
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew thiocyanate-free ruthenium(II) sensitizers with different pyrid-2-yl tetrazolate ligands for dye-sensitized solar cells†
Alessia
Colombo
a,
Claudia
Dragonetti
*ab,
Mirko
Magni
a,
Daniela
Meroni
a,
Renato
Ugo
a,
Gabriele
Marotta
c,
Maria
Grazia Lobello
c,
Paolo
Salvatori
cd and
Filippo
De Angelis
*c
aDipartimento di Chimica dell'Università degli Studi di Milano, UdR-INSTM, Italy. E-mail: claudia.dragonetti@unimi.it
bISTM-CNR, via Golgi 19, I-20133, Milano, Italy
cComputational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), Istituto CNR di Scienze e Tecnologie Molecolari, Via Elce di Sotto 8, I-06213, Perugia, Italy. E-mail: filippo@thch.unipg.it
dD3-Computation, Istituto Italiano di Tecnologia, Via Morego 30, I-16163 Genova, Italy
First published on 18th May 2015
Abstract
The synthesis of two new thiocyanate free Ru(II) complexes with different pyrid-2-yl tetrazolate ligands is reported, along with their electrochemical, spectroscopic and theoretical characterization. The corresponding dye-sensitized solar cell devices were prepared, leading to 3.4% conversion efficiency, promising data, considering the simplicity of the ligands and the high chemical stability of the complexes.
Introduction
Although tetrazoles have been known for over a century,1 there is a continuous and considerable interest in this field.2–4 Important developments include the patented use of tetrazole in air bags5 and pharmaceutical applications.6Since the first tetrazole complex was discovered by Bladin,1 tetrazole functional groups have been extensively used in various fields of chemistry such as medicinal chemistry and materials science. Owing to their multiple N-donor atoms and various coordination modes, tetrazole and its derivatives have potential importance as functional ligands in coordination chemistry and crystal engineering.7–11 To the best of our knowledge, pyrid-2-yl tetrazolate ligands have been used in Ru dyes for DSSCs only in one work, recently reported by some of us,12 although they have been used in the last decade for the preparation of luminescent organometallic complexes for OLEDs,13,14 as ligands for polymeric coordination materials15 and lanthanide ions, as contrast agents.16
The use of renewable energy sources instead of fossil fuel is a necessity for humanity. The Sun is the greenest and cheapest source of energy.17,18 The world's energy challenges can be overcome by harnessing the Sun's power with photovoltaic technologies. Dye-sensitized solar cells (DSSCs) have been extensively studied as sunlight-to-electricity conversion systems19,20 with power conversion efficiencies exceeding the value of 12%.21
The photosensitizer dye plays a key role in DSSCs, absorbing the solar light and promoting, at the interface between a mesoporous high bandgap semiconductor and a charge transport layer (the redox couple, for example I−/I3−), the formation of an electron–hole pair, which is then separated, transported, and collected at the electrodes. The semiconductor used is very often TiO2, in fact between its various uses22,23 nowadays, TiO2 nanoparticles have found their principal application in DSSCs. Very recently, the development of perovskite-sensitized solar cells24 appears to have overcome some limits of classical DSSCs, leading to solid-state solar cells with efficiencies of more than 19%.25
The most commonly used photosensitizers in solution-processed classical DSSCs are 2,2′-bipyridine (bpy) Ru(II) complexes with two thiocyanate (NCS) ligands, such as cis-di(thiocyanato)bis(bpy-4,4′-dicarboxylate)ruthenium(II) (N3 and N719).26,27 A limitation of such Ru(II) complexes is the presence of monodentate NCS ancillary ligands that can be easily replaced by other competing ligands in the electrolyte, yielding less efficient species. Often, 4-tert-butylpyridine is used as an additive in the electrolyte solution,28,29 but NCS-based Ru dyes30 may release a thiocyanate ligand by ligand exchange with 4-tert-butylpyridine or with the solvent. The efficiency of N719-4-tert-butylpyridine complex compared to N719 in a similar DSSC is 50% lower due to ca. 30 nm blue shift in its optical absorption spectrum.31 Clearly it is crucial to develop thiocyanate-free Ru sensitizers.
Cyclometallated Ru complexes are an alternative for classical NCS-Ru dyes in DSSCs. In fact cyclometallating ligands give stability to complexes often used for organic light-emitting diodes (OLEDs)32–34 or organic light-emitting electrochemical cells.35–39 Graetzel,40 Berlinguette,41,42 van Koten43 and our team44–46 have studied Ru complexes with cyclometallated ligands for DSSCs. In addition to cyclometallated ligands other bidentate ligands were used instead of thiocyanates, for example oxyquinolate,47,48 pyridyl pyrazole, pyridyl triazole,49–52 and dipyrrinate.53,54
The stronger electron withdrawing character of the tetrazolate ligand with respect to phenylpyridine can increase the oxidation potential of Ru(II) dyes containing this ligand, a tool for the facile regeneration of the oxidized sensitizers by the redox couple. This observation, together with our previous work,12 prompted us to synthesize thiocyanate-free Ru dyes with different pyrid-2-yl tetrazolate ligands for DSSCs (see Fig. 1).
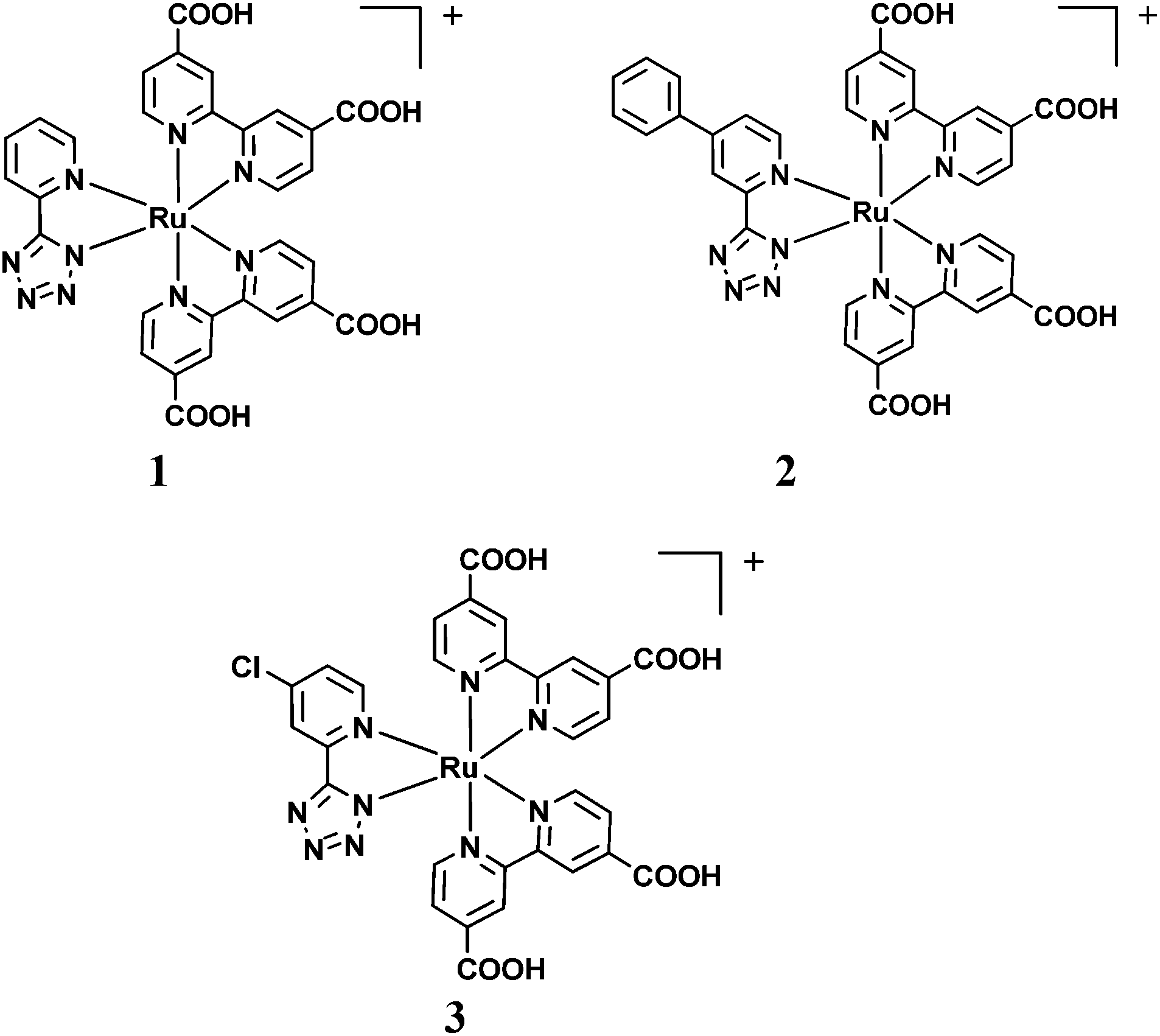 | ||
| Fig. 1 Molecular structures of the investigated dyes, 1,122 and 3. | ||
The aim of this work is to study the effects of different R substituents on the spectroscopic and electrochemical behavior of bis-heteroleptic Ru complexes of general formula [Ru(5-(4-R-2-pyridyl)tetrazolate)(dcbpy)2]+ and their application as sensitizers in DSSCs (dcbpy = 2,2′-bipyridine-4,4′-dicarboxylic acid). Complex 2 was synthesized by introducing on the pyrid-2-yl tetrazolate ligand a mesomeric π-delocalizing group, R = Ph, while complex 3 was functionalized with an electron withdrawing substituent, R = Cl (see Fig. 1). The introduction of the Ph and Cl substituents should serve respectively (i) to increase the absorptivity of dye 2 with respect to the complex 1, exploiting the higher π-delocalization offered by the phenyl group, in order to improve the light harvesting capability of the sensitized photoanode; (ii) to increase the driving force for the dye regeneration by the redox mediator (in this work I−/I3− couple) making the Ru-based dye more difficult to oxidize by introducing the electron withdrawing Cl atom used to positively shift the oxidation half-wave potential, E1/2(dye+/dye), of the complex. These two features should render 2 and 3 as more efficient sensitizers than the already published parent complex 1.
Experimental
Materials
All reagents were purchased from Sigma-Aldrich and were used without further purification. Reactions requiring anhydrous conditions were performed under argon. 1H NMR spectra were recorded at 400 MHz using a Bruker AVANCE-400 instrument. Chemical shifts (δ) for 1H spectra are expressed in ppm relative to internal Me4Si as standard. Signals were abbreviated as s, singlet; d, doublet; dd, doublet of doublets, triplet; q, quartet; m, multiplet. Mass spectra were recorded using a FT-ICR Mass Spectrometer APEX II and Xmass software (Bruker Daltonics) – 4.7 Magnet and Autospec Fission Spectrometer (FAB ionization). Electronic absorption spectra were recorded using a Jasco V-530 spectrophotometer. Thin layer chromatography (TLC) was carried out with pre-coated Merck F254 silica gel plates. Flash chromatography (FC) was carried out with Macherey-Nagel silica gel 60 (230–400 mesh).Complex 1 was synthesized as reported by some of us in a previous work.12 The ruthenium precursor, [RuCl2(dcbpy)2] (dcbpy = 4,4′-dicarboxylic acid-2,2′-bipyridine) was prepared according to the literature.55
The new dyes were synthesized involving two steps: (i) preparation of the Ru(II) precursor, [RuCl2(dcbpy)2]; (ii) substitution of the two chloride ions in [RuCl2(dcbpy)2] with 5-(4-R-2-pyridyl)tetrazole ligand in methanol [R = Phenyl (2), Cl (3)].
Synthesis of 5-(4-phenyl-2-pyridyl)tetrazole
5-(4-Phenyl-2-pyridyl)tetrazole was obtained, following a procedure reported in the literature,56 by refluxing 4-phenyl-2-pyridinecarbonitrile (1.66 mmol, 300.0 mg), sodium azide (1.83 mmol, 119.0 mg) and zinc bromide (1.66 mmol, 373.8 mg) in 4 mL of water for 24 h under a nitrogen atmosphere. After cooling, NaOH (4.15 mmol, 166.0 mg) was added and then the mixture was filtered and acidified with HCl (37% weight) until a precipitate was formed. Finally, the white solid was filtered and washed with water (277.9 mg, 75% yield).1H-NMR (400 MHz; DMSO) δ ppm: 8.87 (d, J = 5.14 Hz, 1H), 8.48 (s, 1H), 7.98 (d, J = 5.14 Hz, 1H), 7.94 (d, J = 7.03 Hz, 2H), 7.59 (d, J = 7.03, 2H), 7.61–7.53 (m, 1H).
Anal. Calcd for C12H9N5: C, 64.56; H, 4.06; N, 31.37; found C, 64.60; H, 4.05; N, 31.39. FAB+: 224 m/z.
Synthesis of the complex [Ru(5-(4-phenyl-2-pyridyl)tetrazolate)(dcbpy)2]+Cl− (2)
In a two-neck flask containing [RuCl2(dcbpy)2] (0.07 mmol, 45.0 mg) in methanol (20 mL), 5-(4-phenyl-2-pyridyl)tetrazole ligand (0.14 mmol, 30.3 mg) was added and the mixture was heated at reflux for 18 h under a nitrogen atmosphere. The obtained orange/red suspension was filtered and the filtrate was dried in vacuo. Then it was washed with CH2Cl2 to remove the free ligand which is in excess and finally was dried to obtain an orange solid (56.9 mg, 48% yield).1H-NMR (400 MHz; MeOD) δ ppm: 9.13 (d, J = 6.19 Hz, 2H), 9.09 (d, J = 6.19 Hz, 2H), 8.79 (s, 1H), 8.15 (d, J = 5.69 Hz 1H), 8.09 (d, J = 5.69 Hz 1H), 8.08 (d, J = 5.69 Hz, 1H), 7.99 (s, 1H), 7.97–7.94 (m, 2H), 7.89–7.85 (m, 4H), 7.71 (s, 1H), 7.67 (d, J = 5.69 Hz, 1H), 7.59–7.54 (m, 3H). Anal. Calcd for C36H24ClN9O8Ru: C, 51.04; H, 2.86; N, 14.88. Found C, 51.08; H, 2.84; N, 14.90. FAB+: 812 m/z.
Synthesis of 5-(4-chloro-2-pyridyl)tetrazole
5-(4-Chloro-2-pyridyl)tetrazole was obtained, following a procedure reported in the literature,56 by refluxing 4-chloro-2-pyridinecarbonitrile (3.60 mmol, 500.0 mg), sodium azide (3.97 mmol, 257.9 mg) and zinc bromide (3.60 mmol, 810.7 mg) in 7 mL of water for 24 h under a nitrogen atmosphere. After cooling, NaOH (9.0 mmol, 360.0 mg) was added and then the mixture was filtered and acidified with HCl (37% weight) until a precipitate was formed. Finally, the white solid was filtered and washed with water (503.2 mg, 77% yield).1H-NMR (400 MHz; DMSO) δ ppm: 8.78 (d, J = 5.20 Hz, 1H), 8.27 (s, 1H), 7.80 (d, J = 5.20 Hz, 1H). Anal. Calcd for C6H4ClN5: C, 39.69; H, 2.22; N, 38.57; found C, 36.71; H, 2.21, N, 38.6. FAB+: 182 m/z.
Synthesis of the complex [Ru(5-(4-chloro-2-pyridyl)tetrazolate)(dcbpy)2]+Cl− (3)
In a two-neck flask containing [RuCl2(dcbpy)2] (0.07 mmol, 45.0 mg) in methanol (20 mL), 5-(4-chloro-2-pyridyl)tetrazole ligand (0.14 mmol, 24.7 mg) was added and the mixture was heated at reflux for 18 h under a nitrogen atmosphere. The obtained orange/red suspension was filtered and the filtrate was dried under vacuum. Then it was washed with CH2Cl2 to remove the free ligand in excess and finally was dried to obtain an orange solid (50.7 mg, 45% yield).1H-NMR (400 MHz; MeOD) δ ppm: 9.05 (s, 2H), 9.00 (d, J = 6.56 Hz, 2H), 8.74 (d, J = 2.14 Hz, 1H), 8.17 (d, J = 5.8 Hz, 1H), 8.13 (d, J = 5.8 Hz, 1H), 8.03–7.98 (m, 4H), 7.90 (dd, J = 5.8, J = 2.14 Hz, 1H), 7.85 (d, J = 5.8 Hz, 1H), 7.61 (d, J = 5.8 Hz, 1H), 7.49(dd, J = 5.8 Hz, J = 2.14 Hz, 1H). Anal. Calcd for C30H19Cl2N9O8Ru: C, 44.73; H, 2.38; N, 8.80; found C, 44.72; H, 2.39; N, 8.78. FAB+: 770 m/z.
Computational details
All the calculations have been performed by the GAUSSIAN 09 program.57 The molecular structure of the fully protonated complexes were optimized in a vacuum using the B3LYP exchange–correlation functional58 and a 3-21G* basis set.59 TDDFT calculations of the lowest singlet–singlet excitations were performed in DMSO solution on the structure optimized in a vacuum and using a DGDZVP basis set.60 The C-PCM model61–63 was employed to introduce solvent effects in TDDFT calculations, as implemented in G09. To simulate the optical spectra, the 70 lowest spin-allowed singlet–singlet transitions were computed on the ground state geometry. Transition energies and oscillator strengths were interpolated by a Gaussian convolution with a σ value of 0.15 eV.Electrochemical measurements
Staircase cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments were carried out in a three-electrode minicell containing 2 ml of a well deaerated solution (by bubbling nitrogen) of 1–3 compounds dissolved in N,N-dimethyl-formamide (DMF, Sigma Aldrich, anhydrous, ≥99.8%) in the presence of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6, Fluka, ≥98.0%) as the supporting electrolyte. Measurements were performed using an Autolab PGSTAT128N potentiostat/galvanostat (EcoChemie, The Netherlands) managed by a PC with GPES software; a glassy carbon disk (Metrohm, geometric area 0.071 cm2) was used as the working electrode, in combination with a platinum sheet and an aqueous calomel (SCE) as the counter and the operating reference electrodes respectively. The SCE electrode was inserted into a glass jacket, ending with a porous frit, filled with the same electrolyte used in the cell (DMF + TBAPF6 0.1 M) in order to minimize the leakage of water and additional chloride ions in the working solution. An instrumental compensation of the resistance (i.e. positive feedback) was carefully performed in order to minimize the ohmic drop between the working and the reference electrodes. The recorded potentials were subsequently referred to as the intersolvental redox reference couple Fc+/Fc (ferrocenium/ferrocene)64 used as an external standard (ca. 10−3 M) at the end of the daily measures (E1/2(Fc+/Fc) ≈ 0.48 V(SCE) under our reaction conditions).The experimental parameters for the differential pulse measures were the following: 5 s of equilibration time at the starting potential, 0.05 s modulation time, 0.05 V modulation amplitude, 0.1 s interval time, 0.005 V step potential (i.e. 0.05 V s−1 scan rate); the staircase CVs were registered at a scan rate ranging between 0.05 and 2 V s−1 and 0.001 V as the step potential. Half-wave potentials of the dyes, E1/2(dye+/dye), were derived by two subsequent and independent measures from the corresponding DPV patterns as E1/2(dye+/dye) = EII Ox + ΔE/2, where EII Ox is the oxidation peak potential attributed to the ruthenium-centred process (see the Results and discussion section for more details) and ΔE is the modulation amplitude.65
Solar cell fabrication and characterization
FTO glass (TEC-15, 2.2 mm thickness, Solaronix) was used for transparent conducting electrodes. The substrate was first cleaned in an ultrasonic bath using a detergent solution, acetone and ethanol respectively (each step 15 min long). The FTO glass plates were immersed into a 40 mM aqueous TiCl4 solution at 70 °C for 30 min and washed with water and ethanol. A double layer of TiO2 paste (transparent + scattering layer, Dyesol) was spread on the FTO glass plates by using a doctor blade. The TiO2 layer was treated in an ethanol chamber and dried for 5 min at 120 °C. The TiO2 coated electrodes (active area 0.2 cm2) were gradually heated under air flow at 325 °C for 5 min, at 375 °C for 5 min, at 450 °C for 15 min, and 500 °C for 15 min. After the sintering process, the TiO2 film was treated with 40 mM TiCl4 solution, then rinsed with water and ethanol. The electrodes were heated at 500 °C for 30 min and after cooling (80 °C) were immersed for 20 h into sensitizing baths. These consisted of EtOH/DMSO (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solutions of the dyes in 0.2 mM concentration with 0.01 M 3a,7a-dihydroxy-5b-cholic acid (CDCA) added. Counter electrodes were prepared by coating with a drop of H2PtCl6 solution (2 mg of Pt in 1 mL of ethanol) on a FTO plate (TEC 15/2.2 mm thickness, Solaronix) and heating at 400 °C for 15 min. The TiO2 sensitized photoanodes and Pt counter electrodes were assembled in a sealed sandwich-type cell by a hot-melt ionomer film (Surlyn, 25 lm thickness, Dyesol). The electrolyte solution consisted of 1-butyl-3-methylimidazolium iodide, iodine, guanidinium thiocyanate, and tert-butylpyridine in valeronitrile/acetonitrile (IOLITECH ES-004-HP) with LiI 0.1 M added. The electrolyte solution was introduced by vacuum backfilling. Then, the hole was sealed by using an additional Surlyn patch and a cover glass and finally a conductive Ag-based paint was deposited at the electrical contacts. Photovoltaic measurements were recorded by means of AM 1.5 solar simulator equipped with a xenon lamp (LOT-ORIEL LS 0106). The power of the incoming radiation, set at 100 mW cm−2, was checked by a pyranometer. J–V curves were obtained by applying an external bias to the cell and measuring the generated photocurrent with a Keithley model 2400 digital source-meter, under the control of dedicated LabTracer 2.0 software. A black shading mask was employed to avoid the overestimation of the measured parameters.
:1) solutions of the dyes in 0.2 mM concentration with 0.01 M 3a,7a-dihydroxy-5b-cholic acid (CDCA) added. Counter electrodes were prepared by coating with a drop of H2PtCl6 solution (2 mg of Pt in 1 mL of ethanol) on a FTO plate (TEC 15/2.2 mm thickness, Solaronix) and heating at 400 °C for 15 min. The TiO2 sensitized photoanodes and Pt counter electrodes were assembled in a sealed sandwich-type cell by a hot-melt ionomer film (Surlyn, 25 lm thickness, Dyesol). The electrolyte solution consisted of 1-butyl-3-methylimidazolium iodide, iodine, guanidinium thiocyanate, and tert-butylpyridine in valeronitrile/acetonitrile (IOLITECH ES-004-HP) with LiI 0.1 M added. The electrolyte solution was introduced by vacuum backfilling. Then, the hole was sealed by using an additional Surlyn patch and a cover glass and finally a conductive Ag-based paint was deposited at the electrical contacts. Photovoltaic measurements were recorded by means of AM 1.5 solar simulator equipped with a xenon lamp (LOT-ORIEL LS 0106). The power of the incoming radiation, set at 100 mW cm−2, was checked by a pyranometer. J–V curves were obtained by applying an external bias to the cell and measuring the generated photocurrent with a Keithley model 2400 digital source-meter, under the control of dedicated LabTracer 2.0 software. A black shading mask was employed to avoid the overestimation of the measured parameters.
Results and discussion
Synthesis
Complex 1 was synthesized as reported by some of us in a previous work.12 The ruthenium precursor, [RuCl2(dcbpy)2], was prepared according to the literature.55The new dyes were synthesized involving two steps: (i) preparation of the Ru(II) precursor, [RuCl2(dcbpy)2]; (ii) substitution of the two chloride ions in [RuCl2(dcbpy)2] with 5-(4-R-2-pyridyl)tetrazole ligand in methanol [R = Phenyl (2), Cl (3)] (Scheme 1).
 | ||
| Scheme 1 Synthesis of investigated dyes. | ||
The chemical stability of the complexes in acetonitrile solution was tested at reflux temperature (82 °C) for 24 h with a 100:1 (mol:mol) excess of 4-tert-butylpyridine (a commonly used additive in DSSC electrolytes) with respect to the Ru complex.
For comparison an analogous test was performed on the N719 complex. This procedure was inspired by previous studies by Lund et al.30,66 investigating the stability of commonly employed ruthenium dyes, such as N719 and Z907, toward substitution of the thiocyanate ligands with pyridine-based additives or solvent molecules, both in homogeneous solutions and in colloidal mixtures of dyed TiO2 nanoparticles. In later studies,31,67 they correlated the dye degradation to the device's efficiency losses, finding that high temperatures (∼80 °C) lead to performance decay, even in the dark, due to ligand exchange reactions. In line with previous results, FAB+ mass spectrometry confirmed that N719 underwent, at least partially, substitution of NCS ligands with 4-tert-butylpyridine, whereas our Ru complexes were inert to the presence of the competitive ligand (see ESI†).
Furthermore it has to be noted that pyrid-2-yl pirazolate (or triazolate) Ru-based dyes have shown promising stability in working devices under 1000 h-1 Sun light soaking, at 60 °C.49,51 These results therefore suggest that the newly designed dyes could show possible improvements concerning the stability compared to the thiocyanate-bearing ruthenium sensitizers.
Electrochemical and optical characterization
The electrochemical and optical properties of our complexes were investigated.In Table 1 the main absorption bands of the sensitizers are listed, while in Fig. 2 a comparison between the experimental and calculated absorption spectra in DMSO solution is reported.
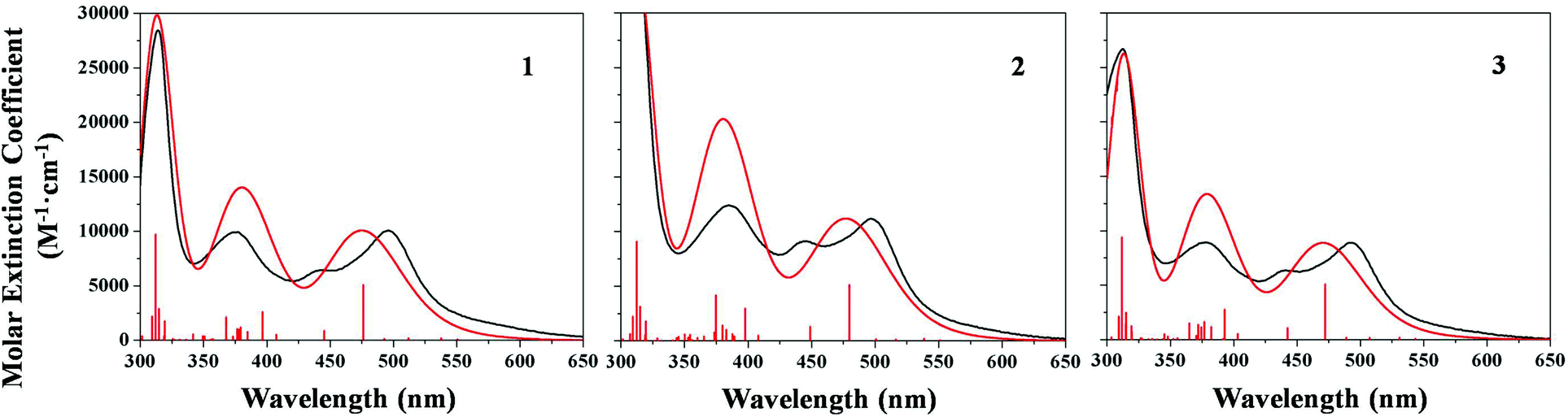 | ||
| Fig. 2 Comparison between experimental (black line) and calculated (red line) UV-vis absorption spectra of ruthenium complexes in DMSO solution. Intensity of the calculated spectra has been rescaled to match the experimental one. Red vertical lines represent the calculated excitation energies. | ||
| Dye | Wavelength (nm)/ε (×103, M−1 cm−1) |
|---|---|
| 1 | 496/10.1 |
| 440/6.5 | |
| 377/9.9 | |
| 2 | 497/11.2 |
| 444/9.1 | |
| 385/12.4 | |
| 3 | 492/8.9 |
| 441/6.4 | |
| 378/8.9 |
The UV-Vis absorption spectra of all the three compounds are essentially characterized by three main bands, localized at about 490, 440 and 380 nm. Complex 2 presents the most promising optical properties, having the highest extinction coefficient (11200 M−1 cm−1 for the lowest energy absorption band), due to the extended π-conjugation guaranteed by the phenyl group. The calculated absorption spectra are in good agreement with the experimental ones, with discrepancies of less than 0.12 eV for the main absorption band in the visible region. Also the experimental trend of the absorption maxima, showing a red shift moving from 3 to 1 and 2, is well reproduced by TDDFT at the employed calculation level (see ESI†). The experimental shoulder at about 440 nm is not observed in the calculated spectra, even if a transition essentially coincident with the absorption maxima is calculated. The main absorption bands in the visible region are characterized by MLCT transitions for all the complexes. HOMO orbitals are essentially Ru-centered, mixed with the tetrazole, and exhibit slight differences in their energy levels, depending on the substituents on the pyrid-2-yl tetrazolate ligand. LUMO and LUMO+1 are localized on the carboxylated bipyridine, and are essentially isoenergetic along the series (see Fig. 3 and ESI†).
 | ||
| Fig. 3 Schematic representation of the energy levels of the investigated complexes. Isodensity surface plots (isodensity contour: 0.035) of HOMO and LUMO molecular orbitals are also shown. | ||
The electrochemical characterization of complexes 1–3 (see Fig. 4) shows only small variations along the series according to the spectroscopic results. In particular the anodic window is characterized by two peaks clearly different in terms of current. The more intense signal at ca. 0.5 V vs. Fc+/Fc can be associated with the electrochemical oxidation of Cl− counterion, characterized by a significant higher diffusion coefficient with respect to the complex cation which is in turn oxidized only at a more positive potential due to the electron withdrawing nature of the tetrazole-containing ligand which influences the electron transfer tentatively related to a Ru-based reaction.
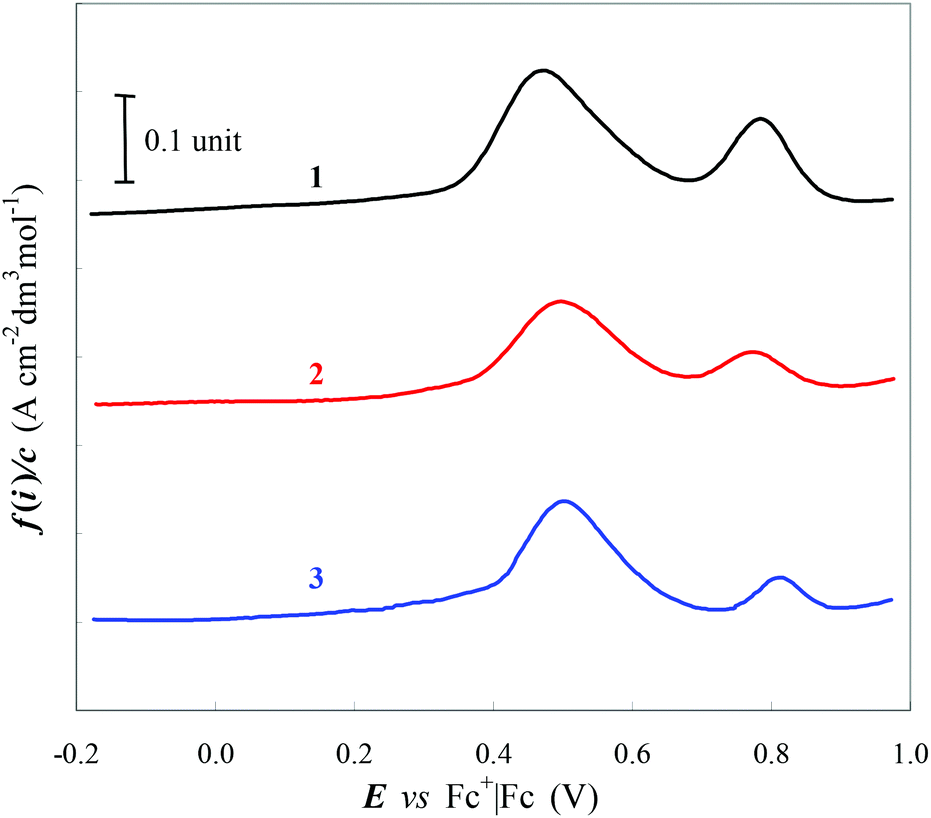 | ||
| Fig. 4 Synopsis of DPV characteristics for complexes 1–3 in DMF + TBAPF6 0.1 M. Sample concentration ca. 8 × 10−4 M; glassy carbon electrode (0.071 cm2); potential scan rate 0.05 V s−1. | ||
The attribution of the two voltammetric signals was corroborated by different DPV tests (see ESI†). First of all we verified the coincidence in terms of both position and current intensity of the peak invariably recorded at ca. 0.5 V vs. Fc+/Fc in the solutions of complexes 1–3 with that obtained by adding to a blank solution (DMF + TBAPF6) an equimolar amount of TBACl which was chosen as an external source of chloride ions. This first correspondence was validated by recording voltammograms obtained at increasing amount of Cl− (spiking TBACl solution) added as the internal standard to solutions containing Ru complexes.
As anticipated the more positive oxidation peak at ca. 0.8 V vs. Fc+/Fc is related to the metal-centered electron transfer process. In quite perfect qualitative agreement with theoretical HOMO energy values (Fig. 3) the half-wave potential is essentially invariant for 1 and 2 compounds, being E1/2(dye+/dye) = 0.78 V vs. Fc+/Fc, while it is E1/2(dye+/dye) = 0.81 V vs. Fc+/Fc for 3 in DMF solution. The slight positive shift of 0.03 V for complex 3 can be attributed to the electron withdrawing nature of the halogen atom on the pyrid-2-yl tetrazolate ligand, which can affect the electronic properties of the metal center. The positive displacement induced by the Cl atom on the E1/2 of complex 3 is significantly lower with respect to the effect induced by the same substitution on a ferrocene molecule (0.03 V vs. 0.14 V,68 respectively). The related lower stabilization of the HOMO level for complex 3 can be tentatively attributed to the distance of the halogen atom from the redox site directly involved in the electron transfer, largely centered on the Ru atom and tetrazole ring as indicated by computational results. The E1/2 of complexes 1–3 are invariably more positive than the value of the actual active couple I2˙−/I− (E1/2(I2˙−/I−) ≈ 0.17 V vs. Fc+/Fc in acetonitrile,69,70 obtained considering E1/2(Fc+/Fc) = 0.63 V vs. NHE) involved in the dye reduction in iodine-based electrolyte in DSSCs. This results in a driving force of more than 0.6 V for the dye regeneration process. As a consequence the increase of 0.03 V in the half-wave potential of complex 3 with respect to its analogues 1 and 2 is probably too small to cause perceivable improvement in the regeneration process occurring in DSSC (see below).
Photovoltaic data
The ruthenium complexes were finally tested in working devices. All the devices have been realized by using a double TiO2 layer, a transparent and a scattering one. In our previous work12 we demonstrated that the use of the CDCA coadsorbent (0.01 M) and the LiI electrolyte additive (0.1 M) was beneficial for the cell performance of dye 1, so we employed the same experimental conditions for the test of the new dyes. The photovoltaic data of realized devices are shown in Table 2 and in Fig. 5. A comparison with the reference N719 dye, measured in analogous fabrication conditions is also reported.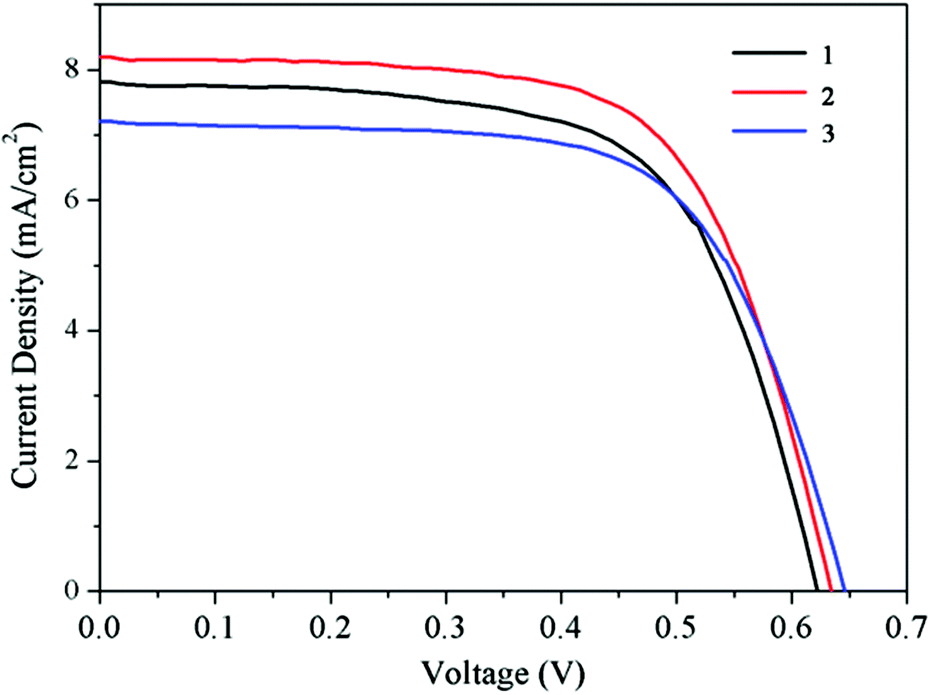 | ||
| Fig. 5 J–V curves of DSSCs sensitized with dye 1 (black line), dye 2 (red line) and dye 3 (blue line), respectively. 100 mW cm−2 AM 1.5 G illumination. | ||
| Dye | J sc (mA cm−2) | V oc (mV) | FF | η (%) |
|---|---|---|---|---|
| 1 | 7.81 | 623 | 0.64 | 3.1 |
| 2 | 8.20 | 635 | 0.65 | 3.4 |
| 3 | 7.21 | 647 | 0.65 | 3.0 |
| N719 | 13.88 | 752 | 0.62 | 6.5 |
The three investigated sensitizers show comparable efficiencies, of more than 3%, while the reference N719 dye performs better (6.5% efficiency). The main differences in the photovoltaic parameters are in the smaller photocurrent, Jsc, values and are probably due to the different light harvesting capabilities of the tetrazolate-based sensitizers with respect to the benchmark N719. All the new thiocyanate-free sensitizers show in fact less extended absorption spectra (Table 1 and Fig. 2) with respect to N719 dye (535 nm, 14700 M−1 cm−1 for the main absorption band in the visible region).71 The Jsc values of the new dyes are about 40% lower compared to the N719-based devices, and this reflects well the discrepancy in the light absorption.
Focusing the photovoltaic data analysis on the three tetrazole-containing sensitizers 1–3, the results show that once again short-circuit current is the only cell parameter significantly modified along the series of nominally identical cells in which the only variation is related to the nature of the dye. In fact Jsc exhibits a 14% maximum variation (from 7.21 mA cm−2 with dye 3 to 8.20 mA cm−2 with dye 2) compared to 4% and 1% for Voc and FF respectively (Table 2). This is in good agreement with expectation since both Voc and FF are primarily affected by the electrolyte composition (e.g. redox couple, additives, solvent) that in contrast was kept constant in all measurements. Moreover photocurrent values, and also efficiencies, follow the trend of the absorption maxima of the dyes (Tables 2 and 1 respectively). As anticipated computational results show some electronic structural similarities between dyes 1–3; in particular LUMOs of the three tetrazole-based sensitizers have essentially isoenergetic levels (Fig. 3) and are mainly localized on the carboxylated bipyridine moiety which is directly involved in the anchoring process to the TiO2 surface. This allows us to tentatively consider the comparable efficiency of the electron injection process occurring from the excited charge-separated state of dyes 1–3 to the conduction band of the semiconductor, being induced by the same thermodynamic driving force. Coherently the higher Jsc for the cell sensitized with 2 (and the related higher overall conversion efficiency of 3.4%) can be attributed, with a certain reasonableness, to the increased light harvesting related to the higher extinction coefficient of the more π-delocalized dye 2 with respect to its structural analogues 1 and 3. These results suggest that the photovoltaic performances of this class of easy-to-prepare thiocyanate-free dyes were improved by introducing a π-delocalizing substituent (i.e. a phenyl ring) and could potentially be further improved by extending the π-conjugation on the pyridyl-tetrazolate ligand, and thus enhance the light harvesting ability of the sensitizers.
Conclusions
A remarkable increase of ca. 10% of the overall cell conversion efficiency was obtained by simply functionalizing the pyrid-2-yl tetrazolate ligand of complex 1 with a phenyl group achieving ca. 3.4% efficiency with the new bis-heteroleptic Ru dye 2. The use of the highly π-delocalized 5-(4-phenyl-pyrid-2-yl)tetrazole increases the extinction coefficient of the corresponding complex 2 improving the solar light harvesting of the sensitized photoanode and enhancing the DSSCs efficiency. Surely the absorptivity of the sensitizer is only one of the numerous factors that define the cell efficiency. Nevertheless (i) the isoenergetic LUMO levels of dyes 1–3, and as a consequence the supposed comparable electron injection efficiency into TiO2 film, and (ii) the almost complete invariance of cell parameters except for the photocurrent in DSSCs differing only for the sensitizer nature, make plausible the hypothesis that the better efficiency obtained with dye 2 is mainly related to its higher π-delocalized electronic system and as a consequence to its improved light harvesting.In conclusion our work confirms the great potential of tetrazole Ru complexes as convenient DSSC sensitizers and gives useful guidelines for future work in this field. Computational, spectroscopic and electrochemical results suggest that the functionalization of the pyrid-2-yl-tetrazole scaffold with a π-conjugated group having also an electron releasing character could further improve performances of this class of thiocyanate-free dyes achieving a better absorption spectrum, which should combine higher extinction coefficient (due to a more π-extended system) and red-shifted absorption (due to destabilization of the HOMO energy level).
Acknowledgements
This work was supported by MIUR (FIRB 2004: RBPR05JH2P). G. M., P.S., M.G.L., and F.D.A. are also thankful for FP7-ENERGY-2010 Project 261920 “ESCORT” for financial support. We deeply thank Arianna Valsecchi for experimental help.Notes and references
- I. J. Bladin, Ber. Dtsch. Chem. Ges., 1885, 18, 1544 CrossRef.
- F. R. Benson, in Heterocyclic Compounds, ed. R. C. Elderfield, John Wiley & Sons, Inc., New York, 1967, vol. 8, pp. 1–104 Search PubMed.
- R. N. Butler, Adv. Heterocycl. Chem., 1977, 21, 323 CrossRef CAS.
- R. N. Butler, Comprehensive Heterocyclic Chemistry, ed. K. T. Potts, Pergamon Press, New York, 1984, vol. 5, pp. 791–838 Search PubMed.
- D. R. Poole and P. C. Kwong (Automotive Systems Laboratory, Inc.), PCT Int. Appl, WO 9500, 462, 1995 Search PubMed.
- S. J. Wittenberger, Org. Prep. Proced. Int., 1994, 26, 499 CrossRef CAS.
- V. A. Ostrovskii and A. O. Koren, Heterocycles, 2000, 53, 1421 CrossRef CAS.
- M. M. Harding and G. Mokdsi, Curr. Med. Chem., 2000, 7, 1289 CrossRef CAS PubMed.
- H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC.
- L. Ma, Y.-C. Qiu, G. Peng, J.-B. Cai and H. Deng, Eur. J. Inorg. Chem., 2011, 23, 3446 CrossRef.
- Y. Chia, B. Tonga and P. T. Chouc, Coord. Chem. Rev., 2014, 28, 11 Search PubMed.
- C. Dragonetti, A. Colombo, M. Magni, P. Mussini, F. Nisic, D. Roberto, R. Ugo, A. Valore, A. Valsecchi, P. Salvatori, M. G. Lobello and F. De Angelis, Inorg. Chem., 2013, 52, 10723 CrossRef CAS PubMed.
- M. V. Werrett, D. Chartrand, J. D. Gale, G. S. Hanan, J. G. MacLellan, M. Massi, S. Muzzioli, P. Raiteri, B. W. Skelton, M. Silberstein and S. Stagni, Inorg. Chem., 2011, 50, 1229 CrossRef CAS PubMed.
- C. Femoni, S. Muzzioli, A. Palazzi, S. Stagni, S. Zacchini, F. Monti, G. Accorsi, M. Bolognesi, N. Armaroli, M. Massi, G. Valenti and M. Marcaccio, Dalton Trans., 2013, 42, 997 RSC.
- G. Aromì, L. A. Barriosa, O. Roubeaub and P. Gameza, Coord. Chem. Rev., 2011, 255, 485 CrossRef.
- A. Facchetti, A. Abbotto, L. Beverina, S. Bradamante, P. Mariani, C. L. Stern, T. J. Marks, A. Vacca and G. A. Pagani, Chem. Commun., 2004, 1770 RSC.
- M. K. Nazeeruddin, E. Baranoff and M. Graetzel, Sol. Energy, 2011, 85, 1172 CrossRef CAS.
- J.-H. Yum, E. Baranoff, S. Wenger, M. K. Nazeeruddin and M. Graetzel, Energy Environ. Sci., 2011, 4, 842 CAS.
- B. O'Regan and M. Greatzel, Nature, 1991, 353, 737 CrossRef.
- Q. Wang, S. Ito, M. Graetzel, F. Fabregat-Santiago, I. Mora-Sero, J. Bisquert, T. Bessho and H. J. Imai, Phys. Chem. B, 2006, 110, 25210 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Graetzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- G. Maino, D. Meroni, V. Pifferi, L. Falciola, G. Soliveri, G. Cappelletti and S. Ardizzone, J. Nanopart. Res., 2013, 15, 2087 CrossRef.
- A. Antonello, G. Soliveri, D. Meroni, G. Cappelletti and S. Ardizzone, Catal. Today, 2014, 230, 35 CrossRef CAS.
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. Song, H. S. Duan, Z. Hong, J. You, Y. Yang and Y. Liu, Science, 2014, 345, 542 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Graetzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- G. Boschloo, L. Haggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144 CrossRef CAS PubMed.
- H. Kusama, Y. Konishi, H. Sugihara and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 80, 167 CrossRef CAS.
- P. T. Nguyen, R. Degn, H. T. Nguyen and T. Lund, Sol. Energy Mater. Sol. Cells, 2009, 93, 1939 CrossRef CAS.
- A. R. Andersen, J. Halme, T. Lund, M. I. Asghar, P. T. Nguyen, K. Miettunen, E. Kemppainen and O. Albrektsen, J. Phys. Chem. C, 2011, 115, 15598 CAS.
- R. C. Evans, P. Douglas and C. Winscom, J. Coord. Chem. Rev., 2006, 250, 2093 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS.
- W. Y. Wong and C. L. Ho, Coord. Chem. Rev., 2009, 253, 1709 CrossRef CAS.
- E. Zysman-Colman, J. D. Slinker, J. B. Parker, G. G. Malliaras and S. Bernhard, Chem. Mater., 2008, 20, 388 CrossRef CAS.
- H. J. Bolink, L. Cappelli, E. Coronado, M. Graetzel, E. Ortí, R. D. Costa, M. Viruela and M. K. Nazeeruddin, J. Am. Chem. Soc., 2006, 128, 14786 CrossRef CAS PubMed.
- W. Y. Wong, G. J. Zhou, X. M. Yu, H. S. Kwok and Z. Lin, Adv. Funct. Mater., 2007, 17, 315 CrossRef CAS.
- E. Margapoti, V. Shukla, A. Valore, A. Sharma, C. Dragonetti, C. Kitts, D. Roberto, M. Murgia, R. Ugo and M. Muccini, J. Phys. Chem. C, 2009, 113, 12517 CAS.
- E. Margapoti, M. Muccini, A. Sharma, A. Colombo, C. Dragonetti, D. Roberto and A. Valore, Dalton Trans., 2012, 41, 9227 RSC.
- T. Bessho, E. Yoneda, J. H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Graetzel, J. Am. Chem. Soc., 2009, 131, 5930 CrossRef CAS PubMed.
- P. G. Bomben, K. D. Theriault and C. P. Berlinguette, Eur. J. Inorg. Chem., 2011, 11, 1806 CrossRef.
- K. C. D. Robson, B. D. Koivisto, A. Yella, B. Sporinova, M. K. Nazeeruddin, T. Baumgartner, M. Graetzel and C. P. Berlinguette, Inorg. Chem., 2011, 50, 5494 CrossRef CAS PubMed.
- S. H. Wadman, J. M. Kroon, K. Bakker, R. W. A. Havenith, G. P. M. van Klin and G. van Koten, Organometallics, 2010, 29, 1569 CrossRef CAS.
- C. Dragonetti, A. Valore, A. Colombo, D. Roberto, V. Trifiletti, N. Manfredi, M. M. Salamone, R. Ruffo and A. Abbotto, J. Organomet. Chem., 2012, 714, 88 CrossRef CAS.
- A. Abbotto, C. Coluccini, E. Dell'Orto, N. Manfredi, V. Trifiletti, M. M. Salamone, R. Ruffo, M. Acciarri, A. Colombo, C. Dragonetti, S. Ordanini, D. Roberto and A. Valore, Dalton Trans., 2012, 41, 11731 RSC.
- A. Colombo, C. Dragonetti, A. Valore, C. Coluccini, N. Manfredi and A. Abbotto, Polyhedron, 2014, 82, 50 Search PubMed.
- H. C. Zhao, J. P. Harney, Y.-T. Huang, J.-H. Yum, M. K. Nazeeruddin, M. Graetzel, M.-K. Tsai and J. Rochford, Inorg. Chem., 2012, 51, 51 CrossRef PubMed.
- C. Dragonetti, A. Valore, A. Colombo, M. Magni, P. Mussini, D. Roberto, R. Ugo, A. Valsecchi, V. Trifiletti, N. Manfredi and A. Abbotto, Inorg. Chim. Acta, 2013, 405, 98 CrossRef CAS.
- K. L. Wu, H. C. Hsu, K. Chen, Y. Chi, M. K. Chung, W. H. Liu and P. T. Chou, Chem. Commun., 2010, 46, 5124 RSC.
- K. L. Wu, C. H. Li, Y. Chi, J. N. Clifford, L. Cabau, E. Palomares, Y. M. Cheng, H. A. Pan and P. T. Chou, J. Am. Chem. Soc., 2012, 134, 7488 CrossRef CAS PubMed.
- K.-L. Wu, W.-P. Ku, S.-W. Wang, A. Yella, Y. Chi, S.-H. Liu, P.-T. Chou, M. K. Nazeeruddin and M. Graetzel, Adv. Funct. Mater., 2013, 23, 2285 CrossRef CAS.
- S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, J. Kübel, B. Dietzek, C. P. Berlinguette and U. S. Schubert, Inorg. Chem., 2014, 53, 1637 CrossRef CAS PubMed.
- G. Li, P. G. Bomben, K. C. D. Robson, S. I. Gorelsky, C. P. Berlinguette and M. Shatruk, Chem. Commun., 2012, 48, 8790 RSC.
- G. Li, K. Hu, K. C. D. Robson, S. I. Gorelsky, G. J. Meyer, C. P. Berlinguette and M. Shatruk, Chem. – Eur. J., 2015, 21, 2173 CrossRef CAS PubMed.
- X.-H. Wu, S. Wang, Y. Guo, Z.-Y. Xie, L. Han and Z.-H. Jiang, Chin. J. Chem., 2008, 26, 1939 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian, Inc., Wallingford, CT, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS.
- S. Mierts, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117 Search PubMed.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461 CrossRef.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods. Fundamentals and Applications, Wiley, 2nd edn, 1994 Search PubMed.
- H. T. Nguyen, H. M. Ta and T. Lund, Sol. Energy Mater. Sol. Cells, 2007, 91, 1934 CrossRef CAS.
- P. T. Nguyen, A. R. Andersen, E. M. Skou and T. Lund, Sol. Energy Mater. Sol. Cells, 2010, 94, 1582 CrossRef.
- D. A. Khobragade, S. G. Mahamulkar, L. Pospisil, I. Cisarova, L. Rulisek and U. Jahn, Chem. – Eur. J., 2012, 18, 12267 CrossRef CAS PubMed.
- G. Boschloo, E. A. Gibson and A. Hagfeldt, J. Phys. Chem. Lett., 2011, 2, 3016 CrossRef CAS.
- K. C. D. Robson, P. G. Bomben and C. P. Berlinguette, Dalton Trans., 2012, 41, 7814 RSC.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Grätzel, Inorg. Chem., 1999, 38, 6298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and differential pulse voltammograms. See DOI: 10.1039/c5dt01216c |
| This journal is © The Royal Society of Chemistry 2015 |