH2]2+ state. Both multi-component53 and covalently linked dyads50–52 have explored in this fashion.
Curiously, despite the well-developed ‘wire-like’ properties of the alkynyl moiety, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–, capable of promoting electron transfer between remote sites, and synthetic compatibility with polypyridyl-based ligand scaffolds,54–56 there have been no reports to date of this fragment being used in the design of photoactive water oxidation dyads. As an initial step towards the design of alkyne-bridged photosensitizer-water oxidation complex dyads we have been drawn to the elementary [Ru(H2O)(bpy′)(tpy′)]2+ family of water oxidation catalysts.23 Substitution of both the 2,2′-bipyridine (bpy) and 2,2′:6′,2′′-terpyridine (tpy) ligands with a range of electron donating and withdrawing groups has been achieved, with general conclusions being that 4′-substitution of the tpy ligand by donor groups (OEt, OMe) gave enhanced catalytic activity, albeit with lower catalyst stability (Fig. 1).34,57,58
C–, capable of promoting electron transfer between remote sites, and synthetic compatibility with polypyridyl-based ligand scaffolds,54–56 there have been no reports to date of this fragment being used in the design of photoactive water oxidation dyads. As an initial step towards the design of alkyne-bridged photosensitizer-water oxidation complex dyads we have been drawn to the elementary [Ru(H2O)(bpy′)(tpy′)]2+ family of water oxidation catalysts.23 Substitution of both the 2,2′-bipyridine (bpy) and 2,2′:6′,2′′-terpyridine (tpy) ligands with a range of electron donating and withdrawing groups has been achieved, with general conclusions being that 4′-substitution of the tpy ligand by donor groups (OEt, OMe) gave enhanced catalytic activity, albeit with lower catalyst stability (Fig. 1).34,57,58
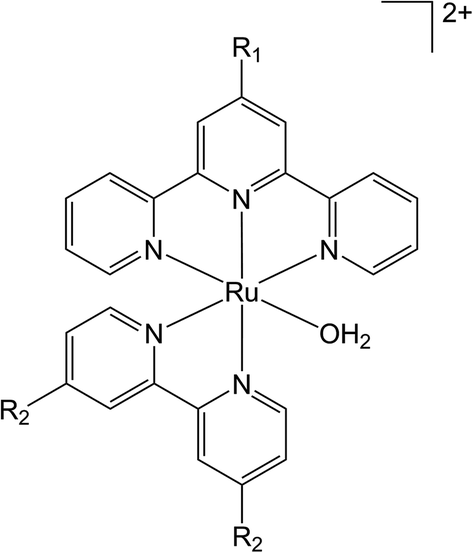 | ||
| Fig. 1 Substitution pattern of the ligands demonstrated by Yagi and Berlinguette et al. where R1 = H, EtO, MeO, Me, Cl or COOH and R2 = H, OMe, COOH, Cl. | ||
Here we describe the preparation and characterisation of 4-arylethynyl-2,2′:6′,2′′-terpyridine ligands Ln, and the complexes [RuCl(bpy)(Ln)]PF6. The behaviour of [RuCl(bpy)(Ln)]PF6 and the analogous aqua complex [Ru(H2O)(bpy)(Ln)]2+ (prepared in situ by chloride ion abstraction) as oxidation photocatalysts was assayed through the [Ru(bpy)3]2+ sensitized photocatalytic oxidation of 4-methoxybenzyl alcohol.7,11 The comparable catalytic response of the alkynyl substituted derivatives with the parent system [Ru(H2O)(bpy)(tpy)](OTf)2 indicates that the introduction of the ethynyl moiety to the tpy ligand scaffold is not detrimental to the catalytic process, opening avenues for further development of covalently linked sensitizer-catalyst dyads.
Experimental section
The compounds 4-ethynyl-nitrobenzene,59 Pd(PPh3)4,60 4′-(phenylethynyl)-2,2′:6′,2′′-terpyridine (L1)61 and 4′-[[(trifluoromethyl)sulfonyl]oxy]-2,2′:6′,2′′-terpyridine (tpyOTf)62 were synthesised by literature procedures. The compounds 4-ethynyl toluene, bpy, LiCl, N-ethyl morpholine and RuCl3·3H2O were purchased and used as received. All glassware was over dried (120 °C), nitrogen environments were created through a high purity nitrogen line and dry solvents were reagent grade.4′-(Methoxyphenylethynyl)-2,2′:6′,2′′-terpyridine (L2)
Triethylamine (7 mL) was added to a THF solution (20 mL) of tpyOTf (250 mg, 0.65 mmol) and 4-ethynyl anisole (86 mg, 0.65 mmol). The mixture was degassed by three freeze–pump–thaw cycles before Pd(PPh3)4 (75 mg, 0.065 mmol) was added. The solution was heated at reflux overnight in the dark, after which time the solvent was removed, the residue extracted in dichloromethane, and the extracts filtered. The filtrate was passed down a silica column initially with neat CH2Cl2 then CH2Cl2:acetonitrile (1:1) to elute the product. The fraction containing the product was taken to dryness and washed with methanol and dried, giving a white solid. Yield: 191 mg (81%). ES-MS: m/z 364 [M + H]+. 1H NMR(CDCl3): δ 8.71 (ddd (J = 5, 2, 1 Hz), 2H), 8.61 (dt (J = 8, 1 Hz), 2H), 8.55 (s, 2H), 7.86 (td (J = 8, 2 Hz), 2H), 7.51 (d (J = 8 Hz), 2H), 7.34 (ddd (J = 8, 5, 1 Hz), 2H), 6.90 (d (J = 8 Hz), 2H), 2.16 (s, 3H) ppm. 13C{1H} NMR (CDCl3): δ. 160.2, 155.9, 155.5, 149.2, 137.0, 133.9, 124.0, 122.7, 121.3, 114.6, 114.3, 94.2, 86.6, 55.3 ppm. Anal. Calc. C24H17N3O·0.1CH2Cl2: C, 77.83; H, 4.66; N, 11.30%. Found: C, 77.58; H, 4.35; N, 11.26%.
4′-(Tolylethynyl)-2,2′:6′,2′′-terpyridine (L3)
The same procedure as for L2 except 4-ethynyl toluene was used in place of 4-ethynyl anisole. Crystals were grown by the slow evaporation of a chloroform solution. Yield: 178 mg (79%). ES-MS: m/z 348 [M + H]+. 1H NMR (CDCl3): δ 8.71 (ddd (J = 5, 2, 1 Hz), 2H), 8.60 (dt (J = 8, 1 Hz), 2H) 8.56 (s, 1H) 7.84 (td (J = 8, 2 Hz), 2H) 7.47 (d (J = 8 Hz), 2H), 7.33 (ddd (J = 8, 5, 1 Hz) 2H) 7.18 (d (J = 8 Hz), 2H) 2.38 (s, 3H) ppm. 13C NMR (CDCl3): δ 155.82, 155.56, 149.26, 139.43, 136.96, 133.74, 131.98, 129.36, 124.06, 122.87, 121.31, 119.52, 94.22, 87.11, 21.71 ppm. Anal. Calc. C24H17N3: C, 82.97; H, 4.93; N, 12.09%. Found: C, 82.83; H, 4.95; N, 12.23%.4′-(Nitrophenylethynyl)-2,2′:6′,2′′-terpyridine (L4)
The same procedure as for L2 except 4-ethynyl nitrobenzene was used in place of 4-ethynyl anisole. Yield: 159 mg (65%). ESMS: m/z 379 [M + H]+. 1H NMR (CDCl3): δ 8.72 (ddd (J = 5, 2, 1 Hz), 2H), 8.63 (dt (J = 8, 1 Hz), 2H) 8.60 (s, 2H) 8.25 (d (J = 8 Hz), 2H) 7.88 (td (J = 8, 2 Hz), 2H), 7.71 (d (J = 8 Hz), 2H) 7.37 (ddd (J = 8, 5, 1 Hz), 2H) ppm. 13C NMR (CDCl3): δ 155.6, 155.2, 149.1, 147.5, 137.1, 132.6, 129.1, 124.2, 123.7, 122.9, 121.3, 92.0, 91.1 ppm. Anal. Calc. for C23H14N4O2: C, 73.01; H, 3.73; N, 14.81%. Found: C, 72.86; H, 3.77; N, 14.74%.[RuCl(bpy)(L1)]PF6 ([1Cl]PF6)
A solution of L1 (200 mg, 0.60 mmol) and RuCl3·3H2O (156 mg, 0.60 mmol) in ethanol (30 mL) was heated at reflux for 3 h. and allowed to cool. The solution was filtered, and the precipitate collected washed with water, ethanol, diethyl ether and finally chloroform giving a brown powder, presumably RuCl3(L1). A suspension of the brown powder, bpy (94 mg, 0.60 mmol), LiCl (25 mg, 0.60 mmol), N-ethylmorpholine (0.3 mL) and methanol (20 mL) was heated at reflux for 3 h. before being allowed to cool and filtered. The filtrate was dried under vacuum, extracted into dichloromethane and filtered. The solvent was removed from the filtrate, the resulting residue redissoved in methanol and NH4PF6 (196 mg, 1.20 mmol) was added to cause precipitation. The solvent was removed to give a purple solid which was purified on a neutral alumina column, eluting with acetonitrile:CH2Cl2 (1:1). The purple fraction was collected and the solvent removed to give a purple solid. Crystals were grown by vapour diffusion of diethyl ether into an acetonitrile solution. Yield: 226 mg (49%). ES-MS: m/z 626 [M]+. 1H NMR (CD3CN): δ 10.30 (dd (J = 6, 0.8 Hz), 1H), 8.62–8.60 (m, 3H), 8.31–8.28 (m, 4H), 8.01 (ddd (J = 8, 6, 1 Hz), 1H), 7.72–7.64 (m, 7H), 7.53–7.49 (m, 3H), 7.26–7.24 (m, 3H), 6.93 (ddd (J = 7, 6, 1 Hz), 1H) ppm. 13C NMR (CD3CN): δ 158.48, 158.11, 157.89, 155.99, 152.30, 152.13, 151.90, 136.93, 136.76, 135.71, 131.90, 129.84, 128.94, 127.35, 126.94, 126.06, 123.98, 123.59, 123.48, 123.20, 121.64, 95.73, 86.73 ppm. Anal. Calc. for C33H23ClF6N5PRu: C, 51.40; H, 3.01; N, 9.08%. Found: C, 51.37; H, 2.93; N, 8.97%.
[RuCl(bpy)(L2)]PF6 ([2Cl]PF6)
The same procedure as for 1Cl except L2 was used in place of L1. Crystals were grown by vapour diffusion of diethyl ether into an acetonitrile solution. Yield: 288 mg (60%). ES-MS: m/z 656 [M]+. 1H NMR (CD3CN): δ 10.26 (dd (J = 6, 0.8 Hz), 1H), 8.61 (dt (J = 8, 1 Hz), 1H), 8.59 (s, 1H), 8.34 (dt (J = 8, 0.8 Hz), 2H), 8.30–8.28 (m, 2H), 8.00 (ddd (J = 8, 6, 1 Hz), 2H), 7.82 (td (J = 8, 2 Hz), 2H), 7.69–7.67 (m, 4H), 7.62 (d (J = 8 Hz), 4H), 7.30–7.27 (m, 4H), 7.05 (d (J = 8 Hz), 2H), 6.95 (ddd (J = 7, 6, 1 Hz), 1H), 3.89 (s, 2H) ppm. 13C NMR (CD3CN): δ 161.98, 159.49, 159.18, 158.81, 156.99, 153.14, 137.96, 137.66, 136.63, 134.61, 128.97, 128.31, 127.02, 124.70, 124.55, 124.41, 124.16, 115.58, 114.43, 97.21, 86.68, 56.25 ppm. Anal. Calc. for C34H25ClF6N5OPRu: C, 50.98; H, 3.15; N, 8.74%. Found: C, 50.93; H, 3.07; N, 8.74%.[RuCl(bpy)(L3)]PF6 ([3Cl]PF6)
The same procedure as for 1Cl except L3 was used in place of L1. Crystals were grown by vapour diffusion of diethyl ether into an acetonitrile solution. Yield: 52 mg (55%). ES-MS: m/z 639 [M]+. 1H NMR (CD3CN): δ 10.27 (dd (J = 6, 0.8 Hz), 1H), 8.59 (dt (J = 8, 1 Hz), 1H), 8.57 (s, 2H), 8.29 (m, 4H), 7.99 (ddd (J = 8, 6, 1 Hz), 1H), 7.74 (td (J = 8, 2 Hz), 2H), 7.67 (m, 3H) 7.54 (dt (J = 8, 2 Hz), 2H), 7.31 (d (J = 8 Hz), 2H), 7.25 (m, 3H), 6.92 (ddd (J = 7, 6, 1 Hz), 2H) ppm. 13C NMR (CD3CN): δ 159.46, 159.12, 158.83, 156.97, 153.30, 153.11, 152.91, 141.52, 137.92, 137.69, 136.65, 132.83, 130.59, 128.63, 128.31, 127.90, 127.02, 124.85, 124.55, 124.43, 124.16 ppm. Anal. Calc. for C34H25ClF6N5PRu: C, 52.02; H, 3.21; N, 8.92%. Found: C, 51.92; H, 3.15; N, 8.82%.[RuCl(bpy)(L4)]PF6 ([4Cl]PF6)
The same procedure as for 1Cl except L4 was used in place of L1. Crystals were grown by vapour diffusion of diethyl ether into an acetonitrile solution. Yield: 43 mg (43%). ES-MS: m/z 671 [M]+. 1H NMR (CD3CN): δ 10.23 (dd (J = 6, 0.8 Hz), 1H), 8.65 (s, 2H), 8.61 (dt (J = 8, 1 Hz), 1H), 8.38 (dt (J = 8, 0.8 Hz), 2H), 8.33 (d (J = 8 Hz), 2H) 8.30 (m, 2H), 7.99 (ddd (J = 8, 6, 1 Hz), 1H), 7.88 (m, 4H) 7.68 (m, 3H), 7.30 (m, 3H), 6.94 (ddd (J = 7, 6, 1 Hz), 1H) ppm. 13C NMR (CD3CN): δ 159.36, 159.16, 159.03, 156.82, 153.32, 153.15, 153.00, 138.13, 137.87, 136.82, 133.85, 129.27, 128.49, 127.96, 127.05, 125.08, 125.04, 124.65, 124.47, 124.22, 93.03, 90.77 ppm. Anal. Calc. for C33H22F6RuN6P: C, 48.57; H, 2.72; N, 10.30%. Found: C, 48.63; H, 2.60; N, 10.37%.X-ray crystallography
The X-ray single crystal data for L3, [3Cl]PF6·CH3CN·C4H10O and [4Cl]PF6·2CH3CN have been collected at 120.0 K on a Bruker SMART CCD 6000 diffractometer (graphite monochromator, λMoKα, λ = 0.71073 Å) equipped with a Cryostream (Oxford Cryosystems) open-flow nitrogen cryostat. The structures were solved by direct method and refined by full-matrix least squares on F2 for all data using Olex263a and SHELXTL63b software. All non-disordered non-hydrogen atoms were refined anisotropically, hydrogen atoms were refined freely in the structures of L3 and [4Cl]PF6·2CH3CN and were placed in calculated positions in the structure [3Cl]PF6·CH3CN·C4H10O. Disordered PF6 anion and solvent molecules were refined with fixed site occupancy factor (SOF) in isotropic mode. Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as the following supplementary publications: CCDC-1046731–1046733.Instrumentation
Microanalyses were performed by Elemental Analysis Service, London Metropolitan University, UK. NMR spectra were recorded on a Bruker Avance (1H 400.13, 13C 100.61 MHz) Electrospray ionization mass spectra were recorded using Thermo Quest Finnigan Trace MS-Trace GC or Waters Micromass LCT spectrometers. Listed peaks correspond to the most abundant isotopomer; assignments were made by a comparison of observed spectra and simulated ion patterns. Raman spectra were collected on a Horiba Jobin Yvon LabRAM HR confocal Raman microscope, equipped with a peltier-cooled CCD and 50× LWD objective lens, running with a frequency doubled Nd:YAG at 532 nm. Laser power at the sample <1 mW. UV-Visible absorbance spectra were recorded on an Agilent Technologies Cary 5000 UV-vis-NIR spectrophotometer from solutions in acetonitrile for the complexes [1–4Cl]PF6 at 10−5 M and acetone:water (9:1) for complexes [1–4H2O]PF6 at 10−5 M. Electrochemical analyses of the complexes [1–4Cl]PF6 were carried out using an EcoChemie Autolab PGSTAT-30 potentiostat, with platinum working, platinum counter and platinum pseudo reference electrodes, from solutions in acetonitrile containing 0.1 M supporting electrolyte (tetrabutylammonium hexafluorophosphate, TBAPF6), scan rate = 100 mV s−1. The ferrocene/ferricenium couple was used as the internal reference. Spectroelectrochemical measurements of the complexes [1–4Cl]PF6 were made in an OTTLE cell of Hartl design from acetonitrile solutions containing 0.1 M TBAPF6 electrolyte. The cell was fitted into the sample compartment of the Agilent Technologies Cary 5000 UV-vis-NIR, and electrolysis in the cell was performed with a PalmSens EmStat2 potentiometer.
Photocatalytic measurements were performed in 5 mL of degased H2O solution, at pH (0.10 M phosphate buffer) with 0.02 mM Rucat, 10 mM 4-methoxy benzyl alcohol, 0.4 mM [Ru(bpy)3](PF6)2 and 20 mM [Co(NH3)5Cl]Cl2, irradiated by a xenon lamp (300 W) with UV filter for 24 hours. In order to maintain solubility of the chloro-complexes ([1–4Cl]PF6) it was necessary to add 0.5 mL of acetone per 5 mL solution. Stock solutions of [1–4H2O]PF6 were prepared by refluxing a measured quantity of [1–4Cl]PF6 in an acetone:water (1:1) with silver triflate. After removing the residual AgCl by filtration, the filtrate concentration was then adjusted by the addition of water. To determine the ratio of product to substrate solution was extracted three times with CH2Cl2, the extracts collected, and taken to dryness. 1H NMR was used to measure the relative integrals of the methoxy proton signals of 4-methoxybenzyl alcohol (substrate) and 4-methoxybenzaldehyde (product).
Results and discussion
Synthesis
Three new 4-ethynyl substituted tpy based ligands (L2, L3 and L4) were synthesised by cross-coupling tpyOTf with the respective alkyne (4-ethynyl anisole, 4-ethynyl toluene and 4-ethynyl nitrobenzene) under ‘copper free’ Sonogashira conditions (Scheme 1). Ligand L1 has been previously reported from a similar synthetic method.14,64 Isolated yields ranged from 65 to 81%, the yields being lower for more electron withdrawing substituents.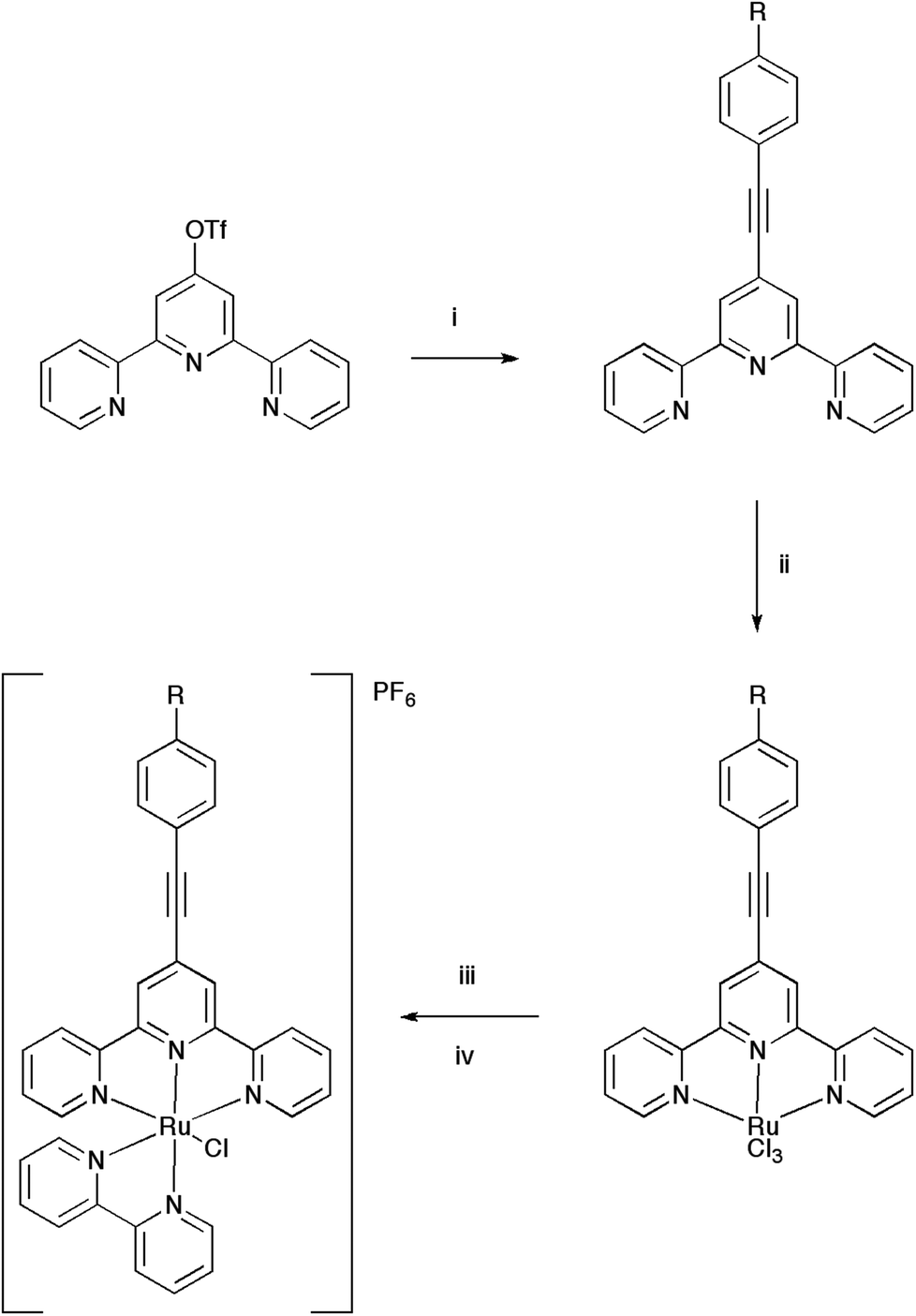 | ||
| Scheme 1 Synthesis schemes for the synthesis of the ligands Ln and ruthenium complexes ([1–4Cl]PF6). (i) Pd(PPh3)4, NEt3, THF, HCCC6H4R-4 (R = H (L1), OMe (L2), Me (L3), NO2 (L4)); (ii) RuCl3·3H2O, EtOH; (iii) bpy, LiCl, ethylmorpholine, MeOH; (iv) NH4PF6, MeOH. | ||
The ligands L1–L4 were reacted with ruthenium trichloride to form their respective RuCl3Ln complexes, which were not characterised but in turn reacted with bpy, LiCl and NH4PF6, catalysed with 4-ethylmorpholine to form the chloro-complexes ([RuCl(bpy)Ln]PF6, [1–4Cl]PF6) (Scheme 1). Attempts were also made to synthesise chloro-complexes from 4′-ethynyl- or 4′-trimethylsilylethynyl-2,2′:6′,2′′-terpyridine, however, intractable mixtures were formed during the course of the reaction. The purification of the chloro-complexes [1–4Cl]PF6 was significantly improved by separating the desired heteroleptic product from [Ru(bpy)3]2+ typically formed as a by-product by extracting the crude reaction mixture into dichloromethane and filtering prior to anion metathesis with NH4PF6 and column chromatography.
Molecular structures
The ligand L3 crystallises in a space group P21/c with one molecule per unit cell (Fig. 2). The structure shows an ethynyl toluene fragment attached to a tpy scaffold at the 4-position, which is consistent with the chemical reaction scheme and NMR and mass spectroscopy data collected from the compound. The structure shows the nitrogen atoms of the tpy ligand directed to maximise intramolecular hydrogen bonding, as is common with tpy derivatives. The C18–C23 phenylene ring lies out of the plane defined by the terpyridine moiety (C7–C8–C18–C23 = 27.01(11)°) whilst the C8–(C16–C17 midpoint)–C18 angle is bent from linearity by only 4.5°. The long-short-long bond length alternation along the C8–C16–C17–C18 fragment (1.433(4), 1.203(5), 1.434(4) Å) clearly showing that the triple bond remains localised to C16–C17.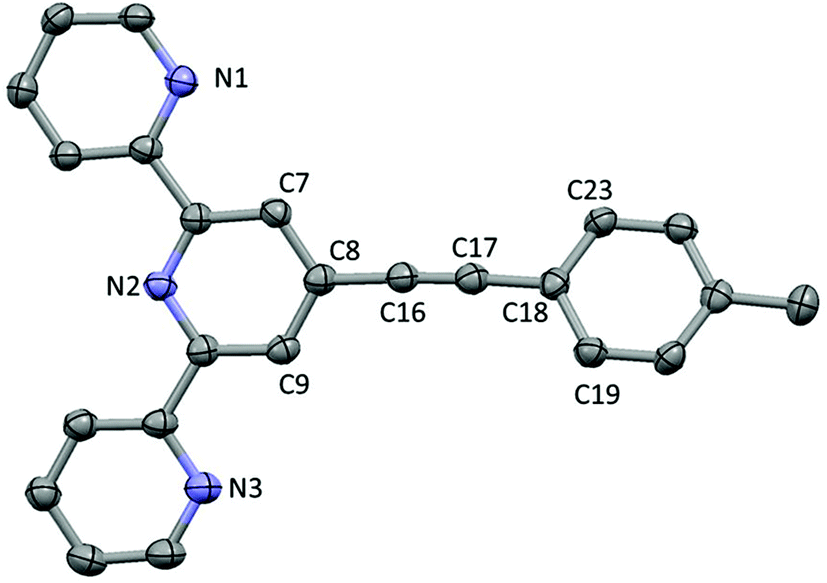 | ||
| Fig. 2 Crystal structure of L3, hydrogen atoms removed for clarity. | ||
The complex [3Cl]PF6 crystallises in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z′ = 2; selected bond lengths and angles are given in the ESI.† The two crystallographically distinct cations differ by the relative orientation of the tolyl moiety with respect to the approximate plane of the tpy ligand (the corresponding torsion angles are 3.6° [3Cla]+ and 27.1° [3Clb]+). The ruthenium atom of cation has octahedral coordination, the chloride is located trans to the axial N4 atom of the bpy ligand (Fig. 3). The Ru–N bond lengths consist of Ru(1)–N(1) (2.073(3)), –N(2) (1.963(3)), –N(3) (2.066(3)), –N(4) (2.035(3)) and –N(5) (2.082(3) Å) with a Ru(1)–Cl(1) bond length of 2.4093(10) Å, within experimental error similar to those in other reported [RuCl(bpy)(tpy)]+ complexes.65–69 The angles at the ruthenium centre are very similar between the both cationic complexes in the unit cell. The cations have identical ethynyl bond lengths C18–C17 (1.196(7) Å in [3Cla]+ and 1.197(7) Å [3Clb]+).
with Z′ = 2; selected bond lengths and angles are given in the ESI.† The two crystallographically distinct cations differ by the relative orientation of the tolyl moiety with respect to the approximate plane of the tpy ligand (the corresponding torsion angles are 3.6° [3Cla]+ and 27.1° [3Clb]+). The ruthenium atom of cation has octahedral coordination, the chloride is located trans to the axial N4 atom of the bpy ligand (Fig. 3). The Ru–N bond lengths consist of Ru(1)–N(1) (2.073(3)), –N(2) (1.963(3)), –N(3) (2.066(3)), –N(4) (2.035(3)) and –N(5) (2.082(3) Å) with a Ru(1)–Cl(1) bond length of 2.4093(10) Å, within experimental error similar to those in other reported [RuCl(bpy)(tpy)]+ complexes.65–69 The angles at the ruthenium centre are very similar between the both cationic complexes in the unit cell. The cations have identical ethynyl bond lengths C18–C17 (1.196(7) Å in [3Cla]+ and 1.197(7) Å [3Clb]+).
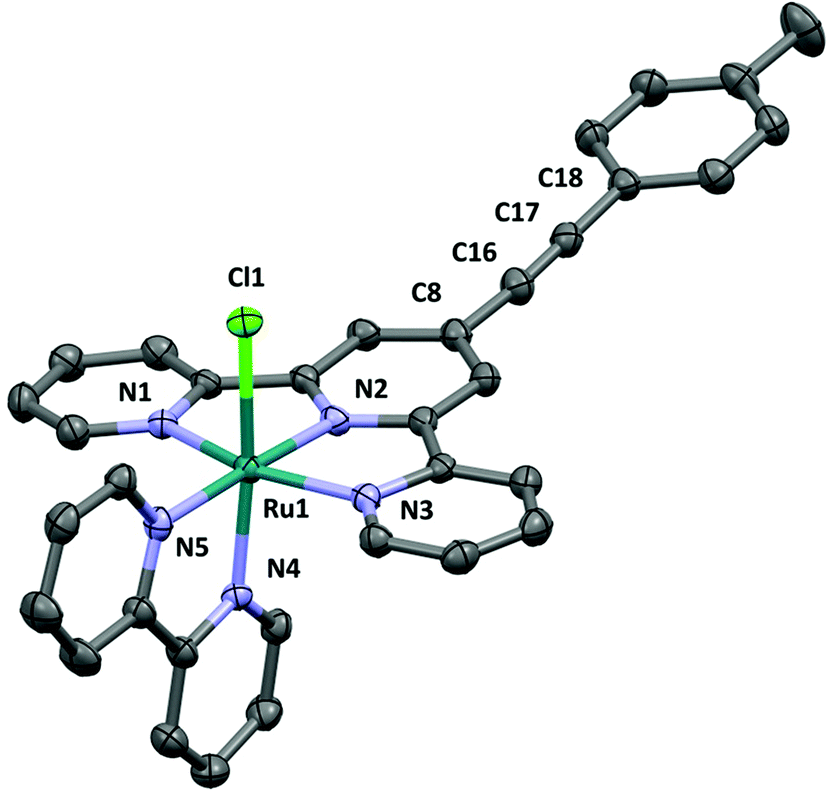 | ||
| Fig. 3 The cation (a) in the crystal structure of [3Cl]PF6·CH3CN·C5H10O. Hydrogen atoms removed for clarity. | ||
Unlike [3Cl]PF6, Z′ = 1 for [4Cl]PF6 (Fig. 4), and selected bond lengths and angles are given in the ESI.† The ethynyl substituted tpy ligand in [4Cl]+ adopts a planar conformation similar to that found in [3Cla]+. Most of the bond lengths to the ruthenium centre are very similar to those in [3Cl]+ (See ESI†). The coordination geometry at ruthenium, Ru–N (Ru(1)–N(1) (2.071 (3)), –N(2) (1.950 (3)), –N(3) (2.060 (3)), –N(4) (2.027 (3)), –N(5) (2.088 (3) Å)) and Ru(1)–Cl(1) (2.3927 (8) Å) bond lengths are identical to those of [3Cl]+, suggesting there is little structure influence at the metal centre brought about by the introduction of the nitro group.
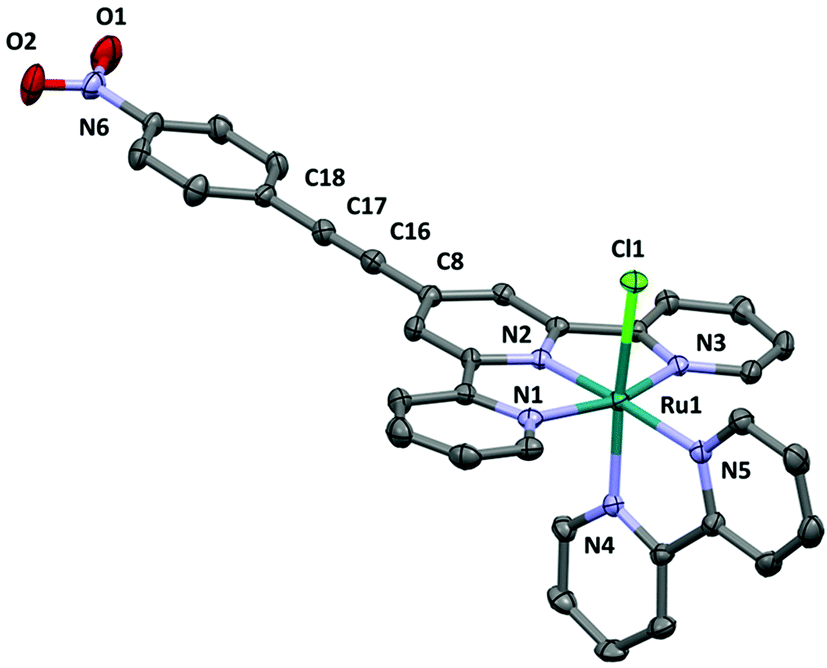 | ||
| Fig. 4 The cation in the crystal structure of [4Cl]PF6·2CH3CN. Hydrogen atoms removed for clarity. | ||
All three structures contain extending planar aromatic systems and not surprisingly in crystal these planar moieties are arranged parallel to each other at the distances (3.6–3.9 Å) typical to π⋯π interaction. Interestingly the triple bonds and nitro-group (in the structure [4Cl]PF6) are also sandwiched between aromatic rings and such arrangement is probably also attractive and additionally stabilizes the crystals.
Computational
A brief investigation was performed using ab initio calculations to study the electronic structure of [1–4Cl]+ and the related complexes [Ru(H2O)(bpy)Ln]2+ [1–4H2O]2+, which complement earlier studies of the parent systems [RuCl(bpy)(tpy)]+ and [Ru(H2O)(bpy)(tpy)]2+.70 Initial geometries for [1–4Cl]+ cations were based on the crystallographic structures of [3Cl]+ and [4Cl]+, while for the initial geometries of [1–4H2O]2+ dications, the H2O–Ru distances and angles were based on the crystallographic data available for [Ru(H2O)(bpy)(tpy)](PF6)2.71 Full optimisations were performed using density functional theory (DFT) as part of the Gaussian09 package.72 Frequency and time-dependent (TD) calculations were performed on optimised ground-state structures, and results were displayed using GaussView.73 All calculations were carried out at the B3LYP level employing an SDD basis set. The calculated frequencies were scaled by 0.96174 to account for the anharmonicity of the vibrational modes. The assignments of the spectra were made using each level and the MAD values determined for all of the assigned peaks (see ESI†). An unambiguous assignment of vibrational modes from visual comparison of spectra was possible for most absorption features. TD-DFT calculations were carried out in an acetonitrile solvent field using the SCRF-PCM method which creates the solvent cavity via a set of overlapping spheres.75 Geometry optimisations were not carried out in a solvent field for reasons of computational expense; however, correlation between the experimental results and the TD DFT calculations which include the solvent and gas-phase optimised geometries was found to be better than for calculations where solvent contributions were completely neglected.A comparison of [1–4Cl]+ cation HOMO energy levels shows almost no change across the series, despite the significant differences in the electron donating (OMe, Me) and withdrawing (NO2) nature of the phenylene substituents (Table 1). This is explained by the HOMO being 70% ruthenium in nature with small contributions from the bpy and tpy ligands; only for [4Cl]+ is there a 13% contribution from the ethynyl bond (see ESI†). Whilst there is similarly little change in the LUMO energy level across the partial series [1–3Cl]+ (Table 1), the introduction of the NO2 group in [4Cl]+ causes a significant lowering of the LUMO energy by ca. 0.8 eV. For [1–3Cl]+ the LUMO orbital is π* in nature and 75% is localised on the terpyridyl fragment with <10% contribution from the ethynyl group and almost none from the phenylene ring. However, in the case of [4Cl]+ the nitrophenylene π* orbitals lie lower than the tpy π* and from the LUMO (Fig. 5). The tpy π* orbital is found 0.82 eV higher in energy and forms the LUMO+1. Broadly similar behaviour was observed for the aqua complexes [1–4H2O]2+. The substitution of the chloride ligand by water, and the resulting increase in positive charge on the complex results in an overall lowering of orbital energies (Table 1). The HOMO levels of the aqua complexes span a slightly larger range of energies than the chloride analogues, but still differ by less than 0.17 eV. In the case of the unoccupied orbitals, the lowering of the orbital energies associated with the complex serves to limit the nitrophenyl based LUMO and tpy π* LUMO+1 energy gap, and the difference of the LUMO energies between [1H2O]2+ and [4H2O]2+ is reduced to 0.578 eV.
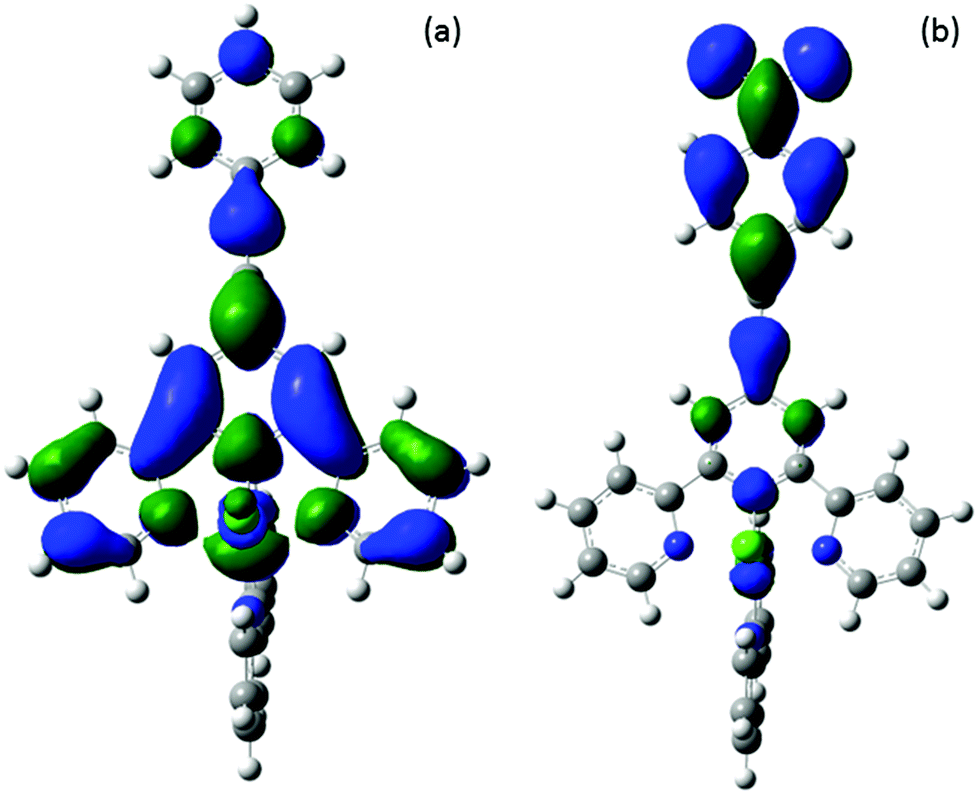 | ||
| Fig. 5 Plots of the LUMO for (a) [1Cl]+ and (b) [4Cl]+. | ||
| Complex | HOMO energy (eV) | LUMO energy (eV) | ||
|---|---|---|---|---|
| Cl | H2O | Cl | H2O | |
| 1 | −5.676 | −6.103 | −2.830 | −3.207 |
| 2 | −5.658 | −6.038 | −2.789 | −3.166 |
| 3 | −5.667 | −6.134 | −2.807 | −3.225 |
| 4 | −5.730 | −6.205 | −3.629 | −3.785 |
Electronic spectroscopy
The electronic absorption spectra of the complexes [1–4Cl]+ feature π → π* transitions at wavelengths shorter than 400 nm and MLCT bands between 600–450 nm which are most clearly resolved for [1Cl]+ and [3Cl]+ (Fig. 6, Table 2).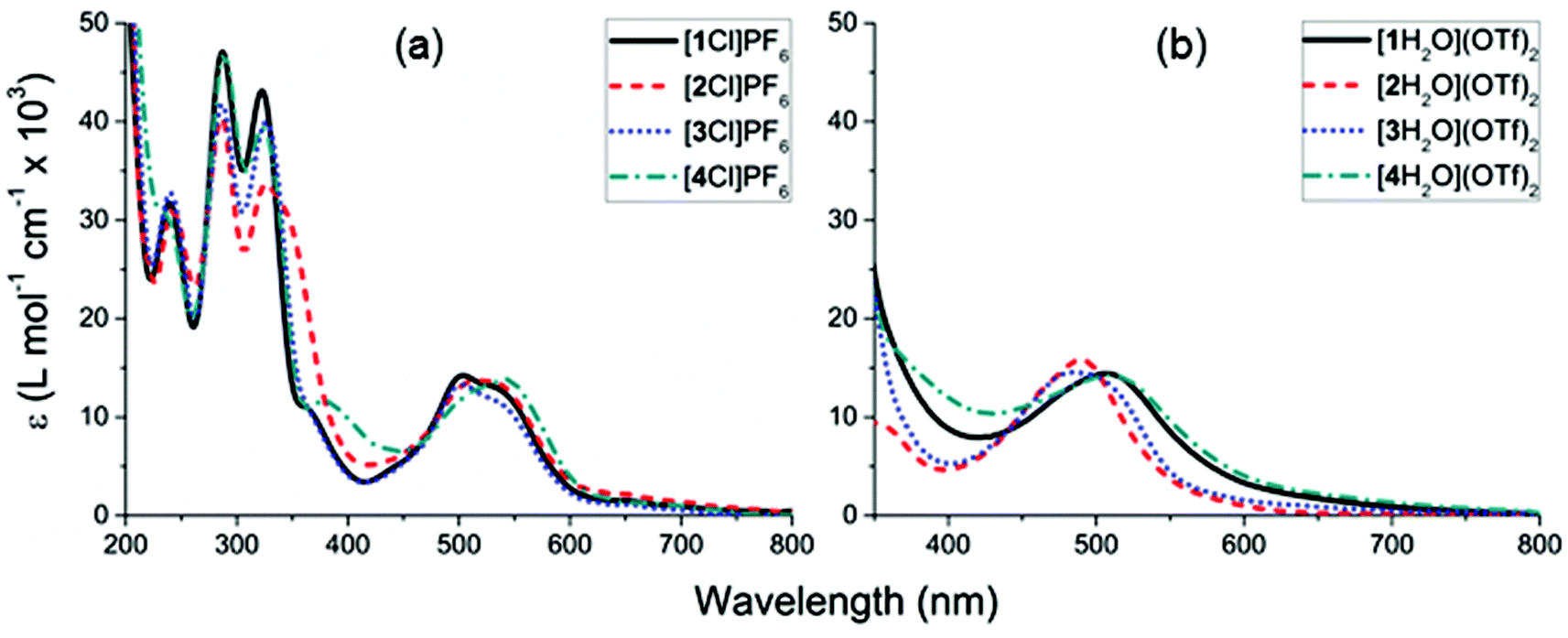 | ||
| Fig. 6 Electronic absorbance spectra of: (a) complexes [1–4Cl]PF6 in acetonitrile, (b) complexes [1–4H2O](TfO)2 in an acetone:water (9:1) solution. | ||
:water (9:1) solution
| Complex | λ MLCT/nm (ε/L cm−1 mol−1 × 103) | |
|---|---|---|
| Cl | H2O | |
| 1 | 541 (12.4), 499 (13.8) | 506 (12.8) br |
| 2 | 522 (13.5) br | 490 (15.9) br |
| 3 | 535 (10.4), 505 (13.2) | 487 (15.1) br |
| 4 | 541 (13.8) br | 510 (11.8) br |
Based on time-dependent density functional theory (TD-DFT) calculations of complexes [1–4Cl]+ (see ESI† for further details) the higher energy feature of the MLCT absorption envelope (ca. 500 nm) consists of a mixed transition between the pseudo ‘t2g’ 3d orbitals of the ruthenium (HOMO, HOMO−1 and HOMO−2), which are heavily metal centred and only in the case of the HOMO−1 carry contribution (7–21%) from the ethynyl-phenylene moiety, to low energy π* orbitals (LUMO, LUMO+1, LUMO+2, as well as LUMO+3 in the case of [4Cl]+). These unoccupied orbitals are mainly localised on the bpy and tpy moieties with moderate (12–14%) contributions from the ethynyl-phenylene (–CCC6H4R) moiety to the LUMO in the case of [1–3Cl]+. However, in the case of [4Cl]+ the nitro groups leads to significant orbital reordering and mixing, with nitrophenylacetylide fragment comprising the LUMO (90%) and contributing together with the tpy fragment to the LUMO+1 (tpy 72%; CCC6H4NO2 19%). The lower energy component of the band envelope is attributed by TD DFT calculations to transitions between HOMO−1 → LUMO and HOMO → LUMO+2 (Fig. 7). Plots of these orbitals show that whilst the HOMO → LUMO+2 transition is largely MLCT in character and localised on the metal 3d and {Ru(bpy)(tpy)} fragments, the HOMO−1 → LUMO transition admixes a degree of intra ligand π–π* character from the arylethynylterpyridyl ligand, which becomes more extensively associated with the nitrophenyl group in the case of [4Cl]+.
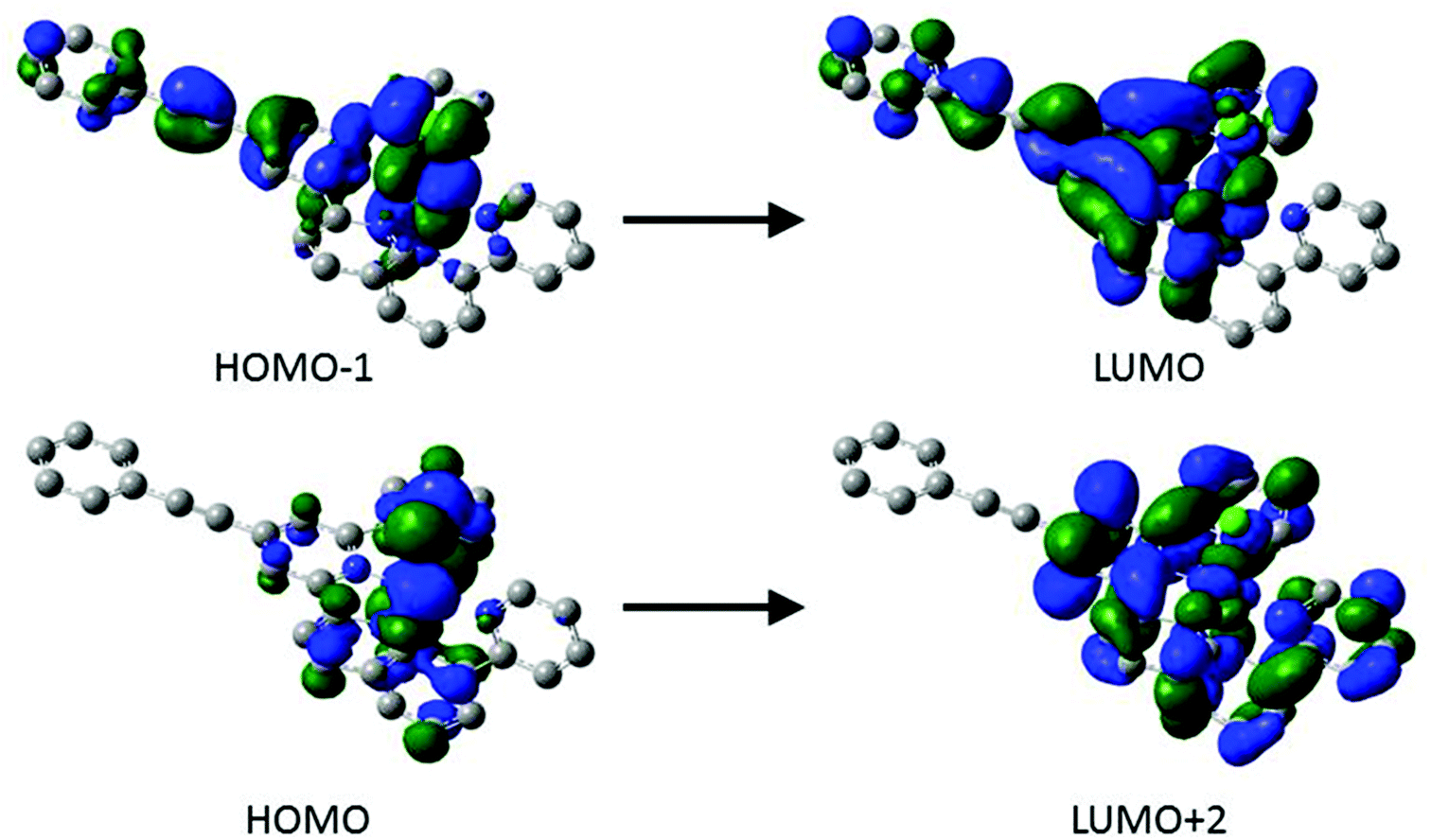 | ||
| Fig. 7 Plots of the important orbital involved in the transitions responsible for the absorbance at 541 nm of [1Cl]+. | ||
The implications of low lying electronic transitions involving the ethynyl phenylene fragment in the chloride complexes [1–4Cl]+ prompted consideration of the analogous aqua complexes, which are the key active species in the oxidative chemistry described in the Introduction. Solutions containing the aqua complexes [Ru(H2O)(bpy)Ln]2+ ([1–4H2O](OTf)2) were prepared from [1–4Cl]PF6 by treatment with AgOTf in refluxing acetone:water (50:50) solution (5 h). Solutions were then filtered to remove precipitated AgCl, solvents removed and the residue re-dissolved in the desired solvent.
Once the chloride was removed to form the complexes [1–4H2O](OTf)2 the solution changed from purple to orange with the MLCT band envelope blue-shifted relative to the chloride analogue (Fig. 6). However, the apparent band maxima of these spectra remain significantly red-shifted compared to that of the parent complex [Ru(H2O)(bpy)(tpy)]2+ (λmax = 476 nm),76 likely due to the extended conjugation between the terpyridyl and CCC6H4R moieties. The lowest energy visible absorption band envelope is rather broad, which could either be attributed to a mixture of aqua and chloride species present in the in the solution or multiple transitions within the envelope. Based on TD-DFT calculations (see ESI†) the aqua complexes each feature two relatively intense transitions (i.e. of oscillator strength >0.1) between 530–450 nm. As with the chloro analogues, these transitions are of essentially MLCT character and are similarly comprised.
Vibrational spectroscopy
Resonance Raman (RR) spectroscopy allows selective enhancement of modes within a chromophore when the excitation wavelength (λex, here 532 nm) is coincident with the chromophore absorption. The Raman spectrum of each of the complexes [1–4Cl]PF6 was collected using λex = 532 nm, a wavelength chosen to probe the lower energy component of the MLCT absorption envelope and for which TD-DFT calculations indicated involved orbitals with the greatest contribution from the ethynyl phenylene moiety (Fig. 8). Although many of the vibrations observed in the RR spectra associated with tpy and bpy based aromatic modes (622–1604 cm−1) remained unchanged despite the variation in phenylene substitution, the lowest energy vibrational modes associated with the most delocalised parts of the ethynyl substituted terpyridine ligand were found to be more sensitive to the nature of the substituent (582–553 cm−1).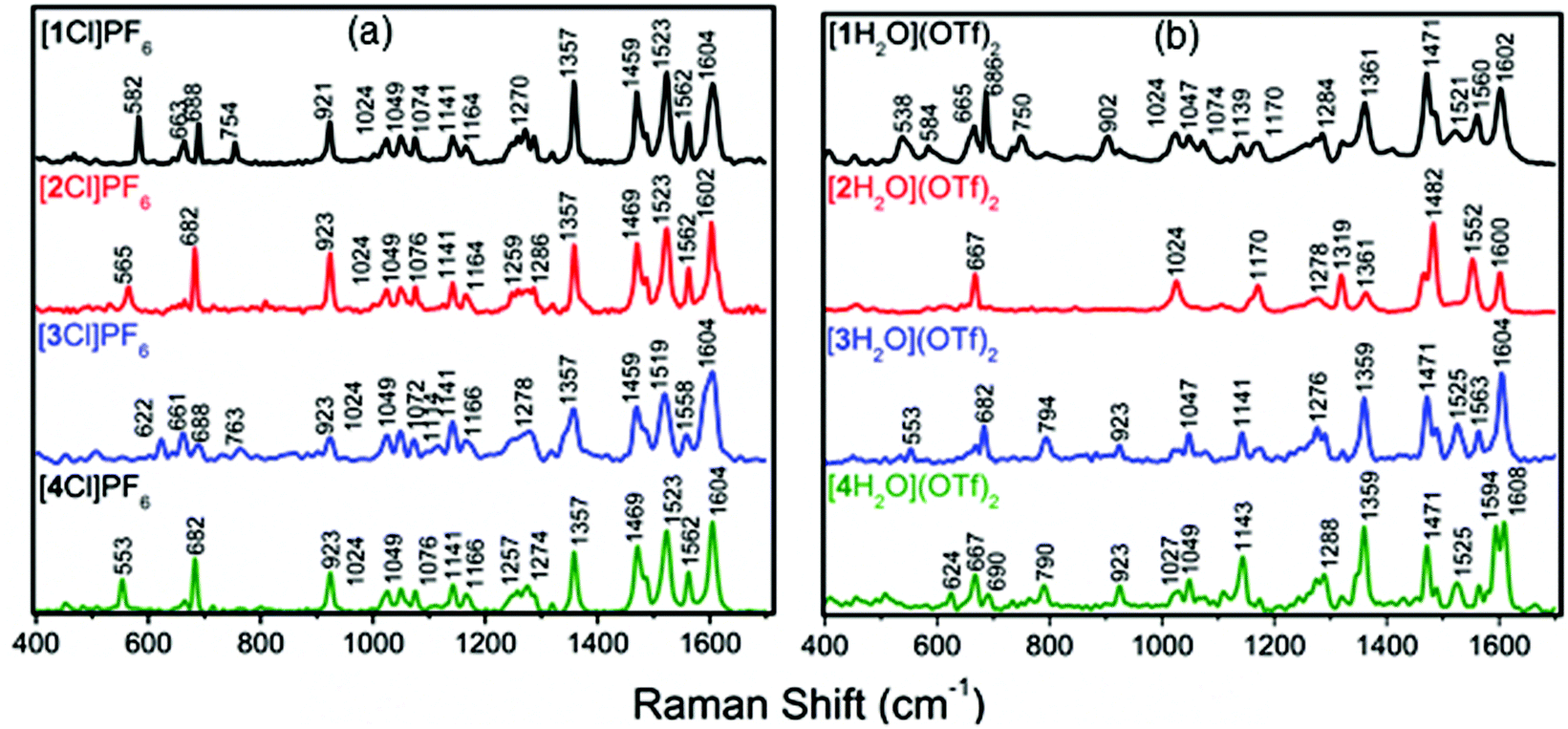 | ||
| Fig. 8 RR using and excitation of 532 nm. (a) chloro-complexes [1–4Cl]PF6 in acetonitrile and (b) aqua complexes [1–4H2O](OTf)2 in an acetone:water (9:1) solution. | ||
RR spectra of the aqua complexes [1–4H2O](OTf)2 were generally very similar to those of the chloride complexes, although the blue shifted λMLCT resulted in less resonant enhancement by the 532 nm laser resulting in lower relative intensities of many of the bands. Nevertheless, the key symmetric in plane stretches of the aryleneethylene substituted terpyridine ligand bands were clearly apparent near 1350 and 1600 cm−1 in each case (Fig. 8). No vibrational modes associated with a coordinated triflate were observed which suggests the triflate counter ion was not coordinating to the metal centre.
Electrochemistry and spectroelectrochemistry
Each of the chloro-complexes ([1–4Cl]PF6) display a single almost fully electrochemically reversible oxidation wave at 0.44–0.46 V (vs. ferrocene) associated with the Ru(II)/(III) couple, albeit with some evidence for border-line slow electron transfer based on the behaviour of the peak-current ratios with scan rate, and a single similarly near reversible reduction wave associated with the reduction of one of the ligands (Table 3). The oxidation potential is similar to that of [RuCl(bpy)(tpy)]+ (E1/2Ox = 0.45 V),76 suggesting that the addition of –CCC6H4R moiety has little effect on the ruthenium centre, which is also supported by <0.02 V potential difference in oxidation potential across the series.
| Complex | E 1/2 (V vs. Fc/Fc+) | ΔEp (mV) |
|---|---|---|
| [1Cl]PF6 | 0.45 | 75 |
| −1.69 | 92 | |
| [2Cl]PF6 | 0.44 | 93 |
| −1.74 | 96 | |
| [3Cl]PF6 | 0.46 | 77 |
| −1.73 | 124 | |
| [4Cl]PF6 | 0.46 | 72 |
| −1.61 | 78 |
Given the well-behaved solution redox chemistry, spectroelectrochemistry was employed to observe the oxidised species [1Cl]2+ as a representative example (Fig. 9).63,77 Upon oxidation of [1Cl]+ the MLCT band envelope is quenched while the π → π* transitions (<400 nm) are only slightly affected, confirming that the electron is being removed from the metal centre, and therefore the assignment of the oxidation to a formal Ru(II/III) process. The original spectrum was fully recovered on back-reduction, confirming both the assignment of the spectrum to [1Cl]2+ and the chemical stability of this complex under these conditions.
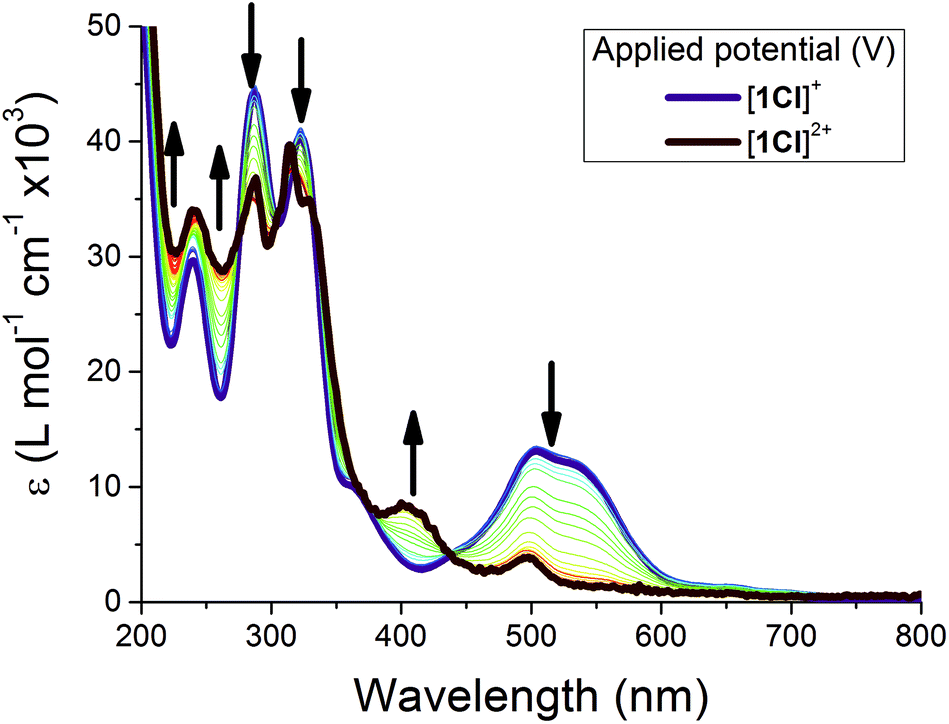 | ||
| Fig. 9 UV-visible SEC spectra of [1Cl]PF6, arrows indicate spectral changes upon oxidation. | ||
Photocatalytic behaviour
As Rocha et al. and Kojima et al. have demonstrated, the oxidation of benzyl alcohols is a convenient means of testing the activity of mononuclear ruthenium oxidation catalysts mediated by the photosensitized oxidation of the aqua complex resting state to the active oxo-form, [{RuII}–OH2]2+ → [{RuIV}O]2+.51,78,79 Here, a comparison was made between the chloro-complexes [1–4Cl]PF6 and in situ generated [1–4H2O](OTf)2 complexes as catalysts for the oxidation of 4-methoxybenzyl alcohol. Reactions were conducted in 5 mL of degassed H2O solution (also containing 0.5 mL acetone in the case of [1–4Cl]PF6 for reasons of solubility), at pH = 6.8 (0.10 M phosphate buffer) with 0.02 mM Rucat, 10 mM 4-methoxy benzyl alcohol, 0.4 mM [Ru(bpy)3](PF6)2 as photosensitizer and 20 mM [Co(NH3)5Cl]Cl2 as a sacrificial oxidant to re-cycle the sensitizer. Illumination was provided by a xenon lamp (300 W) with UV filter for 24 h, after which time no further reaction progress was detected. To determine the ratio of product to substrate, after reaction the solution was extracted with dichloromethane three times, the extracts collected and the solvent removed. Integration of the methoxy proton signals (1H NMR) of 4-methoxybenzyl alcohol (substrate) and 4-methoxybenzaldehyde (product) was used to assess the extent of reaction. The relatively high level of error associated with these measurements prevented valid determination of the initial rates; however, this technique proved sufficient for determining catalytic activity across the series with TONs 269(20)–396(8) being determined (Table 4). Reactions conducted in the absence of substrate, catalyst, oxidant or light, gave no detectible quantities of product. In the absence of [Ru(bpy)3](PF6)2, photo oxidation was detected, with catalytic TONs approximately half that of those run in the presence of the additional sensitizer, although variation in results between runs were increased. It is conceivable that photodecomposition of the heteroleptic catalyst may result in the formation of other catalytically active species such as RuO2 nanoparticles;80,81 however, the exact determination of this is beyond the scope of this report but remains a topic for future investigation.
Under the same conditions, the TONs for the aqua-complexes [1–4H2O](OTf)2 were determined to be 150–309. Accounting for the error in the measurements this is similar to [Ru(H2O)(bpy)(tpy)](OTf)2 (TON = 217 (30)) measured under the same conditions.51,78,79 Although the unavoidably high error in the measurements prevented any relationship being drawn between the electron-withdrawing effects of the ethynyl-phenyl substituents, this report clearly demonstrates these compounds remain catalytically active with the ethnyl moiety neither decreasing activity markedly nor leading to excessive photodecomposition.
Conclusion
Three new tpy ligands (L2–L3) and four new heteroleptic complexes ([1–4Cl]PF6) containing ethynyl phenylene substituted moieties have been synthesised and characterised. The electronic absorption and RR spectra show that the ethynyl phenylene moiety has some effect on the excited state of both the chloro and aqua complexes by tuning the composition of the LUMO, whilst the HOMO remains largely metal centred, which is in agreement with electrochemical, spectroelectrochemical and computational results. Finally, through the photocatalytic measurements it has been possible to show that the catalytic oxidation of 4-methoxybenzylalcohol to 4-methoxybenzaldehyde can be performed using either the chloro- or aqua-catalysts and that the addition of ethynyl phenyl moieties to the 4-position of the tpy ligand does not have a detrimental effect on the catalytic behaviour of these complexes, potentially allowing a host of other functionalities to be added to these complexes via an alkyne without the risk of adversely affecting the catalysis.Acknowledgements
We gratefully acknowledge the EPSRC for funding this research. PJL held and EPSRC Leadership Fellowship and currently holds an ARC Future Fellowship [FT120100073].References
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- R. Tran, J. Kern, J. Hattne, S. Koroidov, J. Hellmich, R. Alonso-Mori, N. K. Sauter, U. Bergmann, J. Messinger, A. Zouni, J. Yano and V. K. Yachandra, Philos. Trans. R. Soc. London, Ser. B, 2014, 369 Search PubMed.
- G. W. Brudvig, Water oxidation chemistry of photosystem II, 2008 Search PubMed.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2000, 101, 21–36 CrossRef PubMed.
- X. Sala, I. Romero, M. Rodríguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- D. K. Bediako, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 3662–3674 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Surendranath, M. Dincǎ and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2007, 20, 35–54 CrossRef.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 CAS.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 2014, 645–659 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- T. Wada, K. Tsuge and K. Tanaka, Inorg. Chem., 2000, 40, 329–337 CrossRef PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed.
- H.-W. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed.
- S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- J. J. Concepcion, M.-K. Tsai, J. T. Muckerman and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 1545–1557 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- R. Cao, W. Lai and P. Du, Energy Environ. Sci., 2012, 5, 8134–8157 CAS.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2007, 130, 210–217 CrossRef PubMed.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC.
- N. Kaveevivitchai, R. Zong, H.-W. Tseng, R. Chitta and R. P. Thummel, Inorg. Chem., 2012, 51, 2930–2939 CrossRef CAS PubMed.
- Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845–8850 CrossRef CAS PubMed.
- Z. F. Chen, J. J. Concepcion, J. W. Jurss and T. J. Meyer, J. Am. Chem. Soc., 2009, 131, 15580–15581 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion and T. J. Meyer, Dalton Trans., 2011, 40, 3789–3792 RSC.
- D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094–16106 CrossRef CAS PubMed.
- M. Yoshida, S. Masaoka, J. Abe and K. Sakai, Chem. – Asian J., 2010, 5, 2369–2378 CrossRef CAS PubMed.
- M. Yoshida and S. Masaoka, Res. Chem. Intermed., 2014, 40, 3169–3182 CrossRef CAS.
- J. Kiyota, J. Yokoyama, M. Yoshida, S. Masaoka and K. Sakai, Chem. Lett., 2010, 39, 1146–1148 CrossRef CAS.
- Z. Chen, J. J. Concepcion, J. F. Hull, P. G. Hoertz and T. J. Meyer, Dalton Trans., 2010, 39, 6950–6952 RSC.
- J. J. Concepcion, J. W. Jurss, P. G. Hoertz and T. J. Meyer, Angew. Chem., Int. Ed., 2009, 48, 9473–9476 CrossRef CAS PubMed.
- T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 2000, 39, 1479–1482 CrossRef CAS.
- S. Roeser, P. Farràs, F. Bozoglian, M. Martínez-Belmonte, J. Benet-Buchholz and A. Llobet, ChemSusChem, 2011, 4, 153–153 CrossRef PubMed.
- N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721–10724 CrossRef CAS PubMed.
- L. Duan, Y. Xu, L. Tong and L. Sun, ChemSusChem, 2011, 4, 238–244 CAS.
- L. Kohler, N. Kaveevivitchai, R. Zong and R. P. Thummel, Inorg. Chem., 2013, 53, 912–921 CrossRef PubMed.
- D. L. Ashford, W. Song, J. J. Concepcion, C. R. K. Glasson, M. K. Brennaman, M. R. Norris, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 19189–19198 CrossRef CAS PubMed.
- M. T. Vagnini, A. L. Smeigh, J. D. Blakemore, S. W. Eaton, N. D. Schley, F. D'Souza, R. H. Crabtree, G. W. Brudvig, D. T. Co and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15651–15656 CrossRef CAS PubMed.
- A. M. Lopez, M. Natali, E. Pizzolato, C. Chiorboli, M. Bonchio, A. Sartorel and F. Scandola, Phys. Chem. Chem. Phys., 2014, 16, 12000–12007 RSC.
- E. A. Karlsson, B.-L. Lee, R.-Z. Liao, T. Åkermark, M. D. Kärkäs, V. S. Becerril, P. E. M. Siegbahn, X. Zou, M. Abrahamsson and B. Åkermark, ChemPlusChem, 2014, 79, 936–950 CrossRef CAS PubMed.
- A. C. Benniston and A. Harriman, Mater. Today, 2008, 11, 26–34 CrossRef CAS.
- W. Chen, F. N. Rein and R. C. Rocha, Angew. Chem., Int. Ed., 2009, 48, 9672–9675 CrossRef CAS PubMed.
- W. Chen, F. N. Rein, B. L. Scott and R. C. Rocha, Chem. – Eur. J., 2011, 17, 5595–5604 CrossRef CAS PubMed.
- D. Chao and W.-F. Fu, Chem. Commun., 2013, 49, 3872–3874 RSC.
- S. Ohzu, T. Ishizuka, Y. Hirai, S. Fukuzumi and T. Kojima, Chem. – Eur. J., 2013, 19, 1563–1567 CrossRef CAS PubMed.
- S. Fraysse, C. Coudret and J.-P. Launay, J. Am. Chem. Soc., 2003, 125, 5880–5888 CrossRef CAS PubMed.
- S. A. Vail, P. J. Krawczuk, D. M. Guldi, A. Palkar, L. Echegoyen, J. P. C. Tomé, M. A. Fazio and D. I. Schuster, Chem. – Eur. J., 2005, 11, 3375–3388 CrossRef CAS PubMed.
- A. Harriman and R. Ziessel, Chem. Commun., 1996, 1707–1716 RSC.
- M. Yagi, S. Tajima, M. Komi and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed.
- M. Stein, R. Berger, W. Seichter, J. Hulliger and E. Weber, J. Fluorine Chem., 2012, 135, 231–239 CrossRef CAS PubMed.
- S. O. Mihigo, W. Mammo, M. Bezabih, K. Andrae-Marobela and B. M. Abegaz, Bioorg. Med. Chem., 2010, 18, 2464–2473 CrossRef CAS PubMed.
- P. Du, J. Schneider, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2008, 47, 69–77 CrossRef CAS PubMed.
- K. T. Potts and D. Konwar, J. Org. Chem., 1991, 56, 4815–4816 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
- A. C. Benniston, G. Chapman, A. Harriman, M. Mehrabi and C. A. Sams, Inorg. Chem., 2004, 43, 4227–4233 CrossRef CAS PubMed.
- A. Rilak, I. Bratsos, E. Zangrando, J. Kljun, I. Turel, Z. D. Bugarcic and E. Alessio, Inorg. Chem., 2014, 53, 6113–6126 CrossRef CAS PubMed.
- C. M. Hartshorn, K. A. Maxwell, P. S. White, J. M. DeSimone and T. J. Meyer, Inorg. Chem., 2001, 40, 601–606 CrossRef CAS.
- W. Chen, F. N. Rein, B. L. Scott and R. C. Rocha, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m510–m511 CAS.
- G. Sathyaraj, M. Kiruthika, T. Weyhermueller and B. U. Nair, Dalton Trans., 2012, 41, 8460–8471 RSC.
- D. Oyama, M. Kido, A. Orita and T. Takase, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, M1117–U1942 CAS.
- E. Jakubikova, W. Chen, D. M. Dattelbaum, F. N. Rein, R. C. Rocha, R. L. Martin and E. R. Batista, Inorg. Chem., 2009, 48, 10720–10725 CrossRef CAS PubMed.
- X.-J. Yang, F. Drepper, B. Wu, W.-H. Sun, W. Haehnel and C. Janiak, Dalton Trans., 2005, 256–267 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima and O. K. Y. Honda, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, A.1 edn, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, Version 5 edn, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- R. D. Johnson, NIST Computational Chemistry Comparison and Benchmark Database, April 2010 edn, 2010 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. Benaltabef, S. B. R. Degallo, M. E. Folquer and N. E. Katz, Inorg. Chim. Acta, 1991, 188, 67–70 CrossRef CAS.
- P. J. Low and S. Bock, Electrochim. Acta, 2013, 110, 681–692 CrossRef CAS PubMed.
- W. Chen, F. N. Rein and R. C. Rocha, Angew. Chem., Int. Ed., 2009, 48, 9672–9675 CrossRef CAS PubMed.
- S. Ohzu, T. Ishizuka, Y. Hirai, S. Fukuzumi and T. Kojima, Chem. – Eur. J., 2013, 19, 1563–1567 CrossRef CAS PubMed.
- J. P. Collin and J. P. Sauvage, Inorg. Chem., 1986, 25, 135–141 CrossRef CAS.
- K. Nagoshi, S. Yamashita, M. Yagi and M. Kaneko, J. Mol. Catal. A: Chem., 1999, 144, 71–76 CrossRef CAS.
- D. C. Ware, P. A. Lay, H. Taube, M. H. Chou and C. Creutz, Inorg. Synth., 2007, 299–306 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information files (CIF) for compounds L3, [3Cl]PF6·CH3CN·C4H10O, and [4Cl]PF6·2CH3CN. Selected bond parameters, TD-DFT data and vibrational mode assignments. CCDC 1046731–1046733. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01278c |
| This journal is © The Royal Society of Chemistry 2015 |