
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous phase selective detection of 2,4,6-trinitrophenol using a fluorescent metal–organic framework with a pendant recognition site†
Sanjog S.
Nagarkar
,
Aamod V.
Desai
,
Partha
Samanta
and
Sujit K.
Ghosh
 *
*
Indian Institute of Science Education and Research (IISER), Pune, Dr. Homi Bhabha Road, Pashan, Pune, 411008 India. E-mail: sghosh@iiserpune.ac.in; Web: http://www.iiserpune.ac.in/~sghosh/ Fax: +91 20 2589 8022; Tel: +91 20 2590 8076
First published on 6th March 2015
Abstract
Prompt and selective detection of nitro explosives in the aqueous phase is in high demand to meet homeland security and environmental concerns. Herein we report the chemically stable porous metal organic framework UiO-68@NH2 with a pendant recognition site for selective detection of the nitro-aromatic explosive TNP in the aqueous phase. The pendant Lewis basic amine moieties are expected to selectively interact with TNP via electrostatic interactions and act as recognition sites for TNP. The MOF can detect the presence of TNP in water at a concentration as low as 0.4 ppm with a response time of a few seconds. In addition, both excitation and emission wavelengths of the MOF are in the visible region. The high selectivity was observed even in the presence of competing nitro analytes in the aqueous phase. The quenching constant for TNP was found to be 5.8 × 104 M−1 which is 23 times higher than that for TNT and for RDX, demonstrating superior and selective quenching ability. This unprecedented selectivity is ascribed to electron-transfer and energy-transfer mechanisms as well as electrostatic interactions between TNP and the MOF. An MOF-coated paper strip that we prepared demonstrated fast and selective response to TNP in water, which represents a first step towards a practical application.
Introduction
The selective and sensitive detection of lethal explosive materials is decisive for homeland security, anti-terrorist operations and civilian safety.1 The most common compounds used as explosives are nitroaromatics such as 2,4,6-trinitrotoluene (TNT), 2,4-dinitrotoluene (2,4-DNT), 2,6-dinitrotoluene (2,6-DNT), 2,4,6-trinitrophenol (TNP), aliphatic nitro compounds such as 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX), and explosive tag (molecule to be added to legally manufactured explosives) like 2,3-dimethyl-2,3-dinitrobutane (DMNB). Among these, TNP has higher explosive power in similar class of explosive compounds.2 Apart from its explosive nature, TNP causes skin/eye irritation, headaches, anaemia, liver injury, and male infertility.3 Despite these issues, TNP is widely used in the fireworks, leather, and dye industries.4 Because of its high water solubility, TNP can easily lead to contamination of nearby soil and aquatic systems, posing serious health hazards.2 Furthermore, owing to its highly electron-deficient nature, TNP does not easily biodegrade. Curiously, few efforts have been devoted to the selective detection of TNP. As a consequence, the development of materials for efficient sensing of TNP as well as constant monitoring of soil and aquatic systems are in high demand. However, it is very difficult to selectively detect TNP in the presence of competing nitro analytes.5 Fluorescence-based chemosensors have high sensitivity, portability, short response times, low cost and compatibility in both solid and solution states, and have thus attracted greater attention for in-field use than the current sophisticated detection systems.6 Numerous luminescent materials have been studied as fluorescence-based sensors of explosives, but their multi-step processing, toxicity and lack of control over molecular organization limit their use.1aMetal–organic frameworks (MOFs) have recently emerged as promising materials for gas storage/separation, chemical sensing, heterogeneous catalysis, biomedicine, magnetism, clean energy technology and optoelectronics.7 Especially, as fluorescence-based chemical sensors, MOFs offer great promise by virtue of their crystalline nature, permanent porosity, designable/modifiable pores, systematically tuneable band gaps and electronic structures.8 The fluorescence sensing performance of MOFs is determined by interactions between the target analyte and MOF backbone. The porosity allows host–guest interactions between analyte and the MOF matrix while the designable pore size/shape yields a molecular sieving effect. The pendant recognition sites allow selective interaction with adsorbate via open metal sites, hydrogen bonding, Mulliken-type interactions, Lewis acid/base sites, etc., and thus can selectively accumulate the targeted analyte in the MOF matrix, giving rise to improved sensing performance, which is also called a pre-concentration effect.8b Additionally, immobilization of organic ligands in the rigid MOF framework leads to stronger emissions. Most importantly, the near limitless combinations of organic ligands and metal centres allow tuning of the valence and/or conduction bands, and in turn of the band gap, which is crucial for sensing applications. Owing to these advantages, a variety of MOFs have been designed and studied for detection of cations, anions, small molecules, explosives and biomolecules.9
Despite recent advances, there are few reports about the use of fluorescent MOFs for the selective and sensitive detection of TNP in the presence of other nitro analytes, and there are hardly any reports of such compounds exhibiting superior selectivity in aqueous systems.9,10 The UiO (University of Oslo) series porous MOFs are composed of less toxic Zr(IV) metal ions and are chemically stable, making them the most suitable candidates for sensing.11 Recently, we reported a chemically stable porous zirconium-based fluorescent MOF for selective detection of TNP in the aqueous phase.10b We achieved highly selective detection of TNP in the aqueous phase by deploying a (Lewis) basic pyridyl functionality in a three-dimensional (3D) MOF matrix. We reasoned that a similarly Lewis basic amine functionality can also be a promising recognition site for selective detection of TNP owing to its possible ionic and hydrogen-bonding interactions with TNP.12 We thus chose the chemically stable porous Zr(IV)-based MOF Zr6O4(OH)4(L)6 (1, UiO-68@NH2, L = 2′-amino-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylate) with a pendant amine functionality for selective sensing of TNP in the aqueous phase (Scheme 1).13 The MOF has large pores, with dimensions of 11 and 22 Å, which are expected to allow easy diffusion and concentration of analytes and facile host–guest interactions. The guest-accessible Lewis basic amine functionalities can act as recognition sites for TNP via ionic and hydrogen-bonding interactions. Also excitation and emission wavelengths of MOF 1 are in the visible region, making it an ideal material to study TNP sensing performance.
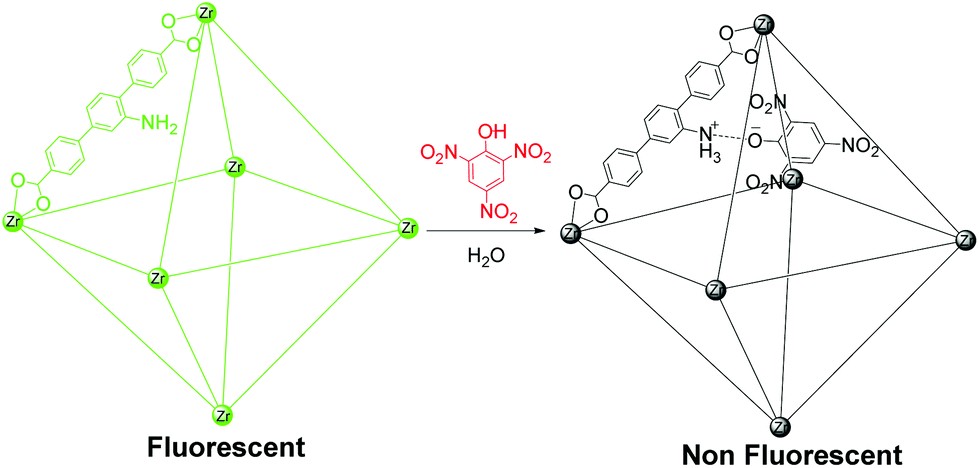 | ||
| Scheme 1 Fluorescent MOF 1 based sensor for highly selective nitro explosive detection in aqueous phase. | ||
Experimental section
Caution: TNT, RDX and TNP are highly explosive and should be handled carefully and in small amounts. The explosives should be handled as dilute solutions and with safety measures to avoid an explosion.Materials
TNT and RDX were provided by HEMRL Pune (India). TNP, 2,4-DNT, 2,6-DNT, and DMNB were purchased from Aldrich, while 1,3-dinitrobenzene (1,3-DNB) and nitrobenzene (NB) were purchased from a local company. All the chemicals were used as received. Dry solvents were used during complete analysis and were obtained locally.Physical measurements
1H NMR was recorded using a 400 MHz Jeol ECS-400 Instrument. Thermogravimetric analyses were recorded on a PerkinElmer STA 6000 TGA analyser under N2 atmosphere with a heating rate of 10 °C min−1. Powder X-ray diffraction patterns (PXRD) were obtained from a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation (λ = 1.5406 Å) with a tube voltage of 40 kV and current of 40 mA between 2θ values of 5 and 40°. The FT-IR spectra were recorded using a NICOLET 6700 FT-IR spectrophotometer with a KBr pellet between 400 and 4000 cm−1. Fluorescence measurements were taken using a Horiba FluoroMax 4 with a stirring attachment. UV-Vis measurements were taken using Chemito SPECTRASCAN UV-2600.Synthesis of UiO-68@NH2 (1)
The ligand 2′-amino-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylic acid (LH2) was synthesized using the procedure previously reported (Fig. S1†).13a For synthesis of 1, ZrCl4 (24 mg) and LH2 (56 mg) were dissolved in N,N-dimethylformamide (DMF, 3 mL) in a Teflon-lined Parr stainless steel vessel (17 mL). The vessel was sealed and placed in an oven and heated at 120 °C for 16 h. After cooling to room temperature, the crystalline product was isolated by filtration, and the solid was washed with DMF.Activation of UiO-68@NH2 (1′)
The occluded solvent and starting material in the MOF was then exchanged with MeOH by dipping it in MeOH for 3 days, with the MeOH refreshed every 24 h. The guest-free porous MOF (1′) was obtained by heating the MeOH-exchanged MOF at 130 °C under vacuum for 24 h, and was then used for fluorescence measurements.Photophysical study
In a typical experiment, 1 mg of 1′ was weighed and added to a cuvette containing 2 mL of water and stirred. Upon excitation at 395 nm, the fluorescence response of 1′ (0.5 mg mL−1) dispersed in water was measured in situ in the 410–780 nm range and the corresponding fluorescence intensity was monitored at 500 nm. For fluorescence titration, emission was recorded upon incremental addition of freshly prepared analyte solutions (1 mM or saturated). To maintain homogeneity, the solution was stirred at a constant rate during the experiment.Results and discussion
Synthesis
The MOF 1 was synthesized by a solvothermal reaction between ZrCl4 and LH2 in DMF at 120 °C.13 The similarity of the as-synthesized and simulated PXRD patterns confirmed the successful formation of 1 (Fig. S2†). The MOF 1, composed of a hexameric Zr node [Zr6O4(OH)4(CO2)12] connected at regular intervals by organic ligand L forming 3D structure. The decrease in the ν(COO)as frequency from 1685 cm−1 to 1608 cm−1 in the FT-IR spectra of LH2 and MOF 1, respectively, also supported the bond formation (Fig. S3†). Thermogravimetric analysis (TGA) of as-synthesized MOF 1 showed ∼60% weight loss below 300 °C, ascribed to loss of DMF and starting material present in the free volume of the MOF matrix (Fig. S4†). The DMF and starting material were removed by the solvent exchange method reported previously to obtain the activated form 1′. The thermogravimetric analysis of activated MOF 1′ did not show any weight loss below 300 °C confirming the successful removal of the guest (Fig. S4†). The activated MOF remains stable even after removal of the guest as confirmed by PXRD analysis (Fig. S2†). The larger pore window has dimensions of 22 Å while the smaller pore has dimensions of 11 Å, which enables concentration of nitro analytes in the MOF matrix (Table S1†). The pores are decorated with guest-accessible amine moieties, which can act as recognition sites for TNP, giving rise to an efficient quenching response.Photophysical studies
The photoluminescence spectra of 1′ in water exhibit strong emission peaks at 500 nm upon excitation at 395 nm at room temperature (Fig. 1a and S5†). To explore the potential application of 1′ to detect TNP in aqueous medium, fluorescence-quenching titration was performed by gradual addition of a solution of aqueous TNP. As anticipated, the incremental addition of TNP resulted in fast and efficient fluorescence quenching of 86% (Fig. 1). The 1′ can recognize TNP at as low as 0.4 ppm concentration, which is comparable to/or better than previous MOF reports.10a We also checked the quenching ability of other competing nitro analytes such as TNT, RDX, 2,4-DNT, 2,6-DNT, DNB, NB, and DMNB in water (Fig. 1b and S6–S12†). Compared to TNP, the competing nitro analytes showed small effects on the emission intensity of 1′ (Fig. 1b). This result clearly demonstrates the high selectivity of 1′ towards TNP compared to potentially interfering nitro analytes. To further quantify the quenching efficiency, Stern–Volmer (SV) plots of the relative luminescent intensities (I0/I) of all the nitro analytes were compared, where I0 and I are the intensities in the absence and in the presence of the respective nitro analytes (Fig. 2). All the nitro analytes except TNP yielded linear SV plots. Interestingly, a linear SV plot resulted for low concentrations of TNP, but a slight non-linear feature was produced at a higher concentration. This non-linear feature in the SV plot suggests a consolidation of both dynamic as well as static quenching and/or the occurrence of an energy-transfer phenomenon between 1′ and TNP.14 The quenching constant for TNP was calculated from the SV plot to be 5.8 × 104 M−1. The observed quenching constant for TNP is equivalent to those of organic polymer based probes and is ∼23 times higher than those of its aromatic (TNT) and aliphatic (RDX) tri-nitro analogues, demonstrating the superior detection performance.15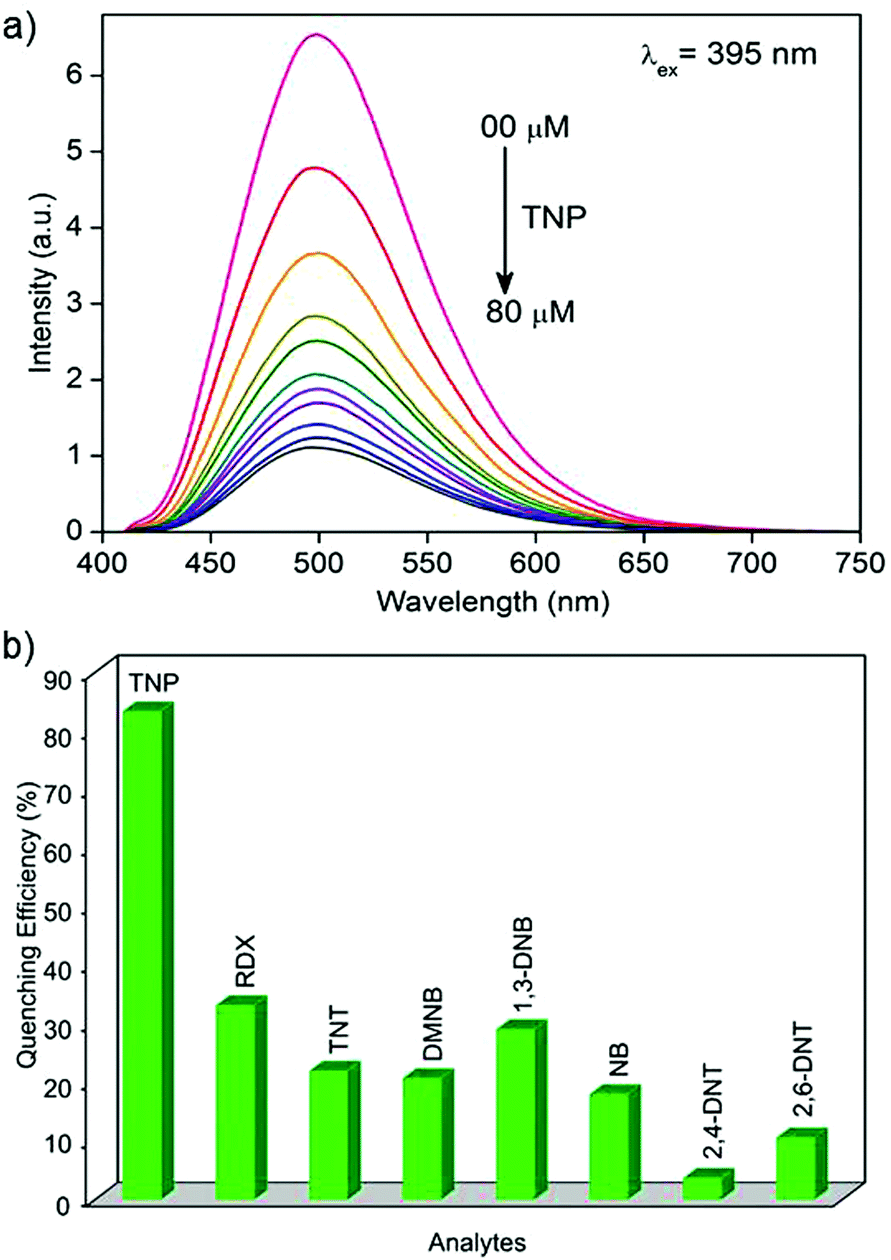 | ||
| Fig. 1 (a) Decrease in fluorescence intensity of 1′ dispersed in water upon incremental addition of aqueous TNP solution. (b) Comparison of fluorescence quenching efficiency of different nitro analytes towards 1′ in water. | ||
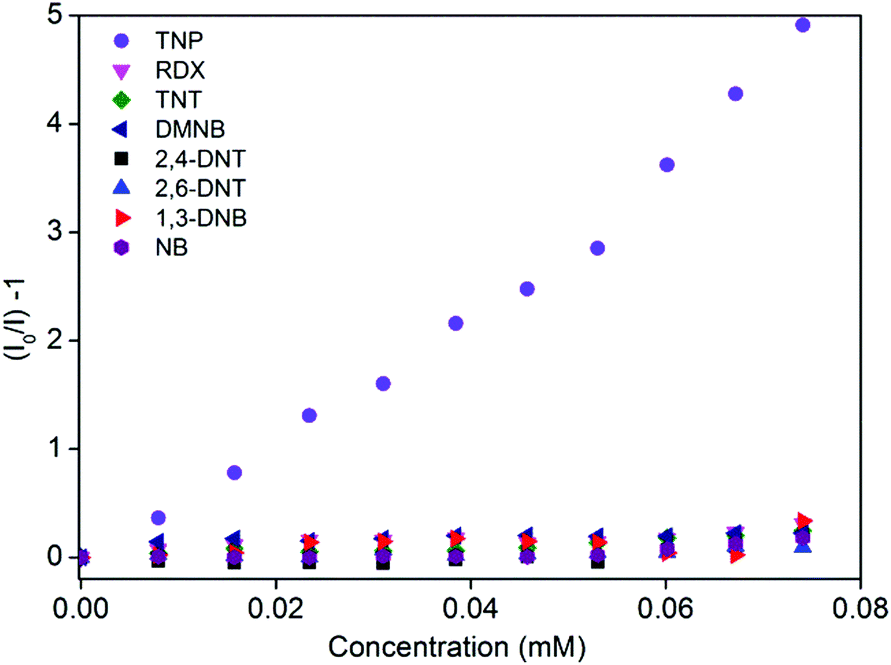 | ||
| Fig. 2 Stern–Volmer plots for nitro analytes added to 1′ in water. | ||
Investigation of possible quenching mechanism
To gain more insight into the superior sensing ability of 1′, we sought to examine the electronic properties of both the MOF and nitro analytes. Fluorescence quenching by electron transfer from the conduction band (CB) of the MOF to LUMO orbitals of the electron-deficient nitro analyte is a well-established quenching mechanism.16 The lower the LUMO energy, the higher is the electron accepting ability of the nitro analyte and thus the higher is the efficiency of fluorescence quenching. The effective fluorescence quenching by TNP is in good agreement with its LUMO energy being lower than that of any of the other nitro analytes, as calculated by density functional theory at the B3LYP/6-31G* level (Fig. S13†). However, the fluorescence quenching performances of the other nitro analytes are not in accordance with their LUMO energy trend, indicating the simultaneous presence of other quenching mechanisms in addition to electron transfer. The resonance energy transfer is another effective fluorescence quenching mechanism. The non-linear SV plot supports the presence of long-range resonance energy transfer. The effectiveness of the energy transfer heavily depends on the extent of spectral overlap between the emission of the fluorophore and the absorption spectrum of the analyte. The absorption spectrum of TNP shows a higher extent of overlap with the MOF emission than do the other competing nitro analytes (Fig. 3). This observation is in good agreement with the observed higher quenching efficiency obtained for TNP than for the other nitro analytes. Thus, it is apparent that in the case of TNP, both electron- and energy-transfer mechanisms are operational while other nitro analytes utilize only an electron-transfer mechanism.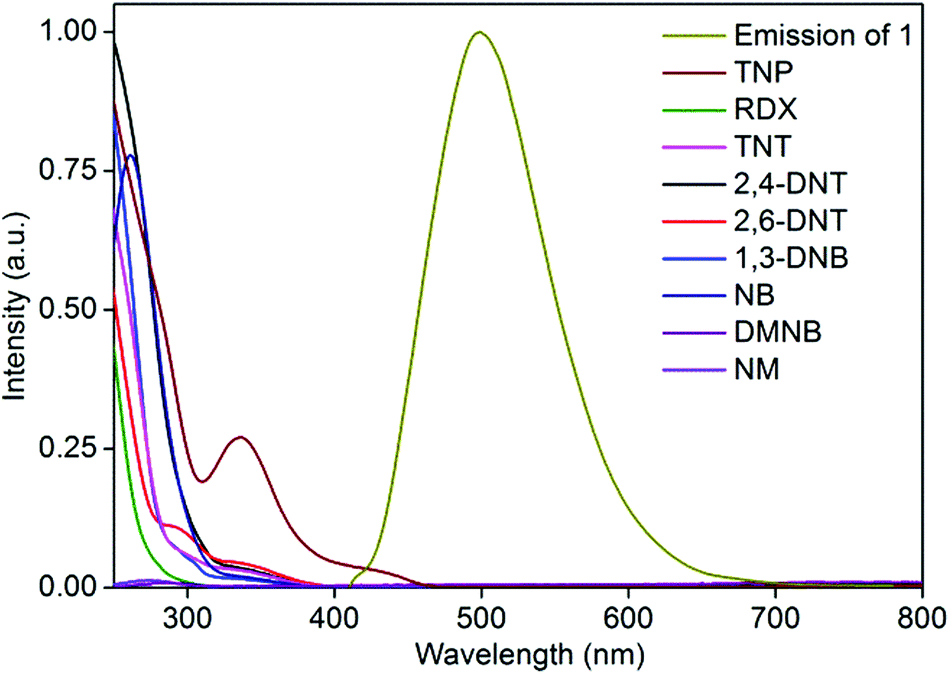 | ||
| Fig. 3 Extent of spectral overlap between absorption spectra of nitro analytes and emission spectrum of 1′ in water. | ||
TNP is known to interact with Lewis basic sites owing to its acidic phenolic proton. To probe the role of the pendant Lewis basic free amine present in 1 in the observed selectivity, fluorescence quenching titrations were performed with 2,4-dinitrophenol (2,4-DNP) and 4-nitrophenol (NP). The fluorescence quenching performance of the phenolic analytes correlates with the acidity of the phenolic protons: TNP > 2,4-DNP > NP. This trend indicates the presence of an electrostatic interaction between TNP and the pendant amine functionality, similar to that which occurs for the pyridyl functionalized MOF reported earlier (Fig. S14–16†).12 This result supports the observed selectivity for TNP. The highly acidic TNP selectively and strongly interacts with the pendant Lewis basic amine group via ionic and hydrogen-bonding interactions, leading to an amplified quenching response. Such interactions are absent for the other nitro analytes, resulting in the low quenching effect. Thus the Lewis basic amine functionality acts as a recognition site for TNP, and a combination of electron-transfer and energy-transfer quenching mechanisms gives rise to the unprecedented selectivity for TNP in the aqueous phase. The solution pH in the range of 4.5 to 7 has little effect on MOF fluorescence; this pH range is that observed for the solutions to which TNP, 2,4-DNP, and NP were added.
It is highly desirable in applied scenarios to be able to selectively detect TNP in the presence of other competing nitro analytes in aqueous systems. To assess the selectivity of 1′ for TNP in the presence of competing nitro analytes, we designed a competitive fluorescence quenching assay. Initially, the fluorescence spectrum of 1′ dispersed in water was recorded. An aqueous TNT solution was then added in two equal portions (total 40 μL) to the above solution to allow effective access to the pendant free Lewis basic amine sites, and the fluorescence response was recorded. No significant fluorescence quenching was observed upon addition of the aqueous TNT solution (Fig. 4). However, when the same quantity of the aqueous TNP solution (40 μL in two equal portions) was added to the above solution, significant fluorescence quenching was observed. The trend repeated even in subsequent cycles in which TNT and TNP were added. A similar phenomenon was observed in competitive assays with other nitro analytes, demonstrating unprecedented selectivity of 1′ for TNP in the presence of competing nitro analytes (Fig. 4).
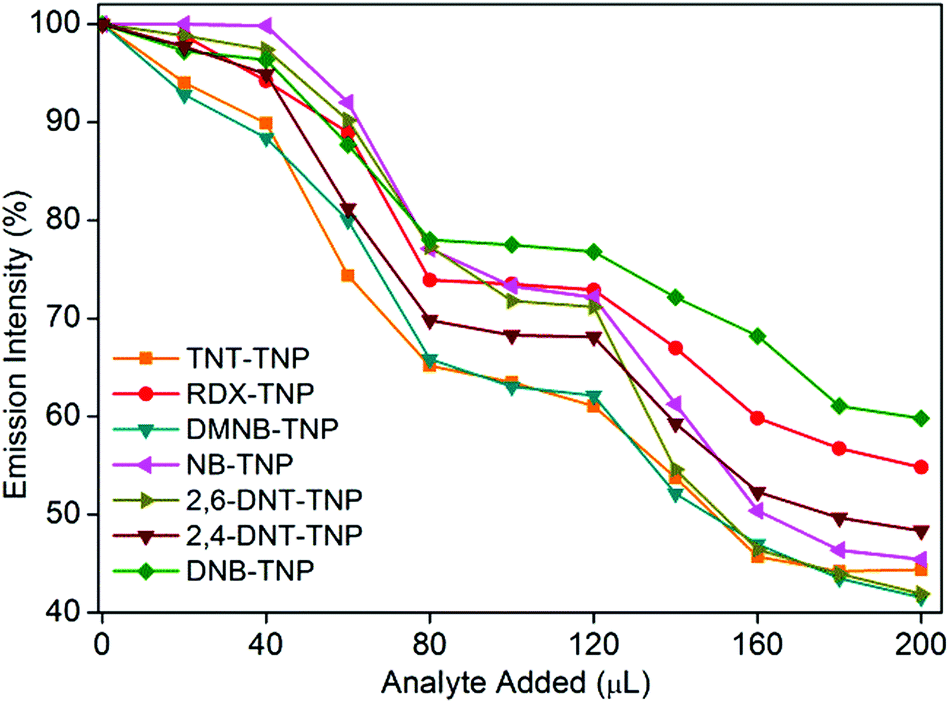 | ||
| Fig. 4 Competitive fluorescence quenching efficiency of TNP upon addition of other competing nitro analytes followed by TNP to 1′. | ||
MOF-based paper strip
For in-field aqueous phase detection of explosives, the paper strip method comes in handy. We prepared MOF 1′-coated black paper strips for in-field aqueous phase detection of TNP. The pristine strip showed good fluorescence under UV illumination. The individual strips were then partially dipped in aqueous solutions of nitro analytes (Fig. 5a). The part of the strip dipped in the TNP solution showed significant fluorescence quenching when visualized under UV light (Fig. 5b strip A) while the unexposed part of the same strip still showed good fluorescence. All the other strips dipped in competing nitro analytes showed negligible fluorescence quenching responses (Fig. 5b strip B–H). Thus the MOF 1′-coated paper strip provides an efficient way to trace the presence of TNP in aqueous systems and demonstrates the potential of 1′ for real-time in-field sensing applications. | ||
| Fig. 5 (a) MOF 1-coated paper strip for detection of explosives in the aqueous phase. (b) Response of MOF-coated paper strips towards various nitro analytes under UV light (A = TNP, B = TNT, C = RDX, D = 2,4-DNT, E = DNB, F = 2,6-DNT, G = NB, H = DMNB). | ||
Conclusions
We have developed a chemically stable porous fluorescent MOF with a Lewis basic free amine functionality as a pendant recognition site for the selective and sensitive detection of TNP in the aqueous phase. Both the excitation and emission wavelengths of the MOF are in the visible region. The MOF can within a few seconds detect TNP at a concentration as low as 0.4 ppm. The quenching constant for TNP was found to be 5.8 × 104 M−1 which is 23 times higher than that for TNT and for RDX. Importantly, the high selectivity is observed even in the presence of competing nitro analytes. The combination of electron- and energy-transfer mechanisms along with selective recognition sites is credited for the high selectivity of 1′ for TNP. An MOF-based paper strip provides an effective and efficient way to detect TNP in the aqueous phase in-field for security and environmental monitoring applications.Acknowledgements
S.S.N. thanks CSIR, India for research fellowship. A.V.D. thanks IISER, Pune for research fellowship. This work was funded by IISER, Pune, DST (GAP/DST/CHE-12-0083). HEMRL, Pune is acknowledged for TNT and RDX samples.Notes and references
- (a) Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Castero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC; (b) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543 RSC; (c) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Soc. Rev., 2007, 36, 1339 CrossRef PubMed.
- (a) N. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2010, 48, 5007 RSC; (b) Y. Peng, A.-J. Zhang, M. Dong and Y.-W. Wang, Chem. Commun., 2011, 47, 4505 RSC; (c) M. Dong, Y.-W. Wang, A.-J. Zhang and Y. Peng, Chem. – Asian J., 2013, 8, 1321 CrossRef CAS PubMed.
- J. F. Wyman, M. P. Serve, D. W. Honson, L. H. Lee and D. E. Uddin, J. Toxicol. Environ. Health, 1992, 37, 313 CrossRef CAS PubMed.
- J. Akhavan, The Chemistry of Explosives, Royal Society of Chemistry, Cambridge, 2004, p. 4 Search PubMed.
- B. Xu, X. Wu, H. Li, H. Tong and L. Wang, Macromolecules, 2011, 44, 5089 CrossRef CAS.
- (a) L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993 RSC; (b) Y. Xin, Q. Wang, T. Liu, L. Wang, J. Li and Y. Fang, Lab Chip, 2012, 12, 4821 RSC.
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (b) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed; (c) R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042 CrossRef CAS PubMed; (d) A. Aijaz, P. Lama and P. K. Bharadwaj, Inorg. Chem., 2010, 49, 5883 CrossRef CAS PubMed; (e) S. S. Nagarkar, S. M. Unni, A. Sharma, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2638 CrossRef CAS PubMed; (f) R. Liang, L. Shen, F. Jing, W. Wu, N. Qin, R. Lin and L. Wu, Appl. Catal., B, 2015, 162, 245 CrossRef CAS PubMed; (g) L. Shen, S. Liang, W. Wu, R. Lianga and L. Wu, Dalton Trans., 2013, 42, 13649 RSC.
- (a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed; (b) R. Xiong, K. Odbadrakh, A. Michalkova, J. P. Luna, T. Petrova, D. J. Keffer, D. M. Nicholson, M. A. Fuentes-Cabrera, J. P. Lewis and J. Leszczynski, Sens. Actuators, B, 2010, 148, 459 CrossRef CAS PubMed.
- (a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed; (b) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed; (c) B. Joarder, A. V. Desai, P. Samanta, S. Mukherjee and S. K. Ghosh, Chem. – Eur. J., 2015, 21, 965 CrossRef CAS PubMed; (d) D. K. Singha, S. Bhattacharya, P. Majee, S. K. Mondal, M. Kumar and P. Mahata, J. Mater. Chem. A, 2014, 2, 20908 RSC; (e) J. Ye, L. Zhao, R. F. Bogale, Y. Gao, X. Wang, X. Qian, S. Guo, J. Zhao and G. Ning, Chem. – Eur. J., 2015, 21, 2029 CrossRef CAS PubMed; (f) S. S. Nagarkar, T. Saha, A. V. Desai, P. Talukdar and S. K. Ghosh, Sci. Rep., 2014 DOI:10.1038/srep07053.
- (a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (b) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915 RSC; (c) X. Z. Song, S. Y. Song, S. N. Zhao, Z. M. Hao, M. Zhu, X. Meng, L. L. Wu and H. J. Zhang, Adv. Funct. Mater., 2014, 24, 4034 CrossRef CAS.
- (a) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed; (b) M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632 CrossRef CAS; (c) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurine, P. Couvreur, G. Ferey, R. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- G. He, H. Peng, T. Liu, M. Yang, Y. Zhang and Y. Fang, J. Mater. Chem., 2009, 19, 7347 RSC.
- (a) A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643 CrossRef CAS PubMed; (b) H.-L. Jiang, D. Feng, T.-F. Liu, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 14690 CrossRef CAS PubMed.
- (a) W. Wu, S. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, Macromol. Rapid Commun., 2012, 33, 164 CrossRef CAS PubMed; (b) D. Zhao and T. M. Swager, Macromolecules, 2005, 38, 9377 CrossRef CAS; (c) K. Acharya and P. S. Mukherjee, Chem. Commun., 2014, 50, 15788 RSC.
- (a) A. Saxena, M. Fujiki, R. Rai and G. Kwak, Chem. Mater., 2005, 17, 2181 CrossRef CAS; (b) J. C. Sanchez, A. G. DiPasquale, A. L. Rheingold and W. C. Trogler, Chem. Mater., 2007, 19, 6459 CrossRef CAS.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed; (b) S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: For details of MOF synthesis, PXRD patterns and photo physical studies. See DOI: 10.1039/c5dt00397k |
| This journal is © The Royal Society of Chemistry 2015 |