DOI:
10.1039/C5DT01080B
(Paper)
Dalton Trans., 2015,
44, 11717-11724
16-Electron pentadienyl- and cyclopentadienyl-ruthenium half-sandwich complexes with bis(imidazol-2-imine) ligands and their use in catalytic transfer hydrogenation†
Received
18th March 2015
, Accepted 2nd April 2015
First published on 7th April 2015
Abstract
Bis(η5-2,4-dimethylpentadienyl)ruthenium(II), [(η5-C7H11)2Ru] (1, “open ruthenocene”), which has become accessible in high yield and large quantities via an isoprene-derived diallyl ruthenium(IV) complex, can be converted into the protonated open ruthenocene 2 by treatment with HBF4 and subsequently into the protonated half-open ruthenocene 3 by reaction with cyclopentadiene. The electronic structure of 3 was studied by DFT methods, revealing that the CH-agostic complex [(η5-C5H5)Ru{(1-4η)-C7H12-η2-C5,H5}]BF4 (A) represents the global minimum, which is 3.7 kcal mol−1 lower in energy than the hydride complex [(η5-C5H5)RuH(η5-C7H11)]BF4 (B). 2 and 3 were treated with the ligands N,N′-bis(1,3,4,5-tetramethylimidazolin-2-ylidene)-1,2-ethanediamine (BLMe) and N,N′-bis(1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene)-1,2-ethanediamine (BLiPr) to afford the cationic 16-electron pentadienyl and cyclopentadienyl complexes [(η5-C7H11)Ru(BLR)]BF4 (4a, R = Me; 4b, R = iPr) and [(η5-C5H5)Ru(BLR)]BF4 (5a, R = Me; 5b, R = iPr). All complexes catalyse the transfer hydrogenation of acetophenone in isopropanol, and the most active complex 4a in this reaction was employed for the hydrogenation of a broader range of aliphatic and aromatic ketones.
Introduction
Imidazolin-2-imine ligands such as BLMe and BLiPr,1,2 and related guanidine-type ligands,3 have found widespread use in organometallic and coordination chemistry and proved to be suitable ancillary ligands for applications in homogeneous catalysis.4 Because the imidazole moiety can efficiently stabilise a positive charge (Scheme 1),5 these diimine species are highly basic and may serve as strong N-donor ligands towards transition metals, as was demonstrated e.g. by the isolation of stable 16-electron half-sandwich complexes of the type [(η5-C5Me5)Ru(BLR)]+,6,7 [(η7-C7H7)Mo(BLR)]+,8 and [(η6-arene)Ru(BLR)]2+ (R = Me, iPr; arene = cymene, benzene).9 The mono- and dicationic cyclopentadienyl- and arene-ruthenium complexes, which are isolable even in the presence of chloride counterions,6,9 were employed as catalysts for the transfer hydrogenation of acetophenone, with isopropanol serving as the hydrogen source.9
 |
| | Scheme 1 Mesomeric structures for the bis(imidazolin-2-imine) ligands BLR (R = Me, iPr). | |
To vary the half-sandwich ruthenium moiety in these coordinatively unsaturated and catalytically active complexes,10 we chose bis(η5-2,4-dimethylpentadienyl)ruthenium(II), [(η5-C7H11)2Ru] (1, “open ruthenocene”),11 as a suitable starting material, since this compound has recently become conveniently accessible in high yield and large quantities via an isoprene-derived diallyl ruthenium(IV) complex.12–14 Previously, a related protocol had directly furnished the protonated open ruthenocene 2,15 which was also obtained by treatment of 1 with HBF4 (Scheme 2).16 It was also briefly mentioned that the reaction of 2 with cyclopentadiene affords the corresponding cationic Cp complex 3, but to the best of our knowledge, no experimental details were published.17 Alternatively, 3 can be prepared by the reaction of the half-open ruthenocene (η5-2,4-dimethylpentadienyl)(η5-cyclopentadienyl)ruthenium(II), [(η5-C5H5)Ru(η5-C7H11)], with HBF4.18 Complex 2 served as a versatile starting material for the preparation of numerous half-sandwich 2,4-dimethylpentadienyl-ruthenium complexes by displacement of the diene ligand,12,16,19,20 and consequently we expected that both 2 and 3 would readily react with the diimine ligands BLR (R = Me, iPr) to afford complexes of type 4 and 5, respectively (Scheme 2).21 Thus, the preparation and characterisation of these complexes and their use in hydrogen transfer catalysis is described herein. In addition, an electronic structure calculation of 3 together with full experimental details for its preparation from 2 are reported.
 |
| | Scheme 2 Preparation of pentadienyl and cyclopentadienyl ruthenium half-sandwich complexes from protonated open and half-open ruthenocenes. | |
Results and discussion
Preparation and electronic structure of the half-open ruthenocene 3
The open ruthenocene 1 was treated with HBF4·Et2O in diethyl ether solution as previously described to afford 2 as a yellow crystalline solid in quantitative yield.16 Dropwise addition of freshly distilled cyclopentadiene to an acetone solution of 2 at −75 to −80 °C gave 3 as a pale yellow solid in 80% yield after stirring at room temperature and precipitation with diethyl ether (Scheme 2).17 It should be noted that this method is generally applicable and provides e.g. the corresponding methyl- and tert-butylcyclopentadienyl complexes by reaction with the corresponding cyclopentadiene derivatives.21 The NMR spectroscopic data of 3 are identical to those reported previously, with the 1H NMR spectrum displaying signals at ca. −10.1, −0.3 and 3.0 ppm in the temperature range between −10 and −80 °C,17,18 which can be assigned to the RuH and to the endo- and exo-CH2 hydrogen atoms of an agostic ruthenium species (see the ESI†). Accordingly, methyl rotation, i.e. H exchange within an agostic CH3 group, is frozen out in this temperature range on the NMR time scale, and a detailed dynamic NMR study derived a barrier of ΔG‡ = 12.6 kcal mol−1 for this process. In contrast, 1,5-H transfer cannot be frozen out (vide supra), and an upper limit of ca. 7.2 kcal mol−1 was estimated for the barrier of the latter process.17,22
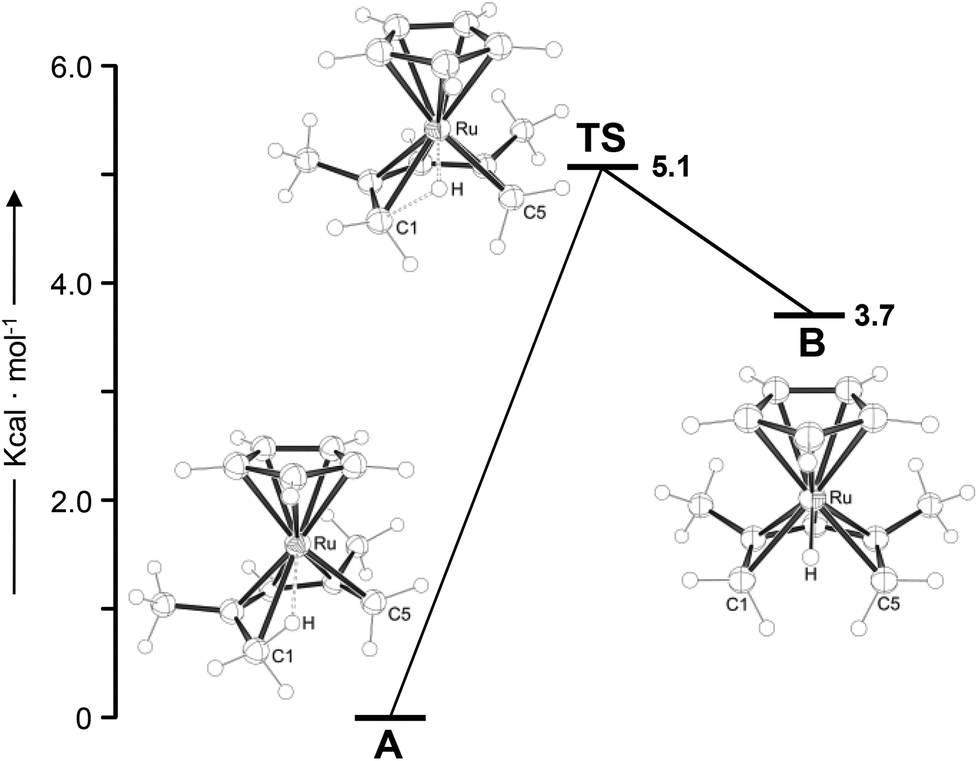
To support the experimental findings and to further characterise the electronic structure of the protonated half-open ruthenocene, DFT calculations were carried out for the cation in 3 at the B3LYP/6-311G(d,p) level of theory. Two minimum structures A and B were located, which are connected by a low-energy transition state TS (Fig. 1). The global minimum A represents an agostic ruthenium complex [(η5-C5H5)Ru{(1-4η)-C7H12-η2-C5,H5)}]BF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 with Ru–C5 and Ru–H5 distances of 2.38 and 1.85 Å.24 The agostic C–H bond is elongated (1.18 Å), and the C4–C5 bond is significantly longer than the other C–C bonds (1.49 vs. 1.42, 1.44 and 1.41 Å) within the pentadienyl ligand. Structure B is 3.7 kcal mol−1 higher in energy and represents the hydride complex [(η5-C5H5)RuH(η5-C7H11)] with a Ru–H bond of 1.57 Å. The pentadienyl ligand is symmetrically bound, with long C1–H and C5–H distances of ca. 2.20 Å. A and B are connected by a transition state (TS) that is 5.1 and 1.4 kcal mol−1 higher in energy than A and B, respectively. Naturally, B can be regarded as an intermediate in the intraligand 1,5-H transfer from C1 to C5, and the low calculated barrier for this process is in agreement with the observation that this process is not frozen out in an accessible temperature range on the NMR time scale.17
23 with Ru–C5 and Ru–H5 distances of 2.38 and 1.85 Å.24 The agostic C–H bond is elongated (1.18 Å), and the C4–C5 bond is significantly longer than the other C–C bonds (1.49 vs. 1.42, 1.44 and 1.41 Å) within the pentadienyl ligand. Structure B is 3.7 kcal mol−1 higher in energy and represents the hydride complex [(η5-C5H5)RuH(η5-C7H11)] with a Ru–H bond of 1.57 Å. The pentadienyl ligand is symmetrically bound, with long C1–H and C5–H distances of ca. 2.20 Å. A and B are connected by a transition state (TS) that is 5.1 and 1.4 kcal mol−1 higher in energy than A and B, respectively. Naturally, B can be regarded as an intermediate in the intraligand 1,5-H transfer from C1 to C5, and the low calculated barrier for this process is in agreement with the observation that this process is not frozen out in an accessible temperature range on the NMR time scale.17
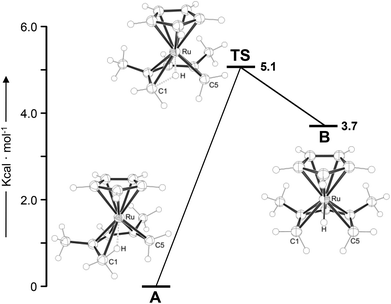 |
| | Fig. 1 Potential-energy profile for 1,5-intraligand H transfer in the protonated half-open ruthenocene 3. Values correspond to ΔG298 K = B3LYP/6-311G(d,p) Gibbs free energies at 298 K. For values derived with the B97-D and M06 functionals, see ESI.† TS = transition state. | |
Preparation of pentadienyl and cyclopentadienyl ruthenium bis(imidazolin-2-imine) complexes
The reaction of the protonated open and half-open ruthenocenes 2 and 3 with the ligands BLMe and BLiPr in acetonitrile afforded the half-sandwich pentadienyl and cyclopentadienyl complexes [(η5-C7H11)Ru(BLR)]BF4 (4a, R = Me; 4b, R = iPr) and [(η5-C5H5)Ru(BLR)]BF4 (5a, R = Me; 5b, R = iPr) as purple solids in good yield (ca. 80%, Scheme 2). These reactions are likely to proceed via the cationic tris(acetonitrile) complexes [(η5-C7H11)Ru(CH3CN)3]+ and [(η5-C5H5)Ru(CH3CN)3]+,19,25 which are conveniently accessible from 2 and 3.21 NMR spectra recorded in acetone-d6 gave no indication for the formation of stable solvate complexes with tightly bound acetonitrile or acetone ligands. The 1H NMR spectra of complexes 4 show the expected signals for the 2,4-dimethylpentadienyl ligand, with a singlet at ca. 4.9 ppm for the 3-CH and doublets at ca. 2.0 and −0.9 ppm (JH,H = 2.5 Hz) for the exo- and endo-1,5-CH hydrogen atoms. The 1H NMR spectra of complexes 5 display a singlet at ca. 3.52 ppm for the C5H5 moieties, which is at higher field than usually observed for cyclopentadienyl-ruthenium half-sandwich complexes and could be ascribed to shielding by the imidazole rings.24 Resonances assigned to the BLMe and BLiPr ligands in 4 and 5 are similar to those reported for the corresponding [(η5-C5Me5)Ru(BLR)]+ ions.6
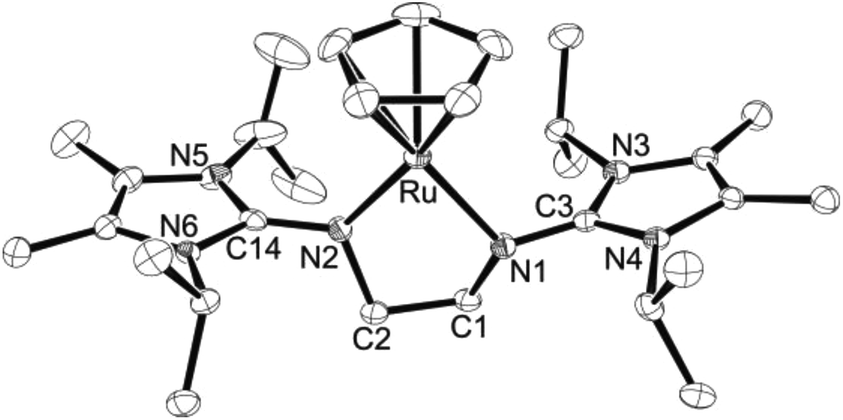
The crystal structures of 4a·THF, 4b and 5b were determined by X-ray diffraction analyses, confirming in all cases the formation of 16-electron half-sandwich complexes with two-legged piano-stool (pseudo-trigonal planar) geometries around the ruthenium atoms (Fig. 2–4). Interestingly, two different polymorphs of 4a·THF (A and B) were characterised, which both crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . While the metal–carbon and metal–nitrogen bond lengths are similar (Table 1), the structures differ in the orientation of the pentadienyl ligand, which renders the molecules either almost Cs- (A, Fig. 2, top) or C1-symmetric (B, Fig. 2, bottom). These conformations are similar to those found in bis(pentadienyl) complexes, which may exhibit symmetric syn- and anti-eclipsed or asymmetric gauche-eclipsed conformations.26 The latter orientation is also found for the two independent molecules in 4b (Fig. 3, only one cation shown). It should be noted that 4a and 4b represent, to the best of our knowledge, the first structurally characterised half-sandwich pentadienyl ruthenium complexes with a (formal) electron count of 16, and previously described pentadienyl species are usually 18-electron complexes with common three-legged piano-stool (pseudo-tetrahedral) geometries.16,19,27,285a and 5b are also very rare examples of 16-electron half-sandwich ruthenium complexes bearing the (η5-C5H5)Ru unit, with [(η5-C5H5)Ru(tmeda)][BArF4] and [(η5-C5H5)Ru(dppe)][GaI4] representing the only previously characterised systems (tmeda = Me2NCH2CH2NMe2, dppe = Ph2PCH2CH2PPh2).29,30 In contrast, a significantly larger number of 16-electron complexes are known that contain the sterically more demanding (η5-C5Me5)Ru fragment.31–33
. While the metal–carbon and metal–nitrogen bond lengths are similar (Table 1), the structures differ in the orientation of the pentadienyl ligand, which renders the molecules either almost Cs- (A, Fig. 2, top) or C1-symmetric (B, Fig. 2, bottom). These conformations are similar to those found in bis(pentadienyl) complexes, which may exhibit symmetric syn- and anti-eclipsed or asymmetric gauche-eclipsed conformations.26 The latter orientation is also found for the two independent molecules in 4b (Fig. 3, only one cation shown). It should be noted that 4a and 4b represent, to the best of our knowledge, the first structurally characterised half-sandwich pentadienyl ruthenium complexes with a (formal) electron count of 16, and previously described pentadienyl species are usually 18-electron complexes with common three-legged piano-stool (pseudo-tetrahedral) geometries.16,19,27,285a and 5b are also very rare examples of 16-electron half-sandwich ruthenium complexes bearing the (η5-C5H5)Ru unit, with [(η5-C5H5)Ru(tmeda)][BArF4] and [(η5-C5H5)Ru(dppe)][GaI4] representing the only previously characterised systems (tmeda = Me2NCH2CH2NMe2, dppe = Ph2PCH2CH2PPh2).29,30 In contrast, a significantly larger number of 16-electron complexes are known that contain the sterically more demanding (η5-C5Me5)Ru fragment.31–33
 |
| | Fig. 2 ORTEP diagram of the cation in 4a·THF in polymorphs A (top) and B (bottom) with thermal displacement parameters drawn at 50% probability (see Table 1 for bond lengths and angles). | |
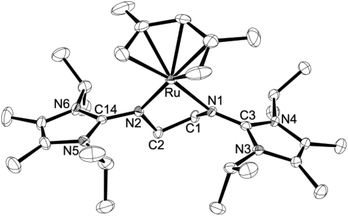 |
| | Fig. 3 ORTEP diagram of the cation in 4b (only one of the two independent cations is shown) with thermal displacement parameters drawn at 50% probability (see Table 1 for bond lengths and angles). | |
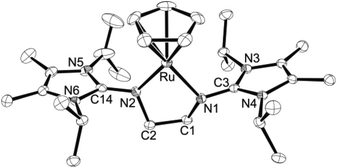 |
| | Fig. 4 ORTEP diagram of the cation in 5b with thermal displacement parameters drawn at 50% probability (see Table 1 for bond lengths and angles). | |
Table 1 Selected bond lengths (Å) and angles (°) for pentadienyl and cyclopentadienyl ruthenium bis(imidazolin-2-imine) complexes
| |
4a·THF (polymorph A) |
4a·THF (polymorph B) |
4ba |
5b
|
|
The asymmetric unit contains two independent molecules with very similar structural parameters; the values of molecule 1 are shown.
Angle between the RuN2 and imidazole ring planes.
ρ = 2a/(b + c), with a, b, and c representing the exo- (a) and endocyclic (b, c) bonds within the CN3 guanidine moiety.
|
| Ru–N1 |
2.0529(17) |
2.1262(12) |
2.123(2) |
2.0601(15) |
| Ru–N2 |
2.0627(17) |
2.0155(12) |
2.009(2) |
2.0423(15) |
| Ru–C |
2.128(2)–2.159(2) |
2.0928(14)–2.1850(14) |
2.104(3)–2.153(3) |
2.1135(19)–2.1569(19) |
| C3–N1 |
1.344(3) |
1.3266(18) |
1.336(3) |
1.348(2) |
| C3–N3 |
1.358(3) |
1.3620(18) |
1.360(3) |
1.362(2) |
| C3–N4 |
1.351(3) |
1.3554(18) |
1.360(3) |
1.355(2) |
| C10/C14–N2 |
1.359(3) |
1.3530(18) |
1.362(3) |
1.353(2) |
| C10/C14–N5 |
1.345(3) |
1.3472(18) |
1.348(3) |
1.351(2) |
| C10/C14–N6 |
1.351(3) |
1.3528(18) |
1.355(3) |
1.362(2) |
| N1–Ru–N2 |
78.52(7) |
77.79(5) |
78.85(8) |
77.31(6) |
| (N–Ru–N)/(C3N2)b |
69.5; 86.7 |
62.8; 69.9 |
79.1; 78.4 |
83.9; 82.6 |
|
ρ
|
0.992; 1.008 |
0.976; 1.002 |
0.982; 1.008 |
0.992; 0.997 |
Similarly to the previously reported complexes [(η5-C5Me5)Ru(BLR)]+ and [(η6-arene)Ru(BLR)]2+,6,9 the stability of 4 and 5 can be ascribed to the strong π-electron-releasing ability of the BLR ligands as illustrated by the ylidic mesomeric structure in Scheme 1, which consistently affords short Ru–N bond lengths in the range 2.01–2.13 Å (Table 1). Charge separation and delocalisation can also be clearly deduced from the observation of large dihedral angles between the imidazole and N–Ru–N planes, which rules out the possibility of any substantial π-interaction between the imidazole rings and the nitrogen atoms. As a consequence, the three C–N bond lengths within the imidazolin-2-imine (or guanidine) CN3 unit are almost equal, and this structural feature can be illustrated by the parameter ρ = 2a/(b + c), with a, b, and c representing the exo- (a) and endocyclic (b, c) bonds.34 In agreement with efficient charge delocalisation, ρ values between 0.98 and 1.01 are found (Table 1).
Catalytic transfer hydrogenation of ketones
Catalytic transfer hydrogenation has become a standard method for the reduction of ketones to secondary alcohols,35 with half-sandwich ruthenium complexes representing the most widely studied catalyst systems.36 Coordinatively unsaturated species are considered as important reaction intermediates, and therefore, the complexes 4 and 5 can be expected to serve as hydrogen transfer catalysts. The hydrogenation of acetophenone was chosen as a test reaction, and the activity of the complexes 4a, 4b, 5a and 5b was studied in boiling 2-propanol (b.p. = 82 °C) with 1 mol% catalyst loading and potassium hydroxide (10 mol%) as the base. The reaction progress was monitored by gas chromatography (GC), and the results are summarized in Table 2. The cyclopentadienyl complexes 5a and 5b display activities similar to those previously reported for the related complexes [(η5-C5Me5)Ru(BLR)]X and [(η6-arene)Ru(BLR)]X2 (X = Cl or weakly-coordinating counterion),9 whereas the pentadienyl complexes 4a and 4b show higher activities. The most active complex 4a was further employed as a catalyst under the same conditions for the reduction of a range of aliphatic and aromatic ketones to the corresponding secondary alcohols. In all cases, full conversion was achieved in less than one hour (Table 3).
Table 2 Catalytic transfer hydrogenation of acetophenonea
| Substrate |
Catalyst |
Time [h] |
Conversion [%] |
TOF [h−1] |
|
Conditions: 0.02 mmol cat., 0.2 mmol KOH, 10 mL iPrOH, 2 mmol acetophenone, temperature: 82 °C, the formation of 2-phenylethanol is monitored by gas chromatography.
|
| Acetophenone |
4a
|
0.5 |
99 |
198 |
| Acetophenone |
4b
|
0.75 |
98 |
131 |
| Acetophenone |
5a
|
3 |
86 |
29 |
| Acetophenone |
5b
|
4 |
100 |
25 |
Table 3 Catalytic transfer hydrogenation of various ketones with 4aa
| Substrate |
Catalyst |
Time [min] |
Conversion [%] |
TOF [h−1] |
|
Conditions: 0.02 mmol cat., 0.2 mmol KOH, 10 mL iPrOH, 2 mmol ketone, temperature: 82 °C, the formation of the respective secondary alcohol is monitored by gas chromatography.
|
| 3-Pentanone |
4a
|
40 |
100 |
150 |
| 2-Methylbutanone |
4a
|
60 |
99 |
99 |
| Cyclohexanone |
4a
|
10 |
100 |
600 |
| Cycloheptanone |
4a
|
40 |
97 |
146 |
| 4-Heptanone |
4a
|
60 |
96 |
96 |
|
p-Chloroacetophenone |
4a
|
30 |
100 |
200 |
| Benzophenone |
4a
|
60 |
98 |
98 |
| 1-Acetonaphthone |
4a
|
20 |
96 |
288 |
Conclusions
This paper provides further evidence for the ability of the bis(imidazolin-2-imine) ligands BLMe and BLiPr to stabilise coordinatively unsaturated transition metal complexes, furnishing the complexes [(η5-C7H11)Ru(BLR)]BF4 (4a, R = Me; 4b, R = iPr) and [(η5-C5H5)Ru(BLR)]BF4 (5a, R = Me; 5b, R = iPr) as rare examples of 16-electron ruthenium complexes containing pentadienyl- or cyclopentadienyl (C5H5) ligands. These complexes were prepared from bis(η5-2,4-dimethylpentadienyl)ruthenium(II), [(η5-C7H11)2Ru] (1, “open ruthenocene”) via the protonated open or half-open ruthenocenes 2 and 3, and this route provides a general and convenient pathway to the important class of pentadienyl and cyclopentadienyl ruthenium half-sandwich complexes.
Experimental
General
All operations were performed in an atmosphere of dry argon using Schlenk and vacuum techniques. All solvents were purified by standard methods and distilled prior to use. 1H and 13C NMR spectra were recorded on a Bruker DPX 400 device. The chemical shifts are given in ppm relative to tetramethylsilane (TMS) as internal standard. The spin coupling patterns are indicated as s (singlet), d (doublet), m (multiplet), sept (septet) and br (broad, for unresolved signals). Elemental analyses (C, H, N) were measured on an Elementar Vario EL III CHNS elemental analyser. 1,132,14,16 BLMe,6 and BLiPr,6 were prepared according to published procedures.
[(η5-C5H5)Ru{(1-4η)-C7H12-η2-C5,H5}]BF4 (3, “protonated half-open ruthenocene”).
A solution of the protonated open ruthenocene 2 (190 mg, 0.50 mmol) in acetone (7.5 mL) was cooled to −80 °C. A solution of freshly distilled cyclopentadiene (42 μL, 0.50 mmol) in acetone (7.5 mL) was added dropwise at −75 to −80 °C over a period of 20 min. The solution was allowed to warm to room temperature, concentrated to ca. 3 mL, and the product was precipitated with diethyl ether (15 mL). The supernatant solution was removed via syringe, and the precipitate was washed with diethyl ether (3 × 5 mL). The precipitate was dried in vacuo to yield the product as a pale yellow solid (138 mg, 0.40 mmol, 80%). The spectroscopic data are identical to those previously published.17,18 A variable-temperature NMR study is shown in the ESI.†
[(η5-C7H11)Ru(BLMe)]BF4 (4a).
A solution of BLMe (82.2 mg, 0.27 mmol) in acetonitrile (5 mL) was added dropwise to an orange suspension of 2 (100.0 mg, 0.26 mmol) in acetonitrile (10 mL) at room temperature. During the addition of the ligand, the reaction mixture turned purple and the solution was stirred overnight. The solvent was removed under reduced pressure. The residue was dissolved in 5 mL THF and the product precipitated with n-hexane (30 mL). Filtration, washing with n-hexane (2 × 10 mL) and drying in vacuo afforded the product as a purple solid (123.2 mg, 81%). Anal. Calcd for C23H39BF4N6Ru: C, 47.02; H, 6.69; N, 14.31. Found: C, 46.92; H, 6.80; N, 13.70. 1H NMR (400 MHz, CD2Cl2, 25 °C): δ = 4.92 (s, 1 H, CH), 3.57 (s, 12 H, NCH3), 2.81 (s, 4 H, C2H4), 2.19 (s, 12 H, BLMe-CCH3), 2.00 (d, JH,H = 2.5 Hz, 2 H, CH2-exo), 1.49 (s, 6 H, C7H11–CH3), −0.86 (d, JH,H = 2.5 Hz, 2 H, CH2-endo) ppm. 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C): δ = 155.9 (NCN), 119.6 (NCCH3), 87.0 (C7H11–CCH3), 83.7 (CH), 56.3 (C2H4), 38.8 (CH2), 31.2 (NCH3), 24.8 (C7H11–CH3), 9.0 (BLMe-CCH3) ppm.
[(η5-C7H11)Ru(BLiPr)]BF4 (4b).
A solution of BLiPr (112.5 mg, 0.27 mmol) in acetonitrile (5 mL) was added dropwise to an orange suspension of 2 (100.0 mg, 0.26 mmol) in acetonitrile (10 mL) at room temperature. During the addition of the ligand the reaction mixture turned purple and the solution was stirred overnight. The solvent was removed under reduced pressure. The residue was dissolved in 5 mL THF and the product precipitated with n-hexane (30 mL). Filtration, washing with n-hexane (2 × 10 mL) and drying in vacuo afforded the product as a purple solid (144.6 mg, 81%). Anal. Calc. for C31H55BF4N6Ru: C, 53.22; H, 7.92; N, 12.01. Found: C, 53.26; H, 8.12; N, 12.28. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ = 5.35 (sept, 3JH,H = 7.1 Hz, 4 H, NCH,), 5.01 (s, 1 H, CH), 2.78 (s, 4 H, C2H4), 2.29 (s, 12 H, BLiPr-CCH3), 2.01 (d, JH,H = 2.5 Hz, 2 H, CH2-exo), 1.60 (d, 12 H, CHCH3, 3JH,H = 7.1 Hz), 1.60 (s, 6 H, C7H11–CH3), 1.43 (d, 12 H, CHCH3, 3JH,H = 7.1 Hz), −0.95 (d, JH,H = 2.5 Hz, 2 H, CH2-endo) ppm. 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ = 155.7 (NCN), 120.6 (NCCH3), 87.2 (C7H11–CCH3), 85.6 (CH), 58.9 (C2H4), 48.9 (NCH), 36.6 (CH2), 24.7 (C7H11–CCH3), 22.7 (CHCH3), 21.8 (CHCH3), 10.4 (BLiPr-CCH3) ppm.
[(η5-C5H5)Ru(BLMe)]BF4 (5a).
A solution of BLMe (54.8 mg, 0.18 mmol) in acetonitrile (5 mL) was added dropwise to an orange suspension of 3 (59.6 mg, 0.17 mmol) in acetonitrile (10 mL) at room temperature. During the addition of the ligand, the reaction mixture turned purple and the solution was stirred overnight. The solvent was removed under reduced pressure. The residue was dissolved in 5 mL THF and the product precipitated with n-hexane (30 mL). Filtration, washing with n-hexane (2 × 10 mL) and drying in vacuo afforded the product as a purple solid (63.0 mg, 65%). Anal. Calcd for C25H35BF4N6Ru: C, 45.25; H, 5.97; N, 15.08. Found: C, 45.15; H, 6.13; N, 14.44. 1H NMR (400 MHz, acetone-d6, 25 °C): δ = 3.71 (s, 12 H, NCH3), 3.53 (s, 5 H, C5H5), 2.74 (s, 4 H, C2H4), 2.28 (s, 12 H, CCH3) ppm. 13C NMR (100 MHz, acetone-d6, 25 °C): δ = 158.0 (NCN), 120.0 (NCCH3), 58.3 (C5H5), 55.6 (C2H4), 31.6 (NCH3), 8.7 (CCH3) ppm.
[(η5-C5H5)Ru(BLiPr)]BF4 (5b).
A solution of BLiPr (100.0 mg, 0.24 mmol) in acetonitrile (5 mL) was added dropwise to an orange suspension of 3 (80.3 mg, 0.23 mmol) in acetonitrile (10 mL) at room temperature. During the addition of the ligand the reaction mixture turned purple, and the solution was stirred overnight. The solvent was removed under reduced pressure. The residue was dissolved in 5 mL THF and the product precipitated with n-hexane (30 mL). Filtration, washing with n-hexane (2 × 10 mL) and drying in vacuo afforded the product as a purple solid (128.2 mg, 83%). Anal. Calcd for C29H49BF4N6Ru: C, 52.02; H, 7.38; N, 12.55. Found: C, 51.98; H, 7.59; N, 12.40. 1H NMR (400 MHz, acetone-d6, 25 °C): δ = 5.40 (sept, 3JH,H = 7.1 Hz, 4 H, NCH), 3.52 (s, 5 H, C5H5), 2.72 (s, 4 H, C2H4), 2.41 (s, 12 H, CCH3), 1.68 (d, 12 H, CHCH3, 3JH,H = 7.1 Hz), 1.56 (d, 12 H, CHCH3, 3JH,H = 7.1 Hz), ppm. 13C NMR (100 MHz, acetone-d6, 25 °C): δ = 156.9 (NCN), 121.1 (NCCH3), 58.3 (C5H5), 57.8 (C2H4), 49.6 (NCH), 22.1 (CHCH3), 21.7 (CHCH3), 10.2 (CCH3) ppm.
General procedure for transfer hydrogenation
Potassium hydroxide (11.2 mg, 0.2 mmol) was added to the catalyst solution (0.02 mmol, 1 mol%) in 2-propanol (10 mL) at room temperature. The solution was heated to 81 °C, and the ketone (2 mmol, 0.2 M) was added. Approximately 0.1 mL samples were regularly taken and filtered through the short silica gel column by using diethyl ether as a solvent. The reaction progress was monitored by gas chromatography (GC).
Single-crystal X-ray structure determinations
Numerical data are summarized in Table 4. Crystals were mounted in inert oil on glass fibres. Intensity data were recorded at low temperature on an Oxford Diffraction Xcalibur E diffractometer using monochromated Mo Kα radiation. Absorption corrections were performed on the basis of multi-scans. Structures were refined anisotropically on F2 using the program SHELXL-97.37 Methyls were refined as idealized rigid groups allowed to rotate but not tip; other hydrogens were included using a riding model starting from calculated positions. Exceptions and special features: For both polymorphs of 4a, the hydrogens at C17, C19 and C21 were refined freely (but with DFIX distance restraints). The THF of 4a (polymorph A) and the methylene bridge (C41–C42) of 4b are disordered over two positions. The refinements were stabilized using similarity restraints, but the dimensions of disordered groups should be interpreted with caution. For 5b, the tetrafluoroborate anions lie with their B atoms on crystallographic twofold axes.
Table 4 Crystallographic dataa
| |
4a·THF (polym. A) |
4a·THF (polym. B) |
4b
|
5b
|
|
CCDC 1054781–1054784 contain the supplementary crystallographic data for this paper.
|
| Empirical formula |
C27H47BF4N6ORu |
C27H47BF4N6ORu |
C31H55BF4N6Ru |
C29H49BF4N6Ru |
| Formula weight |
659.59 |
659.59 |
699.69 |
669.62 |
| Space group |
P |
P |
P21/c |
P2/c |
|
a [Å] |
8.0534(2) |
7.4989(5) |
15.7948(2) |
18.4902(4) |
|
b [Å] |
13.6581(4) |
11.8261(4) |
14.0426(4) |
11.6717(2) |
|
c [Å] |
14.1481(4) |
17.6250(8) |
31.4970(9) |
15.5756(2) |
|
α [°] |
100.097(2) |
100.627(3) |
90 |
90 |
|
β [°] |
97.841(2) |
95.959(4) |
99.176(2) |
110.188(2) |
|
γ [°] |
96.840(2) |
97.082(4) |
90 |
90 |
|
V [Å3] |
1501.38(7) |
1511.62(13) |
6896.6(3) |
3154.89(10) |
|
Z
|
2 |
2 |
8 |
4 |
|
T [K] |
100(2) |
100(2) |
100(2) |
100(2) |
|
λ [Å] |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
ρ
calc[g cm–3] |
1.459 |
1.449 |
1.348 |
1.410 |
|
μ [mm–1] |
0.58 |
0.58 |
0.51 |
0.55 |
| Reflections collected |
42715 |
60750 |
128239 |
94761 |
| Independent reflections |
7446 [Rint = 0.052] |
9616 [Rint = 0.041] |
17068 [Rint = 0.036] |
6935 [Rint = 0.053] |
| Goodness of fit on F2 |
0.89 |
0.94 |
1.11 |
0.89 |
|
R(Fo) [I > 2σ(I)] |
0.0313 |
0.0258 |
0.0418 |
0.0260 |
| wR (Fo2) (all refl.) |
0.0621 |
0.0554 |
0.0971 |
0.0546 |
| Δρ [e Å–3] |
0.63/−0.57 |
0.74/−0.63 |
1.10/−0.99 |
0.46/−0.52 |
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through grant Ta 189/6-3. We thank Dr C. G. Daniliuc and Dr C. G. Hrib for recording the X-ray data.
Notes and references
- X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116 CrossRef CAS PubMed.
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793 CrossRef CAS PubMed.
-
(a) O. Bienemann, A. Hoffmann and S. Herres-Pawlis, Rev. Inorg. Chem., 2011, 31, 83 CrossRef CAS;
(b) I. dos Santos Vieira and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2012, 765 CrossRef CAS PubMed;
(c) H.-J. Himmel, Z. Anorg. Allg. Chem., 2013, 639, 1940 CrossRef CAS PubMed;
(d)
J. Sundermeyer, V. Raab, E. Gaoutchenova, U. Garrelts, N. Abacilar and K. Harms, in Activating Unreactive Substrates, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 17–37 Search PubMed.
-
(a) J. Börner, U. Flörke, T. Glöge, T. Bannenberg, M. Tamm, M. D. Jones, A. Döring, D. Kuckling and S. Herres-Pawlis, J. Mol. Catal. A: Chem., 2010, 316, 139 CrossRef PubMed;
(b) T. Glöge, D. Petrovic, C. G. Hrib, C. Daniliuc, E. Herdtweck, P. G. Jones and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2303 CrossRef PubMed;
(c) D. Petrovic, L. M. R. Hill, P. G. Jones, W. B. Tolman and M. Tamm, Dalton Trans., 2008, 887 RSC;
(d) D. Petrovic, T. Bannenberg, S. Randoll, P. G. Jones and M. Tamm, Dalton Trans., 2007, 2812 RSC;
(e) S. Randoll, P. G. Jones and M. Tamm, Organometallics, 2008, 27, 3232 CrossRef CAS;
(f) T. K. Panda, C. G. Hrib, P. G. Jones, J. Jenter, P. W. Roesky and M. Tamm, Eur. J. Inorg. Chem., 2008, 4270 CrossRef CAS PubMed;
(g) J. Bogojeski, R. Jelić, D. Petrović, E. Herdtweck, P. G. Jones, M. Tamm and Ž. D. Bugarčić, Dalton Trans., 2011, 40, 6515 RSC;
(h) J. Volbeda, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2014, 422, 158 CrossRef CAS PubMed;
(i) S.-A. Filimon, C. G. Hrib, S. Randoll, I. Neda, P. G. Jones and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 691 CrossRef CAS PubMed.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523 CAS.
- D. Petrovic, T. Glöge, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2007, 3472 CrossRef CAS PubMed.
- T. Glöge, D. Petrovic, C. G. Hrib, P. G. Jones and M. Tamm, Z. Naturforsch., B: Chem. Sci., 2008, 63, 1155 Search PubMed.
- D. Petrovic, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Organometallics, 2008, 27, 778 CrossRef CAS.
- T. Glöge, D. Petrovic, C. Hrib, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2009, 4538 CrossRef PubMed.
-
(a) V. N. Sapunov, R. Schmid, K. Kirchner and H. Nagashima, Coord. Chem. Rev., 2003, 238–239, 363 CrossRef CAS;
(b) H. Nagashima, H. Kondo, T. Hayashida, Y. Yamaguchi, M. Gondo, S. Masuda, K. Miyazaki, K. Matsubara and K. Kirchner, Coord. Chem. Rev., 2003, 245, 177 CrossRef CAS;
(c) M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Eur. J. Inorg. Chem., 2004, 17 CrossRef PubMed;
(d) U. Koelle, Chem. Rev., 1998, 98, 1313 CrossRef CAS PubMed;
(e) T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599 CrossRef CAS PubMed;
(f) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630 CrossRef CAS PubMed;
(g) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067 CrossRef CAS PubMed;
(h) H. Werner, Organometallics, 2005, 24, 1036 CrossRef CAS.
- R. D. Ernst and L. Stahl, Organometallics, 1983, 2, 1229 CrossRef.
- A. Bauer, U. Englert, S. Geyser, F. Podewils and A. Salzer, Organometallics, 2000, 19, 5471 CrossRef CAS.
-
A. Salzer, A. Bauer, S. Geyser and F. Podewils, in Inorganic Syntheses, ed. J. R. Shapley, John Wiley & Sons, Inc, 2004, pp. 59–65 Search PubMed.
-
A. Salzer, A. Bauer and F. Podewils, in Synthetic Methods of Organometallic and Inorganic Chemistry, ed. W. A. Herrmann, Thieme, Stuttgart, 2000, vol. 9, pp. 45–50 Search PubMed.
- D. N. Cox and R. Roulet, J. Chem. Soc., Chem. Commun., 1988, 951 RSC.
- T. D. Newbound, L. Stahl, M. L. Ziegler and R. D. Ernst, Organometallics, 1990, 9, 2962 CrossRef CAS.
- D. N. Cox and R. Roulet, J. Chem. Soc., Chem. Commun., 1989, 175 RSC.
- R. Gleiter, I. Hyla-Kryspin, M. L. Ziegler, G. Sergeson, J. C. Green, L. Stahl and R. D. Ernst, Organometallics, 1989, 8, 298 CrossRef CAS.
- T. Lumini, D. N. Cox, R. Roulet and K. Schenk, J. Organomet. Chem., 1992, 434, 363 CrossRef CAS.
-
(a) D. N. Cox, T. Lumini and R. Roulet, J. Organomet. Chem., 1992, 438, 195 CrossRef CAS;
(b) A. Doppiu and A. Salzer, Eur. J. Inorg. Chem., 2004, 2244 CrossRef CAS PubMed;
(c) T. Lumini, D. N. Cox, R. Roulet, G. Chapuis and F. Nicolo, Helv. Chim. Acta, 1990, 73, 1931 CrossRef CAS PubMed.
-
N. Langenscheidt-Dabringhausen, Open Ruthenocene: A versatile Entry into Ruthenium(II) Chemistry for Synthesis and Catalysis, Dissertation, RWTH Aachen, 2009 Search PubMed.
- H. W. Bosch, H.-U. Hund, D. Nietlispach and A. Salzer, Organometallics, 1992, 11, 2087 CrossRef CAS.
- A. Salzer, Pure Appl. Chem., 1999, 71, 1557 CrossRef CAS.
- M. E. van der Boom, M. A. Iron, O. Atasoylu, L. J. Shimon, H. Rozenberg, Y. Ben-David, L. Konstantinovski, J. M. Martin and D. Milstein, Inorg. Chim. Acta, 2004, 357, 1854 CrossRef CAS PubMed.
-
(a) T. P. Gill and K. R. Mann, Organometallics, 1982, 1, 485 CrossRef CAS;
(b) B. M. Trost and C. M. Older, Organometallics, 2002, 21, 2544 CrossRef CAS;
(c) E. P. Kündig and F. R. Monnier, Adv. Synth.
Catal., 2004, 346, 901 Search PubMed.
-
(a) R. D. Ernst, Chem. Rev., 1988, 88, 1255 CrossRef CAS;
(b) R. D. Ernst, Acc. Chem. Res., 1985, 18, 56 CrossRef CAS;
(c) L. Stahl and R. D. Ernst, Adv. Organomet. Chem., 2007, 55, 137 Search PubMed.
- J. R. Bleeke and D. J. Rauscher, Organometallics, 1988, 7, 2328 CrossRef CAS.
- A. Reyna-Madrigal, A. Moreno-Gurrola, O. Perez-Camacho, M. E. Navarro-Clemente, P. Juárez-Saavedra, M. A. Leyva-Ramirez, A. M. Arif, R. D. Ernst and M. A. Paz-Sandoval, Organometallics, 2012, 31, 7125 CrossRef CAS.
- C. Gemel, J. C. Huffman, K. G. Caulton, K. Mauthner and K. Kirchner, J. Organomet. Chem., 2000, 593–594, 342 CrossRef CAS.
- N. D. Coombs, A. Stasch and S. Aldridge, Inorg. Chim. Acta, 2008, 361, 449 CrossRef CAS PubMed.
-
(a) T. D. Johnson, K. Folting, W. E. Streib, J. D. Martin, J. C. Huffmann, S. A. Jackson, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1995, 34, 488 CrossRef CAS;
(b) Y. Yamaguchi and H. Nagashima, Organometallics, 2000, 19, 725 CrossRef CAS;
(c) H. Kondo, Y. Yamaguchi and H. Nagashima, Chem. Commun., 2000, 1075 RSC;
(d) H. Kondo, Y. Yamaguchi and H. Nagashima, J. Am. Chem. Soc., 2001, 123, 500 CrossRef CAS;
(e) J.-i. Terasawa, H. Kondo, T. Matsumoto, K. Kirchner, Y. Motoyama and H. Nagashima, Organometallics, 2005, 24, 2713 CrossRef CAS.
-
(a) C. Gemel, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 1997, 16, 5601 CrossRef CAS;
(b) C. Gemel, V. N. Sapunov, K. Mereiter, M. Ferencic, R. Schmid and K. Kirchner, Inorg. Chim. Acta, 1999, 286, 114 CrossRef CAS;
(c) C. Gemel, A. LaPenée, K. Mauthner, K. Mereiter, R. Schmid and K. Kirchner, Monatsh. Chem., 1997, 128, 1189 CrossRef CAS.
-
(a) M. J. Tenorio, K. Mereiter, M. C. Puerta and P. Valerga, J. Am. Chem. Soc., 2000, 122, 11230 CrossRef;
(b) H. Aneetha, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, V. N. Sapunov, R. Schmid, K. Kirchner and K. Mereiter, Organometallics, 2002, 21, 5334 CrossRef CAS.
-
(a) A. Neuba, R. Haase, M. Bernard, U. Flörke and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2008, 634, 2511 CrossRef CAS PubMed;
(b) J. Börner, U. Flörke, K. Huber, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362 CrossRef PubMed.
-
(a) S. Manzini, J. A. Fernández-Salas and S. P. Nolan, Acc. Chem. Res., 2014, 47, 3089 CrossRef CAS PubMed;
(b) P. E. Sues, K. Z. Demmans and R. H. Morris, Dalton Trans., 2014, 43, 7650 RSC;
(c) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729 CrossRef CAS PubMed;
(d) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC;
(e) W. Baratta and P. Rigo, Eur. J. Inorg. Chem., 2008, 4041 CrossRef CAS PubMed;
(f) C. Wang, X. Wu and J. Xiao, Chem. – Asian J., 2008, 3, 1750 CrossRef CAS PubMed;
(g) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef CAS PubMed;
(h) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226 RSC;
(i) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC;
(j) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS PubMed;
(k) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS;
(l) R. Noyori and T. Ohkuma, Pure Appl. Chem., 1999, 71, 1493 CrossRef CAS;
(m) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
-
(a) S.-H. Lee and G. I. Nikonov, ChemCatChem, 2015, 7, 107 CrossRef CAS PubMed;
(b) O. Dayan, S. Demirmen and N. Özdemir, Polyhedron, 2015, 85, 926 CrossRef CAS PubMed;
(c) F. Ok, M. Aydemir, F. Durap and A. Baysal, Appl. Organomet. Chem., 2014, 28, 38 CrossRef CAS PubMed;
(d) N. Şahin, ARKIVOC, 2014, 2015, 20 CrossRef;
(e) R. Aznar, A. Grabulosa, A. Mannu, G. Muller, D. Sainz, V. Moreno, M. Font-Bardia, T. Calvet and J. Lorenzo, Organometallics, 2013, 32, 2344 CrossRef CAS;
(f) S. Dayan, N. Ozpozan Kalaycioglu, J.-C. Daran, A. Labande and R. Poli, Eur. J. Inorg. Chem., 2013, 3224 CrossRef CAS PubMed;
(g) J. DePasquale, M. Kumar, M. Zeller and E. T. Papish, Organometallics, 2013, 32, 966 CrossRef CAS;
(h) D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2013, 723, 26 CrossRef CAS PubMed;
(i) O. Prakash, K. N. Sharma, H. Joshi, P. L. Gupta and A. K. Singh, Dalton Trans., 2013, 42, 8736 RSC;
(j) F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2013, 32, 3595 CrossRef CAS;
(k) C. Aliende, M. Pérez-Manrique, F. A. Jalón, B. R. Manzano, A. M. Rodríguez and G. Espino, Organometallics, 2012, 31, 6106 CrossRef CAS;
(l) C. G. Arena, A. Cuzzola and D. Drommi, Polyhedron, 2012, 48, 221 CrossRef CAS PubMed;
(m) O. Dayan, N. Özdemir, Z. Şerbetci, M. Dinçer, B. Çetinkaya and O. Büyükgüngör, Inorg. Chim. Acta, 2012, 392, 246 CrossRef CAS PubMed;
(n) B. T. Kırkar, H. Türkmen, İ. Kani and B. Çetinkaya, Tetrahedron, 2012, 68, 8655 CrossRef PubMed;
(o) V. Parekh, J. A. Ramsden and M. Wills, Catal. Sci. Technol., 2012, 2, 406 RSC;
(p) H. Türkmen, İ. Kani and B. Çetinkaya, Eur. J. Inorg. Chem., 2012, 4494 CrossRef PubMed;
(q) F. E. Fernández, M. C. Puerta and P. Valerga, Organometallics, 2011, 30, 5793 CrossRef;
(r) S. Horn, C. Gandolfi and M. Albrecht, Eur. J. Inorg. Chem., 2011, 2863 CrossRef CAS PubMed;
(s) M. Kanthak, A. Aniol, M. Nestola, K. Merz, I. M. Oppel and G. Dyker, Organometallics, 2011, 30, 215 CrossRef CAS;
(t) W. Lackner-Warton, S. Tanaka, C. M. Standfest-Hauser, Ö. Öztopcu, J.-C. Hsieh, K. Mereiter and K. Kirchner, Polyhedron, 2010, 29, 3097 CrossRef CAS PubMed;
(u) P. Singh and A. K. Singh, Organometallics, 2010, 29, 6433 CrossRef CAS;
(v) M. Yadav, A. K. Singh, R. Pandey and D. S. Pandey, J. Organomet. Chem., 2010, 695, 841 CrossRef CAS PubMed;
(w) M. C. Carrión, F. Sepúlveda, F. A. Jalón, B. R. Manzano and A. M. Rodríguez, Organometallics, 2009, 28, 3822 CrossRef;
(x) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2009, 131, 3593 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected NMR spectra and computational details. CCDC 1054781–1054784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C5DT01080B |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence