
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring excited states of Pt(II) diimine catecholates for photoinduced charge separation†
Paul A.
Scattergood
a,
Patricia
Jesus
b,
Harry
Adams
a,
Milan
Delor
a,
Igor V.
Sazanovich
ac,
Hugh D.
Burrows
b,
Carlos
Serpa
*b and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: Julia.Weinstein@sheffield.ac.uk
bCentro de Química de Coimbra and Chemistry Department, University of Coimbra, P3004-535, Coimbra, Portugal. E-mail: serpasoa@ci.uc.pt
cLaser for Science Facility, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
First published on 23rd January 2015
Abstract
The intense absorption in the red part of the visible range, and the presence of a lowest charge-transfer excited state, render Platinum(II) diimine catecholates potentially promising candidates for light-driven applications. Here, we test their potential as sensitisers in dye-sensitised solar cells and apply, for the first time, the sensitive method of photoacoustic calorimetry (PAC) to determine the efficiency of electron injection in the semiconductor from a photoexcited Pt(II) complex. Pt(II) catecholates containing 2,2′-bipyridine-4,4′-di-carboxylic acid (dcbpy) have been prepared from their parent iso-propyl ester derivatives, complexes of 2,2′-bipyridine-4,4′-di-C(O)OiPr, (COOiPr)2bpy, and their photophysical and electrochemical properties studied. Modifying diimine Pt(II) catecholates with carboxylic acid functionality has allowed for the anchoring of these complexes to thin film TiO2, where steric bulk of the complexes (3,5-ditBu-catechol vs. catechol) has been found to significantly influence the extent of monolayer surface coverage. Dye-sensitised solar cells using Pt(dcbpy)(tBu2Cat), 1a, and Pt(dcbpy)(pCat), 2a, as sensitisers, have been assembled, and photovoltaic measurements performed. The observed low, 0.02–0.07%, device efficiency of such DSSCs is attributed at least in part to the short excited state lifetime of the sensitisers, inherent to this class of complexes. The lifetime of the charge-transfer ML/LLCT excited state in Pt((COOiPr)2bpy)(3,5-di-tBu-catechol) was determined as 250 ps by picosecond time-resolved infrared spectroscopy, TRIR. The measured increase in device efficiency for 2a over 1a is consistent with a similar increase in the quantum yield of charge separation (where the complex acts as a donor and the semiconductor as an acceptor) determined by PAC, and is also proportional to the increased surface loading achieved with 2a. It is concluded that the relative efficiency of devices sensitised with these particular Pt(II) species is governed by the degree of surface coverage. Overall, this work demonstrates the use of Pt(diimine)(catecholate) complexes as potential photosensitizers in solar cells, and the first application of photoacoustic calorimetry to Pt(II) complexes in general.
Introduction
Catechol ligands, members of the dioxolene family, have been extensively utilised within the coordination chemistry of various metal ions such as Ru(II),1–8 Os(II)4,9,10 and Re(I).11–13 The most attractive feature of catechol ligands is their rich electrochemistry, with facile and reversible oxidation to semiquinone and quinone forms, together with the strong interplay between catechol and metal-based orbitals frequently resulting in transition metal complexes which display ‘non-innocent’ behaviour.14,15 Whilst transition metal catecholates have been widely studied, Pt(II) catecholate chemistry has been rather less explored.16–25 Previously reported Pt(II) catecholates commonly feature α,α′-diimines such as substituted bipyridines or phenanthrolines, in conjunction with simple dioxolenes, for example pyrocatechol, 3,5-di-tert-butyl catechol or 4-tert-butyl catechol. These Pt(II)-centred complexes are characterised by the broad, moderately intense, low-energy electronic absorption, being predominantly of a ligand-to-ligand charge transfer nature (LL'CT).16,17 Previous investigations have shown the aforementioned electronic transition to involve a HOMO which is primarily catechol-based and a diimine localised LUMO, with ligand substitution having a profound effect upon the electronic and photophysical properties of the complex as a whole.16,21 Catechol ligands have also been utilised to move the lowest energy electronic absorption band of metal complexes into the strategically important near infra-red (NIR) region, with the first oxidation potential being typically between −0.2 and +0.2 V vs. Fc/Fc+.The tuneable nature of both the photophysical and electrochemical properties of Pt(II) catecholates makes them potential candidates for use within inorganic dye-sensitised solar cells (DSSC's). These photoelectrochemical devices are centred around a light absorbing ‘dye’ molecule anchored to a thin film semi-conductor surface, commonly TiO2.26,27 The operational principle of these photovoltaic cells involves the injection of electrons from the photo-excited dye into the conduction band of TiO2, thereby producing electrical current, with the oxidised dye being regenerated through electron transfer from a redox electrolyte, commonly I2/I3−. The ability to control and fine tune the electronic properties of the dye is of great importance in ensuring efficient electron injection; in the close matching of electrochemical potentials with those of the redox electrolyte and in tailoring the optical absorbance to cover as much of the solar emission spectrum as possible – particularly the NIR region. Whilst substituted polypyridyl-containing Ru(II) complexes have dominated DSSC research to date, a limited range of Pt(II)-centred systems have been explored. For example, previous reports28–30 introduce a range of diimine Pt(II) dithiolates, with these complexes being anchored onto TiO2 surfaces through the bipyridine ligands appended with carboxylic acid functionality. These studies take advantage of the easily tuneable Mixed Metal–Ligand-to-Ligand charge transfer (MMLL'CT') transition, with the complexes being subsequently incorporated in DSSCs to give overall device efficiencies (η) typically between 0.1–1.1%. Another report31 describes a range of terpyridine-containing alkynyl-based Pt(II) sensitisers. These systems were found to exhibit intense optical absorption in the visible region and relatively long-lived charge transfer excited states, with overall device efficiencies reaching η ≈ 3.6%.
To the best of our knowledge, the potential of Pt(II) catecholates to function as sensitisers within DSSCs remains unexplored. Here, we introduce two new catecholate-based sensitising dyes and investigate their electrochemical and photophysical properties. The uptake of these species onto TiO2 is evaluated, together with an assessment of their suitability as sensitisers through incorporation within DSSCs and the analysis of current–voltage data.
Electron injection from the dye in its excited electronic state into the conduction band of TiO2 with the formation of a charge-separated state between the oxidised dye and the electron within the conduction band of the semiconductor is one of the most important processes in DSSCs.32 The parameter of particular importance is the quantum yield of electron injection, yet it is the quantity which is the most difficult to determine directly. This value is often estimated from the initial amplitude of fast transient absorption signals;29,33 or determined in absolute terms through the kinetics of the dye emission component quenched by electron injection process,34,35 assuming that electron injection competes only with the decay of the excited state. We employ here an alternative approach, which allows one to directly determine the yields of injection – namely, photoacoustic calorimetry (PAC).36,37 This calorimetric technique monitors the photo-induced pressure waves that result from heat deposition processes. Fast piezoelectric transducers are used to evaluate the magnitude of the detected pressure waves which depend upon the non-radiative processes occurring after light absorption, and on the thermoelastic properties of the materials under study. From the relation between the energy absorbed and the heat released the quantum yields of non-radiative processes can be obtained. In the present application, PAC is used for the detection of pressure waves generated upon conversion of excitation energy into heat as the photoexcited dye injects an electron into the conduction band of the semiconductor. Whilst PAC is commonly employed to study the thermodynamics of photoinduced processes in solution,38 we utilise a front-face set-up to follow electron injection at the TiO2–dye–solvent interface and to ultimately determine the quantum yield of electron injection. Here, the yield of electron injection is estimated by measuring the yield of formation of the long-lived charge-separation between the oxidised sensitizer, and the semiconductor (ϕcs).
Previously, we have demonstrated a successful use of PAC for determining the energy of the charge separated state (ECS) formed between the oxidised dye and the electron within the conduction band.36,37 We now report new transition metal complexes which intensely absorb light in the visible region of the spectrum, investigate their photophysical properties, and evaluate their potential as dyes for DSSCs using the advanced PAC technique.
Experimental
cis-Pt(DMSO)2Cl2 was synthesised following a previously reported procedure.39Synthesis of (COOiPr)2bpy
2,2′-Bipyridine-4,4′-dicarboxylic acid, hereafter dcbpy, (350 mg, 1.433 mmol) was suspended in SOCl2 (12 ml) and heated to 85 °C for 48 h. The solvent was removed to leave pale yellow solids which were completely purged of excess SOCl2 under a stream of dry nitrogen for a further hour. The thus obtained acyl chloride functionalised intermediate was then re-dissolved in deaerated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) toluene–iPrOH (25 ml) and heated to 80 °C for 21 h. Removal of the solvent gave pale yellow coloured solids which were subsequently re-dissolved in CHCl3 (75 ml) and washed with 2 × 75 ml saturated aqueous NaHCO3 solution. The aqueous phases were extracted with a further 1 × 30 ml portion of CHCl3. The combined organic layers were washed with 2 × 50 ml H2O, dried over MgSO4, filtered and the solvent removed to yield the product as an off-white coloured solid. Yield = 379 mg, 81%. 1H NMR (CDCl3, 400 MHz): 1.42 (d, J = 6.28 Hz, 12H), 5.32 (sept, J = 6.28 Hz, 2H), 7.90 (dd, J = 1.32, 4.82 Hz, 2H), 8.86 (d, J = 4.88 Hz, 2H), 8.92 (d, J = 1.12 Hz, 2H). EI-MS: m/z = 328 (M+, 34%), 269 (M+ − OiPr, 42%), 242 (M+ − COOiPr, 100%).
:1 (v/v) toluene–iPrOH (25 ml) and heated to 80 °C for 21 h. Removal of the solvent gave pale yellow coloured solids which were subsequently re-dissolved in CHCl3 (75 ml) and washed with 2 × 75 ml saturated aqueous NaHCO3 solution. The aqueous phases were extracted with a further 1 × 30 ml portion of CHCl3. The combined organic layers were washed with 2 × 50 ml H2O, dried over MgSO4, filtered and the solvent removed to yield the product as an off-white coloured solid. Yield = 379 mg, 81%. 1H NMR (CDCl3, 400 MHz): 1.42 (d, J = 6.28 Hz, 12H), 5.32 (sept, J = 6.28 Hz, 2H), 7.90 (dd, J = 1.32, 4.82 Hz, 2H), 8.86 (d, J = 4.88 Hz, 2H), 8.92 (d, J = 1.12 Hz, 2H). EI-MS: m/z = 328 (M+, 34%), 269 (M+ − OiPr, 42%), 242 (M+ − COOiPr, 100%).
Synthesis of {(COOiPr)2bpy}PtCl2
2,2′-Bipyridine-4,4′-diisopropylacetate (COOiPr)2bpy (334 mg, 1.017 mmol) and Pt(DMSO)2Cl2 (423 mg, 1.001 mmol) were suspended in deaerated EtOH (40 ml) and heated to 65 °C in the dark for 20 h. The mixture was allowed to cool to room temperature, with the resultant bright orange coloured precipitate being collected by filtration. The solids were washed with EtOH followed by Et2O and dried in vacuo. Yield = 519 mg, 87%. 1H NMR (CDCl3, 400 MHz): 1.48 (d, J = 6.28 Hz, 12H), 5.37 (sept, J = 6.28 Hz, 2H), 8.05 (dd, J = 1.72, 6.05 Hz, 2H), 8.57 (d, J = 1.52 Hz, 2H), 9.73 (d, J = 6.00 Hz, 2H). EI-MS: m/z = 594 (M+, 55%), 508 (M+ − COOiPr, 16%), 328 (M+ − PtCl2, 14%), 242 (M+ − PtCl2COOiPr, 100%). Anal. Calc. for C18H20N2O4Cl2Pt (%): C 36.37, H 3.39, N 4.71, Cl 11.93. Found (%): C 36.19, H 3.31, N 4.57, Cl 11.63.Synthesis of {(COOiPr)2bpy}Pt(tBu2Cat) (1)
3,5-Di-tert-butyl catechol (93 mg, 0.418 mmol) and tBuOK (87 mg, 0.775 mmol) were dissolved in dry, deaerated THF (20 ml) and stirred rapidly at room temperature for 45 min. The deep blue coloured solution was then transferred via cannula to {(COOiPr)2bpy}PtCl2 (222 mg, 0.373 mmol) in dry, deaerated THF (20 ml). The resulting reaction mixture was heated to 75 °C in the dark for 48 h. The solvent was removed and the residue purified by column chromatography (SiO2, slow gradient, 0.1% MeOH–DCM to 0.4% MeOH–DCM). After complete removal of all yellow coloured material the product was collected from a deep blue fraction. Removal of the solvent afforded the title complex as a dark blue coloured solid. Yield = 237 mg, 85%. 1H NMR (CDCl3, 400 MHz): 1.32 (s, 9H), 1.46 (d, J = 6.52 Hz, 6H), 1.48 (d, J = 6.56 Hz, 6H), 1.52 (s, 9H), 5.30–5.44 (m, 2H), 6.47 (d, J = 2.08 Hz, 1H), 6.74 (d, J = 2.12 Hz, 1H), 7.96 (dd, J = 1.64, 6.00 Hz, 1H), 8.04 (dd, J = 1.64, 6.00 Hz, 1H), 8.39 (d, J = 1.16 Hz, 1H), 8.47 (d, J = 1.28 Hz, 1H), 9.61 (d, J = 5.96 Hz, 1H), 9.72 (d, J = 5.96 Hz, 1H). MALDI-MS: m/z = 743.2 (M+).Synthesis of {(COOiPr)2bpy}Pt(pCat) (2)
Pyrocatechol (59 mg, 0.535 mmol) and tBuOK (80 mg, 0.712 mmol) were added to dry, deaerated THF (40 ml) and stirred rapidly at room temperature for 45 min. The mixture was then transferred via cannula to ((COOiPr)2bpy)PtCl2 (250 mg, 0.420 mmol) and heated to 75 °C in the dark for 60 h. (A deep blue-green colour evolved over the first 30 min.) The solvent was removed and the residue purified by column chromatography (SiO2, slow gradient, 0.2% MeOH–DCM to 1.1% MeOH–DCM). After complete removal of all yellow and orange coloured material the product was obtained from a deep blue fraction. Removal of the solvent afforded the title complex as a dark blue solid. Yield = 141 mg, 53%. 1H NMR (CDCl3, 400 MHz): 1.49 (d, J = 6.24 Hz, 12H), 5.32 (sept, J = 6.28 Hz, 2H), 5.79–5.85 (m, 2H), 5.87–5.94 (m, 2H), 7.49 (dd, J = 1.00, 5.64 Hz, 2H), 7.68 (d, J = 1.04 Hz, 2H), 8.70 (d, J = 5.24 Hz, 2H). MALDI-MS: m/z = 631.0 (M+).Synthesis of (dcbpy)Pt(tBu2Cat) (1a)
KOH (98 mg, 1.746 mmol) was dissolved in 1:1 (v/v) THF–H2O (16 ml). The solution was deaerated by bubbling argon for 15 min and then transferred via cannula to (1) (45 mg, 0.060 mmol). The resulting deep blue coloured solution was stirred at room temperature in the dark for 3 days. The reaction mixture was acidified (pH 4) through the dropwise addition of 0.1 M HCl, yielding a fine precipitate which was subsequently collected by centrifugation. Purification was achieved by thorough washing with H2O, DCM and Et2O to leave fine dark blue solids which were dried in vacuo. The product was found to be fully soluble in DMF and DMSO only. Yield = 22 mg, 55%. 1H NMR (d6-DMSO, 400 MHz): 1.22 (s, 9H), 1.46 (s, 9H), 6.28 (d, J = 1.92 Hz, 1H), 6.50 (d, J = 2.08 Hz, 1H), 8.12 (dd, J = 1.72, 5.96 Hz, 1H), 8.21 (dd, J = 1.68, 5.88 Hz, 1H), 8.97 – 9.00 (m, 2H), 9.38 – 9.43 (m, 2H), 14.26 (s broad, 2H). MALDI-MS: m/z = 659.4 (M+).
Synthesis of (dcbpy)Pt(pCat) (2a)
KOH (129 mg, 2.299 mmol) was dissolved in 1:1 (v/v) THF–H2O (16 ml) and deaerated through argon bubbling for 15 min. The basic solution was then transferred via cannula to (2) (45 mg, 0.071 mmol) and stirred at room temperature in the dark for 3 days. The deep red-purple coloured solution was acidified (pH 4) through the dropwise addition of 0.1 M HCl, yielding a fine precipitate which was collected by centrifugation. Purification was effected by thorough washing with H2O, DCM and Et2O. The resultant dark blue coloured solids were dried in vacuo. The product was found to be fully soluble only in DMF and DMSO. Yield = 34 mg, 87%. 1H NMR (d6-DMSO, 400 MHz): 6.17–6.24 (m, 2H), 6.43–6.50 (m, 2H), 8.10 (dd, J = 1.60, 5.92 Hz, 2H), 8.87 (d, J = 1.44 Hz, 2H), 9.29 (d, J = 5.92 Hz, 2H), 14.24 (s, broad, 2H). MALDI-MS: m/z = 547.1 (M+).
General information
All synthetic manipulations were carried out under an atmosphere of argon using standard Schlenk line techniques. Dry solvents were obtained from a Grubbs solvent purification column. Deaeration of solvents was performed through the vigorous bubbling of argon for a period of at least 15 minutes.The PAC experiments measure the pressure wave generated upon conversion of excitation energy into heat, as the electronically excited dye evolves to a long lived charge separated state with the electron located in the conduction band of the semiconductor, and the formation of the dye cation. PAC experiments separate several processes which can contribute to heat deposition into the medium by their characteristic lifetimes. A 2.25 MHz transducer employed in the experiments described here allows one to investigate processes associated with the deposition of heat operating in three “time regimes”:
1) For the processes occurring in the time frame shorter than 10 ns, an integrated amount of heat deposited is recorded;
2) For the processes occurring in the time range between ∼10 ns and ∼500 ns the kinetics of the heat deposition can be resolved;
3) For the processes occurring on a timescale longer than ∼500 ns, no detection of heat deposition is possible.
TiO2 films were immersed in the solutions of the dyes in DMF (50 μM), and UV-Vis absorption spectra taken at selected time intervals, from 20 s to 210 min. The amount of adsorbed dye was monitored as a function of time by measuring the absorbance of the dye-modified TiO2 films at the wavelength of maximum absorption using a Shimadzu UV-2100 UV-Vis spectrophotometer. Excess dye was removed by rinsing with DMF followed by ether or ethanol before the absorption spectrum was registered. For the de-adsorption studies, the films were in contact with a few drops of NaOH (0.1 M) and then immersed in DMF (3 mL). The total amount of the dye adsorbed on the TiO2 films was determined through the subsequent measurement of a UV-Vis absorption spectra and use of the solution-phase extinction coefficient.
After cooling, the TiO2 electrode was further treated with a 40 mM TiCl4 aqueous solution at 70 °C for 30 min and then calcined at 500 °C for 30 min. When the temperature was reduced to 80 °C, the TiO2 electrode was immersed into a freshly prepared dye solution for a period of 12 h in the dark. The electrode was rinsed and dried. A thin Pt layer (Pt-Catalyst T/SP Solaronix) was deposited onto transparent conducting glass by the ‘doctor-blade’ technique and used as the counter electrode. The two electrodes were sealed together using polymer film (25 μm, Solaronix) in a sandwich-type cell. Electrolyte was introduced through holes drilled in the counter electrode, subsequently sealed with microscope slides and polymer film to avoid leakage of the electrolyte solution.
The current–voltage (J–V) curve were measured by a Metrohm Autolab PGSTAT 10 electrochemical analyser using a 150 W Solar Simulator (100 mW cm−2, AM 1.5 G) (Newport Oriel) for illumination. The electrolyte used for performance measurements consists of 0.6 M 1-butyl-3-methylimidazolium iodide, 0.03 M iodine, 0.10 M guanidinium thiocyanate, 0.50 M tert-butyl pyridine in a solution of acetonitrile–valeronitrile (85:15 (v/v)). The DSSCs were masked to expose only the active area (1 cm2) under irradiation for all measurements.
Results and discussion
Synthesis
The ester-functionalised diimine Pt(II) catecholates 1 and 2 were prepared via a ‘two-pot’ method, whereby the appropriate catechol was first deprotonated with tBuOK in anhydrous THF prior to complexation with {(COOiPr)2bpy}PtCl2. It was noted that carrying out the complexation reaction in alcoholic solvents resulted in loss of the diisopropylacetate functionality through base mediated trans-esterification. For example, performing the reaction in MeOH produced {(COOMe)2bpy}Pt(catechol), with trans-esterification being rapid and quantitative. The resultant Pt(II) catecholate products were readily purified by column chromatography, giving very dark blue coloured solids found to be soluble in the majority of common laboratory solvents. Conversion of 1 and 2 to the corresponding carboxylic acid-containing species (1a and 2a) was effected through KOH mediated hydrolysis in a THF–H2O solvent system.40 The final ‘dye’ molecules 1a and 2a were found to be sparingly soluble in all solvents apart from DMF and DMSO (Fig. 1). | ||
| Fig. 1 Structures of the Pt(II) diimine complexes investigated in this study. 1, {(COOiPr)2bpy}Pt(tBu2Cat) and 2{(COOiPr)2bpy}Pt(pCat) are the ester-functionalised parent complexes, the corresponding acid-containing 1a and 2a are investigated herein as sensitisers for DSSCs. | ||
X-ray crystallography
Single crystals were grown for the dichloride precursor {(COOiPr)2bpy}PtCl2 and Pt(II) catecholate 2 (Fig. 2 and 3). The crystal structure of {(COOiPr)2bpy}PtCl2 reveals the Pt(II) centre to be in a distorted square planar coordination environment, with the bond angle between the two chelating nitrogen atoms and the metal centre N(1)–Pt–N(2) being 80.76°. The 4,4′-substituted-2,2′-bipyridine ligand is close to being planar, with a dihedral angle between the two pyridyl rings of 3.45°. This value is consistent with that measured previously for similar diimine Pt(II) complexes.17 The crystal structure of 2 (Fig. 3) also shows the expected distorted square planar geometry as a result of the two bidentate ligands, with N(1)–Pt–N(2) and O(1)–Pt–O(2) being 80.97° and 84.22° respectively. The torsion angle between the two aforementioned planes is determined to be 2.17°, revealing the complex to be essentially planar. Both 2 and {(COOiPr)2bpy}PtCl2 stack in dimer-like ‘head-to-tail’ pairs, facilitated by short Pt⋯Pt contacts,41 with the inter-metal distances measured to be 3.56 Å and 3.52 Å respectively. In the former instance, these metal–metal interactions appear to be supported by additional π–π interactions between the bipyridyl and pyrocatechol ligands within each pair. | ||
| Fig. 2 X-ray crystal structure of {(COOiPr)2bpy}PtCl2. Brilliant orange coloured crystals suitable for X-ray diffraction 0.43 × 0.12 × 0.12 mm3 were grown from the slow vapour diffusion of Et2O into a CH2Cl2 solution of the dichloride complex. Selected bond lengths (Å) and angles (°): Pt(1)–N(1) 2.005(4), Pt(1)–N(2) 2.010(4), Pt(1)–Cl(5) 2.2947(13), Pt(1)–Cl(6) 2.2968(12), N(1)–Pt(1)–N(2) 80.76(16), Cl(5)–Pt(1)–Cl(6) 89.36(5), N(1)–Pt(1)–Cl(6) 94.77(12), N(2)–Pt(1)–Cl(5) 95.08(12). CCDC 1028955. | ||
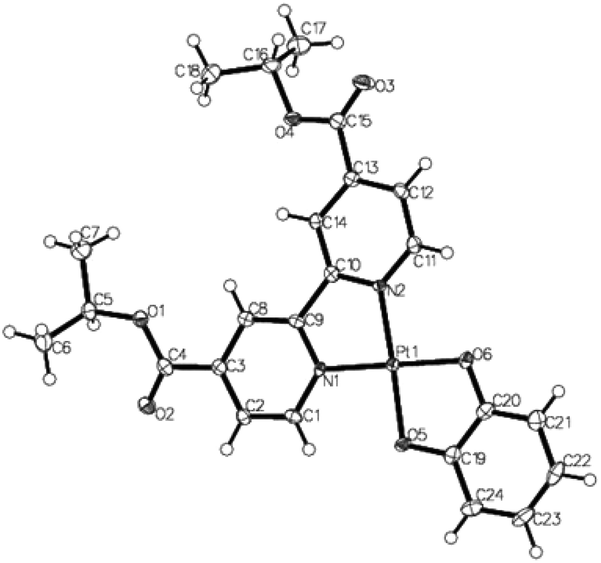 | ||
| Fig. 3 X-ray crystal structure of 2. Deep blue coloured crystals were grown from the slow evaporation of a mixed toluene–CH2Cl2 solution at room temperature. Selected bond lengths (Å) and angles (°): Pt(1)–N(1) 1.983(3), Pt(1)–N(2) 1.994(3), Pt(1)–O(5) 1.986(2), Pt(1)–O(6) 2.005(2), N(1)–Pt(1)–N(2) 95.68(11), O(5)–Pt(1)–O(6) 84.22(10), N(1)–Pt(1)–O(5) 95.68(11), N(2)–Pt(1)–O(6) 99.16(11). CCDC 1028956. | ||
Electronic absorption spectroscopy
UV-Vis-NIR electronic absorption spectra for DMF solutions of complexes 1, 1a, 2, 2a and the dichloride precursor {(COOiPr)2bpy}PtCl2 are shown in Fig. 4. All complexes show an intense absorption band in the region of 300 nm, assigned to π–π* intra-ligand transitions localised upon the bipyridine moiety. The spectrum recorded for {(COOiPr)2bpy}PtCl2 reveals an additional, less intense band centered at 415 nm. This transition is attributed to a metal-to-ligand charge transfer (MLCT), a feature observed in (diimine)PtCl2 type complexes reported previously, where the transition has been more stringently described as a ‘charge transfer-to-diimine’ owing to the chloride ligands making an appreciable contribution to the HOMO.42 The absorption spectra of the catecholates are dominated by a broad, low-energy band which extends into the NIR region. This band is characteristic of Pt(II) catecholates, with its’ absence in the spectrum of {(COOiPr)2bpy}PtCl2 allowing primary assignment to a ligand-to-ligand charge transfer (LL'CT).16,17,20,43 However, since the electronic transition between catechol and diimine ligands is known to also involve orbital contributions from the metal centre, the process may be more accurately labelled as a mixed metal–ligand-to-ligand charge transfer (MMLL'CT).21 It is noted that the change in catechol ligand between 1 and 2 is manifest in the electronic absorption spectra. The inclusion of tertiary butyl substituents in 1 is seen to shift the maximum of the low energy CT band by approximately 60 nm below that observed for 2. This is attributed to the weakly electron-donating alkyl groups destabilising the catechol based HOMO, thus reducing the HOMO–LUMO energy gap and the energy of the corresponding transition. It is also observed that hydrolysis of the ester groups results in a shift of the MMLL'CT band to a higher energy in both instances, being approximately 1300 cm−1 and 1200 cm−1 above that observed for the parent esters of 1a and 2a respectively. | ||
| Fig. 4 Ground state electronic absorption spectra for DMF solutions of Pt(II) catecholates 1, 2, 1a, 2a and the dichloride precursor {(COOiPr)2bpy}PtCl2, recorded at room temperature. | ||
Solvatochromism of the absorption spectra
The CT nature of the lowest energy absorption band is further verified through solvatochromism. Previously, CT transitions in Pt(II) catecholates have been shown to display negative solvatochromism; namely, the shift of the MMLL'CT band maximum to lower energy with decreasing solvent polarity.16,20 This behaviour is clearly exemplified for 1 in Fig. 5, where electronic absorption spectra recorded in a range of solvents are displayed. The red-shift of absorption maxima from 673 nm in MeCN to 768 nm in CHCl3 is indicative of an inversion, or decrease in magnitude, of the dipole moment upon formation of the catechol-to-diimine CT state.44,45 The inset within Fig. 5 illustrates the linear correlation between absorption energy and solvent polarity parameter derived for Pt(II) dimine complexes,46 with a solvatochromic shift (slope) of 4665 cm−1 (0.578 eV), consistent with a CT-type transition. | ||
| Fig. 5 Ground state electronic absorption spectra of 1 recorded in a variety of solvents, exemplifying the negative solvatochromic behaviour of the lowest energy absorption band. The solvent polarity parameters46 are shown within brackets. The inset shows the linear correlation between the energy of the CT state and solvent polarity, with a slope (solvatochromic shift) of 4665 cm−1 (0.578 eV). | ||
Electrochemistry
Cyclic voltammograms were recorded for CH2Cl2 solutions of 1, 2 and the dichloride precursor {(COOiPr)2bpy}PtCl2 across the potential range +1.2 to −2.2 V vs. Fc/Fc+ (Fig. 6). | ||
| Fig. 6 Cyclic voltammograms for 1.8 mM CH2Cl2 solutions of {(COOiPr)2bpy}PtCl2 (top), 1 (middle) and 2 (bottom). Scans shown were recorded at 100 mV s−1 at room temperature with 0.2 M NBu4PF6 as supporting electrolyte. All potentials are quoted against Fc/Fc+. | ||
| Reduction/V | Oxidation/V | τ, ps | |
|---|---|---|---|
| a Electrochemically quasi-reversible process. b Chemically irreversible under the experimental conditions. c From ref. 16. | |||
| {(COOiPr)2bpy}PtCl2 | −1.26 (99) | ||
| −1.87 (91) | |||
| 1 | −1.37 (92) | −0.03 (95) | |
| −1.98 (131)a | +0.93b | 250 | |
| 2 | −1.39 (95) | +0.11 (124)a | |
| −2.00 (113)a | +1.05b | ||
| Pt(bpyam)(tBu2cat) | −1.59c | 420 | |
| Pt(tBubpy)(tBu2cat) | −1.81c | 630 | |
The occurrence of these electrochemical features in all complexes allow the processes to be assigned to the sequential reduction of the bipyridine fragment. The influence of the weakly electron-withdrawing ester functionality may be quantified through comparison with similar Pt(II) catecholates where an unsubstituted 2,2′-bipyridine analogue of 1 gives a first reduction potential at −1.74 V and a 4,4′-di-tert-butyl-2,2′-bipyridine version a more cathodic reduction at −1.82 V.47 It is concluded that for the complexes studied here, both the LUMO and LUMO+1 are localised mainly upon the bipyridine moiety.
Picosecond time-resolved infrared spectroscopy
The lifetime of the excited state of 1 in fluid solution was obtained by TRIR spectroscopy, in the detection range from 1400 to 1800 cm−1. Excitation of a solution of 1 in CH2Cl2 with 400 nm, ∼50 fs laser pulse lead to instantaneous bleaching of the ground state IR absorption bands, and the formation of transient signals across the fingerprint region (Fig. 7). | ||
| Fig. 7 Picosecond time-resolved infrared spectroscopic data of 1 mM solution of 1 in CH2Cl2 at r.t. following 400 nm, ∼50 fs excitation. Top: spectra at selected delay times after the excitation pulse; bottom: kinetic traces at selected frequencies (symbols) and the global fit to the data (solid lines). | ||
The early time dynamics (before 5 ps) are difficult to resolve due to very broad signals around 1600 cm−1, which for similar complexes were previously assigned to vibrationally hot water molecules associated with the catechol moiety.24 After ca. 5 ps, the spectral dynamics can be satisfactorily described with the 250 (±50) ps lifetime, using global fit analysis. The corresponding kinetic traces and fitting curves are shown in Fig. 7. The observed spectral features are very similar to those observed by us previously for other Pt(diimine)(catecholate) compounds16,24 and are consistent with the ML/LLCT assignment of the excited state detected. The 250 ps value of the lifetime of the charge-transfer state in 1 forms a trend with the 420 ps and 630 ps lifetimes for the excited states of Pt(4,4′-(C(O)NEt2)2bpy)(tBu2Cat), and Pt(4,4′-tBu2bpy)(tBu2Cat), resp.16 In this series of compounds, which share the same donor ligand, tBu2Cat, the lifetime systematically increases with a decrease in electron-accepting ability of the diimine ligand, in accordance with the energy gap law.
Dye adsorption on TiO2
Thin films (1.2 μm) of nanocrystalline TiO2 deposited on glass slides were immersed in DMF solutions of dyes 1a and 2a. The resulting intense blue-purple colouration imparted on the TiO2 films affirmed successful dye anchorage, with the corresponding UV-Visible absorption spectra of the surface adsorbed dyes being shown in Fig. 8. The spectra of the surface anchored dyes clearly show a broad band in the region of 575–650 nm and 500–575 nm for 1a and 2a respectively. These absorbance features correspond to the MMLL'CT bands observed in the solution phase spectra, although being notably broadened and blue-shifted in both instances. These spectra highlight one of the most favourable attributes of Pt(II) catecholates – their ability to harvest photons from across a large low-energy part of the visible region.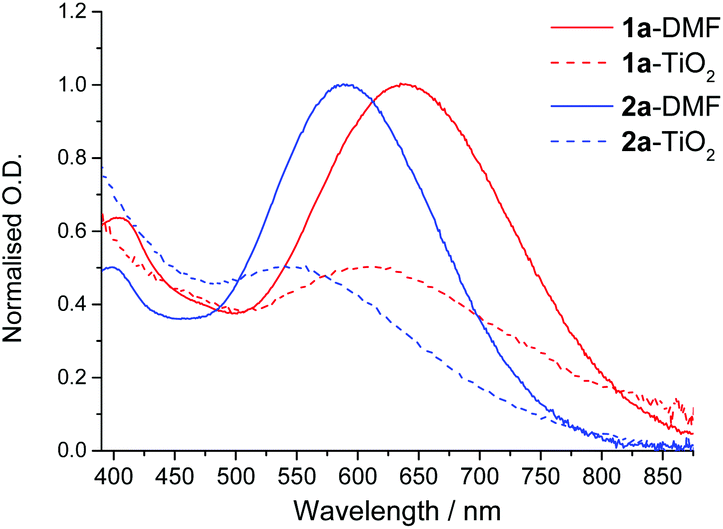 | ||
| Fig. 8 UV-Visible electronic absorption spectra of 1a and 2a in DMF solutions (solid line), and when anchored onto thin film of TiO2 (dashed line). Optical densities have been normalised at the absorbance maximum of the lowest energy band: solution OD = 1.0, adsorbed dye OD = 0.5. | ||
Dye loadings were investigated by monitoring the progress of dye adsorption: UV-Vis absorption spectra of the sensitised TiO2 films were collected as a function of immersion time, with the surface being rinsed with DMF and Et2O prior to each measurement to remove the non-adsorbed dye. Typical dye solution concentrations of 50 μM were employed so as to minimise dye agglomeration and reduce surface aggregation. The dye uptake plots for 1a and 2a are shown in Fig. 9. Both curves reveal the very rapid initial uptake of the dyes, with approximately 50% of the maximum observed surface coverage being achieved within the first 25 min immersion time. After this period the rate of dye uptake decreases in both instances. Monolayer surface coverage is determined to be reached when the rate of change of surface adsorbed dye concentration reaches zero, being observed here after approximately 175 min immersion time for both dyes. Desorption of the dyes with NaOH and use of the solution phase extinction coefficient allows monolayer surface loading to be defined as 0.5 × 10−8 and 1.2 × 10−8 mol cm−2 for 1a and 2a respectively. Allowing for a consideration of the thickness (1.2 μm) of the films used within this study, the magnitude of this loading is comparable with that which has been reported for Pt(II) thiolate dyes,28,29 although with longer immersion times being required due to the lower dye solution concentrations utilised here.48
 | ||
| Fig. 9 Dye uptake curves for 1a and 2a onto TiO2 film of thickness 1.2 μm from DMF solutions (50 μM) at room temperature. The curves are plotted as a function of immersion time. Maximum dye loading at monolayer surface coverage is determined to be approximately 0.5 × 10−8 and 1.2 × 10−8 mol cm−2 for 1a and 2a respectively. | ||
With the mode and position of surface attachment being identical for both dyes, the higher loading achieved with 2a is attributed directly to the planar nature of this Pt(II) catecholate. Favourable π–π stacking interactions between the pyrocatechol entities, together with short Pt⋯Pt contacts, are likely to facilitate the close packing of the dye when adsorbed on a semi-conductor surface. The inclusion of tertiary butyl substituents in 1a affords the dye with a significantly greater steric bulk, thus hindering close surface packing and reducing the maximum possible monolayer surface loading by a factor of approx. 2.5.
Photoacoustic calorimetry (PAC)
MnTPPS adsorbed onto a TiO2 film was employed as a photoacoustic reference, and is defined as a dye which converts all absorbed energy into heat on a time-scale much shorter than that of the time-resolution of acoustic detection.36 After subtraction of an almost negligible background signal, sample and reference PAC signals were compared, with no mis-match in phase, indicating that the release of heat from the sample also occurs on a time-scale faster than the acoustic detection time-resolution. Table 2 shows the values of photoacoustic wave maxima obtained from PAC performed on 1a and 2a anchored to thin film TiO2, where ϕ denotes the fraction of heat released by the sample under study relative to the photoacoustic reference. The maxima of the photoacoustic waves were observed to be independent of the excitation laser pulse energy, indicating that no bi-photonic processes were occurring under the laser fluences used. From the experimentally obtained ϕ values, the quantum yield of formation of the long-lived charge separation (ϕCS) between the electron injected in the semiconductor and the oxidised dye can be calculated through eqn (1), where Ehν is the energy of excitation and Ecs is the energy of the charge separated state. Ecs itself may be determined from eqn (2). We assume a negligible change in the first (catechol-based) oxidation potential upon hydrolysis of the ester functionality, therefore the measured potentials for the parent ester compounds 1 and 2 (Table 1) are used in eqn (2) and the following analysis. | (1) |
| Ecs = E(D/D+) − Ecb | (2) |
| Dye 1a | Dye 2a | |||||||
|---|---|---|---|---|---|---|---|---|
| Relative laser intensity/% | Reference/mV | Sample/mV | ϕ | ϕ cs | Reference/mV | Sample/mV | ϕ | ϕ cs |
| 100 | 71 | 58 | 0.92 | 69 | 63 | 0.81 | ||
| 84 | 63 | 48 | 0.92 | 61 | 56 | 0.77 | ||
| 52 | 48 | 37 | 0.86 | 46 | 40 | 0.78 | ||
| 26 | 35 | 28 | 0.93 | 33 | 31 | 0.79 | ||
| Average ϕ: 0.91 | 0.26 | Average ϕ: 0.79 | 0.56 | |||||
The PAC results for (1) and (2) showed signals only for the fast (<10 ns) heat deposition, which were assigned to charge injection and also account for the fast recombination of the injected electron with the oxidized sensitizer that occurs on a time scale less than 10 ns. No signal on the longer timescale was detected, therefore the energy not released as heat is assigned to the formation of the long-lived, >500 ns, charge separated state between the sensitizer S+ and the semiconductor (TiO2˙−). We stress that this way of estimating the yield of electron injection, through the yield of long-lived charge-separation, does not reflect the instantaneous injection only, but also takes into account the recombination processes on the fast, <10 ns, timescale. Thus the value quoted here is the yield of electron transfer to the semiconductor from the photoexcited sensitizer, which remains after 10 ns time.
Here, we consider that dye sensitised TiO2 electrodes exhibit an exponential density of states extending into the TiO2 band gap. Since a low excitation light intensity leads to a low density of photoinjected electrons, the majority of these electrons will occupy states with energies close to the quasi-Fermi level.49 Under such condition, the calculation of Ecs through eqn (2) with a single value is acceptable. Thus, using a value of −0.34 V (vs. SCE) for the energy of the TiO2 conduction band (Ecb)50 we obtained values of 0.26 and 0.56 for the yields of long-lived dye-TiO2 charge separation for 1a and 2a, respectively. A control experiment on the dye N719 (cis-bis(isothiocyanato)-bis(2,2′-bipyridyl-4,4′-dicarboxylato)-ruthenium(II)) performed under identical experimental conditions37 yielded a 100% quantum yield of the formation of the charge separated state (assuming Ecs of 1.05 eV37,51 for this dye), which is consistent with the previous results.52
It has been reported that the energy of the conduction band (Ecb) for thin film anatase TiO2 depends upon the nature and composition of a contiguous electrolyte solution,53 namely upon the presence of cations in the non-aqueous electrolytes,54 in addition to the chemical nature of the sensitiser itself.55 In this work, PAC studies were performed in neat acetonitrile, and it is therefore possible that the effective position of the conduction band may differ from the value considered above. Values as negative as −2 V (vs. SCE) have been reported for TiO2 in contact with acetonitrile.53 As a result, the values for the yield of formation of charge separated states estimated using PAC technique should be considered an upper limit.
The observed low dye-to-semi-conductor electron injection yields in sensitised TiO2 films (in contact with MeCN) may be accounted for by a short excited state lifetime of the dyes. Results presented previously56 have illustrated that a complete sensitised solar cell (including electrolyte) exhibits an approx. 20-fold retardation in the rate of electron injection in comparison to a dye-sensitised TiO2 film. The addition of a redox electrolyte is, therefore, likely to reduce the electron injection efficiency further,57 and therefore the quantum yields of electron injection determined through PAC (Table 2) may be substantially reduced upon incorporation of the dye into a complete functioning DSSC.
Photocurrent–voltage characteristics of Pt-Dye/TiO2 electrodes
Photovoltaic device performance parameters were obtained for cells sensitised with dyes 1a and 2a (Table 3) with accompanying current–voltage curves generated under illumination with AM 1.5 G (100 mW cm−2) simulated sunlight displayed in Fig. 10. It is apparent that cells sensitised with these particular Pt(II) complexes operate with a poor overall efficiency (η), being somewhat lower than other devices sensitised with Pt(II) diimine-based dyes (η ≈ 0.1–3%)28,30 and significantly below that achievable with the archetypal N719 dye.58 Photovoltaic characteristics for cells sensitised with N719 under identical experimental conditions to those employed for 1a and 2a yield an overall device efficiency of 6% (Jsc = 14.6 mA cm−2; Voc = 0.67 V; FF = 0.61). These values are typical for cells employing high-efficiency dyes48 and are used for comparison with the Pt(II) dyes investigated here. The low efficiency of Pt(II) catecholates as sensitisers may be explained by the short excited state lifetime. Both ourselves16 and others59 have shown the lifetime of the charge separated state in Pt(II)(diimine)(catechol) complexes, where an electron resides on the bipyridine fragment, to be on the picosecond time scale; the value of 250 ps obtained for 1 using TRIR is consistent with the previous observations for related complexes. As electron injection occurs from this excited state, the relatively short lifetime and rapid intramolecular charge recombination are expected to out-compete charge injection. Additionally, the low first oxidation potential associated with these Pt(II) catecholates is likely to hinder the efficient regeneration of the oxidised dye through electron transfer from the redox electrolyte, with recombination of injected electrons with the oxidised dye occurring more rapidly.28 These features are akin to those reported for other Pt(II)-centred sensitisers, particularly those which absorb photons from across the low-energy portion of the solar spectrum.29 The performance of 1a and 2a with respect to N719 could also be decreased by a lower absorption of light: it is estimated that 5 μm thick TiO2 films dyed with N719 absorb in excess of 90% of incident light at the wavelength of maximum absorption, compared with around 60% for the films containing either 1a or 2a. | ||
| Fig. 10 Current–voltage curves for solar cells sensitised with dyes 1a and 2a. Measurements were taken under illumination with AM 1.5 G simulated solar radiation, mask area 1 cm2. | ||
| Dye | J sc/mA cm−2 | V oc/V | FF | η/% |
|---|---|---|---|---|
| 1a | 0.14 | 0.37 | 0.47 | 0.02 |
| 2a | 0.31 | 0.38 | 0.55 | 0.07 |
Devices containing 2a yield a higher photocurrent density and greater overall device efficiency than those employing 1a (Table 3). Since the chemical and photophysical properties of the two Pt(II) catecholates are very similar, and the optical densities used are also comparable, the difference in device performance may be attributed directly to the degree of surface loading.
The magnitude of the improvement in efficiency upon moving from 1a to 2a is consistent with the 2.5-fold increase in monolayer surface coverage achievable in the latter instance. This conclusion is consistent with the data obtained by PAC, where the yield of formation of charge separation was found to be approximately 2.5 times greater for 2a than 1a anchored on a semiconductor.
Conclusion
Pt(II) diimine catecholates which intensely absorb in the red part of the visible spectrum are potentially promising candidates for light-driven applications. Here, we test their potential as sensitisers in dye-sensitised solar cells, and correlate this property with the efficiency of charge separation, evaluated by photoacoustic calorimetry (PAC). Two 2,2′-bipyridine-4,4′-di-carboxylic acid-containing Pt(II) catecholates have been prepared from their parent iso-propyl ester derivatives, and their photophysical and electrochemical properties studied. Solution phase electronic absorption spectra show intense charge transfer bands which extend into the NIR region, the energy of which may be readily tuned through substituent variation upon the diimine and catechol ligands. Modifying diimine Pt(II) catecholates with carboxylic acid functionality has allowed for the successful anchoring of these complexes to thin film TiO2, where steric bulk of the donor ligands (tBu-catechol vs. catechol) has been found to significantly influence the extent of monolayer surface coverage. In this instance, 2a bearing an unsubstituted cathecol shows a 2.5-fold increase in surface loading over 1a, which contains a bulkier tBu-catecholate ligand. Photovoltaic measurements on solar cells sensitised with 1a and 2a have allowed for an assessment as to the suitability of these complexes as sensitisers. The observed 0.02–0.07% device efficiency is attributed to the short excited state lifetime and rapid charge recombination dynamics inherent to this class of complex. Specifically, the lifetime of the charge-transfer ML/LLCT excited sate in the parent complex 1 was determined as 250 ps by time-resolved infrared spectroscopy, TRIR. The measured increase in device efficiency for 2a over 1a is consistent with a similar increase in the quantum yield of the long-lived charge separated state (where the complex acts as a donor and the semiconductor as an acceptor) as determined by Photo-Acoustic Calorimetry, and is also matching the magnitude of the increased surface loading achieved with 2a. It is concluded that the relative efficiency of devices sensitised with these particular Pt(II) species is governed by the degree of surface coverage. Overall, this work demonstrates for the first time the use of Pt(diimine)(catecholate) complexes as potential photo-sensitizers in solar cells: the intense absorption in the visible/NIR part of the spectrum is a strong advantage of these dyes, whilst the short excited state lifetime can in the future be addressed by modifying the diimine ligands involved.Acknowledgements
We thank the EPSRC, E-Futures DTC, the STFC, the University of Sheffield, and the Coimbra Chemistry Centre from the FCT (project PEst-OE/QUI/UI0313/2014) for financial support. We are also grateful for the receipt of a Santander research mobility award (JAW and PAS), and to Johnson Matthey for the generous loan of Pt(II) salts. We thank Prof. M. Towrie and Prof. M. Ward for fruitful collaboration. The research leading to the results presented herein has received funding from the European Community's FP7 under grant agreement number 228334 (Laser Lab Europe II). PJ and CS acknowledge support for project PTDC/QUI/QUI/099730/2008 from Fundação para a Ciência e a Tecnologia (Portugal) through the COMPETE programme and from FEDER (European Union).References
- M. Haga, E. S. Dodsworth and A. B. P. Lever, Inorg. Chem., 1986, 25, 447–453 CrossRef CAS.
- S. Bhattacharya and C. G. Pierpont, Inorg. Chem., 1994, 33, 6038–6042 CrossRef CAS.
- M. R. Churchill, K. M. Keil, B. P. Gilmartin, J. J. Schuster, J. B. Keister and T. S. Janik, Inorg. Chem., 2001, 40, 4361–4367 CrossRef CAS PubMed.
- S. Bhattacharya, S. R. Boone, G. A. Fox and C. G. Pierpont, J. Am. Chem. Soc., 1990, 112, 1088–1096 CrossRef CAS.
- A. M. Barthram, R. L. Cleary, J. C. Jeffery, S. M. Couchman and M. D. Ward, Inorg. Chim. Acta, 1998, 267, 1–5 CrossRef CAS.
- P. R. Auburn, E. S. Dodsworth, M. Haga, W. Liu, W. A. Nevin and A. B. P. Lever, Inorg. Chem., 1991, 30, 3502–3512 CrossRef CAS.
- S. Patra, B. Sarkar, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2003, 42, 6469–6473 CrossRef CAS PubMed.
- A. P. Meacham, K. L. Druce, Z. R. Bell, M. D. Ward, J. B. Keister and A. B. P. Lever, Inorg. Chem., 2003, 42, 7887–7896 CrossRef CAS PubMed.
- A. M. Barthram, Z. R. Reeves, J. C. Jeffery and M. D. Ward, J. Chem. Soc., Dalton Trans., 2000, 3162–3169 RSC.
- M. Haga, K. Isobe, S. R. Boone and C. G. Pierpont, Inorg. Chem., 1990, 29, 3795–3799 CrossRef CAS.
- F. Hartl and A. Vlcek, Inorg. Chem., 1996, 35, 1257–1265 CrossRef CAS PubMed.
- G. A. Abakumov, V. K. Cherkasov, K. G. Shalnova, I. A. Teplova and G. A. Razuvaev, J. Organomet. Chem., 1982, 236, 333–341 CrossRef CAS.
- K. A. M. Creber and J. K. S. Wan, J. Am. Chem. Soc., 1981, 103, 2102–2104 CrossRef.
- M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- W. P. Griffith, Transition Met. Chem., 1993, 18, 250–256 CrossRef CAS.
- J. Best, I. V. Sazanovich, H. Adams, R. D. Bennett, E. S. Davies, A. J. H. M. Meijer, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, M. D. Ward and J. A. Weinstein, Inorg. Chem., 2010, 49, 10041–10056 CrossRef CAS PubMed.
- N. M. Shavaleev, E. S. Davies, H. Adams, J. Best and J. A. Weinstein, Inorg. Chem., 2008, 47, 1532–1547 CrossRef CAS PubMed.
- B. Hirani, J. Li, P. I. Djurovich, M. Yousufuddin, J. Oxgaard, P. Persson, S. R. Wilson, R. Bau, W. A. Goddard and M. E. Thompson, Inorg. Chem., 2007, 46, 3865–3875 CrossRef CAS PubMed.
- W. Paw and R. Eisenberg, Inorg. Chem., 1997, 36, 2287–2293 CrossRef CAS PubMed.
- S. S. Kamath, V. Uma and T. S. Srivastava, Inorg. Chim. Acta, 1989, 166, 91–98 CrossRef CAS.
- J. A. Weinstein, M. T. Tierney, E. S. Davies, K. Base, A. A. Robeiro and M. W. Grinstaff, Inorg. Chem., 2006, 45, 4544–4555 CrossRef CAS PubMed.
- V. Anbalagan and T. S. Srivastava, Polyhedron, 2004, 23, 3173–3183 CrossRef CAS.
- P. L. Hill, L. Y. Lee, T. R. Younkin, S. D. Orth and L. McElwee-White, Inorg. Chem., 1997, 36, 5655–5657 CrossRef CAS.
- N. Deibel, D. Schweinfurth, S. Hohloch, M. Delor, I. V. Sazanovich, M. Towrie, J. A. Weinstein and B. Sarkar, Inorg. Chem., 2014, 53, 1021–1031 CrossRef CAS PubMed.
- N. Deibel, D. Schweinfurth, J. Fiedler, S. Zalis and B. Sarkar, Dalton Trans., 2011, 40, 9925–9934 RSC.
- M. Gratzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef CAS.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed.
- A. Islam, H. Sugihara, K. Hara, L. P. Singh, R. Katoh, M. Yanagida, Y. Takahashi, S. Murata, H. Arakawa and G. Fujihashi, Inorg. Chem., 2001, 40, 5371–5380 CrossRef CAS PubMed.
- E. A. M. Geary, N. Hirata, J. Clifford, J. R. Durrant, S. Parsons, A. Dawson, J. Yellowlees and N. Robertson, Dalton Trans., 2003, 3757–3762 RSC.
- E. A. M. Geary, L. J. Yellowlees, L. A. Jack, I. D. H. Oswald, S. Parsons, N. Hirata, J. R. Durrant and N. Robertson, Inorg. Chem., 2005, 44, 242–250 CrossRef CAS PubMed.
- E. C.-H. Kwok, M.-Y. Chan, K. M.-C. Wong, W. H. Lam and V. W.-W. Yam, Chem. – Eur. J., 2010, 16, 12244–12254 CrossRef CAS PubMed.
- L. G. Arnaut, M. Barroso and C. Serpa, Applied Photochemistry, ed. R. Evans, H. D. Burrows and P. Douglas, Springer-Verlag, Dordrecht, 2013, pp. 267–304 Search PubMed.
- R. Katoh, A. Furube, T. Yoshihara, K. Hara, G. Fujihashi, S. Takano, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 4818–4822 CrossRef CAS.
- R. Eichberger and F. Willig, Chem. Phys., 1990, 141, 159–173 CrossRef CAS.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- C. Serpa, J. Schabauer, A. P. Piedada, C. J. P. Monteiro, M. M. Pereira, P. Douglas, H. D. Burrows and L. G. Arnaut, J. Am. Chem. Soc., 2008, 130, 8876–8877 CrossRef CAS PubMed.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22, 13318–13327 RSC.
- T. Gensch and C. Viappiani, Photochem. Photobiol. Sci., 2003, 2, 699–721 CAS.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Inorg. Chem., 1972, 11, 1280–1284 CrossRef CAS.
- P. Jarosz, P. Du, J. Schneider, S.-H. Lee, D. McCamant and R. Eisenberg, Inorg. Chem., 2009, 48, 9653–9663 CrossRef CAS PubMed.
- J. J. Novoa, G. Aullon, P. Alemany and S. Alvarez, J. Am. Chem. Soc., 1995, 117, 7169–7171 CrossRef CAS.
- I. V. Sazanovich, M. A. H. Alamiry, J. Best, R. D. Bennett, O. V. Bouganov, E. S. Davies, V. P. Grivin, A. J. H. M. Meijer, V. F. Plyusnin, K. L. Ronayne, A. H. Shelton, S. A. Tikhomirov, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 10432–10445 CrossRef CAS PubMed.
- S. Shukla, S. S. Kamath and T. S. Srivastava, J. Photochem. Photobiol., A, 1989, 47, 287–298 CrossRef CAS.
- T. R. Miller and I. G. Dance, J. Am. Chem. Soc., 1973, 95, 6970–6979 CrossRef CAS.
- D. M. Manuta and A. J. Lees, Inorg. Chem., 1986, 25, 3212–3218 CrossRef CAS.
- S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS.
- J. Best, PhD thesis, University of Sheffield, 2009.
- C.-R. Lee, H.-S. Kim, I.-H. Jang, J.-H. Im and N.-G. Park, ACS Appl. Mater. Interfaces, 2011, 3, 1953–1957 CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Gratzel, J. Nelson, X. Li, N. L. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225–5233 CrossRef CAS PubMed.
- S. K. Haram, Handbook of Electrochemistry, ed. C. G. Zoski, Elsevier, Amsterdam, 1st edn, 2007 Search PubMed.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C. H. Fischer and M. Gratzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS PubMed.
- R. Katoh, A. Huijser, K. Hara, T. J. Savenije and L. D. A. Siebbeles, J. Phys. Chem. C, 2007, 111, 10741–10746 CAS.
- J. E. Moser, Dye Sensitised Solar Cells, ed. K. Kalyanasundaram, EPFL Press, Lausanne, 1st edn, 2010 Search PubMed.
- H. Wang and L. M. Peter, J. Phys. Chem. C, 2012, 116, 10468–10475 CAS.
- T. Moehl, H. N. Tsao, K.-L. Wu, H.-C. Hsu, Y. Chi, E. Ronca, F. D. Angelis, M. K. Nazeeruddin and M. Gratzel, Chem. Mater., 2013, 25, 4497–4502 CrossRef CAS.
- S. A. Haque, E. Palomares, B. M. Cho, A. N. M. Green, N. Hirata, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 3456–3462 CrossRef CAS PubMed.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 Search PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Vlachopoulos and M. Gratzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- J. Yang, D. K. Kersi, L. J. Giles, B. W. Stein, C. Feng, C. R. Tichnell, D. A. Schultz and M. L. Kirk, Inorg. Chem., 2014, 53, 4791–4793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1028955 and 1028956. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03466j |
| This journal is © The Royal Society of Chemistry 2015 |