
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and (spectro)electrochemistry of mixed-valent diferrocenyl–dihydrothiopyran derivatives†
Konrad
Kowalski
*a,
Rafał
Karpowicz
a,
Grzegorz
Mlostoń
b,
Dominique
Miesel
c,
Alexander
Hildebrandt
c,
Heinrich
Lang
c,
Rafał
Czerwieniec
d and
Bruno
Therrien
e
aFaculty of Chemistry, Department of Organic Chemistry, University of Łódź, Tamka 12, PL-91403 Łódź, Poland. E-mail: kondor15@wp.pl
bFaculty of Chemistry, Department of Organic and Applied Chemistry, University of Łódź, Tamka 12, PL-91403 Łódź, Poland
cTechnische Universität Chemnitz, Faculty of Natural Sciences, Institute of Chemistry, Department of Inorganic Chemistry, D-09107 Chemnitz, Germany
dUniversität Regensburg, Institut für Physikalische und Theoretische Chemie, Universitätsstraße 31, D-93040 Regensburg, Germany
eInstitute of Chemistry, University of Neuchatel, Avenue de Bellevaux 51, CH-2000 Neuchatel, Switzerland
First published on 24th February 2015
Abstract
Three novel diferrocenyl complexes were prepared and characterised. 2,2-Diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran (1, sulphide) was accessible by the hetero-Diels–Alder reaction of diferrocenyl thioketone with 2,3-dimethyl-1,3-butadiene. Stepwise oxidation of 1 gave the respective oxides 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1-oxide (2, sulfoxide) and 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1,1-dioxide (3, sulfone), respectively. The molecular structures of 1 and 3 in the solid state were determined by single crystal X-ray crystallography. The oxidation of sulphide 1 to sulfone 3, plays only a minor role on the overall structure of the two compounds. Electrochemical (cyclic voltammetry (= CV), square wave voltammetry (= SWV)) and spectroelectrochemical (in situ UV-Vis/NIR spectroscopy) studies were carried out. The CV and SWV measurements showed that an increase of the sulphur atom oxidation from −2 in 1 to +2 in 3 causes an anodic shift of the ferrocenyl-based oxidation potentials of about 100 mV. The electrochemical oxidation of 1–3 generates mixed-valent cations 1+–3+. These monooxidised species display low-energy electronic absorption bands between 1000 and 3000 nm assigned to IVCT (= Inter-Valence Charge Transfer) electronic transitions. Accordingly, the mixed-valent cations 1+–3+ are classified as weakly coupled class II systems according to Robin and Day.
Introduction
Recently, mixed-valent (= MV) species have attracted considerable attention in particular in the field of molecular electronics as they offer the possibility to act as model compounds for molecular wires, switches and other electronic building blocks.1–15 Besides these foreseen technological applications MV compounds are used in electron transfer studies and are of key importance in biological systems.16–20Ferrocenyl groups are often used in organometallic chemistry as redox-active terminal units, because ferrocene is thermally stable in its neutral and oxidised form.21 In addition, ferrocenyl/ferrocenium groups in mixed-valent species show an excellent electrochemical reversibility of the Fe(II)/Fe(III) redox couple, i.e. 2,5-diferrocenyl five-membered heterocycles.8,22–32
The electron transfer between the Fe(II)/Fe(III) centres, i.e. from the donor (Fe(II)) to the acceptor (Fe(III)) ion, manifests itself by the appearance of characteristic absorptions, i.e. IVCT (= Inter-Valence Charge Transfer) bands in the near infrared (NIR) spectral range.8,33
Two distinct modes of the electronic communication between the donor and acceptor metal centres in the MV state exist: “through bond” and “through space”.5,6,34 The “through bond” mechanism is characteristic for molecules in which π-conjugated connectivities are linking the two redox-active metal termini,5,6 while the “through space” mechanism requires a close proximity of the interacting centres.34 Depending on the degree of the electronic communication, three classes of MV complexes are distinguished according to the Robin and Day classification: non-coupled (class I), weakly-coupled (class II) and fully delocalized (class III) systems.35 Based on the linking group constitution, five structural types of dinuclear transition-metal compounds are known (types A–E), as schematically shown in Fig. 1.
 | ||
| Fig. 1 Classification of homobimetallic mixed-valence (= MV) metal complexes. M represents a metal containing redox centre. In type A molecules the centres are connected by a conjugated bridge consisting of C,C double or triple bonds, or aromatic moieties.5,6 In type B molecules the M termini are directly connected to each other,36 while in type D and E species the metal centres are separated by an aliphatic37,38 or heteroatom fragment, respectively.37–39 Type C molecules represent a special variety of type A compounds, in which the two M termini in the mixed-valent species interact most likely “through space”.34 | ||
Among the type A and type B molecules (Fig. 1), the respective diferrocenyl-functionalized systems have been extensively studied,5,6,36,40–42 whereas compounds of structural type D remain almost unexplored.37,43 The simplest representative of the Fc-based class D molecules is diferrocenylmethane (Fc2CH2, Fc = Fe(η5-C5H4)(η5-C5H5)), however, its mono-oxidised form (Fc2CH2+) does not show any detectable IVCT band.44 On the contrary, such IVCT absorptions were observed for other derivatives, in which the methylene hydrogen atoms were replaced by methyl or ferrocenyl groups,37,44 indicating that the appearance of IVCT transitions is related to the increased bulkiness of the linker unit due to the presence of sterically demanding groups in the latter compounds as compared to the Fc2CH2+ cation. In addition, the strength of the metal–metal interactions in type D compounds featuring CMe2, SiMe2 or GeMe2 bridges, depend on the effective distance between the metal centres.37 It was found that the extent of the electronic coupling in the MV species decreases with an increase of the atomic radius of the bridging atom (e.g. in the order CMe2 > SiMe2 > GeMe2).
In continuation of our works in the area of electron-transfer studies in (multi)ferrocenyl-functionalised organic and organometallic compounds,8,11,22–32,45–49 we herein present the synthesis and characterisation of 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran, 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1-oxide and 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1,1-dioxide. The influence of the electronic and steric effects on the electron transfer between the two ferrocenyl moieties in the respective mixed-valent species is reported. The 3,6-dihydro-2H-thiopyran bridge was chosen, due to its bulkiness and the presence of the oxidisable sulphur atom in a position adjacent to the redox-active ferrocenyl groups. This S-atom reactivity allows us to synthesise three closely related derivatives 1–3 and investigate the influence of the different electron-withdrawing character of the aliphatic bridge on the IVCT properties of the mixed valence species 1+–3+.
Results and discussion
Synthesis
The synthetic methodologies for the preparation of 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran (1, sulphide), 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1-oxide (2, sulfoxide) and 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1,1-dioxide (3, sulfone) are shown in Scheme 1.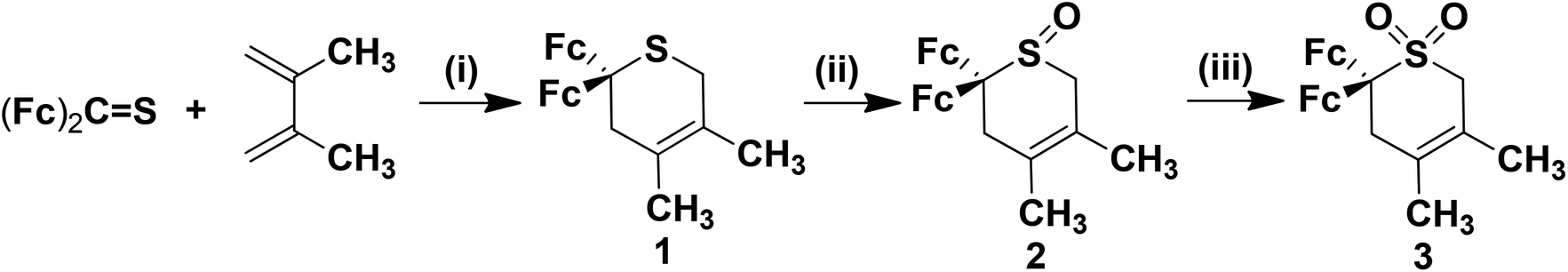 | ||
| Scheme 1 Synthesis of 1–3 (MCPBA = m-chloroperoxybenzoic acid, Fc = ferrocenyl group). (i) 75 °C, 50 h; (ii) 30% H2O2, SeO2, CH3OH, 0 °C to r.t., 10 min; (iii) MCPBA, CH2Cl2, −20 °C, 2 h, then r.t. 24 h and 2nd portion MCPBA at r.t. 24 h. | ||
Sulphide 1 was prepared via the hetero-Diels–Alder cycloaddition of diferrocenylthioketone50,51 with 2,3-dimethyl-1,3-butadiene in a sealed glass-tube at 75 °C (Experimental section).52 After appropriate work-up, compound 1 was obtained as a red solid in 65% yield. Treatment of sulphide 1 with hydrogen peroxide (30%) and selenium dioxide as oxidising reagents53 in methanol gave the respective sulfoxide 2, which was purified by column chromatography in 87% yield (Experimental section). Further oxidation of 2 with m-chloroperoxybenzoic acid (= MCPBA) in dichloromethane produced sulfone 3 at ambient temperature in 21% yield (Scheme 1, Experimental section).54
Compounds 1–3 are red solids soluble in common organic solvents. All three compounds are stable towards air and moisture in the solid state and in solution.
Compounds 1–3 were characterised by IR and NMR (1H, 13C{1H}; for more details see Fig. S1–S3, ESI†) spectroscopy and high-resolution mass spectrometry. The molecular structures of 1 and 3 in the solid state were determined by single crystal X-ray diffraction analysis. Electrochemical investigations were carried out by using cyclic voltammetry (= CV), square wave voltammetry (= SWV) and in situ UV-Vis/NIR spectroelectrochemistry.
X-ray structure determination
Single-crystals of 1 and 3 suitable for a single crystal X-ray diffraction analysis were obtained by slow diffusion of hexane into a dichloromethane solution of 1 and by slow diffusion of hexane into a diethyl ether solution of 3. ORTEP diagrams of 1 and 3 are shown in Fig. 2, while selected bond distances (Å) and angles (°) are listed in Table 1. Crystal and structure refinement data are presented in the ESI (Table S1†).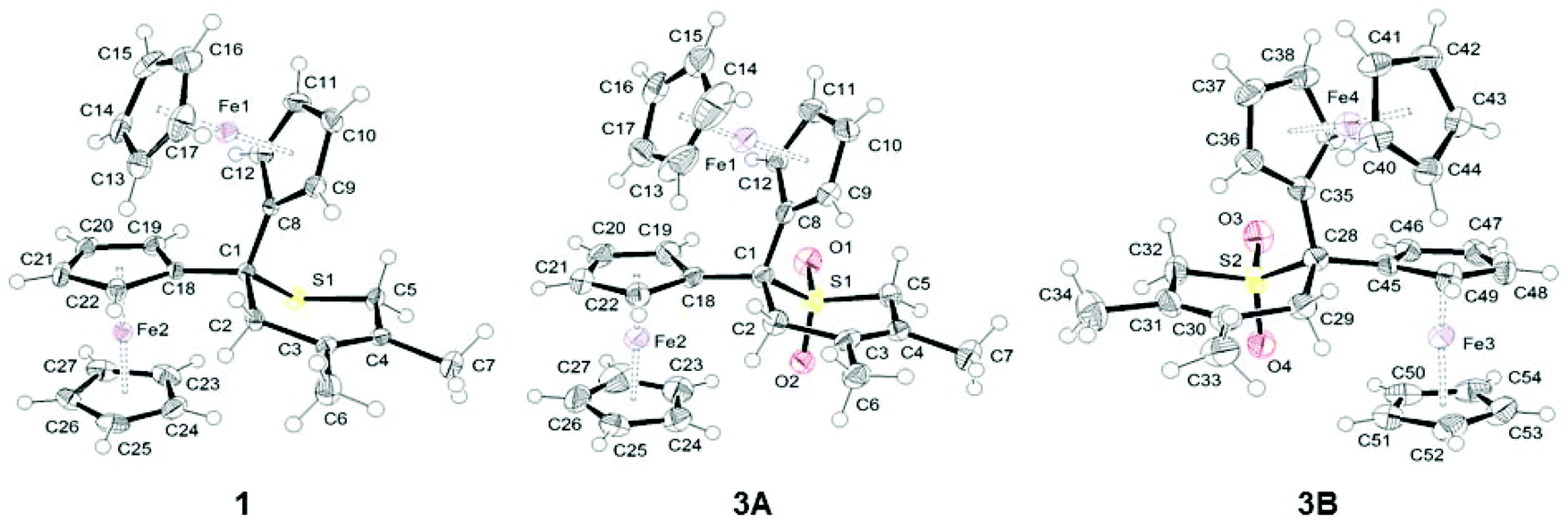 | ||
| Fig. 2 ORTEP diagrams of 1 and 3 (two independent molecules in the crystal, 3A and 3B) at 50% probability level. | ||
| 1 | 3A | 3B | |
|---|---|---|---|
| Bond distances | |||
| Fe⋯Fe | 5.6377(6) | 5.6824(9) | 5.6785(9) |
| S–CFc | 1.850(2) | 1.834(3) | 1.832(3) |
| S–CH2 | 1.799(2) | 1.767(3) | 1.771(3) |
| CFc–CH2 | 1.532(3) | 1.530(4) | 1.542(4) |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
1.339(3) | 1.344(5) | 1.331(5) |
| CFc–CCp | 1.515(3) | 1.527(4) | 1.518(5) |
| CFc–CCp | 1.517(3) | 1.527(4) | 1.526(4) |
| Fe–centroid (Cp–CFc) | 1.657 | 1.656 | 1.653 |
| Fe–centroid (Cp–CFc) | 1.658 | 1.656 | 1.643 |
| Fe–centroid (Cp) | 1.660 | 1.659 | 1.646 |
| Fe–centroid (Cp) | 1.658 | 1.658 | 1.650 |
| Bond angles | |||
| S–CFc–CH2 | 107.91(13) | 104.9(2) | 105.3(2) |
| CFc–S–CH2 | 97.30(10) | 100.65(16) | 101.20(16) |
| CCp–CFc–CCp | 111.20(16) | 112.1(2) | 110.0(3) |
| OSO |
117.81(15) | 117.57(16) | |
Compound 1 crystallizes in the orthorhombic space group Pcab, while complex 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In the crystal packing of 3, two independent molecules (A and B) are observed. The structure analysis of both molecules confirmed the expected structures in which the two ferrocenyl groups are bonded to a sulphide (1) or a sulfone (3) moiety (Fig. 2). All ferrocenyl units show an eclipsed conformation with nearly equivalent distances between Fe and the centroid of the cyclopentadienyl rings (Table 1).
. In the crystal packing of 3, two independent molecules (A and B) are observed. The structure analysis of both molecules confirmed the expected structures in which the two ferrocenyl groups are bonded to a sulphide (1) or a sulfone (3) moiety (Fig. 2). All ferrocenyl units show an eclipsed conformation with nearly equivalent distances between Fe and the centroid of the cyclopentadienyl rings (Table 1).
The oxidation of sulphide 1 to sulfone 3, however, plays only a minor role on the overall structure of the two compounds. A similar behaviour was found for a series of sulphur-containing heterocycles in which the oxidation of the sulphur atom has almost no impact on the geometrical parameters of the heterocycles.54,55 The only bond distances which are somewhat influenced by oxidation are S–CFc and S–CH2 (Table 1), whereby the corresponding bonds in 3, as compared to 1, are shortened by 0.02 Å, as previously observed by Petrov.54,55 In both compounds, the thiopyran ring adopts a half-chair conformation to limit the steric repulsion between the different substituents (Fig. 2). In these carbon-bridged diferrocenyl complexes, the Fe⋯Fe distances are 5.6377(6) (1), 5.6824(9) (3A) and 5.6785(9) Å (3B), respectively. These values are comparable to those found in analogous carbon-bridged diferrocenyl compounds.56 Interestingly, the dihedral angles between the two planes of the covalently bonded cyclopentadienyl rings are quite acute in 1 (61.4°) and molecule 3A (63.1°), while in 3B this dihedral angle is normal at 76.6°.56
UV-Vis spectroscopy, electrochemistry and spectroelectrochemistry
The electronic properties of the ferrocenyl-functionalised compounds 1–3 were studied by using UV-Vis, cyclic (= CV) and square wave voltammetry (= SWV) and in situ UV-Vis/NIR spectroelectrochemistry.Compounds 1–3 show relatively weak absorptions in the visible region and stronger absorptions at higher energies (Fig. 3) as it is characteristic for ferrocene derivatives.57,58 TD-DFT (= Time Dependent Density-Functional Theory) calculations (Fig. 3) performed for 1 predict eight lowest-energy transitions between 450 and 530 nm in which mainly Fe-centred molecular orbitals of 3d-character are involved.58 The low energy spectral features are, thus, assigned to an unresolved series of broad overlapping bands resulting from ferrocene-centred d–d transitions.40,59,60
 | ||
| Fig. 3 UV-Vis absorption spectra of 1–3 in dichloromethane at ambient temperature (1: black solid line; 2: red dashed line; 3: blue dotted line) and TD-DFT calculated electronic transitions for 1 (vertical bars; oscillator strengths are given on the y axis (right)). Contour plots of the frontier orbitals HOMO and LUMO for 1. | ||
The electrochemical measurements (CV and SWV) were performed under an argon atmosphere in dichloromethane solutions containing [Bu4N][B(C6F5)4] (0.1 mol L−1) as supporting electrolyte at a scan rate of 100 mV s−1 at 25 °C. The data of the cyclic voltammetry experiments are summarised in Table 2. All potentials are referenced to the FcH/FcH+ redox couple.61 The voltammograms of 1–3 are shown in Fig. 5.
| Compd. | E °′1 [mV] (ΔEpc [mV]) | E °′2 [mV] (ΔEpc [mV]) | ΔE°′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d [mV] d [mV] |
|---|---|---|---|
| a Potentials vs. FcH/FcH+, scan rate 100 mV s−1 at glassy carbon electrode of a 1.0 mmol L−1 solution in dry dichloromethane; 0.1 mol L−1 [NnBu4][B(C6F5)4] as supporting electrolyte at 25 °C. b E°′ = Formal potential. c ΔEp = difference between the oxidation and the reduction potential. d ΔE°′ = potential difference between the two ferrocenyl-related redox processes (E°′2–E°′1). | |||
| 1 | −90 (73) | 275 (75) | 365 |
| 2 | −15 (75) | 355 (76) | 370 |
| 3 | 5 (73) | 375 (83) | 370 |
As it can be seen from Fig. 5, both ferrocenyl groups in 1–3 can be oxidised separately showing two well-resolved reversible one-electron oxidation steps. The chemical oxidation of the sulphur atom in the neighbouring position to the ferrocenyl units leads to an anodic shift of the both Fc-based oxidation processes, E°′1 from 90 mV (1) over −15 mV (2) to 5 mV (3) and E°2 from 275 mV (1) over 355 mV (2) to 375 (3), respectively. This chemical oxidation process increases the group-electronegativity at the sulfur (for example, see group electronegativity: –SMe = 2.592; –SOMe = 2.841; –SO2Me = 2.998)62 and hence, reduces the electron density at the ferrocenyl groups due to the increased electron withdrawing effect. This trend is also reproduced in the results of the DFT calculations (see below). The Fe-centred occupied frontier orbitals undergo substantial stabilisation upon oxidation of the sulphur atom. For example, the HOMO energy in 3 is about 60 mV smaller than that in non-oxidised 1 (Fig. 4).
 | ||
| Fig. 4 DFT energies of the four lowest occupied orbitals of 1–3 and contour plots of the corresponding Kohn–Sham orbitals for 3. | ||
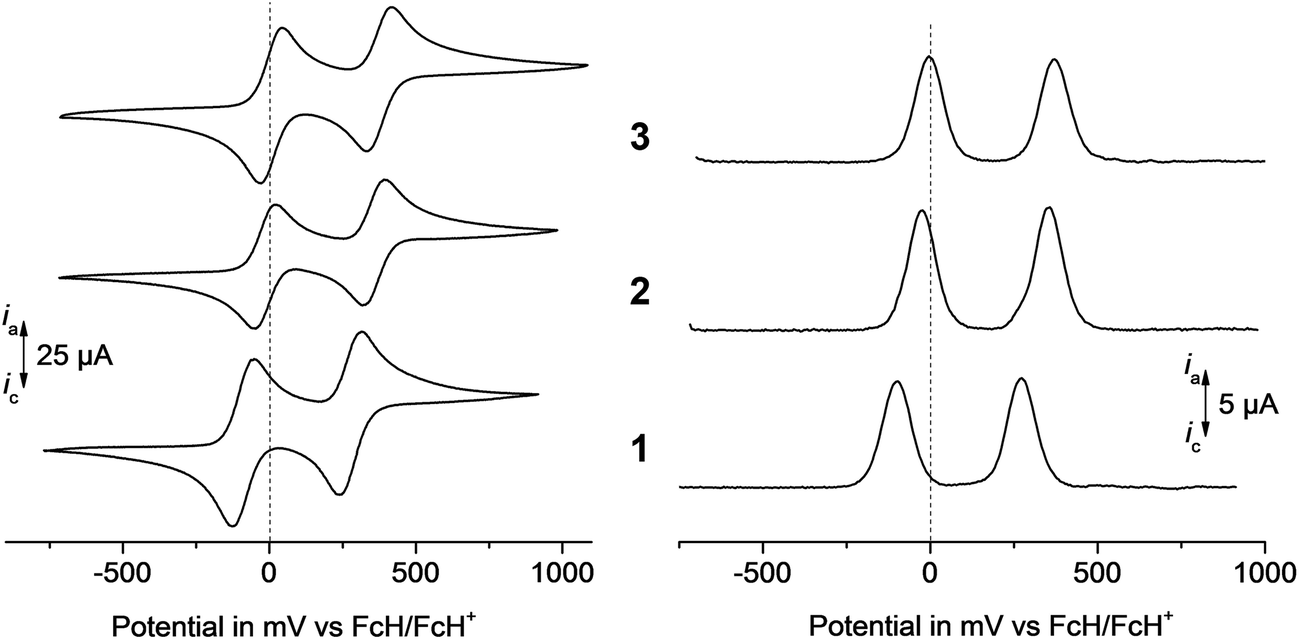 | ||
| Fig. 5 Left: cyclic voltammograms of 1–3, scan rate: 100 mV s−1. Right: square wave voltammograms of 1–3 in dichloromethane solutions (1.0 mmol L−1) at 25 °C, supporting electrolyte 0.1 mol L−1 [Bu4N][B(C6F5)4], working electrode: glassy carbon electrode (surface area 0.031 cm2). | ||
The redox separation between the 1st and the 2nd oxidation processes, however, is not affected by the different degree of the sulphur oxidation and is ca. 370 mV throughout the series. Due to the use of [Bu4N][B(C6F5)4]25,63–69 as weakly coordination counter-ion within the electrolyte, the ion-pairing effects are minimised70–72 and thus, the electrostatic stabilisation forces between the ferrocenyl groups are increased, when compared with diferrocenylmethane measured in [Bu4N][ClO4] (ΔE°′ = 120 mV).73
The UV-Vis/NIR spectroelectrochemical measurements were performed in an OTTLE (= Optically Transparent Thin-Layer Electrochemistry) cell74 using an analyte concentration of 2.0 mmol L−1 and [Bu4N][B(C6F5)4] (0.1 mol L−1) as supporting electrolyte in dichloromethane (1–3) or acetonitrile (2). The UV-Vis/NIR spectra are depicted in Fig. 6 (1), 7 (2), and 8 (3). The spectrum measured in acetonitrile (2) is shown in Fig. S5.†
 | ||
| Fig. 6 Left: UV-Vis/NIR spectra of 1 at 25 °C in dichloromethane (2.0 mmol L−1) at rising potentials (bottom: −200 to 475 mV; top: 475 to 1200 mV vs. Ag/AgCl); supporting electrolyte [Bu4N][B(C6F5)4]. Right: deconvolution of the NIR absorptions of 1+ using three Gaussian shaped bands determined by spectroelectrochemistry in an OTTLE cell. | ||
 | ||
| Fig. 7 Left: UV-Vis/NIR spectra of 2 at 25 °C in dichloromethane (2.0 mmol L−1) at rising potentials (bottom: −200 to 375 mV; top: 375 to 1200 mV vs. Ag/AgCl); supporting electrolyte [Bu4N][B(C6F5)4]. Right: deconvolution of the NIR absorptions of 2+ using three Gaussian shaped bands determined by spectroelectrochemistry in an OTTLE cell. | ||
 | ||
| Fig. 8 Left: UV-Vis/NIR spectra of 3 at 25 °C in dichloromethane (2.0 mmol L−1) at rising potentials (bottom: −200 to 375 mV; top: 375 to 1200 mV vs. Ag/AgCl); supporting electrolyte [Bu4N][B(C6F5)4]. Right: deconvolution of the NIR absorptions of 3+ using three Gaussian shaped bands determined by spectroelectrochemistry in an OTTLE cell. | ||
The appropriate compounds were oxidised by stepwise increasing the potentials (step width 25, 50 and 100 mV). Thus, the studied compounds 1–3 underwent oxidation to the mono-cationic 1+–3+ and di-cationic 12+–32+ species, respectively. After complete oxidation, each sample was reduced at −200 mV to prove the reversibility of the redox processes. The resulting UV-Vis/NIR spectra were identical to those of the starting molecules. During the oxidation of 1–3 a broad band with a very low intensity (εmax = 100 L mol−1 cm−1) between 1000 and 3000 nm appeared (Fig. 6–8). A further increase of the potential resulted in the decrease of this band. Such a behaviour is typically observed for intervalence charge transfer (= IVCT) absorptions.2,13 The experimental spectra can be deconvoluted into three Gaussian-shape bands assigned to an IVCT, a ligand field transition, and a band representing the edge to the higher energy absorptions. The sum of these three Gaussian-shaped bands fits almost exactly with the experimental spectra. The deconvolution reveals the intensity εmax, the full width-at-half-height Δν1/2 and the νmax values for the IVCT component. The solvent polarity change from P = 3.1 (dichloromethane) to P = 5.8 (acetonitrile),75 resulting in a shift of the νmax value from 5250 cm−1 to 7525 cm−1. It is remarked that strong solvatochromic shifts are expected for IVCT absorption bands being of distinct charge transfer character. Thus, the IVCT assignment of the observed NIR absorption features (Fig. 6–8 and Table 3) is further substantiated. The appearance of low energy ligand field transitions is characteristic for ferrocenyl containing compounds as for example demonstrated by UV/Vis-NIR measurements of mono ferrocenyl thiophenes.27 The numerical data derived from the deconvolution procedure is summarised in Table 3. However, the data should be handled with care since the very small extinctions of these bands cause them to be susceptible to errors.
a
| Compd. | ν max (cm−1) (εmax (L mol−1 cm−1)) | Δν1/2 (cm−1) | (Δν1/2)theob (cm−1) |
|---|---|---|---|
| a Measured in dry dichloromethane (DCM) or acetonitrile (ACN) using [Bu4N][B(C6F5)4] (0.1 mol dm−3) as supporting electrolyte at 25 °C. b Values calculated as (Δν1/2)theo = (2310 νmax)1/2 according to the Hush relationships for weakly coupled systems.76 | |||
| 1 + (DCM) | 5200 (100) | 4950 | 3468 |
| 2 + (DCM) | 5250 (100) | 4900 | 3478 |
| 2 + (ACN) | 7525 (60) | 7850 | 4169 |
| 3 + (DCM) | 5300 (95) | 4900 | 3512 |
The Δν1/2 values exceed the theoretical values according to the Hush model, which is a common behaviour for weakly coupled class II systems according to Robin and Day35 and is caused by interactions with the respective solvent. The absorption energy is slightly blue shifted with increasing oxidation state of the sulphur atom (the νmax value increases from 5200 cm−1 for 1+ to 5300 cm−1 determined for 3+). However, these differences are small and lie within the margin of the experimental error of our measurements. Thus, the sulphur oxidation state does not have any significant influence on the electron transfer properties of diferrocenes 1+–3+. Remarkably, for the diferrocenylmethane cation (Fc2CH2+) no IVCT absorptions could be detected, while for the mixed-valent triferrocenylmethane (Fc3CH+) weak charge transfer excitations (εmax = 165 L mol−1 cm−1; Δν1/2 = 3750 cm−1; νmax = 5900 cm−1) were found, possessing similar characteristics as mixed-valent 1+–3+.44
Conclusions
Within this study it was shown that 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran (1) is readily available through a hetero-Diels–Alder cyclo-addition reaction of diferrocenylthioketone with 2,3-dimethyl-1,3-butadiene. Stepwise oxidation of the sulphur atom in 1 afforded the corresponding S-oxides: 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1-oxide (2, sulfoxide) and 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1,1-dioxide (3, sulfone). Electrochemical measurements on 1–3 revealed well separated redox events related to the two ferrocenyl groups. The corresponding E°′1 and E°′2 potentials are shifted anodically in the order of 1 < 2 < 3. This is accounted for the increasing electron-withdrawing effect of the adjacent S, SO, and SO2 fragments on the ferrocenyl groups. Thus, the Fe-centred orbitals in 2 and 3 are more stabilised than in 1. A single crystal X-ray diffraction study of 1 and 3 revealed that the chemical oxidation of the sulphur atom has only a negligible influence on the overall molecule's geometry (bond lengths and angels). Nevertheless, the X-ray analyses have also shown that the Fe–Fe distances in 1 and 3 are shorter than that in Fc2CH2 (5.765 Å).77 This effect is ascribed to the steric hindrance exerted by the 4,5-dimethyl-3,6-dihydro-2H-thiopyran ring. It has been also demonstrated that the cyclic 4,5-dimethyl-3,6-dihydro-2H-thiopyran group enables weak metal–metal electronic interactions in the mono-oxidised species 1+–3+, as confirmed by the appearance of weak IVCT absorptions characteristic for mixed-valent systems (= MV).37 This allows to categorise cations 1+–3+ as weakly coupled class II MV systems according to Robin and Day.35 Moreover, the extent of metal–metal electronic interactions in 1+–3+ does not change significantly with the oxidation state of the sulphur atom (sulphide (1) → sulfoxide (2) → sulfone (3)). In summary, our results demonstrate that sterically demanding 2H-thiopyran-derived bridges enable metal–metal electronic interactions between redox centres in mixed-valent molecular systems. In the studied MV species, most probably, the “through space” mechanism is dominantly operative.Experimental section
General data and reagents
All reactions were carried out under an atmosphere of argon using standard Schlenk techniques. Chromatographic separations were carried out using silica gel 60 (Merck, 230–400 mesh ASTM). Dichloromethane was purified by distillation from CaH2 prior to use and methanol was purified by distillation over magnesium. 2,3-Dimethyl-1,3-butadiene, m-chloroperoxybenzoic acid, 30% hydrogen peroxide and selenium dioxide were purchased from commercial suppliers and were used without further purification.Instruments
1H NMR (600 MHz) and 13C{H} NMR (150 MHz) spectra were recorded with a Bruker Avance III 600 spectrometer operating at 298 K in the Fourier transform mode. Chemical shifts are reported in δ units (ppm) using as residual CDCl3 (1H δ 7.26 ppm, 13C δ 77.00 ppm) as the reference. Infrared spectra were recorded with a FTIR Nexus Nicolet apparatus. Mass spectra were recorded with a Varian 500-MS iT mass spectrometer (ESI) or with a Finnigan Mat95 mass spectrometer (EI). Microanalyses were determined by Analytical Services of the Polish Academy of the Sciences, Łódź. UV-Vis absorption spectra were recorded with a Varian Cary 300 double beam spectrometer.DFT computations and spectroelectrochemical measurements
The geometry optimisations and electronic transition calculations were performed using density-functional theory (= DFT) and time dependent density-functional theory (= TD DFT) with Becke's three parameter functional78 with the non-local Lee–Yang–Parr correlation functional (B3LYP)79 and the standard 6-31G(d,p) basis set as implemented in the Gaussian 09 program package.80Electrochemical measurements of 1.0 mmol L−1 dichloromethane solutions of 1–3 were performed in a dried, argon purged cell at 25 °C with a Radiometer Voltalab PGZ 100 electrochemical workstation interfaced with a personal computer. Dichloromethane solutions (0.1 mol L−1) containing [Bu4N][B(C6F5)4] were used as supporting electrolyte. For the measurements a three electrode cell containing a Pt auxiliary electrode, a glassy carbon working electrode (surface area 0.031 cm2) and an Ag/Ag+ (0.01 mmol L−1 [AgNO3]) reference electrode fixed on a Luggin capillary was applied. The working electrode was pretreated by polishing on a Buehler microcloth first with a 1 micron and then with a 1/4 micron diamond paste. The reference electrode was constructed from a silver wire inserted into a 0.01 mmol L−1 [AgNO3] and 0.1 mol L−1 [Bu4N][B(C6F5)4] acetonitrile solution in a Luggin capillary with a Vycor tip. This Luggin capillary was inserted into a second Luggin capillary containing a 0.1 mol L−1 [Bu4N][B(C6F5)4] dichloromethane solution and a Vycor tip. Experiments under the same conditions showed that all reduction and oxidation potentials were reproducible within 5 mV. Experimental potentials were referenced against an Ag/Ag+ reference electrode but the presented results are referenced against ferrocene as an internal standard as required by IUPAC.61 To achieve this, each experiment was repeated in the presence of 1 mmol L−1 decamethylferrocene (= Fc*). Data were processed on a Microsoft Excel worksheet to set the formal reduction potentials of the FcH/FcH+ couple to 0.0 V. Under our conditions the Fc*/Fc*+ couple was at −619 mV vs. FcH/FcH+ (ΔEp = 60 mV), while the FcH/FcH+ couple itself was at 220 mV vs. Ag/Ag+ (ΔEp = 61 mV).81 Spectroelectrochemical UV-Vis/NIR measurements of 2.0 mmol L−1 solutions of 1–3 in dichloromethane (1–3) or acetonitrile (2) containing 0.1 mol L−1 of [Bu4N][B(C6F5)4] as the supporting electrolyte were performed in an OTTLE (OTTLE = Optically Transparent Thin-Layer Electrochemistry)74 cell with a Varian Cary 5000 spectrophotometer at 25 °C. The values obtained by deconvolution could be reproduced within εmax, 100 L mol−1 cm−1; νmax, 50 cm−1 and Δν1/2, 50 cm−1.
Synthesis of 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran (1)
A mixture of diferrocenyl thioketone (399 mg, 0.96 mmol) and 2,3-dimethyl-1,3-butadiene (8.5 mL) was stirred in a tightly closed glass-tube for 50 h at 75 °C. Afterwards, the reaction mixture was evaporated to dryness and the thus obtained solid was subjected to column chromatography on SiO2 (chloroform–hexane, ratio 1/1 (v/v)). Crystallization from chloroform–hexane gave pure 1 as red crystals in a 65% yield (309 mg).1H NMR (600 MHz, CDCl3): δ = 4.26 (bs, 2H, C5H4), 4.20 (bs, 2H, C5H4), 4.13 (bs, 2H, C5H4), 4.11 (bs, 2H, C5H4), 4.07 (s, 10H, C5H5), 3.00 (s, 2H, CH2), 2.64 (s, 2H, CH2), 1.85 (s, 3H, CH3), 1.74 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 126.6, 124.3, 97.1, 69.0, 66.9, 66.8, 66.7, 66.4, 44.5, 43.3, 31.3, 20.4, 19.4. FTIR (KBr): 3088, 2989, 2911, 2872, 1628, 1443, 1409, 1264, 1106, 1032, 1000, 824, 483 cm−1. MS (ESI): m/z = 496 (M+). MS (EI, 70 eV): m/z = 496 (M+). HRMS: m/z = 496.0614 (Calc. for C27H28SFe2: 496.0610). Anal. Calcd for: C27H28SFe2: C, 65.35; H, 5.69; S, 6.46%. Found: C, 65.10; H, 5.57; S, 6.72%.
Synthesis of 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1-oxide (2)
Hydrogen peroxide (30%, 71 mg) and selenium dioxide (72 mg, 0.65 mmol) in water (1 mL) were subsequently added to a stirred solution of 1 (312 mg, 0.63 mmol) in methanol (10 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 5 min and then for an additional 5 min at ambient temperature. The reaction was quenched with water (∼7 mL) and extracted with dichloromethane. The organic layer was washed with brine, separated and dried over anhydrous MgSO4 and then all volatile materials were evaporated. The residue was subjected to chromatography on SiO2 (diethyl ether–hexane, ratio 3/1 (v/v)) to give pure sulfoxide 2 as red crystals in 87% yield (281 mg).
1H NMR (600 MHz, CDCl3): δ = 4.90–4.89 (m, 1H, C5H4), 4.36–4.35 (m, 1H, C5H4), 4.32–4.31 (m, 1H, C5H4), 4.30–4.29 (m, 1H, C5H4), 4.28 (s, 5H, C5H5), 4.27–4.26 (m, 1H, C5H4), 4.22–4.21 (m, 1H, C5H4), 4.05–4.04 (m, 1H, C5H4), 4.00–3.99 (m, 1H, C5H4), 3.88 (s, 5H, C5H5), 3.18 (d, JH,H = 18.6 Hz, 1H, CH2), 3.01 (d, JH,H = 18.0 Hz, 1H, CH2), 2.97 (d, JH,H = 15.6 Hz, 1H, CH2), 2.50 (d, JH,H = 15.6 Hz, 1H, CH2), 1.84 (s, 3H, CH3), 1.68 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 127.0, 118.6, 94.3, 87.1, 70.1, 69.8, 69.2, 68.9, 68.7, 68.4, 66.9, 66.7, 66.2, 66.1, 59.4, 51.1, 41.6, 20.0, 19.4. FTIR (KBr): 3089, 2917, 2891, 2857, 1629, 1406, 1104, 1053 (s, SO), 1031, 1000, 824, 487 cm−1. MS (ESI): m/z = 535 (MNa+), 513 (MH+). MS (EI, 70 eV): m/z = 512 (M+). HRMS: m/z = 512.0558 (Calc. for C27H28OSFe2: 512.0560). Anal. Calcd for: C27H28OSFe2: C, 63.30; H, 5.51; S, 6.26%. Found: C, 63.23; H, 5.72; S, 6.27%.
Synthesis of 2,2-diferrocenyl-4,5-dimethyl-3,6-dihydro-2H-thiopyran-1,1-dioxide (3)
m-Chloroperoxybenzoic acid (= MCPBA) (45 mg, 0.26 mmol) was added to a stirred solution of sulphoxide 2 (100 mg, 0.19 mmol) in dichloromethane (20 mL) at −20 °C. The resulting reaction mixture was stirred at −20 °C for 2 h and then for an additional 24 h at ambient temperature. Afterwards, the 2nd portion of MCPBA (45 mg, 0.26 mmol) was added and the mixture was stirred at ambient temperature for another 24 h. Subsequently, a saturated NaHCO3 solution was added in a single portion and the resulting mixture was extracted with dichloromethane. The organic layer was washed with brine, separated and dried over anhydrous MgSO4. Then all volatile materials were evaporated. The residue was subjected to preparative TLC on SiO2 (diethyl ether–hexane, ratio 1/1 (v/v) as eluent). Sulfone 3 was obtained as a red solid in 21% (21 mg) yield.1H NMR (600 MHz, CDCl3): δ = 4.47 (bs, 2H, C5H4), 4.24 (s, 4H, C5H4), 4.21–4.20 (pq, JH,H = 1.98 Hz, 1.74 Hz, 2H, C5H4), 4.13 (s, 10H, C5H5), 3.32 (s, 2H, CH2), 3.06 (s, 2H, CH2), 1.91 (s, 3H, CH3), 1.69 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 127.0, 119.9, 89.0, 69.6, 68.7, 67.6, 67.5, 67.2, 62.5, 51.6, 44.5, 19.9, 19.6. FTIR (KBr): 3103, 2918, 2857, 1629, 1305 (s, SO2), 1121(s, SO2), 820, 480 cm−1. MS (ESI): m/z = 551 (MNa+), 528 (M+). MS (EI, 70 eV): m/z = 528 (M+), 464 (M+ − SO2), 462 (M+ − H2SO2). HRMS: m/z = 528.0510 (Calc. for C27H28O2SFe2: 528.0509). Anal. Calcd for: C27H28O2SFe2: C, 61.39; H, 5.34; S, 6.07%. Found: C, 61.20; H, 5.48; S, 5.91%.
Single-crystal X-ray structure analysis of 1 and 3
Red crystals of 1 were obtained by slow evaporation of a chloroform–hexane solution containing 1, while red crystals of 3 were grown by the slow evaporation of a diethyl ether–hexane solution containing 3 at ambient temperature. Data were collected with a Stoe Image Plate Diffraction system equipped with a ϕ circle goniometer using Mo Kα graphite monochromatic radiation (λ = 0.71073 Å) with ϕ range 0–200°. The structures were solved by direct methods applying the program SHELXS-97, while the refinement and all further calculations were carried out with SHELXL-97.82,83 The hydrogen atoms were included in calculated positions and treated as riding atoms using the SHELXL default parameters, except for the N–H hydrogen atoms which were located on the Fourier difference map and refined. The non-hydrogen atoms were refined anisotropically using weighted full-matrix least-square on F2. Crystallographic details for 1 and 3 are summarized in Table S1 (see ESI†).CCDC 1031233 (1) and 1031234 (3) contain the supplementary crystallographic data for this paper.
Acknowledgements
Authors (K. K. and G. M.) thank the National Science Centre (Poland) for financial support (Project Maestro-3; Dec-2012/06/A/ST5/00219) and R. C. thanks the German Federal Ministry of Education and Research (BMBF) for support. The support from the German Academic Exchange Service (DAAD) in the framework of the exchange program “Ostpartnerschaften” is highly appreciated.Notes and references
- S. D. Glover, J. C. Goeltz, B. J. Lear and C. P. Kubiak, Eur. J. Inorg. Chem., 2009, 5, 585–594 CrossRef.
- D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed.
- C. Lapinte, J. Organomet. Chem., 2008, 693, 793–801 CrossRef CAS PubMed.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef CAS PubMed.
- A. Ceccon, S. Santi, L. Orian and A. Bisello, Coord. Chem. Rev., 2004, 248, 683–724 CrossRef CAS PubMed.
- P. Aguirre-Etcheverry and D. O'Hare, Chem. Rev., 2010, 110, 4839–4864 CrossRef CAS PubMed.
- R. F. Winter, Organometallics, 2014, 33, 4517–4536 CrossRef CAS.
- A. Hildebrandt and H. Lang, Organometallics, 2013, 32, 5640–5653 CrossRef CAS.
- K. Costuas and S. Rigaut, Dalton Trans., 2011, 40, 5643–5658 RSC.
- T. Kienz, C. Förster and K. Heinze, Organometallics, 2014, 33, 4803–4812 CrossRef CAS.
- K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H.-J. Krüger, B. Sarkar and W. Kaim, Organometallics, 2009, 28, 4196–4209 CrossRef CAS.
- M. Linseis, S. Záliš, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2012, 134, 16671–16692 CrossRef CAS PubMed.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180, 431–509 CrossRef CAS.
- M. D. Ward, Chem. Soc. Rev., 1995, 121–134 RSC.
- M. Ratner and J. Jortner, Molecular Electronics, Malden, MA, 1997 Search PubMed.
- Electron Transfer in Chemistry and Biology, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed.
- Electron and Proton Transfer in Chemistry and Biology, ed. A. Müller, H. Ratajczak, W. Junge and E. Dieman, Elsevier, New York, 1992 Search PubMed.
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef CAS PubMed.
- M. Pichlmaier, R. F. Winter, M. Zabel and S. Záliš, J. Am. Chem. Soc., 2009, 131, 4892–4903 CrossRef CAS PubMed.
- W. E. Geiger, Organometallics, 2007, 26, 5738–5765 CrossRef CAS.
- A. Hildebrandt, D. Schaarschmidt, R. Claus and H. Lang, Inorg. Chem., 2011, 50, 10623–10632 CrossRef CAS PubMed.
- J. M. Speck, M. Korb, T. Rüffer, A. Hildebrandt and H. Lang, Organometallics, 2014, 33, 4813–4823 CrossRef CAS.
- S. W. Lehrich, A. Hildebrandt, T. Rüffer, M. Korb, P. J. Low and H. Lang, Organometallics, 2014, 33, 4836–4845 CrossRef CAS.
- D. Miesel, A. Hildebrandt, M. Korb, P. J. Low and H. Lang, Organometallics, 2013, 32, 2993–3002 CrossRef CAS.
- U. Pfaff, A. Hildebrandt, M. Korb and H. Lang, Polyhedron, 2015, 86, 2–9 CrossRef CAS PubMed.
- J. M. Speck, R. Claus, A. Hildebrandt, T. Rüffer, E. Erasmus, L. van As, J. C. Swarts and H. Lang, Organometallics, 2012, 31, 6373–6380 CrossRef CAS.
- K. Kaleta, F. Strehler, A. Hildebrandt, T. Beweries, P. Arndt, T. Rüffer, A. Spannenberg, H. Lang and U. Rosenthal, Chem. – Eur. J., 2012, 18, 12672–12680 CrossRef CAS PubMed.
- A. Hildebrandt, U. Pfaff and H. Lang, Rev. Inorg. Chem., 2011, 31, 111–141 CrossRef CAS.
- A. Hildebrandt and H. Lang, Dalton Trans., 2011, 40, 11831–11837 RSC.
- K. Kaleta, A. Hildebrandt, F. Strehler, P. Arndt, H. Jiao, A. Spannenberg, H. Lang and U. Rosenthal, Angew. Chem., Int. Ed., 2011, 50, 11248–11252 CrossRef CAS PubMed.
- A. Hildebrandt, D. Schaarschmidt and H. Lang, Organometallics, 2011, 30, 556–563 CrossRef CAS.
- C. G. Allen and S. N. Hush, Prog. Inorg. Chem., 1967, 8, 357–339 CrossRef.
- P. Mücke, M. Zabel, R. Edge, D. Collison, S. Clément, S. Záliš and R. F. Winter, J. Organomet. Chem., 2011, 696, 3186–3197 CrossRef PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- T.-Y. Dong, C.-H. Huang, C.-K. Chang, H.-C. Hsieh, S.-M. Peng and G.-H. Lee, Organometallics, 1995, 14, 1776–1785 CrossRef CAS.
- S. C. Jones, S. Barlow and D. O'Hare, Chem. – Eur. J., 2005, 11, 4473–4481 CrossRef CAS PubMed.
- J. C. Kotz, C. L. Nivert, J. M. Lieber and R. C. Reed, J. Organomet. Chem., 1975, 91, 87–95 CrossRef CAS.
- D. C. O'Connor Salazar and D. O. Cowan, J. Organomet. Chem., 1991, 408, 227–231 CrossRef.
- A.-C. Ribou, J.-P. Launay, M. L. Sachtleben, H. Li and C. W. Spangler, Inorg. Chem., 1996, 35, 3735–3740 CrossRef CAS PubMed.
- T.-Y. Dong, T.-Y. Lee, S.-H. Lee, G.-H. Lee and S.-M. Peng, Organometallics, 1994, 13, 2337–2348 CrossRef CAS.
- R. J. Weeb, S. J. Geib, D. L. Staley, A. L. Rheingold and D. N. Hendrickson, J. Am. Chem. Soc., 1990, 112, 5031 CrossRef.
- R. Rulkens, A. J. Lough, I. Manners, S. R. Lovelace, C. Grant and W. Geiger, J. Am. Chem. Soc., 1996, 118, 12683–12695 CrossRef CAS.
- F. Delgado-Pena, D. R. Talham and D. O. Cowan, J. Organomet. Chem., 1983, 253, C43–C46 CrossRef CAS.
- U. Pfaff, G. Filipczyk, A. Hildebrandt, M. Korb and H. Lang, Dalton Trans., 2014, 43, 16310–16321 RSC.
- G. Filipczyk, A. Hildebrandt, U. Pfaff, M. Korb, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2014, 4258–4262 CrossRef CAS.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761–6771 CrossRef CAS.
- M. Lohan, F. Justaud, H. Lang and C. Lapinte, Organometallics, 2012, 31, 3565–3574 CrossRef CAS.
- M. Lohan, F. Justaud, T. Roisnel, P. Ecorchard, H. Lang and C. Lapinte, Organometallics, 2010, 29, 4804–4817 CrossRef CAS.
- P. Denifl and B. Bildstein, J. Organomet. Chem., 1993, 453, 53–59 CrossRef CAS.
- D. Enders, E. A. Jonas and T. Klumpen, Eur. J. Org. Chem., 2009, 2149–2162 CrossRef CAS.
- S. D. Larsen, P. V. Fisher, B. E. Libby, R. M. Jensen, S. A. Mizsak and W. Watt, J. Org. Chem., 1996, 61, 4725–4738 CrossRef CAS PubMed.
- J. Drabowicz and M. Mikołajczyk, Synthesis, 1978, 758–759 CrossRef CAS.
- V. A. Petrov, S. Lustig and W. Marshall, J. Fluorine Chem., 2007, 128, 1227–1234 CrossRef CAS PubMed.
- V. A. Petrov and W. Marshall, J. Fluorine Chem., 2007, 128, 729–735 CrossRef CAS PubMed.
- R.-J. Xie, L.-M. Han, Q.-L. Suo, H.-L. Hong and M.-H. Luo, J. Coord. Chem., 2010, 63, 1700–1710 CrossRef CAS.
- Y. Yamaguchi, W. Ding, C. T. Sanderson, M. L. Borden, M. J. Morgan and C. Kutal, Coord. Chem. Rev., 2007, 251, 515–524 CrossRef CAS PubMed.
- S. Barlow and D. O'Hare, Chem. Rev., 1997, 97, 637–670 CrossRef CAS PubMed.
- D. M. Duggan and D. N. Hendrickson, Inorg. Chem., 1975, 14, 955–970 CrossRef CAS.
- K. Kowalski, Ł. Szczupak, J. Skiba, O. S. Abdel-Rahman, R. F. Winter, R. Czerwieniec and B. Therrien, Organometallics, 2014, 33, 4697–4705 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- N. Inamoto and S. Masuda, Chem. Lett., 1982, 11, 1003–1006 CrossRef.
- H. J. Gericke, N. I. Barnard, E. Erasmus, J. C. Swarts, M. J. Cook and M. A. S. Aquino, Inorg. Chim. Acta, 2010, 363, 2222–2232 CrossRef CAS PubMed.
- E. Fourie, J. C. Swarts, D. Lorcy and N. Bellec, Inorg. Chem., 2010, 49, 952–959 CrossRef CAS PubMed.
- J. C. Swarts, A. Nafady, J. H. Roudebush, S. Trupia and W. E. Geiger, Inorg. Chem., 2009, 48, 2156–2165 CrossRef CAS PubMed.
- V. N. Nemykin, G. T. Rohde, C. D. Barrett, R. G. Hadt, J. R. Sabin, G. Reina, P. Galloni and B. Floris, Inorg. Chem., 2010, 49, 7497–7509 CrossRef CAS PubMed.
- V. N. Nemykin, G. T. Rohde, C. D. Barrett, R. G. Hadt, C. Bizzarri, P. Galloni, B. Floris, I. Nowik, R. H. Herber, A. G. Marrani, R. Zanoni and N. M. Loim, J. Am. Chem. Soc., 2009, 131, 14969–14978 CrossRef CAS PubMed.
- E. A. Poppitz, A. Hildebrandt, M. Korb and H. Lang, J. Organomet. Chem., 2014, 752, 133–140 CrossRef CAS PubMed.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Rüffer, P. J. Low and H. Lang, Organometallics, 2013, 32, 6106–6117 CrossRef CAS.
- W. E. Geiger and F. Barrière, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef PubMed.
- F. Barrière, N. Camire, W. E. Geiger, U. T. Mueller-Westerhoff and R. Sanders, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef PubMed.
- G. Ferguson, C. Glidewell, G. Opromolla, C. M. Zakaria and P. Zanello, J. Organomet. Chem., 1996, 517, 183–190 CrossRef CAS.
- M. Krejcik, M. Danek and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- L. R. Snyder, J. Chromatogr. Sci., 1978, 16, 223–234 CAS.
- N. S. Hush, Electrochim. Acta, 1968, 13, 1005–1023 CrossRef CAS.
- R.-J. Xie, L.-M. Han, N. Zhu, H.-L. Hong, Q.-L. Suo and C.-L. Ke, Asian J. Chem., 2013, 25, 197–201 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09W, Version 8.0, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. Nafady and W. E. Geiger, Organometallics, 2008, 27, 5624–5631 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1031233 and 1031234. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00246j |
| This journal is © The Royal Society of Chemistry 2015 |